

Abstract
BACKGROUND AND PURPOSE
Clinical use of cinacalcet in hyperparathyroidism is complicated by its tendency to induce hypocalcaemia, arising partly from activation of calcium-sensing receptors (CaS receptors) in the thyroid and stimulation of calcitonin release. CaS receptor allosteric modulators that selectively bias signalling towards pathways that mediate desired effects [e.g. parathyroid hormone (PTH) suppression] rather than those mediating undesirable effects (e.g. elevated serum calcitonin), may offer better therapies.
EXPERIMENTAL APPROACH
We characterized the ligand-biased profile of novel calcimimetics in HEK293 cells stably expressing human CaS receptors, by monitoring intracellular calcium (Ca2+i) mobilization, inositol phosphate (IP)1 accumulation, ERK1/2 phosphorylation (pERK1/2) and receptor expression.
KEY RESULTS
Phenylalkylamine calcimimetics were biased towards allosteric modulation of Ca2+i mobilization and IP1 accumulation. S,R-calcimimetic B was biased only towards IP1 accumulation. R,R-calcimimetic B and AC-265347 were biased towards IP1 accumulation and pERK1/2. Nor-calcimimetic B was unbiased. In contrast to phenylalkylamines and calcimimetic B analogues, AC-265347 did not promote trafficking of a loss-of-expression, naturally occurring, CaS receptor mutation (G670E).
CONCLUSIONS AND IMPLICATIONS
The ability of R,R-calcimimetic B and AC-265347 to bias signalling towards pERK1/2 and IP1 accumulation may explain their suppression of PTH levels in vivo at concentrations that have no effect on serum calcitonin levels. The demonstration that AC-265347 promotes CaS receptor receptor signalling, but not trafficking reveals a novel profile of ligand-biased modulation at CaS receptors The identification of allosteric modulators that bias CaS receptor signalling towards distinct intracellular pathways provides an opportunity to develop desirable biased signalling profiles in vivo for mediating selective physiological responses.
Tables of Links
| TARGETS |
|---|
| GPCRa |
| CaS receptor |
| Enzymesb |
| ERK1/2 |
| LIGANDS | |
|---|---|
| AC-265347 | IP1, inositol 1-phosphate |
| Calcitonin | PTH, parathyroid hormone |
| Calindol | |
| Cinacalcet |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b).
Introduction
The human calcium-sensing receptor (CaS receptor) is a family C GPCR primarily responsible for the regulation of extracellular calcium (Ca2+o) concentrations in the body. When Ca2+o rises, activation of the CaS receptors expressed in the parathyroid gland suppresses the secretion of parathyroid hormone (PTH). The drop in circulating PTH levels results in reduced renal Ca2+o reabsorption and reduced bone resorption (see Brown, 2013). Additionally, CaS receptor activation in the kidney by elevated serum Ca2+o inhibits Ca2+o reabsorption, leading to enhanced renal Ca2+o excretion independently of changes in PTH (Kantham et al., 2009; Loupy et al., 2012). Elevated serum Ca2+o also decreases bone resorption via CaS receptors expressed on osteoblasts and osteoclasts (see Marie, 2010 for a review), and by stimulation of calcitonin secretion via CaS receptors expressed on thyroid C-cells (Freichel et al., 1996).
The CaS receptor also has non-calciostatic roles. Thus, it mediates the modulation of BP (see Smajilovic et al., 2011 for a review) and protection against vascular calcification (Alam et al., 2009), stimulation of gastrointestinal hormone secretion (Feng et al., 2010; Mace et al., 2012), modulation of electrolyte and water transport in the colon and kidney (reviewed in Macleod, 2013) and modulation of the proliferation and differentiation of numerous cell types, including colonic epithelial cells, keratinocytes, adipocytes and neurones.
Given its ubiquitous expression throughout the body and functionally diverse roles, drugs that target the CaS receptor may have therapeutic application in various clinical contexts. However, these drugs may also produce adverse effects arising from actions in multiple tissues expressing the CaS receptor. Indeed, patients treated with the calcimimetic, cinacalcet ((αR)-(−)-α-methyl-N-[3-[3-[trifluoromethylphenyl]propyl]-1-napthalenemethanamine hydrochloride), a positive allosteric CaS receptor modulator indicated for the treatment of secondary and some forms of primary hyperparathyroidism, have a tendency to develop adverse effects that restrict its use to only severely affected patients. The most problematic adverse effect is hypocalcaemia (Chonchol et al., 2009), likely resulting from both suppressed renal calcium reabsorption induced by CaS receptor activation in the kidney, and calcitonin-mediated inhibition of bone resorption via CaS receptor activation in the thyroid C-cells (Arenas et al., 2013). Thus, novel calcimimetics that selectively stimulate CaS receptor-mediated signalling in the parathyroid gland without affecting CaS receptors in other tissues may have an improved side effect profile and enable treatment of less severe grades of hyperparathyroidism.
One approach to directing desired physiological outcomes of GPCR activation is to selectively target those intracellular signalling pathways that couple to the anticipated effect, while avoiding those that couple to unwanted consequences. Such selectivity can be achieved with a drug that binds to and favours a receptor conformation that preferentially couples to a subset of desired intracellular signalling pathways (Kenakin, 2011). This concept is referred to as ligand-biased signalling, ligand-directed trafficking of receptor stimulus, functional selectivity or biased agonism (Kenakin and Christopoulos, 2013).
The CaS receptor is subject to ligand-biased signalling on a number of levels (Leach et al., 2014). First, it binds multiple endogenous ligands, including Ca2+o, extracellular magnesium (Mg2+o), L-amino acids, polyamines and the glutamyl peptide, γ-glutathione. Ca2+o, spermine and L-phenylalanine have been demonstrated to preferentially activate distinct signalling pathways (Rey et al., 2010; Thomsen et al., 2012a), suggesting that each ligand has the propensity to stabilize a subset of preferred receptor states and subsequently stimulate the repertoire of intracellular signalling proteins that couple to these states. Second, positive allosteric modulators of the CaS receptor, such as cinacalcet, and negative CaS receptor modulators (calcilytics), such as NPS-2143 (2-chloro-6-[(2R)-3-[[1,1-dimethyl-2-(2-naphthalenyl)ethyl]amino-2-hydroxypropoxy]benzonitrile hydrochloride), engender biased allosteric modulation at the CaS receptor, such that they exhibit greater modulation of some pathways over others (Davey et al., 2012; Leach et al., 2013). Third, the ‘natural bias’ of the CaS receptor can be altered in pathophysiological states. This has been demonstrated by naturally occurring mutations in the CaS receptor protein that alter its usual signalling bias (Leach et al., 2012), a switch in CaS receptor signalling from Gi/o to Gs in human breast cancer cells (Mamillapalli et al., 2008), and an autoantibody directed against the CaS receptor in a patient with acquired hypocalciuric hypercalcemia, which potentiated inositol phosphate (IP) accumulation, yet inhibited ERK1/2 phosphorylation (pERK1/2) (Makita et al., 2007). Finally, the complement of intracellular signalling proteins to which the CaS receptor couples differs among cell types, thus, the capacity of the CaS receptor to couple to different signalling pathways depends upon its tissue-specific expression.
Proof-of-concept that tissue-specific effects can be achieved by targeting the CaS receptor with drugs was evident from early experiments with the prototypical calcimimetic, NPS-R568. During the development of the phenylalkylamine calcimimetics (e.g. NPS-R568 and cinacalcet), it was recognized that the natural hypocalcaemic effects of these drugs may be complicated by stimulation of calcitonin release via activation of CaS receptors in the thyroid. Thus, the need to suppress PTH secretion with minimal effects on calcitonin secretion was acknowledged (Fox et al., 1999a,b), but remains suboptimally addressed.
Third-generation agents appear to have enhanced tissue-selective effects. This is evident from studies with the novel dibenzylamine calcimimetic, R,R-calcimimetic B (R-1-(6-methoxy-4′-(trifluoromethyl)-3-biphenylyl)-N-(R)-1-phenylethyl)ethanamine) and the structurally distinct calcimimetic, AC-265347 (1-benzothiazol-2-yl-1-(2,4-dimethyl-phenyl)-ethanol). Both calcimimetics inhibit PTH secretion at concentrations that do not induce calcitonin release in rats (Henley et al., 2011; Ma et al., 2011), demonstrating a means for normalizing serum PTH and calcium levels without causing uncontrolled hypocalcaemia. How these compounds achieve this tissue specificity is unknown, but we hypothesize that it may be a result of ligand-biased allosteric modulation at the CaS receptor. This is based on the fact that distinct intracellular signalling pathways activated by the CaS receptor are responsible for its physiological effects, thus drugs may selectively promote suppression of PTH release by preferentially activating the pathways that couple to that response. For instance, CaS receptor suppression of PTH release is driven by PLC-mediated IP3 production (Brown et al., 1987; Kifor et al., 1997) and pERK1/2 (Corbetta et al., 2002), but there is some evidence that CaS receptor-mediated intracellular calcium (Ca2+i) release is not required for inhibition of PTH from bovine parathyroid cells (Russell et al., 1999). Stimulation of both PLC and Ca2+i mobilization have been linked to the release of calcitonin (McGehee et al., 1997; Liu et al., 2003; Thomsen et al., 2012b), but in rat 6–23 medullary thyroid carcinoma cells, inhibition of pERK1/2 has no effect on Ca2+o-mediated stimulation of calcitonin release (Thomsen et al., 2012b). Thus, drugs that bias CaS receptor signalling towards pERK1/2 may achieve tissue-selective suppression of PTH secretion in the absence of calcitonin release.
To probe the ligand-biased signalling profile(s) required to achieve drug tissue selectivity, pathways that mediate distinct physiological receptor functions should ideally be dissected in systems such as primary or immortalized cells that maintain their physiological function. However, for the CaS receptor, this has been hampered by a lack of relevant cell lines and methods to study, for instance, parathyroid cell function. We have developed techniques to measure signalling in, and PTH release from, primary human parathyroid cells (Mun et al., 2009; Broadhead et al., 2011; Avlani et al., 2013), but performing high-throughput experiments in these cells is at present not possible. Thus, most studies of this nature must rely on recombinant cell systems to investigate CaS receptor signalling in response to agonists and drugs. Nonetheless, recombinant systems can still be used to identify bias and validate whether compounds with desirable in vivo properties have unique pharmacology in vitro, and vice versa.
The current study thus primarily aimed to use a recombinant cell system to determine the potential for structurally distinct calcimimetics to engender ligand-biased signalling and subsequently promote coupling of the CaS receptor to three key signalling pathways that could mediate different physiological effects; accumulation of inositol 1-phosphate (IP1, a stable metabolite of IP3); Ca2+i mobilization and phosphorylation of ERK1/2. Furthermore, we have previously shown that CaS receptor modulators can be biased in their ability to modulate signalling versus trafficking at the CaS receptor (Leach et al., 2013). Therefore, in addition to acute signalling at the CaS receptor, we determined the ability of the calcimimetics to act as pharmaco-chaperones of a naturally occurring mutant CaS receptor, G670E. Differential effects on trafficking versus signalling may have important implications for the treatment of calcium handling disorders caused by mutations in the CaS receptor gene that result in a diverse range of molecular phenotypes.
Methods
Synthesis of calcimimetics
Synthesis of R,R-calcimimetic B (compound 3b – Supporting Information Appendix S1), its diastereoisomer S,R-calcimimetic B (compound 3a – Supporting Information Appendix S1) and nor-calcimimetic B (compound 3c – Supporting Information Appendix S1) was achieved using a two-step procedure derived from described literature (Harrington et al., 2010). Full synthetic details and compound characterization are given in Supporting Information Appendix S1. NPS-R568 and cinacalcet were prepared as described previously (Davey et al., 2012). Calindol was purchased from Tocris Biosciences (Abingdon, UK), whereas AC-265347 was from Sigma-Aldrich (Sydney, Australia).
Cell culture
Generation of FlpIn HEK293 TRex cells (Invitrogen, Carlsbad, CA, USA) stably expressing the human CaS receptor under the control of tetracycline has been described previously (Davey et al., 2012; Leach et al., 2012). Cells were maintained in DMEM with 10% FBS, 200 μg·mL−1 hygromycin B and 5 μg·mL−1 blasticidin.
Optimization of assay conditions
The effect of ambient buffer Ca2+o on allosteric modulation at the CaS receptor has previously been published by us (Davey et al., 2012). Because Ca2+o is both present in the buffer and added as the agonist, assay buffer Ca2+o was optimized to achieve the best possible assay signal while avoiding complications that arise from the presence of physiological Ca2+o concentrations (e.g. signalling desensitization, potentiation of ambient Ca2+o signalling). In this same cell system, Mg2+o is nearly threefold less potent than Ca2+o as a CaS receptor agonist (data not shown). Thus, the presence of 1.18 mM ambient Mg2+o has minimal effect on CaS receptor signalling. Therefore, all assays were performed under low Ca2+o, but physiologically relevant Mg2+o conditions. For concentration–response curves to Ca2+o, data are plotted and analysed without the ambient Ca2+o concentration (i.e. only the added Ca2+o is considered).
Ca2+i mobilization assays
Cells were seeded in a clear 96-well plate coated with poly-D-lysine (50 ng·mL−1) at 80 000 cells per well and incubated overnight in the presence of 100 ng·mL−1 tetracycline. The following day, cells were washed with 200 μL assay buffer (150 mM NaCl, 2.6 mM KCl, 1.18 mM MgCl2, 10 mM D-glucose, 10 mM HEPES, 0.1 mM Ca2+o, 0.5% BSA and 4 mM probenecid at pH 7.4) and loaded with 100 μL Fluo-4 AM (1 μM) for 1 h at 37oC.
Cells were washed again with 200 μL assay buffer prior to the addition of fresh assay buffer. In functional interaction studies between Ca2+o and the calcimimetics, the modulators were co-added with Ca2+o (in all assays measuring agonist-stimulated receptor signalling events, each well was treated with a single agonist and/or modulator concentration). The release of Ca2+i was measured at 37°C using a Flexstation® 1 or 3 (Molecular Devices; Sunnyvale, CA, USA). Fluorescence was detected for 60 s at 485 nm excitation and 525 nm emission, but the peak Ca2+i mobilization response (approximately 12 s after agonist addition) was used for the subsequent determination of the agonist response. We have previously shown that when allosterism at the CaS receptor is quantified in Ca2+i mobilization assays using the potency of Ca2+o obtained by plotting the area under the 60 s Ca2+i mobilization trace, no significant difference in signalling or biased modulation is observed in comparison with parameters derived using the peak Ca2+i mobilization response (Leach et al., 2013). Relative peak fluorescence units were normalized to the fluorescence stimulated by ionomycin to account for differences in cell number and loading efficiency, and further normalized to the maximum response observed for the wild-type (WT) CaS receptor in the absence of modulator.
pERK1/2 assays
Cells were seeded at 80 000 cells per well into a poly-D-lysine-coated (50 μg·mL−1) transparent 96-well plate and grown overnight with 100 ng·mL−1 tetracycline. The following day, cells were washed twice with PBS and serum-free DMEM containing 16 mM HEPES, and 0.1 mM Ca2+o was added to wells. Vehicle or agonist (Ca2+o) with or without modulator were co-added to wells and incubated for 2.5 min (the time determined in prior assays for pERK1/2 to peak) at 37°C. All data were normalized to the response stimulated by 10% FBS and then further normalized to the maximum response stimulated by Ca2+o in the absence of modulator. pERK1/2 was determined using the SureFire pERK1/2 assay kit (kindly donated by Dr Michael Crouch, TGR Biosciences, Adelaide, SA, Australia) employing AlphaScreen technology (PerkinElmer, Boston, MA, USA). All other details are as described previously (Leach et al., 2012; 2013).
IP1 accumulation assays
Following overnight induction of receptor expression with 100 ng·mL−1 tetracycline in a T175 cm2 flask (where appropriate), cells were harvested and resuspended in assay buffer (150 mM NaCl, 2.6 mM KCl, 1.18 mM MgCl2, 10 mM D-glucose, 10 mM HEPES, 0.1 mM Ca2+o, 50 mM LiCl, pH 7.4) at 1.43 × 106 cells per mL. Seven microliters of agonist with or without modulator were added to wells of a 384-well white proxiplate (PerkinElmer) and 7 μL cells (1 × 104 cells) were added to these wells, centrifuged for 1 min at 350× g and incubated at 37°C for 45 min. The IP-One Tb™ assay kit (CisBio Bioassays, Codolet, France) was used to detect myo-IP1, based on FRET between d2-conjugated IP1 and Lumi4™-Tb cryptate conjugated anti-IP1 antibody. These reagents were diluted 1:30 with lysis buffer and 3 μL of each was added to wells following agonist stimulation. Lysates were incubated for 1 h and FRET was detected using an Envision plate reader (PerkinElmer) where emission of Lumi4-Tb cryptate was detected at 620 nm and emission of d2-conjugated IP1 at 665 nm. Results were calculated from the 665 nm/620 nm ratio. Data were normalized to the maximum response stimulated by Ca2+o in the absence of modulator.
Flow cytometry analysis for receptor expression
FlpIn HEK293 TRex cells stably expressing the human WT or G670E mutant CaS receptor were seeded in a 96-well plate at a density of 80 000 cells per well in DMEM containing 100 ng·mL−1 tetracycline and 0.3 or 3.0 μM allosteric modulator and incubated overnight at 37°C. The next day, cells were harvested with PBS supplemented with 0.1 % BSA, 2 mM EDTA and 0.05% NaN3 (washing buffer) and transferred to wells of a 96-well v-bottom plate, centrifuged for 3 min at 350× g, 4°C and resuspended in 100 μL blocking buffer (PBS, 5% BSA, 2 mM EDTA and 0.05% NaN3). Cells were incubated for 30 min in blocking buffer and subsequently incubated for 1 h with an AF647-conjugated 9E10 antibody (made in-house as described later), diluted in blocking buffer at 1 μg·mL−1. Cells were subsequently washed with washing buffer and resuspended in washing buffer with Sytox blue stain (Invitrogen). The fluorescence signal was quantified using a FACS Canto (Becton Dickinson, Franklin Lakes, NJ, USA).
Production of anti-cMyc:AF647 (9E10:AF647)
Supernatant from the 9E10 hybridoma (ATCC Number: CRL-1729) was harvested and antibody purified over a HiTrap protein G sepharose column (GE Life Sciences, Pittsburgh, PA, USA). The purified antibody was coupled to AF647 succinimidyl ester (Invitrogen) using standard protocols. Unincorporated fluor was removed using a 10 kDa MWCO centrifugal concentrator (Merck Millipore, Hessen, Germany). Degree of labelling was determined to be 3.6. The antibody conjugate was validated by titration in flow cytometry. A full description of antibody production, conjugation and validation can be found in the supplementary methods and results.
Data analysis
All non-linear regression analysis was performed using GraphPad Prism® 6 (GraphPad Software, San Diego, CA, USA). Parametric measures of potency, affinity and cooperativity were estimated as logarithms (Christopoulos, 1998). Data of the functional CaS receptor concentration–response curves obtained were fitted as logarithms to the following four-parameter concentration–response curve equation (Equation 2009).
| (1) |
where Y is the response; Bottom and Top represent the bottom and top asymptotes of the curve, respectively; A denotes the agonist concentration (excluding ambient Ca2+o in the buffer); nH (Hill slope) describes the steepness of the curve; and EC50 is the concentration of agonist that gives the mid-point response between Bottom and Top.
For functional interaction experiments between Ca2+o and the allosteric modulators, pEC50 values obtained for each curve in the absence and presence of modulator were fitted to an allosteric ternary complex model (Equation 2013a).
| (2) |
where pEC50 is the negative logarithm of the agonist EC50 in the presence of allosteric modulator; pKB is the negative logarithm of the ‘functional’ dissociation constant of the allosteric modulator determined in signalling assays; αβ is the overall cooperativity between the allosteric modulator and orthosteric agonist; and d is the estimate of the EC50 in the absence of modulator. An extra sum of squares F-test was used to determine whether data obtained in IP1 accumulation, Ca2+i mobilization and pERK1/2 assays were fitted best when the allosteric modulator functional pKB values were shared across the three different pathways. In a second analysis that constrained the functional pKB across datasets (Supporting Information Table S1, Supporting Information Figure S8), an extra sum of squares F-test was used to determine whether the cooperativities among the three pathways differed.
For the ‘cooperativity bias plot’, the pEC50 of Ca2+o in the absence and presence of modulator in IP1 accumulation, Ca2+i mobilization and pERK1/2 assays was first fitted to Equation 2013a and 150 XY coordinates of points that defined the curve that best fit Equation 2013a were determined. Next, the XY coordinates for the different pathways were plotted against one another, with IP1 accumulation or Ca2+i mobilization data on the Y axis against pERK1/2 data on the X axis. XY coordinates corresponding to the effects of 0, 0.003, 0.01, 0.03, 0.1, 0.3, 1, 3 and 10 μM modulator are respresented by symbols on the plots. If the allosteric modulator shows equal cooperativity in the assays, the data points will be coincident and the cooperativity bias plots will overlap with the line of identity. If, however, the modulator exerts greater cooperativity in one of the pathways, the points will fall on either side of this line towards the preferred pathway.
For agonist concentration–response curves in the absence of Ca2+o and Mg2+o, data were fitted as logarithms to an operational model of agonism (Equation 2013b).
| (3) |
where E is the effect (response) stimulated by the allosteric agonist, Em is the maximum response of the system stimulated by the full agonist (Ca2+o), τB is an operational measure of allosteric agonist efficacy, defined as the inverse of the fraction of receptors that must be occupied by agonist to obtain the half-maximal response, [B] is the allosteric agonist concentration and n is the transducer slope.
Results
Rationale for choice of ligands and signalling pathways
The structures of the calcimimetics used in this study are shown in Figure 1. The prototypical phenylalkylamine calcimimetics, cinacalcet and NPS-R568 (3-(2-chlorophenyl)-N-((1R)-1-(3-methoxyphenyl)ethyl)-1-propanamine) have been well characterized in vitro and in vivo (Nemeth et al., 1998; 2004). Calindol ((R)-2-[N-(1-(1-naphthyl)ethyl)aminomethyl]indole) was the most potent calcimimetic identified at the Institut de Chimie des Substances Naturelles (ICSN, France) from a series of diamines based around the structure of NPS-R568 (Kessler et al., 2004). R,R-calcimimetic B was the most potent CaS receptor ligand identified by Amgen in a dibenzylamine series and exhibited ideal in vivo pharmacodynamics. In an IP accumulation assay, R,R-calcimimetic B was estimated to have greater affinity than NPS-R568 (Harrington et al., 2010; Henley et al., 2011). The published synthesis of R,R-calcimimetic B employed a route yielding a diastereomeric ratio (d.r.) of 14:1 of R,R-calcimimetic B and the corresponding S,R-diastereoisomer (S-1-(6-methoxy-4′-(trifluoromethyl)-3-biphenylyl)-N-(R)-1-phenylethyl)ethanamine), respectively, which were then separated via HPLC (Harrington et al., 2010). S,R-calcimimetic B was 100-fold less potent than R,R-calcimimetic B (Harrington et al., 2010), comparable with the stereoselectivity of the R- and S-isomers of NPS-568 and cinacalcet (Hammerland et al., 1998; Nemeth et al., 2004). Given the remarkable difference in potency of the individual diastereoisomers, we sought to isolate and further characterize each one independently. Adapting the synthesis of Harrington et al., we were able to generate a mixture of diastereoisomers with a d.r. of 4:1. These were successfully isolated by either chiral HPLC or preparative layer chromatography (PLC) (see Supporting Informatio Appendix S1 for full synthetic methods). Structurally, the contrasting pharmacological behaviour of each diastereoisomer can be attributed to the spatial orientation of the methyl group adjacent to the biphenyl and amino moieties. With this in mind, it was of interest to evaluate the pharmacological activity of the ‘nor’ calcimimetic B derivative (R-N-((6-methoxy-4′-(trifluoromethyl)-3-biphenylyl)methyl)-1-phenylethanamine), with a methylene group replacing the methyl of interest. This was synthesized in a similar fashion to the R,R- and S,R-calcimimetic B derivatives. AC-265347 was identified in a screen by ACADIA Pharmaceuticals as a potent calcimimetic. It is structurally distinct from the phenylalkylamine calcimimetics and calcimimetic B, and was found to have improved potency over cinacalcet in an IP accumulation assay (Ma et al., 2011).
Figure 1.
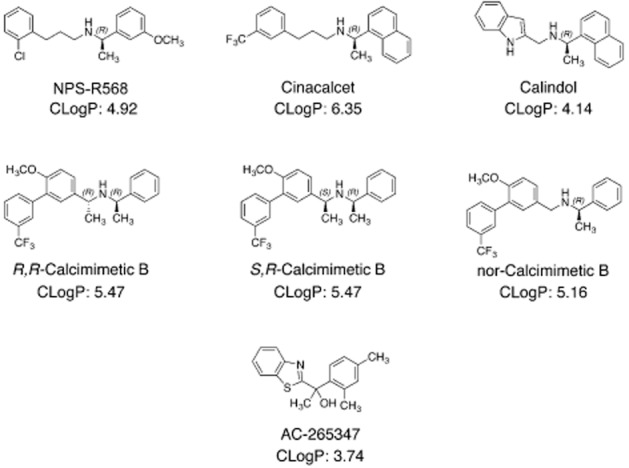
Structure of the CaS receptor allosteric modulators examined in this study. Calculated partition coefficient (CLog P) obtained from PerkinElmer ChemBioDraw software are shown.
We investigated the effects of the calcimimetics in Ca2+i mobilization, IP accumulation and pERK1/2 assays because each of these pathways has been undeniably linked to CaS receptor regulation of PTH release from parathyroid chief cells and/or calcitonin release from thyroid C-cells, as outlined in the Introduction. Although additional pathways are also involved in the regulation of PTH and calcitonin release, we selected those for which assays can be reliably performed in a high-throughput manner to enable robust quantification of allosteric modulation and biased signalling.
Calcimimetics are biased modulators of CaS receptor signalling
To evaluate the extent to which calcimimetics engender ligand-biased modulation at the CaS receptor, we first characterized their ability to potentiate the endogenous agonist, Ca2+o, in IP1 accumulation, Ca2+i mobilization and pERK1/2 assays. These experiments generated Ca2+o concentration–response curves in the absence and presence of the allosteric modulators.
As expected, cinacalcet, NPS-R568, calindol, AC-265347, R,R-calcimimetic B, S,R-calcimimetic B and nor-calcimimetic B, potentiated agonist-mediated activation of the CaS receptor in each assay, demonstrated by a leftward shift in the Ca2+o concentration–response curve, and a consequent increase in Ca2+o potency. In some instances, the calcimimetics elicited a concomitant increase in the baseline response because of potentiation of Ca2+o and Mg2+o in the buffer (Davey et al., 2012) and/or agonist activity. No changes in the maximum response elicited by Ca2+o were observed in the presence of the calcimimetics. Experimental data from IP1 accumulation assays for a representative calcimimetic from each class of compound are shown in Figure 2. Data for all calcimimetics across each pathway are shown in Supporting Information Appendix S3, Supporting Information Figures S1–7.
Figure 2.

Structurally distinct calcimimetics potentiate Ca2+o-mediated receptor activation with different potencies. Ca2+o-mediated IP1 accumulation in the presence of cinacalcet (A), R,R-calcimimetic B (B) and AC-265347 (C), over a range of concentrations (0–10 μM). Data are mean + SEM from at least four independent experiments performed in duplicate.
We have previously demonstrated that both calcimimetics and calcilytics can exhibit biased allosteric modulation via two (albeit related) mechanisms. The first arises from the ability of modulators to bind with distinct affinities to CaS receptor conformations that mediate different signalling pathways (Davey et al., 2012). Divergent affinities indicate that the modulators stabilize distinct receptor states, a requirement of ligand-biased signalling. The second arises from cooperativities between a modulator and the orthosteric agonist that differ at a given receptor state (Davey et al., 2012; Leach et al., 2013). Thus, an allosteric ternary complex model (Equation 2013a) was used to quantify the parameters that governed the activity of the calcimimetics in each assay to estimate the functional affinity (functional pKB) of the modulators and their overall cooperativity (αβ) with Ca2+o (Table 1). An F-test was used to determine whether the functional affinity and/or cooperativity of each calcimimetic differed across signalling assays. However, because functional affinity and cooperativity parameters are correlated in the non-linear regression algorithm, it is sometimes difficult to separate out the two effects. Thus, results of non-linear regression analyses that assumed the binding affinity to be the same or not the same across pathways are presented in Supporting Information Table S1 and Supporting Information Figures.
These analyses established a number of key findings. First, the phenylalkylamine calcimimetics, NPS-R568 and calindol, exhibited ligand-biased modulation that favoured activation of Ca2+i mobilization and IP1 accumulation. This was manifested as a lower functional affinity for the receptor state that coupled to pERK1/2 (Table 2, Figure 3A). Cinacalcet also demonstrated a tendency to modulate pERK1/2 less favourably than the other two pathways (Table 2, Figure 3A and B), but significance for this effect was only reached if its functional affinity was assumed to be the same across pathways (Supporting Information Table S1, Supporting Information Figure S8) and was thus indicative of weaker cooperativity in pERK1/2 assays. Second, S,R-calcimimetic B was biased towards modulation of IP1 accumulation, but showed no preference between Ca2+i mobilization or pERK1/2. Similar to cinacalcet, significance was only reached when its functional affinity was assumed to be the same across pathways (Supporting Information Table S1, Supporting Information Figure S8). Third, nor-calcimimetic B was relatively unbiased in its ability to modulate the three pathways, and its estimated functional affinities and cooperativities were comparable in all three assays. Finally, R,R-calcimimetic B and AC-265347 were biased towards modulation of pERK1/2 and IP1 accumulation, either in terms of functional affinity (Figure 3A) or cooperativity (Figure 3B and Supporting Information Figure S8). The bias arising from AC-265347 can be visualized in Figures 4A–C where the different effects of 0.1 μM AC-265347 on Ca2+o signalling in the three different assays are apparent. The bias engendered by multiple concentrations of AC-265347 can be visualized in the modulator ‘cooperativity bias plot’ as shown in Figure 4D. This illustrates the impact of equivalent AC-265347 concentrations on Ca2+o potency in Ca2+i mobilization or IP1 assays on the Y axis, and pERK1/2 assays on the X axis. If AC-265347 modulated both pathways equally, the data would converge on the line of identity. However, because it modulates one pathway to a greater degree than the other, the data points are distributed away from the line of identity towards the preferred pathway (i.e. towards IP1 over pERK1/2 and towards pERK1/2 over Ca2+i).
Table 2.
Pharmacological parameters that govern calcimimetic agonism at the CaS receptor
| Ca2+i mobilization | IP1 accumulation | |||
|---|---|---|---|---|
| pKB ± SEM (n) | LogτB ± SEM (τB) | pKB ± SEM (n) | LogτB ± SEM (τB) | |
| R,R-Calcimimetic B | 6.77 ± 0.23 (3) | −0.27 ± 0.04 (0.54) | 6.48 ± 0.28 (3) | −0.16 ± 0.06 (0.69) |
| S,R-Calcimimetic B | 5.44 ± 0.29 (3) | −0.10 ± 0.10 (0.79) | 5.89 ± 0.26 (3) | −0.06 ± 0.07 (0.87) |
| nor-calcimimetic B | 6.44 ± 0.14 (3) | −0.10 ± 0.03 (0.79) | 5.61 ± 0.29 (3) | −0.008 ± 0.09 (0.98) |
| AC-265347 | 5.94 ± 0.14 (3) | −0.02 ± 0.14 (0.95) | 6.04 ± 0.18 (3) | 0.08 ± 0.05 (1.1) |
Figure 3.
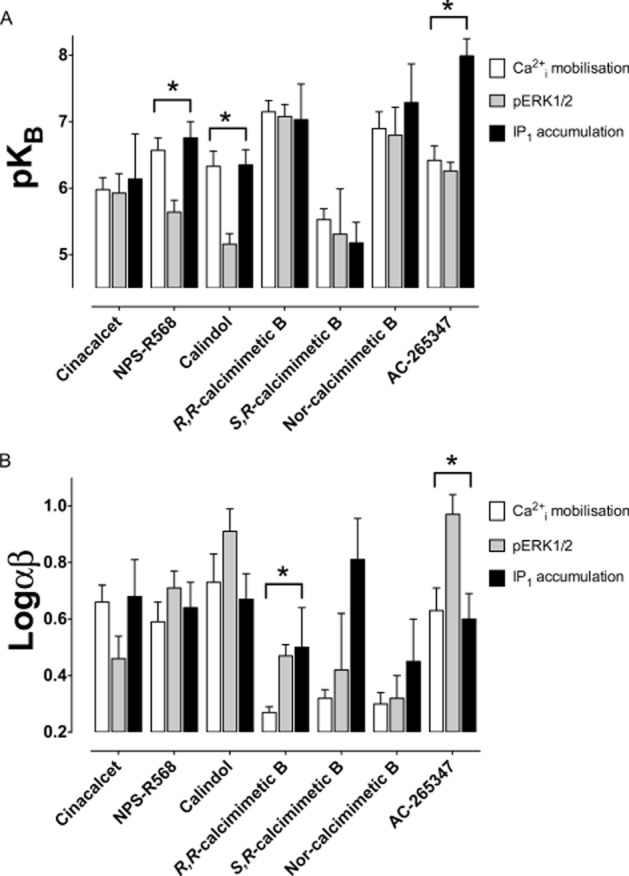
Calcimimetics display distinct functional affinities and/or cooperativities for CaS receptor conformations that couple to different signalling pathways. Modulator functional affinities (functional pKB) and cooperativities (αβ) were determined as described in the Methods, by fitting the Ca2+o pEC50 in the absence and presence of modulator determined in Ca2+i mobilization, pERK1/2 and IP1 accumulation assays to an allosteric ternary complex model (Equation 2013a). The affinity of the modulator was unconstrained in each pathway. Statistical differences shown by asterisks are demonstrated where an F-test determined that the data were fitted best when the modulator affinities and cooperativities were different among the three pathways. Data are mean + SEM from at least four independent experiments performed in duplicate.
Figure 4.

AC-265347 preferentially modulates pERK1/2 and IP1 accumulation over Ca2+i mobilization. Ca2+o-mediated Ca2+i mobilization (A), pERK1/2 (B) and IP1 accumulation (C) with or without 0.1 μM AC-265347. A ‘bias plot’ (D) depicts AC-265347's preferential modulation of pERK1/2 and IP1 accumulation versus Ca2+i mobilization. Ca2+o pEC50 in the absence and presence of modulator was determined in IP1 accumulation, Ca2+i mobilization and pERK1/2 assays and fitted to an allosteric ternary complex model (Equation 2013a) to determine 150 XY coordinates of points that defined the curve that best described the model. The XY coordinates for the different pathways are plotted against one another, with IP1 accumulation or Ca2+i mobilization data on the Y axis against pERK1/2 data on the X axis. Grey and black dashed lines join IP1 accumulation and Ca2+o mobilization XY coordinates, respectively, over a range of concentrations (0–10 μM) of AC-265347. The dotted line represents the line of identity, which is a theoretical representation of how the data would look if the pathways were modulated equally by AC-265347.
Third-generation calcimimetics are agonists at the CaS receptor
In IP1 accumulation and Ca2+i mobilization assays, the calcimimetics stimulated receptor activity in the presence of vehicle (buffer) alone. AC-265347, R,R-calcimimetic B and nor-calcimimetic B also did so in pERK1/2 assays. We previously simulated the effects of cinacalcet on signalling in the presence of an ambient concentration of agonist to reconstruct the experimental conditions under which our Ca2+i mobilization and pERK1/2 assays are undertaken (Davey et al., 2012). These simulations suggested that positive modulation of ambient agonists in the buffer (Ca2+o and Mg2+o) was expected. Accordingly, when we omitted ambient Ca2+o and Mg2+o from the assay buffer, Ca2+i mobilization and IP1 accumulation stimulated by cinacalcet, NPS-R568 and calindol on their own was largely inhibited (Figure 5), indicating that the observed ‘baseline effect’ was primarily due to potentiation of ambient agonist activity. In contrast, AC-265347 and the calcimimetic B analogues retained activity in the absence of ambient Ca2+o and Mg2+o (Figure 5). The effects of omitting only Ca2+o from the buffer can be observed in Supporting Information Figure S9.
Figure 5.

Calcimimetics are agonists at the CaS receptor. Activity of calcimimetics in the absence of ambient Ca2+o and Mg2+o measured in Ca2+i mobilization and IP1 accumulation assays. Data are mean + SEM from three independent experiments performed in triplicate.
We fitted the agonist activity of the calcimimetics (in the absence of Ca2+o and Mg2+o) to the standard operational model of agonism (Equation 2013b) (Black and Leff, 1983) to gain a second estimate of the functional affinity of the modulators at the CaS receptor. These estimates were similar to the affinities estimated for the modulators using the allosteric ternary complex model (Table 2). Of note is the comparable affinity of AC-265347 between Ca2+i mobilization and IP1 assays. This is in contrast to its affinity in ‘potentiation assays’, which were strongly suggestive of a higher affinity for the receptor state that coupled to IP1 accumulation (Table 1, Figure 3A, Supporting Information Figure S7). Thus, the receptor state that mediates direct calcimimetic activation of the CaS receptor may be distinct from the state that modulates Ca2+o activity at the receptor.
Table 1.
Pharmacological parameters that govern the allosteric activity of CaS receptor modulators in Ca2+i mobilization, pERK1/2 and IP1 accumulation assays
| Grouped data analysis | ||||||
|---|---|---|---|---|---|---|
| Ca2+i mobilization | pERK1/2 | IP1 accumulation | ||||
| pKB ± SEM (n) | Logαβ ± SEM (αβ) | pKB ± SEM (n) | Logαβ ± SEM (αβ) | pKB ± SEM (n) | Logαβ ± SEM (αβ) | |
| Cinacalcet | 5.98 ± 0.18 (18)a | 0.66 ± 0.06 (4.6)a | 5.93 ± 0.29 (13)a | 0.46 ± 0.08 (2.9)a | 6.14 ± 0.33 (4) | 0.68 ± 0.13 (4.8) |
| NPS-R568* | 6.57 ± 0.19 (15) | 0.59 ± 0.07 (3.9) | 5.64 ± 0.18 (4) | 0.71 ± 0.06 (5.1) | 6.76 ± 0.24 (4) | 0.64 ± 0.09 (4.3) |
| Calindol* | 6.33 ± 0.23 (4) | 0.73 ± 0.10 (5.4) | 5.16 ± 0.16 (4) | 0.91 ± 0.08 (8.1) | 6.35 ± 0.23 (4) | 0.67 ± 0.09 (4.7) |
| S,R-calcimimetic B | 5.53 ± 0.16 (4) | 0.32 ± 0.03 (2.1) | 5.31 ± 0.68 (3) | 0.42 ± 0.20 (2.6) | 5.18 ± 0.31 (3) | 0.81 ± 0.14 (6.5) |
| R,R-calcimimetic B* | 7.15 ± 0.17 (4) | 0.27 ± 0.02 (1.9) | 7.08 ± 0.18 (4) | 0.47 ± 0.04 (3.0) | 7.03 ± 0.54 (4) | 0.50 ± 0.14 (3.2) |
| Nor-calcimimetic B | 6.90 ± 0.25 (7) | 0.30 ± 0.04 (2.0) | 6.80 ± 0.42 (5) | 0.32 ± 0.08 (2.1) | 7.29 ± 0.58 (4) | 0.45 ± 0.15 (3.0) |
| AC-265347* | 6.42 ± 0.22 (5) | 0.63 ± 0.08 (4.3) | 6.26 ± 0.13 (4) | 0.97 ± 0.07 (9.3) | 7.99 ± 0.26 (4) | 0.60 ± 0.09 (4.0) |
The potency of Ca2+o in the presence of increasing concentrations of modulator was fitted to an allosteric ternary complex model (Equation 2013a) to quantify the equilibrium dissociation constant (pKB) and cooperativity (αβ) of the modulators at the human CaS receptor, using a model in which the binding affinity was not constrained across pathways.
Datasets taken from those used in (Leach et al., 2013).
Significant difference in pKB and/or Logαβ between pathways (P < 0.05, F-test).
Our analysis additionally derived an operational measure of agonism, defined as τB, which reflects both the degree to which the agonist can activate the receptor, and the stimulus–response coupling between the receptor and the intracellular signalling pathway. Interestingly, although Ca2+o is more potent in Ca2+i mobilization than IP1 assays, there was no significant difference in the activity of the modulators in the two assays (P > 0.1 unpaired t-test), indicating that they do not follow the same natural biased profile as the endogenous agonist.
Calcimimetics differentially modulate trafficking of a naturally occurring loss-of-expression mutant
We have previously shown that both calcimimetics and calcilytics are also biased in their abilities to modulate CaS receptor trafficking (Leach et al., 2013). This may have important implications for patients with loss-of-expression CaS receptor mutations that cause disorders of calcium metabolism such as familial hypocalciuric hypercalcaemia and neonatal severe hyperparathyroidism. Thus, to determine the ability of each of the CaS receptor modulators to correct trafficking and signalling of defective CaS receptor mutants, we investigated the consequences of the modulators at the naturally occurring loss-of-expression mutant, G670E (Kobayashi et al., 1997). Expression of this mutant receptor at the cell surface is greatly reduced but its affinity for cinacalcet is unaltered (Leach et al., 2012; 2013). This mutant also signals efficiently in Ca2+i mobilization and pERK1/2 assays (Leach et al., 2012; 2013).
The affinities and cooperativities of AC-265347, cinacalcet, NPS-R568 and calindol were unaltered at the G670E mutation compared with the WT, as assessed in Ca2+i mobilization assays (Table 3). The affinity of the calcimimetic B analogues, however, was reduced approximately 100-fold, although R,R-calcimimetic B and nor-calcimimetic B were still able to bind to the receptor and potentiate Ca2+o-mediated signalling.
Table 3.
Pharmacological properties of CaS receptor modulators at the naturally occurring G670E mutant receptor
| Cell surface expression (% WT) | Ca2+i mobilization | ||||
|---|---|---|---|---|---|
| 0 modulator | 0.3 μM | 3 μM | pKB ± SEM (n) | Logαβ ± SEM (αβ) | |
| Cinacalcet | 12 ± 2 | 74 ± 12 | 152 ± 39 | 6.00 ± 0.19 (7)a | 0.59 ± 0.06 (3.9)a |
| NPS-R568 | 33 ± 9 | 126 ± 37 | 6.61 ± 0.14 (4) | 0.74 ± 0.14 (5.5) | |
| Calindol | 36 ± 7 | 112 ± 28 | 6.33 ± 0.31 (3) | 0.53 ± 0.10 (3.4) | |
| R,R-calcimimetic B | 62 ± 11 | 152 ± 40 | 5.27 ± 0.37 (4) | 0.51 ± 0.12 (3.2) | |
| S,R-calcimimetic B | 12 ± 3 | 14 ± 5 | Not performed | Not performed | |
| nor-calcimimetic B | 28 ± 6 | 91 ± 27 | 6.21 ± 0.23 (3) | 0.42 ± 0.06 (2.6) | |
| AC-265347 | 14 ± 3 | 18 ± 5 | 6.62 ± 0.23 (3) | 0.72 ± 0.10 (5.2) | |
Cell surface expression of the mutant following overnight treatment with modulator was determined by FACS analysis. The potency of Ca2+o in Ca2+i mobilization assays in the presence of increasing concentrations of modulator was fitted to an allosteric ternary complex model (Equation 2013a) to quantify the equilibrium dissociation constant (pKB) and cooperativity (αβ) of the modulators at the G670E mutant.
Datasets taken from those used in (Leach et al., 2013).
Overnight treatment of HEK293 cells with cinacalcet, NPS-R568, calindol, R,R-calcimimetic B and nor-calcimimetic B restored cell surface expression of the G670E mutant (Table 3; Figure 6). S,R-calcimimetic B and AC-265347, however, had no effect on expression. In the case of S,R-calcimimetic B, this was likely due to lower receptor occupancy in comparison to the other calcimimetics because of its reduced functional affinity. The inability of AC-265347 to rescue G670E expression, however, was not due to reduced affinity or to reduced cooperativity, which were comparable with the other calcimimetics. The inability of AC-265347 to restore trafficking may be related to its lower lipophilicity relative to the other compounds. This parameter can be represented by calculated partition coefficient (CLog P, see Figure 1), which for AC-265347 was found to be considerably lower than for the other allosteric modulators tested. Thus, AC-265347 may have a reduced propensity to cross cell membranes to pharmacochaperone misfolded receptors trapped in the endoplasmic reticulum and Golgi compartments.
Figure 6.
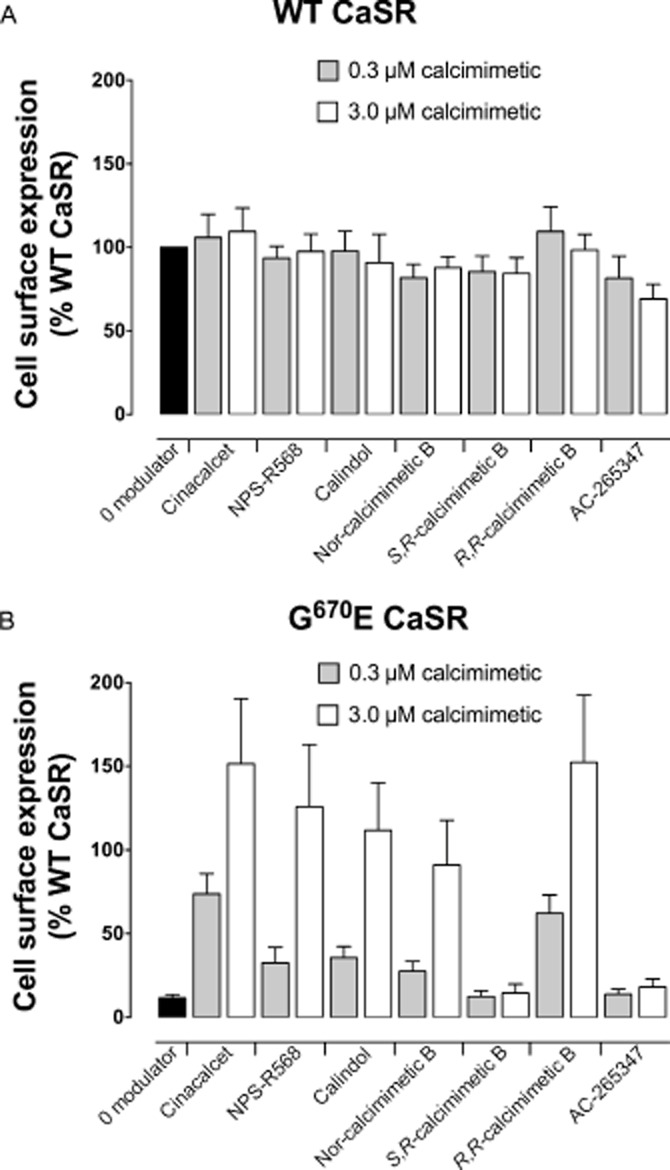
CaS receptor modulators differentially rescue the G670E loss-of-expression mutant. Whereas overnight treatment with the calcimimetics has minimal effect on the expression of the WT CaS receptor in HEK cells, cinacalcet, NPS-R568, calindol, R,R-calcimimetic B and nor-calcimimetic B rescue the expression of the G670E mutant. AC-265347 and S,R-calcimimetic B, however, do not rescue cell surface expression. Data are mean + SEM from at least four independent experiments.
Discussion
The present study evaluated the pharmacological activity of structurally related and diverse calcimimetics across multiple measures of receptor activity, identifying distinct ligand-biased profiles for each compound. Importantly, whereas phenylalkylamine modulators are biased towards Ca2+i mobilization and IP1 accumulation, S,R-calcimimetic B is biased only towards modulation of IP1 accumulation, and nor-calcimimetic B is unbiased. R,R-calcimimetic B and AC-265347 on the other hand are biased towards pERK1/2 and IP1 accumulation. Of note, although Ca2+i mobilization via Gq-coupled receptors typically stems from the PLC-IP pathway, the divergence in bias between Ca2+i and IP1 assays observed herein suggests that CaS receptor-mediated Ca2+i mobilization is also facilitated via alternative mechanisms. This is supported by a number of previous findings. In rat medullary thyroid carcinoma cells, Ca2+o activation of the CaS receptor resulted in Ca2+i influx via ion-gated calcium channels in addition to IP3-mediated calcium mobilization (Thomsen et al., 2012b). Sr2+o, on the other hand, stimulated CaS receptor-mediated PLC/IP3/Ca2+i mobilization, but did not trigger opening of calcium channels in these cells (Thomsen et al., 2012b). Similarly, although both Ca2+o and L-phenylalanine stimulated Ca2+i mobilization in CaS receptor-transfected HEK293 cells, only Ca2+o promoted IP accumulation and diacylglycerol production (Rey et al., 2005). Finally, we recently showed that truncation of the CaS receptor after R866 resulted in a complete inability of the receptor to stimulate Ca2+i mobilization, whereas IP accumulation was reduced, but maintained (Goolam et al., 2014). In the same study, mutations in intracellular loops 2 and 3 greatly impaired IP accumulation, but had a weaker affect on Ca2+i mobilization. These findings suggest Ca2+i mobilization stimulated from the CaS receptor is in part driven via an IP-independent mechanism.
Intriguingly, although AC-265347 is a positive modulator of CaS receptor signalling, it is a neutral modulator of receptor trafficking. These findings build on our earlier studies of prototypical CaS receptor-positive and -negative allosteric modulators that initially identified bias in modulation by these compounds (Davey et al., 2012; Leach et al., 2013).
Ligand-biased signalling by CaS receptor modulators may be driven by ligand-specific stabilization of distinct receptor states that couple preferentially to particular intracellular signalling pathways. This is suggested by the different functional affinities or cooperativities with the endogenous agonist estimated at each pathway. We introduced this concept several years ago (Leach et al., 2007) and have subsequently observed biased allosteric modulation at the M4 muscarinic (Leach et al., 2010), A1 adenosine (Aurelio et al., 2009) and glucagon-like peptide 1 (GLP-1) (Koole et al., 2011) receptors, indicating that pathway selectivity may be achieved with allosteric modulators acting at a number of GPCRs.
AC-265347 exhibited high cooperativity in pERK1/2 assays, maximally enhancing the potency of Ca2+o nearly 10-fold, in comparison with the threefold enhancement in potency observed with cinacalcet. This is consistent with previous findings indicating that AC-265347 is more potent than cinacalcet with respect to IP1 accumulation assays, but has comparable potency with respect to cellular proliferation (Ma et al., 2011). This suggests that AC-265347 exhibits ligand-biased modulation of distinct CaS receptor signalling pathways. pERK1/2 plays a significant role in the suppression of PTH release (Corbetta et al., 2002; Thomsen et al., 2012b), but may be less important for CaS receptor-mediated stimulation of calcitonin release (Thomsen et al., 2012b). Thus, compounds that favour pERK1/2 over Ca2+i mobilization may have reduced propensity to induce calcitonin-dependent hypocalcaemia when compared with cinacalcet. Accordingly, there is pronounced separation (300-fold) in the concentration of S-AC-265347 required to suppress serum PTH levels versus the concentration that reduces serum Ca2+o levels in healthy rats (Ma et al., 2011). Similarly, concentrations of calcimimetic B that maximally inhibit PTH secretion in nephrectomized rats have little effect on calcitonin release or serum Ca2+o levels (Henley et al., 2011). In contrast, cinacalcet concentrations required to maximally suppress PTH secretion also stimulate calcitonin release and reduce serum Ca2+o levels in rats (Nemeth et al., 2004), suggesting less selectivity of cinacalcet for suppression of PTH release. AC-265347 and R,R-calcimimetic B are thus potentially important lead compounds of value in elucidating the roles of pERK1/2 in CaS receptor-mediated regulation of PTH and calcitonin release.
The fact that third–generation, but not phenylalkylamine calcimimetics, are agonists in their own right may also contribute to their parathyroid selectivity. When stimulus–response coupling is strong, for instance in tissues such as the parathyroid glands where CaS receptor expression is high, partial agonist effects will become more pronounced.
The CaS receptor is promiscuous in its coupling to intracellular signalling pathways, and the influence of individual pathways to physiological outcomes such as regulation of hormone release from chief cells of the parathyroid and parafollicular C-cells of the thyroid, and control of ion transport in the kidney, is still being elucidated. Although we have selected to investigate the modulatory effects of calcimimetics on three key signalling pathways that regulate some of the physiological actions of the CaS receptor, these pathways are not exhaustive. For instance, G12/13-mediated cytoskeletal rearrangements are important for CaS receptor-mediated suppression of PTH release (Quinn et al., 2007), but experiments that measure G12/13-mediated membrane ruffling, for instance, are not amenable to high-throughput screening techniques and have subsequently not been included in the present study. Our ongoing work aims to extend these studies to examine activity across multiple pathways in primary cell lines, to establish the link between signalling bias and in vivo pharmacological and physiological calcimimetic effects.
It must also be noted that allosterism may be influenced by the kinetics of ligand binding relative to the different time points underlying response generation in each experiment. Thus, an alternative explanation for the observed bias is that each ligand stabilizes the same state with different kinetics. However, the same direction of bias towards Ca2+i mobilization over pERK1/2 is also observed following preincubation of the CaS receptor with cinacalcet and NPS-R568 for 30 min prior to measurement of agonist-mediated receptor signalling (Davey et al., 2012). Thus, differences in modulator bias in the different assays likely reflect true biased signalling and not an equilibrium artefact. For the detection of agonism, the transient nature of agonist-mediated Ca2+i mobilization, pERK1/2 and indeed many other GPCR signalling responses means signalling will often subside before equilibrium binding can be reached. Thus, the receptor may no longer elicit a response once true equilibrium is obtained. Therefore, it is assumed that one of the most relevant responses for the purpose of detecting receptor signalling and indeed biased signalling is the response elicited upon first exposure of a cell to the activating agonist.
In addition to differences in agonist effects and biased modulation of different signalling pathways, we found that AC-265347, unlike the other calcimimetics tested, was unable to restore the expression of the G670E loss-of-expression mutant. Importantly, this and our previous study have identified unique ligand-biased profiles whereby a drug can positively modulate CaS receptor signalling and trafficking (cinacalcet, NPS-R568, calindol, R,R-calcimimetic B and nor-calcimimetic B), negatively modulate CaS receptor signalling and positively modulate trafficking (NPS-2143) (Leach et al., 2013), or positively modulate signalling without affecting trafficking (AC-265347). The inability of AC-265347 to rescue expression may be due to its lower lipophilicity, which makes it less likely to cross the cell membrane. Thus, compartmentalization of receptors away from the cell surface restricts its access to only a subset of the available receptor pool. This represents an alternative means by which a drug can bias the activity of a receptor, one that is governed by its interaction with receptors that signal (cell surface receptors) versus those that can traffic to the cell surface (intracellular receptors).
The diverse pharmacological profile exhibited by each of the allosteric modulators offers exciting possibilities for their use beyond treatments for secondary hyperparathyroidism. For instance, future identification of pure ‘trafficking modulators’ may be beneficial in disease states where reduced CaS receptor expression has been identified, such as colon cancer (Hizaki et al., 2011; Singh et al., 2012), and primary and secondary hyperparathyroidism (Kifor et al., 1996; Cetani et al., 2000; Yano et al., 2000). Furthermore, drugs may be fine-tuned to the needs of distinct patients carrying naturally occurring CaS receptor mutations, depending on the impact of their mutation on receptor signalling and/or trafficking. The ability to tailor drug therapies to patients harbouring naturally occurring mutations may become an important consideration not just for the CaS receptor, but also for other GPCRs. Indeed, naturally occurring mutations in the GLP-1 receptor, for instance, engender signalling bias, with some mutations altering receptor coupling to only a subset of intracellular signalling pathways (Koole et al., 2011). Thus, a pharmacogenomics approach may be essential for the future treatment of certain patient subtypes.
In conclusion, the current study has characterized structurally diverse calcimimetics and identified distinct ligand-biased signalling engendered by different classes of compounds. Although at present, it is unclear which biased profile will be desirable in different disease states, the identification of biased ligands provides novel tools to probe the in vivo consequences of differentially promoting CaS receptor signalling and trafficking.
Acknowledgments
We thank Dr Michael Crouch for the kind donation of SureFire pERK1/2 assay kits used in this study. This research was supported by National Health and Medical Research Council (NHMRC) of Australia project grant number APP1026962. K. J. G. is a recipient of an NHMRC Overseas Biomedical postdoctoral training fellowship. A. C. and P. M. S. are Principal Research Fellows of the NHMRC.
Glossary
- Ca2+o
extracellular calcium
- Ca2+i
intracellular calcium
- CaS receptors
calcium-sensing receptors
- IP1
inositol 1-phosphate
- Mg2+o
extracellular magnesium
- pERK1/2
phosphorylated ERK1/2
- PTH
parathyroid hormone
Author contributions
A. C., A. D. C., A. E. C. and K. L. planned and coordinated the study; A. E. C., K. J. G. and K. L. performed experimental assays; S. N. M. synthesized calcimimetic B analogues; S. G. B. F. prepared and evaluated the AF647-conjugated 9E10 antibody; A. E. C., S. N. M., K. J. G., P. M. S., P. J. S., A. D. C., A. C. and K. L. wrote the paper.
Conflict of interest
A. E. C., S. N. M., K. J. G., S. G. B. F., P. J. S., P. M. S., A. D.C. and K. L. have nothing to declare. A. C. has previously published work on the CaS receptor in collaboration with researchers from Amgen.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12937
Figure S1 Cinacalcet potentiation of Ca2+o-mediated signalling. (A) Ca2+o stimulation of IP1 accumulation, Ca2+i mobilization and pERK1/2 in the absence and presence of cinacalcet. (B) Non-linear regression analysis of Ca2+o potency in the absence and presence of cinacalcet with an allosteric ternary complex model (Equation 2) where the pKB is constrained between pathways or not (as indicated).
Figure S2 Calindol potentiation of Ca2+o-mediated signalling. (A) Ca2+o stimulation of IP1 accumulation, Ca2+i mobilization and pERK1/2 in the absence and presence of calindol. (B) Non-linear regression analysis of Ca2+o potency in the absence and presence of calindol with an allosteric ternary complex model (Equation 2) where the pKB is constrained between pathways or not (as indicated).
Figure S3 NPS-R568 potentiation of Ca2+o-mediated signalling. (A) Ca2+o stimulation of IP1 accumulation, Ca2+i mobilization and pERK1/2 in the absence and presence of NPS-R568. (B) Non-linear regression analysis of Ca2+o potency in the absence and presence of NPR-R568 with an allosteric ternary complex model (Equation 2) where the pKB is constrained between pathways or not (as indicated).
Figure S4 S,R-calcimimetic B potentiation of Ca2+o-mediated signalling. (A) Ca2+o stimulation of IP1 accumulation, Ca2+i mobilization and pERK1/2 in the absence and presence of S,R-calcimimetic B. (B) Non-linear regression analysis of Ca2+o potency in the absence and presence of S,R-calcimimetic B with an allosteric ternary complex model (Equation 2) where the pKB is constrained between pathways or not (as indicated).
Figure S5 R,R-calcimimetic B potentiation of Ca2+o-mediated signalling. (A) Ca2+o stimulation of IP1 accumulation, Ca2+i mobilization and pERK1/2 in the absence and presence of R,R-calcimimetic B. (B) Non-linear regression analysis of Ca2+o potency in the absence and presence of R,R-calcimimetic B with an allosteric ternary complex model (Equation 2) where the pKB is constrained between pathways or not (as indicated).
Figure S6 nor-calcimimetic B potentiation of Ca2+o-mediated signalling. (A) Ca2+o stimulation of IP1 accumulation, Ca2+i mobilization and pERK1/2 in the absence and presence of nor-calcimimetic B. (B) Non-linear regression analysis of Ca2+o potency in the absence and presence of nor-calcimimetic B with an allosteric ternary complex model (Equation 2) where the pKB is constrained between pathways or not (as indicated).
Figure S7 AC-265347 potentiation of Ca2+o-mediated signalling. (A) Ca2+o stimulation of IP1 accumulation, Ca2+i mobilization and pERK1/2 in the absence and presence of AC-265347. (B) Non-linear regression analysis of Ca2+o potency in the absence and presence of AC-265347 with an allosteric ternary complex model (Equation 2) where the pKB is constrained between pathways or not (as indicated).
Figure S8 Binding affinity and cooperativities of calcimimetics across different pathways. Cooperativities of calcimimetics in Ca2+i mobilization (white bars), pERK1/2 (grey bars) and IP1 accumulation (black bars) assays were determined with the binding affinity constrained to be the same in each assay.
Figure S9 Concentration–response curves to Ca2+o and calcimimetics in the presence of 1.8mM ambient (buffer) Mg2+o but no Ca2+o.
Figure S10 Validation of 9E10:AF647 antibody. (A) Coomassie stained gel imaged on Typhoon Trio (GE Life Sciences) showing purification of 9E10 antibody from biorector hybridoma supernatant. Molecular weight markers are indicated on the left and various purification fractions indicated above the gel. Densitometry on elution fractions 3–6 was performed using ImageJ and these fractions pooled for the subsequent conjugation reaction. Densitometry was consistent with antibody being >90% pure. Staining is consistent with the concentration as determined by A280. (B) Titration of 9E10:AF647 in Cos7 cells stably transfected with either vector control (Vector) or vector containing cMyc tagged calcitonin receptor (cMycCTR). Relative fluorescence intensity corresponds to antibody binding, determined on a FACS CantoII (BD Biosciences), as described in the Supporting Information Appendix S1: supplemental methods.
Table S1 Pharmacological parameters that govern the allosteric activity of CaS receptor modulators in Ca2+i mobilization, pERK1/2 and IP1 accumulation assays. The potency of Ca2+o in the presence of increasing concentrations of modulator was fitted to an allosteric ternary complex model (Equation 2) to quantify the equilibrium dissociation constant (pKB) and cooperativity (αβ) of the modulators at the human CaS receptor, using a model in which the binding affinity was assumed to be the same across pathways.
Appendix S1 Supplemental methods.
References
- Alam MU, Kirton JP, Wilkinson FL, Towers E, Sinha S, Rouhi M, et al. Calcification is associated with loss of functional calcium-sensing receptor in vascular smooth muscle cells. Cardiovasc Res. 2009;81:260–268. doi: 10.1093/cvr/cvn279. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, McGrath JC, et al. The concise guide to PHARMACOLOGY 2013/14: overview. Br J Pharmacol. 2013a;170:1449–1458. [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013b;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas MD, de la Fuente V, Delgado P, Gil MT, Gutierrez P, Ribero J, et al. Pharmacodynamics of cinacalcet over 48 hours in patients with controlled secondary hyperparathyroidism: useful data in clinical practice. J Clin Endocrinol Metab. 2013;98:1718–1725. doi: 10.1210/jc.2012-4003. [DOI] [PubMed] [Google Scholar]
- Aurelio L, Valant C, Flynn BL, Sexton PM, Christopoulos A, Scammells PJ. Allosteric modulators of the adenosine A1 receptor: synthesis and pharmacological evaluation of 4-substituted 2-amino-3-benzoylthiophenes. J Med Chem. 2009;52:4543–4547. doi: 10.1021/jm9002582. [DOI] [PubMed] [Google Scholar]
- Avlani VA, Ma W, Mun HC, Leach K, Delbridge L, Christopoulos A, et al. Calcium-sensing receptor-dependent activation of CREB phosphorylation in HEK-293 cells and human parathyroid cells. Am J Physiol Endocrinol Metab. 2013;304:E1097–E1104. doi: 10.1152/ajpendo.00054.2013. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P. Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Broadhead GK, Mun HC, Avlani VA, Jourdon O, Church WB, Christopoulos A, et al. Allosteric modulation of the calcium-sensing receptor by gamma-glutamyl peptides: inhibition of PTH secretion, suppression of intracellular cAMP levels, and a common mechanism of action with L-amino acids. J Biol Chem. 2011;286:8786–8797. doi: 10.1074/jbc.M110.149724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E, Enyedi P, LeBoff M, Rotberg J, Preston J, Chen C. High extracellular Ca2+ and Mg2+ stimulate accumulation of inositol phosphates in bovine parathyroid cells. FEBS Lett. 1987;218:113–118. doi: 10.1016/0014-5793(87)81029-3. [DOI] [PubMed] [Google Scholar]
- Brown EM. Role of the calcium-sensing receptor in extracellular calcium homeostasis. Best Pract Res Clin Endocrinol Metab. 2013;27:333–343. doi: 10.1016/j.beem.2013.02.006. [DOI] [PubMed] [Google Scholar]
- Cetani F, Picone A, Cerrai P, Vignali E, Borsari S, Pardi E, et al. Parathyroid expression of calcium-sensing receptor protein and in vivo parathyroid hormone-Ca(2+) set-point in patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 2000;85:4789–4794. doi: 10.1210/jcem.85.12.7028. [DOI] [PubMed] [Google Scholar]
- Chonchol M, Locatelli F, Abboud HE, Charytan C, de Francisco AL, Jolly S, et al. A randomized, double-blind, placebo-controlled study to assess the efficacy and safety of cinacalcet HCl in participants with CKD not receiving dialysis. Am J Kidney Dis. 2009;53:197–207. doi: 10.1053/j.ajkd.2008.09.021. [DOI] [PubMed] [Google Scholar]
- Christopoulos A. Assessing the distribution of parameters in models of ligand-receptor interaction: to log or not to log. Trends Pharmacol Sci. 1998;19:351–357. doi: 10.1016/s0165-6147(98)01240-1. [DOI] [PubMed] [Google Scholar]
- Corbetta S, Lania A, Filopanti M, Vicentini L, Ballare E, Spada A. Mitogen-activated protein kinase cascade in human normal and tumoral parathyroid cells. J Clin Endocrinol Metab. 2002;87:2201–2205. doi: 10.1210/jcem.87.5.8492. [DOI] [PubMed] [Google Scholar]
- Davey AE, Leach K, Valant C, Conigrave AD, Sexton PM, Christopoulos A. Positive and negative allosteric modulators promote biased signaling at the calcium-sensing receptor. Endocrinology. 2012;153:1232–1241. doi: 10.1210/en.2011-1426. [DOI] [PubMed] [Google Scholar]
- Feng J, Petersen CD, Coy DH, Jiang JK, Thomas CJ, Pollak MR, et al. Calcium-sensing receptor is a physiologic multimodal chemosensor regulating gastric G-cell growth and gastrin secretion. Proc Natl Acad Sci U S A. 2010;107:17791–17796. doi: 10.1073/pnas.1009078107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox J, Lowe SH, Conklin RL, Petty BA, Nemeth EF. Calcimimetic compound NPS R-568 stimulates calcitonin secretion but selectively targets parathyroid gland Ca(2+) receptor in rats. J Pharmacol Exp Ther. 1999a;290:480–486. [PubMed] [Google Scholar]
- Fox J, Lowe SH, Petty BA, Nemeth EF. NPS R-568: a type II calcimimetic compound that acts on parathyroid cell calcium receptor of rats to reduce plasma levels of parathyroid hormone and calcium. J Pharmacol Exp Ther. 1999b;290:473–479. [PubMed] [Google Scholar]
- Freichel M, Zink-Lorenz A, Holloschi A, Hafner M, Flockerzi V, Raue F. Expression of a calcium-sensing receptor in a human medullary thyroid carcinoma cell line and its contribution to calcitonin secretion. Endocrinology. 1996;137:3842–3848. doi: 10.1210/endo.137.9.8756555. [DOI] [PubMed] [Google Scholar]
- Goolam MA, Ward JH, Avlani VA, Leach K, Christopoulos A, Conigrave AD. Roles of intraloops-2 and -3 and the proximal C-terminus in signalling pathway selection from the human calcium-sensing receptor. FEBS Lett. 2014;588:3340–3346. doi: 10.1016/j.febslet.2014.07.022. [DOI] [PubMed] [Google Scholar]
- Hammerland LG, Garrett JE, Hung BC, Levinthal C, Nemeth EF. Allosteric activation of the Ca2+ receptor expressed in Xenopus laevis oocytes by NPS 467 or NPS 568. Mol Pharmacol. 1998;53:1083–1088. [PubMed] [Google Scholar]
- Harrington PE, St Jean DJ, Jr, Clarine J, Coulter TS, Croghan M, Davenport A, et al. The discovery of an orally efficacious positive allosteric modulator of the calcium sensing receptor containing a dibenzylamine core. Bioorg Med Chem Lett. 2010;20:5544–5547. doi: 10.1016/j.bmcl.2010.07.060. [DOI] [PubMed] [Google Scholar]
- Henley C, 3rd, Yang Y, Davis J, Lu JY, Morony S, Fan W, et al. Discovery of a calcimimetic with differential effects on parathyroid hormone and calcitonin secretion. J Pharmacol Exp Ther. 2011;337:681–691. doi: 10.1124/jpet.110.178681. [DOI] [PubMed] [Google Scholar]
- Hizaki K, Yamamoto H, Taniguchi H, Adachi Y, Nakazawa M, Tanuma T, et al. Epigenetic inactivation of calcium-sensing receptor in colorectal carcinogenesis. Mod Pathol. 2011;24:876–884. doi: 10.1038/modpathol.2011.10. [DOI] [PubMed] [Google Scholar]
- Kantham L, Quinn SJ, Egbuna OI, Baxi K, Butters R, Pang JL, et al. The calcium-sensing receptor (CaS receptor) defends against hypercalcemia independently of its regulation of parathyroid hormone secretion. Am J Physiol Endocrinol Metab. 2009;297:E915–E923. doi: 10.1152/ajpendo.00315.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336:296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2013;12:205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- Kessler A, Faure H, Petrel C, Ruat M, Dauban P, Dodd RH. N2-benzyl-N1-(1-(1-naphthyl)ethyl)-3-phenylpropane-1,2-diamines and conformationally restrained indole analogues: development of calindol as a new calcimimetic acting at the calcium sensing receptor. Bioorg Med Chem Lett. 2004;14:3345–3349. doi: 10.1016/j.bmcl.2004.03.056. [DOI] [PubMed] [Google Scholar]
- Kifor O, Moore FD, Jr, Wang P, Goldstein M, Vassilev P, Kifor I, et al. Reduced immunostaining for the extracellular Ca2+-sensing receptor in primary and uremic secondary hyperparathyroidism. J Clin Endocrinol Metab. 1996;81:1598–1606. doi: 10.1210/jcem.81.4.8636374. [DOI] [PubMed] [Google Scholar]
- Kifor O, Diaz R, Butters R, Brown EM. The Ca2+-sensing receptor (CaR) activates phospholipases C, A2, and D in bovine parathyroid and CaR-transfected, human embryonic kidney (HEK293) cells. J Bone Miner Res. 1997;12:715–725. doi: 10.1359/jbmr.1997.12.5.715. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Tanaka H, Tsuzuki K, Tsuyuki M, Igaki H, Ichinose Y, et al. Two novel missense mutations in calcium-sensing receptor gene associated with neonatal severe hyperparathyroidism. J Clin Endocrinol Metab. 1997;82:2716–2719. doi: 10.1210/jcem.82.8.4135. [DOI] [PubMed] [Google Scholar]
- Koole C, Wootten D, Simms J, Valant C, Miller LJ, Christopoulos A, et al. Polymorphism and ligand dependent changes in human glucagon-like peptide-1 receptor (GLP-1R) function: allosteric rescue of loss of function mutation. Mol Pharmacol. 2011;80:486–497. doi: 10.1124/mol.111.072884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28:382–389. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Leach K, Loiacono RE, Felder CC, McKinzie DL, Mogg A, Shaw DB, et al. Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology. 2010;35:855–869. doi: 10.1038/npp.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K, Wen A, Davey AE, Sexton PM, Conigrave AD, Christopoulos A. Identification of molecular phenotypes and biased signaling induced by naturally occurring mutations of the human calcium-sensing receptor. Endocrinology. 2012;153:4304–4316. doi: 10.1210/en.2012-1449. [DOI] [PubMed] [Google Scholar]
- Leach K, Wen A, Cook AE, Sexton PM, Conigrave AD, Christopoulos A. Impact of clinically relevant mutations on the pharmacoregulation and signaling bias of the calcium-sensing receptor by positive and negative allosteric modulators. Endocrinology. 2013;154:1105–1116. doi: 10.1210/en.2012-1887. [DOI] [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A, Conigrave AD. Engendering biased signalling from the calcium-sensing receptor for the pharmacotherapy of diverse disorders. Br J Pharmacol. 2014;171:1142–1155. doi: 10.1111/bph.12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KP, Russo AF, Hsiung SC, Adlersberg M, Franke TF, Gershon MD, et al. Calcium receptor-induced serotonin secretion by parafollicular cells: role of phosphatidylinositol 3-kinase-dependent signal transduction pathways. J Neurosci. 2003;23:2049–2057. doi: 10.1523/JNEUROSCI.23-06-02049.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loupy A, Ramakrishnan SK, Wootla B, Chambrey R, de la Faille R, Bourgeois S, et al. PTH-independent regulation of blood calcium concentration by the calcium-sensing receptor. J Clin Invest. 2012;122:3355–3367. doi: 10.1172/JCI57407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JN, Owens M, Gustafsson M, Jensen J, Tabatabaei A, Schmelzer K, et al. Characterization of highly efficacious allosteric agonists of the human calcium-sensing receptor. J Pharmacol Exp Ther. 2011;337:275–284. doi: 10.1124/jpet.110.178194. [DOI] [PubMed] [Google Scholar]
- Mace OJ, Schindler M, Patel S. The regulation of K- and L-cell activity by GLUT2 and the calcium-sensing receptor CaS receptor in rat small intestine. J Physiol. 2012;590:2917–2936. doi: 10.1113/jphysiol.2011.223800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod RJ. CaS receptor function in the intestine: hormone secretion, electrolyte absorption and secretion, paracrine non-canonical Wnt signaling and colonic crypt cell proliferation. Best Pract Res Clin Endocrinol Metab. 2013;27:385–402. doi: 10.1016/j.beem.2013.05.005. [DOI] [PubMed] [Google Scholar]
- Makita N, Sato J, Manaka K, Shoji Y, Oishi A, Hashimoto M, et al. An acquired hypocalciuric hypercalcemia autoantibody induces allosteric transition among active human Ca-sensing receptor conformations. Proc Natl Acad Sci U S A. 2007;104:5443–5448. doi: 10.1073/pnas.0701290104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamillapalli R, VanHouten J, Zawalich W, Wysolmerski J. Switching of G-protein usage by the calcium-sensing receptor reverses its effect on parathyroid hormone-related protein secretion in normal versus malignant breast cells. J Biol Chem. 2008;283:24435–24447. doi: 10.1074/jbc.M801738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie PJ. The calcium-sensing receptor in bone cells: a potential therapeutic target in osteoporosis. Bone. 2010;46:571–576. doi: 10.1016/j.bone.2009.07.082. [DOI] [PubMed] [Google Scholar]
- McGehee DS, Aldersberg M, Liu KP, Hsuing S, Heath MJ, Tamir H. Mechanism of extracellular Ca2+ receptor-stimulated hormone release from sheep thyroid parafollicular cells. J Physiol. 1997;502:31–44. doi: 10.1111/j.1469-7793.1997.031bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun HC, Brennan SC, Delbridge L, Wilkinson M, Brown EM, Conigrave AD. Adenomatous human parathyroid cells exhibit impaired sensitivity to L-amino acids. J Clin Endocrinol Metab. 2009;94:3567–3574. doi: 10.1210/jc.2008-2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth EF, Steffey ME, Hammerland LG, Hung BC, Van Wagenen BC, DelMar EG, et al. Calcimimetics with potent and selective activity on the parathyroid calcium receptor. Proc Natl Acad Sci U S A. 1998;95:4040–4045. doi: 10.1073/pnas.95.7.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth EF, Heaton WH, Miller M, Fox J, Balandrin MF, Van Wagenen BC, et al. Pharmacodynamics of the type II calcimimetic compound cinacalcet HCl. J Pharmacol Exp Ther. 2004;308:627–635. doi: 10.1124/jpet.103.057273. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn SJ, Kifor O, Kifor I, Butters RR, Jr, Brown EM. Role of the cytoskeleton in extracellular calcium-regulated PTH release. Biochem Biophys Res Commun. 2007;354:8–13. doi: 10.1016/j.bbrc.2006.12.160. [DOI] [PubMed] [Google Scholar]
- Rey O, Young SH, Yuan J, Slice L, Rozengurt E. Amino acid-stimulated Ca2+ oscillations produced by the Ca2+-sensing receptor are mediated by a phospholipase C/inositol 1,4,5-trisphosphate-independent pathway that requires G12, Rho, filamin-A, and the actin cytoskeleton. J Biol Chem. 2005;280:22875–22882. doi: 10.1074/jbc.M503455200. [DOI] [PubMed] [Google Scholar]
- Rey O, Young SH, Jacamo R, Moyer MP, Rozengurt E. Extracellular calcium sensing receptor stimulation in human colonic epithelial cells induces intracellular calcium oscillations and proliferation inhibition. J Cell Physiol. 2010;225:73–83. doi: 10.1002/jcp.22198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell J, Zhao W, Christ G, Ashok S, Angeletti RH. Ca2+-induced increases in steady-state concentrations of intracellular calcium are not required for inhibition of parathyroid hormone secretion. Mol Cell Biol Res Commun. 1999;1:221–226. doi: 10.1006/mcbr.1999.0135. [DOI] [PubMed] [Google Scholar]
- Singh N, Liu G, Chakrabarty S. Isolation and characterization of calcium sensing receptor null cells: a highly malignant and drug resistant phenotype of colon cancer. Int J Cancer. 2012;132:1996–2005. doi: 10.1002/ijc.27902. [DOI] [PubMed] [Google Scholar]
- Smajilovic S, Yano S, Jabbari R, Tfelt-Hansen J. The calcium-sensing receptor and calcimimetics in blood pressure modulation. Br J Pharmacol. 2011;164:884–893. doi: 10.1111/j.1476-5381.2011.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen AR, Hvidtfeldt M, Brauner-Osborne H. Biased agonism of the calcium-sensing receptor. Cell Calcium. 2012a;51:107–116. doi: 10.1016/j.ceca.2011.11.009. [DOI] [PubMed] [Google Scholar]
- Thomsen AR, Worm J, Jacobsen SE, Stahlhut M, Latta M, Brauner-Osborne H. Strontium is a biased agonist of the calcium-sensing receptor in rat medullary thyroid carcinoma 6–23 cells. J Pharmacol Exp Ther. 2012b;343:638–649. doi: 10.1124/jpet.112.197210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano S, Sugimoto T, Tsukamoto T, Chihara K, Kobayashi A, Kitazawa S, et al. Association of decreased calcium-sensing receptor expression with proliferation of parathyroid cells in secondary hyperparathyroidism. Kidney Int. 2000;58:1980–1986. doi: 10.1111/j.1523-1755.2000.00370.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Cinacalcet potentiation of Ca2+o-mediated signalling. (A) Ca2+o stimulation of IP1 accumulation, Ca2+i mobilization and pERK1/2 in the absence and presence of cinacalcet. (B) Non-linear regression analysis of Ca2+o potency in the absence and presence of cinacalcet with an allosteric ternary complex model (Equation 2) where the pKB is constrained between pathways or not (as indicated).
Figure S2 Calindol potentiation of Ca2+o-mediated signalling. (A) Ca2+o stimulation of IP1 accumulation, Ca2+i mobilization and pERK1/2 in the absence and presence of calindol. (B) Non-linear regression analysis of Ca2+o potency in the absence and presence of calindol with an allosteric ternary complex model (Equation 2) where the pKB is constrained between pathways or not (as indicated).
Figure S3 NPS-R568 potentiation of Ca2+o-mediated signalling. (A) Ca2+o stimulation of IP1 accumulation, Ca2+i mobilization and pERK1/2 in the absence and presence of NPS-R568. (B) Non-linear regression analysis of Ca2+o potency in the absence and presence of NPR-R568 with an allosteric ternary complex model (Equation 2) where the pKB is constrained between pathways or not (as indicated).
Figure S4 S,R-calcimimetic B potentiation of Ca2+o-mediated signalling. (A) Ca2+o stimulation of IP1 accumulation, Ca2+i mobilization and pERK1/2 in the absence and presence of S,R-calcimimetic B. (B) Non-linear regression analysis of Ca2+o potency in the absence and presence of S,R-calcimimetic B with an allosteric ternary complex model (Equation 2) where the pKB is constrained between pathways or not (as indicated).
Figure S5 R,R-calcimimetic B potentiation of Ca2+o-mediated signalling. (A) Ca2+o stimulation of IP1 accumulation, Ca2+i mobilization and pERK1/2 in the absence and presence of R,R-calcimimetic B. (B) Non-linear regression analysis of Ca2+o potency in the absence and presence of R,R-calcimimetic B with an allosteric ternary complex model (Equation 2) where the pKB is constrained between pathways or not (as indicated).
Figure S6 nor-calcimimetic B potentiation of Ca2+o-mediated signalling. (A) Ca2+o stimulation of IP1 accumulation, Ca2+i mobilization and pERK1/2 in the absence and presence of nor-calcimimetic B. (B) Non-linear regression analysis of Ca2+o potency in the absence and presence of nor-calcimimetic B with an allosteric ternary complex model (Equation 2) where the pKB is constrained between pathways or not (as indicated).
Figure S7 AC-265347 potentiation of Ca2+o-mediated signalling. (A) Ca2+o stimulation of IP1 accumulation, Ca2+i mobilization and pERK1/2 in the absence and presence of AC-265347. (B) Non-linear regression analysis of Ca2+o potency in the absence and presence of AC-265347 with an allosteric ternary complex model (Equation 2) where the pKB is constrained between pathways or not (as indicated).
Figure S8 Binding affinity and cooperativities of calcimimetics across different pathways. Cooperativities of calcimimetics in Ca2+i mobilization (white bars), pERK1/2 (grey bars) and IP1 accumulation (black bars) assays were determined with the binding affinity constrained to be the same in each assay.
Figure S9 Concentration–response curves to Ca2+o and calcimimetics in the presence of 1.8mM ambient (buffer) Mg2+o but no Ca2+o.
Figure S10 Validation of 9E10:AF647 antibody. (A) Coomassie stained gel imaged on Typhoon Trio (GE Life Sciences) showing purification of 9E10 antibody from biorector hybridoma supernatant. Molecular weight markers are indicated on the left and various purification fractions indicated above the gel. Densitometry on elution fractions 3–6 was performed using ImageJ and these fractions pooled for the subsequent conjugation reaction. Densitometry was consistent with antibody being >90% pure. Staining is consistent with the concentration as determined by A280. (B) Titration of 9E10:AF647 in Cos7 cells stably transfected with either vector control (Vector) or vector containing cMyc tagged calcitonin receptor (cMycCTR). Relative fluorescence intensity corresponds to antibody binding, determined on a FACS CantoII (BD Biosciences), as described in the Supporting Information Appendix S1: supplemental methods.
Table S1 Pharmacological parameters that govern the allosteric activity of CaS receptor modulators in Ca2+i mobilization, pERK1/2 and IP1 accumulation assays. The potency of Ca2+o in the presence of increasing concentrations of modulator was fitted to an allosteric ternary complex model (Equation 2) to quantify the equilibrium dissociation constant (pKB) and cooperativity (αβ) of the modulators at the human CaS receptor, using a model in which the binding affinity was assumed to be the same across pathways.
Appendix S1 Supplemental methods.