

Abstract
BACKGROUND AND PURPOSE
The aim of this study was to determine the potency and molecular mechanism of action of YM155, a first-in-class survivin inhibitor that is currently under phase I/II clinical investigations, in various drug-resistant breast cancers including the oestrogen receptor positive (ER+) tamoxifen-resistant breast cancer and the caspase-3-deficient breast cancer.
EXPERIMENTAL APPROACH
The potency of YM155 in SK-BR-3, MDA-MB-231, MCF7 and its tamoxifen-resistant sublines, TamR6, TamR7, TamR8, TamC3 and TamC6, were determined by MTT assay. Western blot analysis, flow cytometric analysis, reverse transcription-PCR, fluorescent microscopy and comet assay were used to determine the molecular mechanism of action of YM155 in different breast cancer cell lines.
KEY RESULTS
YM155 was equally potent towards the parental ER+/caspase-3-deficient MCF7 breast cancer cells and its tamoxifen-resistant sublines in vitro. The ER−/HER2+ SK-BR-3 breast cancer cells and the triple-negative/caspase-3-expressing metastatic aggressive MDA-MB-231 breast cancer cells were also sensitive to YM155 with IC50 values in the low nanomolar range. Targeting survivin by YM155 modulated autophagy, induced autophagy-dependent caspase-7 activation and autophagy-dependent DNA damage in breast cancer cells. Interestingly, YM155 also induced XIAP degradation and the degradation of XIAP might play an important role in YM155-induced autophagy in breast cancer cells.
CONCLUSIONS AND IMPLICATIONS
YM155 is a potent survivin inhibitor that has potential for the management of various breast cancer subtypes regardless of the expression of ER, HER2 and caspase-3. Importantly, this study provides new insights into YM155's molecular mechanism of action and therapeutic potential in the treatment of tamoxifen-resistant breast cancer.
Tables of Links
| TARGETS | |
|---|---|
| Steroid hormone receptorsa | Enzymesc |
| Oestrogen receptor (ER) | Caspase-3 |
| Progesterone receptor (PR) | Caspase-7 |
| Catalytic receptorsb | Caspase-8 |
| HER2 | Caspase-9 |
| LIGANDS | |
|---|---|
| Chloroquine | Rapamycin |
| Cycloheximide | Tamoxifen |
| Z-DEVD-FMK |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,b,c).
Introduction
Breast cancer ranks as the most common form of cancer among women. Traditionally, tamoxifen is given to patients with oestrogen receptor positive (ER+) breast cancer as an adjuvant treatment, whereas chemotherapy is given to patients with various breast cancer subtypes including the triple-negative [ER−, progesterone receptor negative (PR−), human EGF receptor 2 negative (HER2−)] breast cancer and the lymph node positive, ER+/HER2+ breast cancer. However, the ER+/tamoxifen-resistant breast cancer is frequently found in patients after prolonged tamoxifen treatment [Encarnacion et al., 1993; Ring and Dowsett, 2004; Early Breast Cancer Trialists' Collaborative Group (EBCTCG), 2005 ]. It has also been demonstrated that caspase-3 down-regulation, which is commonly found in breast cancer cells, affects the response of cancer cells to various chemotherapeutic agents (Devarajan et al., 2002). Thus, it is important to develop novel strategies to target different breast cancer subtypes. It is also of interest to develop a novel targeted therapy that is capable of targeting all breast cancer subtypes simultaneously, rather than just targeting a subpopulation of breast cancer.
Survivin is a member of the inhibitor-of-apoptosis proteins (IAPs) family. It is expressed in many different cancer cell types, but not in the differentiated normal tissue (Ambrosini et al., 1997). In regard to breast cancer, survivin was reported to be frequently overexpressed in invasive primary breast carcinoma (Li et al., 2012). In addition, patients with low survivin expression was shown to have a longer disease-free survival period as compare with patients with high survivin expression level (Yamashita et al., 2007). Therefore, survivin is considered as a promising molecular target for breast cancer therapy.
Sepantronium bromide (YM155) is the first-in-class survivin inhibitor (inhibits survivin gene transcription) (Nakahara et al., 2007; 2011a,b,,; Minematsu et al., 2009; Cheng et al., 2012). While the safety and efficacy of YM155 have already been evaluated in several phase I/II clinical studies (Tolcher et al., 2008; Giaccone et al., 2009; Satoh et al., 2009), its molecular mechanism of action is still unclear. It is also unclear whether this drug is applicable for patients with ER+/tamoxifen-resistant breast cancer. In this study, we found that YM155 is potent in various breast cancer subtypes including the ER+/tamoxifen-resistant breast cancer and the triple-negative metastatic aggressive breast cancer in vitro. Surprisingly, we found that YM155 modulates autophagy instead of caspase-3-dependent apoptosis, which is widely believed to be the function of YM155, and induces autophagy-dependent DNA damage and cell death in breast cancer cells. This study also revealed that down-regulation of X-linked inhibitor of apoptosis protein (XIAP) may play an important role in YM155-modulated autophagy in breast cancer cells.
Methods
Cell lines and cell culture conditions
A series of MCF7-derived ER+/tamoxifen-resistant breast cancer cell lines (TamR6, TamR7, TamR8, TamC3 and TamC6) were used in this study. The cellular and molecular phenotypes of these tamoxifen-resistant breast cancer cell lines have already been characterized in a previous study (Leung et al., 2010). Briefly, the ER+/tamoxifen-resistant TamR6, TamR7 and TamR8 breast cancer cells were created by continuous exposure of MCF7 breast cancer cells to tamoxifen; whereas TamC3 and TamC6 breast cancer cells were created by prolonged culture of MCF7 cells under oestrogen-depleted conditions. MCF7, TamR7 and TamR8 breast cancer cells were cultured in α-MEM containing 5% FBS, penicillin/streptomycin (10 000 u·mL−1 and 10 mg·mL−1, respectively), insulin-transferring selenium (ITS, Roche, Mannheim, Germany) and tamoxifen (5 μM). In contrast, TamC3, TamC6 and TamR6 breast cancer cells were cultured in phenol red-free RPMI containing 5% charcoal-stripped FBS, penicillin/streptomycin (10 000 unit·mL−1 and 10 mg·mL−1, respectively) and ITS (10 mg·L−1). The ER−/HER2+ SK-BR-3 breast cancer cells were cultured in DMEM containing 10% FBS and penicillin/streptomycin (10 000 u·mL−1 and 10 mg·mL−1 respectively). The triple-negative metastatic aggressive MDA-MB-231 breast cancer cells were cultured in RPMI containing 10% FBS and penicillin/streptomycin (10 000 u·mL−1 and 10 mg·mL−1 respectively). All cell lines were shown to be mycoplasma free.
MTT cell viability assay
Cells were seeded onto each well of 96-well plates for 24 h before being treated with either DMSO (control) or YM155 (Selleck, Houston, TX, USA) for 72 h. For combination treatment, cells were pretreated with either 3-methyladenine (3MA, Sigma, St. Louis, MO, USA), chloroquine (CQ, Sigma) or Z-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-fluoromethylketone (Z-DEVD-FMK, Millipore, Billerica, MA, USA) for an hour and then co-treated with YM155 for an additional 72 h. Cell viability was quantified by measuring the absorbance of the solution at 570 nm wavelength by a spectrophotometer. The percentage of viable cells for each treatment group was calculated by adjusting the untreated control group to 100%. Duplicate wells were assayed for each condition and repeated at least three times. The IC50 value resulting from 50% inhibition of cell viability was calculated graphically as a comparison with the growth of the control group.
Lactate dehydrogenase (LDH)-cytotoxicity assay
Breast cancer cells were seeded in each well of 96-well plate. Cells were treated with either DMSO (-ve control) or 2×IC50 YM155 with or without autophagy inhibitors CQ or bafilomycin A1 (BAF, Sigma) for 72 h. Cytotoxicity was determined by LDH-cytotoxicity assay kit II (Abcam, Cambridge, MA, USA) as per the manufacturer's instructions. Cell cytotoxicity was quantified by measuring the absorbance of the solution at 450 nm wavelength by a spectrophotometer. Duplicate wells were assayed for each condition and repeated at least three times. The cytotoxicity index for each treatment group was calculated using the equation (test sample − low control)/(high control − low control) and also by adjusting the untreated control group to 1. ‘Test sample’ represents cells with different treatments (DMSO, YM155 alone, YM155 co-treated with or without CQ or BAF); ‘low control’ represents untreated cells (minimal LDH-value); ‘high control’ represents cells in lysis solution (maximal LDH-value).
Reverse transcription-PCR (RT-PCR) and real-time PCR
Total RNA was extracted using TRIzol® reagent (Invitrogen, Grand Island, NY, USA) and cDNA was synthesized from RNA using the RevertAid H Minus First strand cDNA synthesis kit (Thermo Scientific, Waltham, MA, USA). Expression levels of X-linked inhibitor of apoptosis (XIAP), p62/SQSTM1 and actin transcript were determined by PCR. Quantitative real-time PCR was used to determine the relative expression levels of p62/SQSTM1 and GAPDH in cells treated with or without YM155. The specific primers with the following sequences were used in the study: human XIAP forward primer, 5-CAACACTGGCACGAGCAGGG; human XIAP reverse primer, 5-CATGGCAGGGTTCCTCGGGT; human β-actin forward primer, 5-TACCCCATCGAGCACGGCAT; human β-actin reverse primer, 5-GAAGCAGCCGTGGCCATCTC; human p62/SQSTM1 forward primer, 5-GCACCCCAATGTGATCTGC; human p62/SQSTM1 reverse primer, 5-CGCTACACAAGTCGTAGTCTGG; human p62/SQSTM1 forward primer for real-time PCR, 5-GGAACAGATGGAGTCGGATAAC; human p62/SQSTM1 reverse primer for real-time PCR, 5-TAGGGACTGGAGTTCACCTGTA; human GAPDH forward primer for real-time PCR, 5-GAGTCAACGGATTTGGTCGT; human GAPDH reverse primer for real-time PCR, 5-TTGATTTTGGAGGGATCTCG. Experiments were repeated at least three times.
Gene silencing by siRNA
Target-validated siRNA oligos were transfected into MCF7 and MDA-MB-231 breast cancer cells using Lipofectamine® RNAiMAX reagent (Invitrogen). Briefly, cells were seeded onto 10 cm dishes and cultured overnight in antibiotic-free media. Survivin-specific, XIAP-specific and LC3B-specific siRNA oligomers were purchased from Cell Signaling Technology (Danvers, MA, USA) (cat# 6351, cat# 6446, and cat# 6213). Scramble siRNA oligomers were purchased from Sigma (cat# SIC001). The siRNA oligomers (stock concentration 10 μM, 7.5 μL) were diluted in 100 μL of Opti-MEM® I medium (Invitrogen) without serum and then mixed with 7.5 μL of Lipofectamine RNAiMAX transfection reagent for 20 min at room temperature. Cells were overlaid with the transfection mixture and incubated for various durations.
Western blot analysis
Cells were lysed using Cellytic™ M cell lysis reagent (Sigma) that contained 1 mM PMSF, 1 mM NaF and protease inhibitors cocktail (Roche). Equal amounts of protein were subjected to SDS-PAGE on either a 10 or 12% polyacrylamide gel. The resolved proteins were transferred onto a PVDF membrane (Millipore), which was then exposed to 5% non-fat dried milk in TBS-Tween buffer for an hour at room temperature before incubation overnight at 4°C with primary antibodies. Mouse polyclonal antibodies against human phosphorylated histone H2AX and β-actin were obtained from Millipore (cat# 05-636 and cat# MAB1501); antibodies against caspase-3 were from Origene (Rockville, MD, USA) (cat# TA301776). Rabbit polyclonal antibodies against LC3B were obtained from Origene (cat# TA301543); antibodies against survivin were from R&D system (Minneapolis, MN, USA) (cat# AF886) and those against caspase-7 were from Millipore (cat# AB1999). The PVDF membrane was then washed three times with TBS containing 0.05% Tween-20 before incubation for an hour at room temperature with HRP-conjugated goat antibodies to rabbit (Millipore, cat# AP132P) or mouse (Millipore, cat# AP124P) IgG. Immune complexes were finally detected with chemiluminescence reagents (Millipore). Experiments were repeated at least three times.
Ectopic expression of EGFP-LC3B and detection of autophagosome formation
The autophagosome formation in the YM155-treated breast cancer cells was detected by the localization of exogenous LC3B fused to enhanced green fluorescent protein (EGFP-LC3B). Briefly, the transfection of EGFP-LC3B expression vector (Addgene, plasmid# 11546; Cambridge, MA, USA) was mediated through Lipofectamine LTX and PLUS™ reagent (Invitrogen) as per the manufacturer's protocol. After the indicated treatment, the localization of GFP was then directly observed by fluorescence microscopy and cells expressing multiple punctate EGFP-LC3B-labelled vacuoles were identified for quantification. ImageJ software (National Institutes of Health, USA) was used to count the number of the green fluorescent puncta present in cells.
Monodansylcadaverine (MDC) and LysoTracker® Red staining of acidic vesicular organelles (AVOs)
MDC staining was used to detect the formation of AVOs in the YM155-treated breast cancer cells. Briefly, cells were seeded on the 24-well plates and treated or transfected with transfection reagent alone (vehicle control), scramble siRNA oligos, survivin siRNA oligos or 2×IC50 YM155 for various durations. AVOs were labelled with 0.5 mM MDC in the phenol red-free RPMI at 37°C for 2 h. Then, the cells were washed three times with PBS. AVOs in all cells were observed under a fluorescence microscope (Olympus, IX-71; Tokyo, Japan). ImageJ software was used to calculate the average size of the green fluorescent puncta present in cells. For quantification, 10 000 MDC-stained cells were analysed on a FACSCalibur flow cytometer (excitation wavelength 463 nM, emission wavelength 534 nM) with CellQuest Pro program (BD Bioscience, San Jose, CA, USA). LysoTracker Red staining was also used to detect the formation of AVOs in cells. MCF7 cells were treated with 2×IC50 YM155 for 48 h or transfected with either scramble siRNA or XIAP-specific siRNA oligomers for 72 h. Cells were washed three times with PBS and then stained with LysoTracker Red. AVOs in cells were observed under a fluorescence microscope (Olympus, IX-71). Experiments were repeated at least three times.
Protein stability assay
To measure the rate of p62/SQSTM1 protein degradation, MCF7 cells were treated with DMSO (control), CQ, BAF, rapamycin or YM155 for 24 h and subsequently co-treated with the de novo protein synthesis inhibitor, cycloheximide (CHX, 10 μg·mL−1). Whole-cell extracts were prepared from samples taken at hourly intervals until 5 h post-CHX treatment and the amount of the p62/SQSTM1 protein present in cells was determined by Western blotting. The rate of protein degradation was in relative terms to the control group (0 h post-CHX treatment).
Comet assay
Microscopic slides were gently coated with 100 μL 1% normal melting point (NMP) agarose using a coverslip. The slide was placed on ice for 15 min to allow the agarose to set. After gelling, the coverslips were removed, 25 μL of the cell suspension (contains 105 cells) was gently mixed with 100 μL of 1.5% low melting point (37°C) agarose and pipetted onto the layer of 1% NMP agarose and covered with a coverslip. After 15 min on ice, the coverslips were removed and the slides were lowered into freshly made cold lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, 1% Triton X-100, pH 10) for 30 min. To allow DNA unwinding, the slides were placed into an electrophoresis chamber containing cold alkaline electrophoresis buffer (300 mM NaOH, 1 mM EDTA) for 20 min. Electrophoresis was performed by setting the power supply to 25 V and adjusting the current to 300 mA for 20 min. After electrophoresis, the slides were placed in a freshly made neutralizing buffer (0.4 M Tris, pH 7.5) for 20 min. Cell staining was performed with 10 mL per slide of propidium iodide (20 mg·L−1). The slides were examined with a fluorescence microscope (Nikon, Optiphot-2, Tokyo, Japan) at 20× magnification. Microscopic images of the comets were scored using TriTek CometScore™ Computer Software (Sumeduck, VA, USA). From each sample, one slide was prepared and the images of at least 50 cells from each slide were scored. The tail moment was chosen as our parameter. The major advantage of using the tail moment as an index of DNA damage is that both the amount of the damaged DNA and the distance of the genetic material migration in the tail are represented by a single number. Experiments were repeated at least three times.
Statistical analysis
Each experiment was performed at least three times. Data are presented as mean ± SEM. The significance of difference was evaluated with Student's unpaired two-tailed t-test. P < 0.05 was considered to be statistically significant.
Results
YM155 is effective in targeting various breast cancer subtypes regardless of the expression of ER, HER2 and caspase-3
To determine the effectiveness of the survivin inhibitor YM155 in targeting various types of breast cancer in vitro, IC50 values for YM155 were determined. In this study, the ER+/caspase-3-deficient MCF7 and its tamoxifen-resistant sublines (TamR6, TamR7, TamR8, TamC3 and TamC6) were used. As shown in Table 1, MTT cell viability assay revealed that the IC50 values of YM155 in TamR6, TamR7, TamR8, TamC3 and TamC6 were similar to that in the parental tamoxifen-sensitive MCF7 cells. YM155 was also effective in targeting the ER−/HER2+ SK-BR-3 and the triple-negative (ER−/HER2−/PR−) caspase-3-expressing MDA-MB-231 breast cancer cells at low nanomolar concentrations (Table 1). Taken together, our results revealed that YM155 is effective in reducing cell viability of various breast cancer subtypes regardless of the expression of ER, HER2 and caspase-3. Importantly, YM155 is also effective in reducing cell viability of the ER+/tamoxifen-resistant breast cancer cells.
Table 1.
Characteristics of different human breast cancer cell lines
| Cells | ER status | HER2 status | P53 status | Caspase-3 status | Tamoxifen sensitivity | YM155 IC50 (nM) |
|---|---|---|---|---|---|---|
| MCF7 | ER+ | HER2+ | Wild type | Deficient | Sensitive | 13 ± 6 |
| MCF7-TamR6 | ER+ | NA | Wild type | Deficient | Resistance | 8 ± 6 |
| MCF7-TamR7 | ER+ | HER2+ | Wild type | Deficient | Resistance | 8 ± 3 |
| MCF7-TamR8 | ER+ | HER2+ | Wild type | Deficient | Resistance | 15 ± 6 |
| MCF7-TamC3 | ER+ | HER2- | Wild type | Deficient | Resistance | 6 ± 3 |
| MCF7-TamC6 | ER+ | HER2− | Wild type | Deficient | Resistance | 6 ± 0.1 |
| MDA-MB-231 | ER− | HER2− | Mutant | Expressing | Resistance | 5 ± 1 |
| SK-BR-3 | ER− | HER2+ | Mutant | Expressing | Resistance | 7 ± 0.3 |
Cells were treated with YM155 for 3 days and cell viability was determined by the MTT assay. The IC50 values represent the average of at least five independent experiments.
YM155 induces LC3B-II conversion and caspase-7 activation in breast cancer cells
Because survivin plays important roles in apoptosis and may also modulate autophagy, we investigated whether YM155 induces apoptosis and modulates autophagy in breast cancer cells. Four different breast cancer cell lines, MCF7 (ER+/tamoxifen-sensitive), TamR7 (ER+/tamoxifen-resistant, created by prolonged tamoxifen treatment), TamC3 (ER+/tamoxifen-resistant, created by prolonged estrogen-deprivation) and MDA-MB-231 (ER−/HER2−/PR−/tamoxifen-resistant) were selected for the following molecular investigations. Because caspase-8/-9 and caspase-3/-7 are upstream apoptosis initiators and downstream apoptosis executioners, respectively, we sought to determine possible activation of these caspases in cells treated with YM155. Western blot analysis showed that MCF7 and its sublines (TamR7 and TamC3) used in this study do not express caspase-3, whereas MDA-MB-231 cells express caspase-3 (Figure 1A). Moreover, UV-irradiation was capable of inducing caspase-3 cleavage in MDA-MB-231 cells, demonstrating that MDA-MB-231 cells used in this study are capable of activating caspase-3 in response to pro-apoptotic stimuli (Figure 1B). In our study, YM155 decreased survivin expression in MCF7, TamR7, TamC3 and MDA-MB-231 cells in both concentration- and time-dependent manner as expected (Figure 1C and 1D). The same treatment also induced caspase-9 cleavage in the breast cancer cell lines tested (Figure 1D). Surprisingly, while survivin is believed to inhibit caspase-3 activation through both direct (physical interactions) and indirect (upstream caspase-9 inhibition) mechanisms, YM155 did not induce caspase-3 cleavage in the caspase-3-expressing MDA-MB-231 cells (Figure 1C and 1D). In addition, YM155-induced caspase-7 activation (caspase-7 cleavage) only in TamC3 cells and the activation of caspase-7 could be inhibited by co-incubation of the pan-caspase inhibitor, Z-DEVD-FMK (Figures 1C, 1D and 2A). LC3B-II is a widely used autophagosomal marker in mammalian cells (Kabeya et al., 2000). In contrast to the effect of YM155 on caspase-3 and caspase-7, YM155 increased the amount of LC3B-II present in MCF7, TamR7, TamC3 and MDA-MB-231 cells in both concentration- and time-dependent manner (Figure 1C and 1D). In addition, YM155-induced LC3B-II conversion could be attenuated by co-incubation of 3MA, a compound which is widely used to inhibit autophagy initiation by blocking the formation of autophagosomes (Levine et al., 2008), suggesting that YM155-induced LC3B-II conversion was caused by alteration of the autophagic process (Figure 2B).
Figure 1.
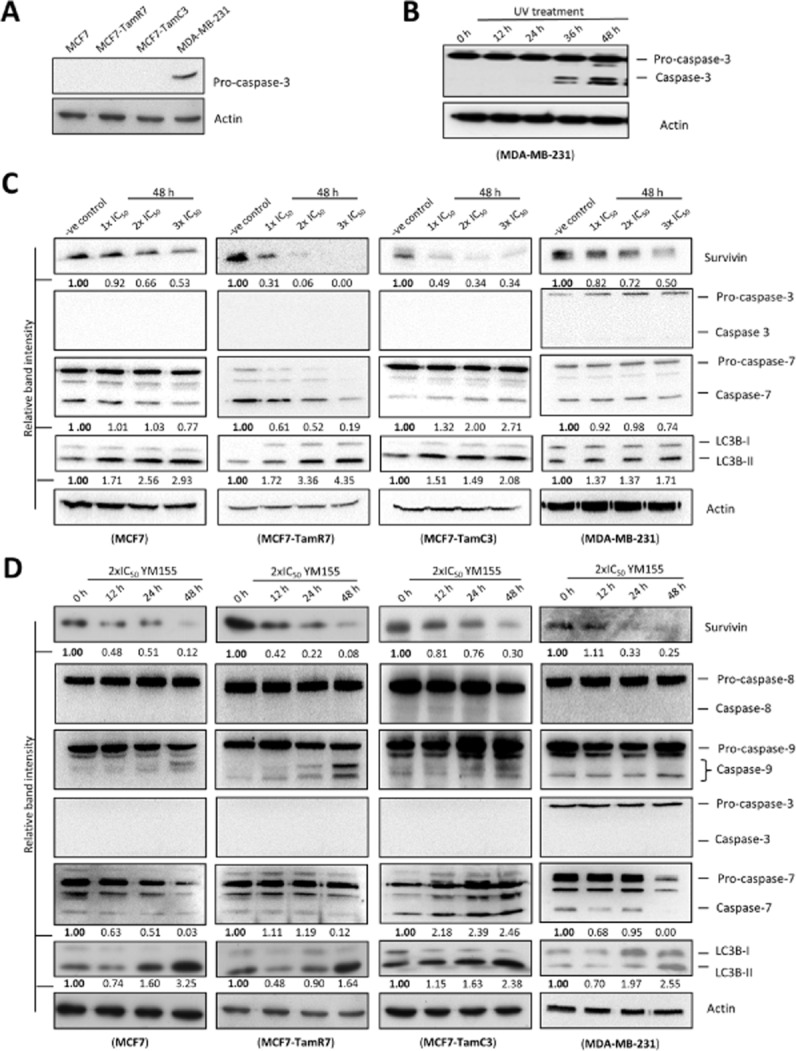
Targeting survivin by YM155 induces LC3B conversion in breast cancer cells. (A) Caspase-3 expression was analysed in MCF7, MCF7-TamR7, MCF7-TamC3 and MDA-MB-231 by Western blotting. Equal protein loading was verified by actin. (B) MDA-MB-231 cells were treated with UV (100 J·m−2) for various durations before being subjected to Western blot analysis with antibodies to caspase-3. Equal protein loading was verified by actin. (C) Breast cancer cells were treated with either DMSO (-ve control) or the indicated concentrations of YM155, and the expression of survivin, LC3B, caspase-3 and caspase-7 were examined by Western blotting. Equal protein loading was verified by actin. The numbers under each blot are intensity of the blot relative to that of the control (-ve control). (D) Breast cancer cells were treated with 2×IC50 of YM155 at the indicated time points. Survivin, LC3B, caspase-8, caspase-9, caspase-3 and caspase-7 expression were examined by Western blotting. Equal protein loading was verified by actin. The numbers under each blot are intensity of the blot relative to that of the control (0 h post-treatment).
Figure 2.
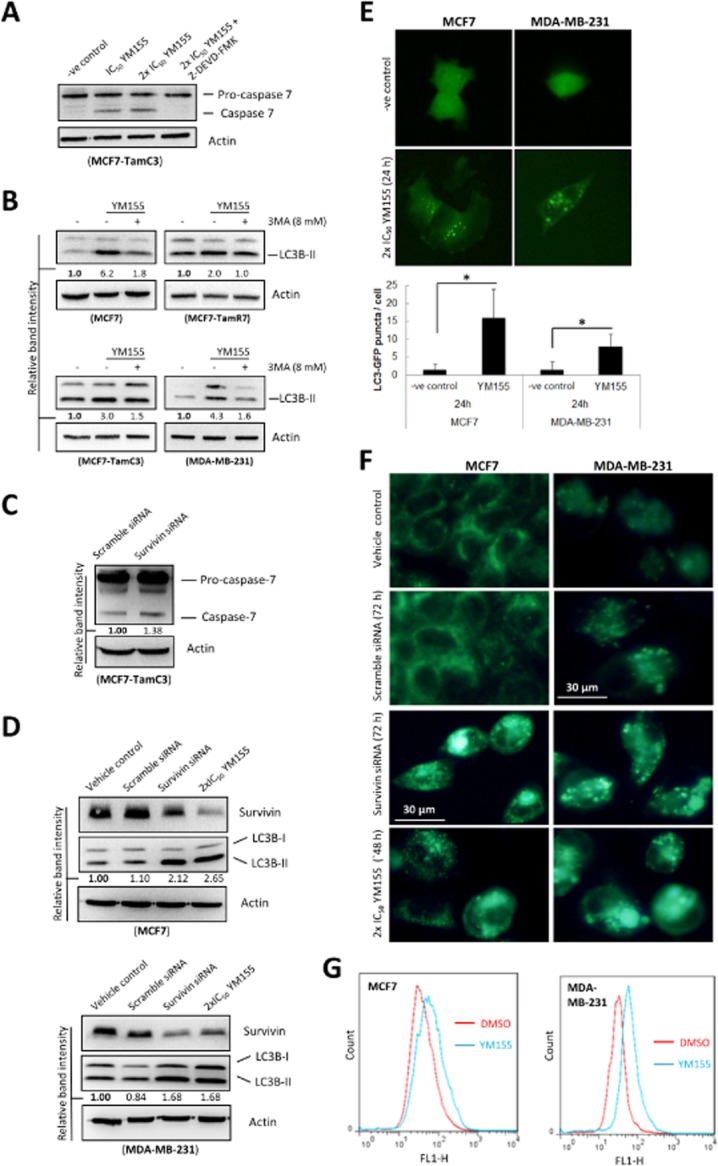
Targeting survivin by YM155 and siRNA induces caspase-7 activation and autophagic vesicle (AVO) formation in breast cancer cells. (A) MCF7-TamC3 cells were treated with either DMSO (-ve control) or YM155 co-treated with or without the pan-caspase inhibitor Z-DEVD-FMK. Caspase-7 expression was examined by Western blotting. Equal protein loading was verified by actin. (B) Breast cancer cells were treated with either DMSO (-ve control) or YM155 co-treated with or without the autophagy inhibitor 3MA. The expression of LC3B was examined by Western blotting. Equal protein loading was verified by actin. The numbers under each blot are intensity of the blot relative to that of the untreated control. (C) MCF7-TamC3 breast cancer cells were transfected with either scrambled siRNA or survivin-specific siRNA for 72 h. Cell lysates were then prepared and subjected to Western blot analysis with antibodies to the indicated proteins. The numbers under each blot are intensity of the blot relative to that of the control (scramble siRNA). (D) MCF7 and MDA-MB-231 breast cancer cells were treated with transfection reagent alone (vehicle control) or transfected with either scrambled siRNA or survivin-specific siRNA for 72 h. Cell lysates were then prepared and subjected to Western blot analysis with antibodies to the indicated proteins. The numbers under each blot are intensity of the blot relative to that of the control (vehicle control). (E) Breast cancer cells expressing the EGFP-tagged LC3B were treated with either DMSO (-ve control) or 2×IC50 YM155 for 24 h. Autophagosomes in cells were observed under a fluorescence microscope. The number of puncta present in cells was analysed by ImageJ software. (F) Breast cancer cells were treated with 2×IC50 YM155, transfection reagent alone (vehicle control) or transfected with either scramble siRNA or survivin-specific siRNA for the indicated durations. Cells were processed by MDC staining. AVOs in cells were observed under a fluorescence microscope. (G) Breast cancer cells were treated with either DMSO or 2×IC50 YM155 for 48 h and then processed by MDC staining. AVOs formation in cells was analyzed by flow cytometry. Red line represents cells treated with DMSO and blue line represents cells treated with 2xIC50 YM155.
To determine whether YM155-induced caspase-7 cleavage and LC3B-II conversion were indeed caused by survivin down-regulation instead of off-target drug effects, survivin-specific siRNA was used to down-regulate survivin expression in breast cancer cells. In consistent with the earlier results, down-regulation of survivin by siRNA induced caspase-7 cleavage and LC3B-II conversion in TamC3 and in all of the tested breast cancer cell lines respectively (Figure 2C and 2D).
YM155 increases the amount of autophagic vesicles and AVOs present in breast cancer cells
Next, we further confirm the effect of YM155 on autophagy modulation in breast cancer cells with ectopic expression of EGFP-tagged LC3B. Fluorescence microscopy revealed that EGFP-LC3B was overexpressed and equally distributed inside the transfected cells under drug-free conditions (Figure 2E, upper panels). In contrast, YM155 induced the formation of EGFP-LC3B punctate in both MCF7 and MDA-MB-231 cells, indicating the formation of autophagosome and/or autolysosome (Figure 2E, lower panels). YM155-treated cells were also stained with MDC to further confirm the formation of autophagic vesicles such as autolysosome. MDC is a fluorescent compound widely used for the detection of AVOs including lysosome and autolysosome (Niemann et al., 2000; Munafo and Colombo, 2001). Increased number and/or size of puncta formed in the MDC-stained cells are considered as the formation of AVOs. Fluorescence microscopy again revealed that YM155 increased both the number and the size of puncta present in the tested breast cancer cells (Figure 2F and Supporting Information Fig. S1). In consistent with the earlier results, down-regulation of survivin by siRNA also increased the amount of the fluorescent punctate dots present in the MDC-stained MCF7 and MDA-MB-231 cells, indicating that YM155-induced AVOs formation was likely caused by survivin down-regulation instead of off-target effects (Figure 2F). Formation of AVOs after YM155 treatment was also quantified by flow cytometry in the MDC-stained cells (Figure 2G). Increased MDC staining was observed in both MCF7 and MDA-MB-231 cells treated with YM155, further confirming that YM155 induced the formation of AVOs in cells. Importantly, down-regulation of LC3B by siRNA attenuated the effect of YM155 on the formation of AVOs in the MDC-stained MCF7 cells, suggesting that YM155-induced AVOs formation was likely caused by the increased formation of autophagic vesicles in the survivin-targeted breast cancer cells (Supporting Information Fig. S2).
YM155 induces biphasic changes in the expression of p62/SQSTM1
Accumulation of LC3B-II and autophagic vesicles/AVOs in the YM155-treated cells (as demonstrated by the Western blot analysis and fluorescence microscopy, respectively) could be caused by either autophagy induction or the blockage of autophagosome/autolysosome maturation and degradation (autophagy inhibition). Thus, it is important to measure autophagic flux as indicated by the expression levels of p62/SQSTM1 in the YM155-treated cells. Rapamycin is a well-known autophagy inducer, whereas CQ inhibits autophagy by inhibiting the acidification of autophagosome and maturation of autolysosome, resulting in the accumulation of autophagic vesicles and the inhibition of autolysosomal protein degradation. As shown in Figure 3A, rapamycin decreased the amount of p62/SQSTM1 protein present in MCF7 cells in a time-dependent manner. In contrast, CQ concurrently increased the amount of both LC3B-II and p62/SQSTM1 protein present in MCF7 cells as expected (Figure 3B). Interestingly, Western blot analysis revealed that YM155 induced biphasic changes in the expression of p62/SQSTM1 in MCF7, TamR7, TamC3 and MDA-MB-231 cells. While YM155 progressively decreased the amount of p62/SQSTM1 protein present in cells at early treatment stages (2, 4, 6 and 8 h), prolonged YM155 treatment (12, 24 and 48 h) induced p62/SQSTM1 protein accumulation in cells in a time-dependent manner (Figure 3C, 3D, 3E and 3F). Furthermore, survivin siRNA was used to confirm that targeting survivin can induce biphasic alterations in the expression of p62/SQSTM1 in breast cancer cells. In consistent with the earlier findings, down-regulation of survivin by siRNA also induced biphasic alterations on the expression of p62/SQSTM1 in MCF7 cells (Figure 3G).
Figure 3.
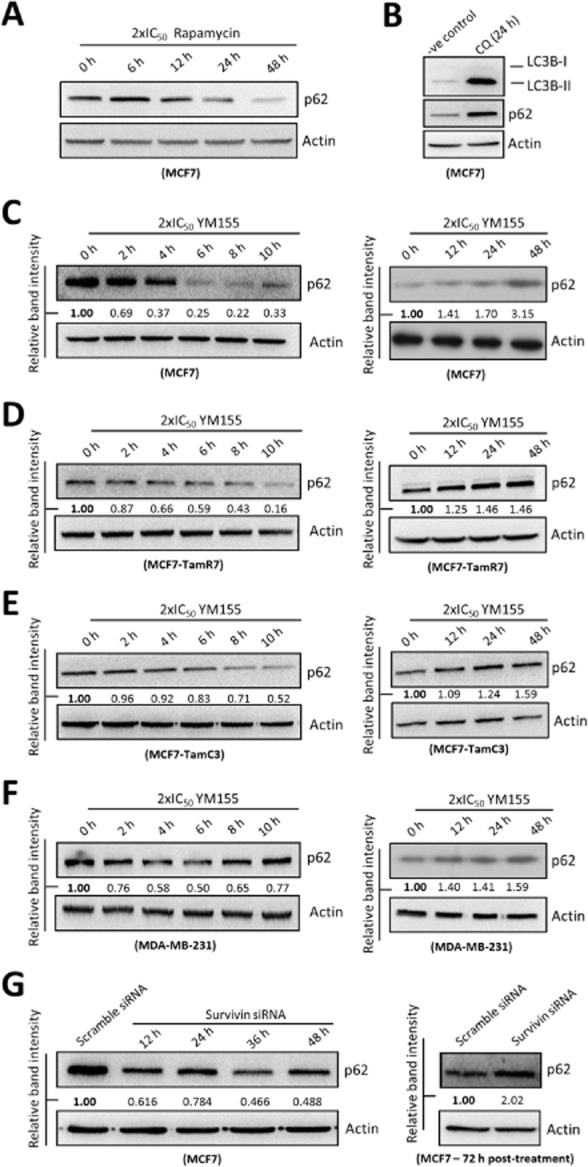
Targeting survivin by YM155 and siRNA induces biphasic expression of p62/SQSTM1 in breast cancer cells. (A) MCF7 cells were treated with 2×IC50 rapamycin for indicated durations. Expression of p62/SQSTM1 was examined by Western blotting. Equal protein loading was verified by actin. (B) MCF7 cells were treated with either DMSO (-ve control) or CQ for 24 h. Expression of p62/SQSTM1 and LC3B was examined by Western blotting. Equal protein loading was verified by actin. (C, D, E and F) Breast cancer cells were treated with 2×IC50 YM155 for indicated durations and expression of p62/SQSTM1 was examined by Western blotting. Equal protein loading was verified by actin. The numbers under each blot are intensity of the blot relative to that of the control (0 h post-treatment). (G) MCF7 cells were transfected with either scramble siRNA or survivin siRNA for indicated durations. Expression of p62/SQSTM1 was examined by Western blotting. Equal protein loading was verified by actin. The numbers under each blot are intensity of the blot relative to that of the control (scramble siRNA).
To determine whether the late phase p62/SQSTM1 protein accumulation in the YM155-treated cells was caused by autophagy inhibition, the rate of p62/SQSTM1 protein clearance/degradation was also investigated using Western blot analysis. Here, incubation of autophagosome/autolysosome maturation inhibitors, CQ and BAF, decreased the rate of p62/SQSTM1 protein degradation, whereas incubation of autophagy inducer, rapamycin, increased the rate of p62/SQSTM1 protein degradation in MCF7 cells as expected (Figure 4A). Surprisingly, while YM155 increased the total amount of p62/SQSTM1 protein present in MCF7 cells at 24 h post-treatment (Figure 3C, right panel), it increased the rate of p62/SQSTM1 protein degradation at the same time point (Figure 4A). In addition, co-incubation of CQ was capable of attenuating YM155-promoted p62/SQSTM1 protein degradation in MCF7 cells (Figure 4A). Interestingly, RT-PCR and quantitative RT-PCR analysis revealed that YM155 up-regulated p62/SQSTM1 gene transcription after 24 h in MCF7 cells (Figure 4B and 4C), indicating that the accumulation of p62/SQSTM1 protein after prolonged YM155 treatment might be caused by up-regulation of p62/SQSTM1 gene transcription instead of autophagy inhibition.
Figure 4.

YM155 concurrently promotes p62/SQSTM1 protein degradation and induces p62/SQSTM1 gene transcription in breast cancer cells. (A) MCF7 cells were treated with DMSO (-ve control), chloroquine, bafilomycin A1, rapamycin, YM155 or YM155 combined with chloroquine for 24 h and subsequently co-treated with the de novo protein synthesis inhibitor, CHX. The rate of p62/SQSTM1 protein degradation in MCF7 cells was examined by Western blotting. Actin was used as an internal control. The numbers under each blot are intensity of the blot relative to that of the control (0 h post-CHX treatment). (B) MCF7 cells were treated either with DMSO (-ve control) or 2×IC50 YM155. Expression of p62/SQSTM1 was examined by RT-PCR. Equal sample loading was verified by actin. The numbers under each band are intensity of the band relative to that of the control (-ve control). (C) MCF7 cells were treated either with DMSO (-ve control) or 2×IC50 YM155. Expression of p62/SQSTM1 was examined by quantitative real-time PCR.
YM155 down-regulates XIAP expression in breast cancer cells
The molecular mechanism by which YM155 or survivin modulates autophagy in cancer cells was seldom investigated. It is widely demonstrated that the protein stability of survivin and XIAP, which is another member of the IAPs family, is inter-regulated in cancer cells. Survivin physically binds to XIAP and formation of the survivin-XIAP complex protects XIAP against protein ubiquitination and proteasomal destruction (Dohi et al., 2004). Interestingly, a recent study reveals that XIAP is capable of inhibiting autophagy and promotes tumourigenesis of colon cancer cells. The same study also reveals that prolonged XIAP inhibition increases p62/SQSTM1 expression level (Huang et al., 2013b). Because survivin stabilizes XIAP and targeting XIAP has been shown to concurrently induce autophagy and increases p62/SQSTM1 expression in colon cancer cells, YM155 might be capable of inducing a survivin-dependent XIAP down-regulation and the reduced XIAP expression might play an important role in YM155-modulated autophagy in breast cancer cells. Thus, investigations were carried out to determine whether YM155 is capable of reducing the amount of XIAP protein present in breast cancer cells. Cells were treated with YM155 and the expression of XIAP was determined by the Western blot analysis and by using RT-PCR. As shown in Figure 5A and 5B, YM155 decreased the amount of XIAP protein present in MCF7, TamR7, TamC3 and MDA-MB-231 cells through a transcription-independent mechanism. To reconfirm that the YM155-induced XIAP reduction was caused by survivin down-regulation instead of off-target effects in breast cancer cells, survivin-specific siRNA was applied. In consistent with the earlier findings, targeting survivin by siRNA also decreased the amount of XIAP protein present in MCF7 and MDA-MB-231 breast cancer cells (Figure 5C). In contrast, ectopic expression of the GFP-tagged survivin increased the amount of XIAP present in MDA-MB-231 cells (Supporting Information Fig. S3). These results reconfirmed that XIAP expression can be regulated by survivin in breast cancer cells and suggested that YM155 decreases XIAP expression possibly through survivin down-regulation.
Figure 5.

YM155 reduces the expression of XIAP in breast cancer cells. (A) Breast cancer cells were treated with either DMSO (-ve control) or the indicated concentrations of YM155 for 48 h. Expression of XIAP was examined by Western blotting. Equal protein loading was verified by actin. (B) Breast cancer cells were treated with either DMSO (-ve control) or the indicated concentrations of YM155 for 48 h. Amount of XIAP transcript present in cells was determined by RT-PCR. Equal DNA loading was verified by actin. The numbers under each band are intensity of the band relative to that of the control (-ve control). (C) MCF7 and MDA-MB-231 breast cancer cells were transfected with either scramble siRNA or survivin-specific siRNA for 72 h or treated with YM155 for 48 h. Expression of various proteins were determined by Western blot analysis. Equal protein loading was verified by actin. (D) Breast cancer cells were treated with either scramble siRNA or XIAP siRNA for 72 h. Expression of various proteins was determined by Western blot analysis. Equal protein loading was verified by actin. The numbers under each blot are intensity of the blot relative to that of the control (scramble siRNA). (E) MCF7 cells were transfected with either scramble siRNA or XIAP siRNA for 72 h or treated with 2×IC50 YM155 for 48 h. Cells were stained with LysoTracker Red and formation of AVOs (red puncta) was determined by fluorescent microscopy. The nucleus was counterstained blue with Hoechst 33342. (F) MCF7 cells were transfected with scramble siRNA, survivin siRNA or XIAP siRNA for 72 h. Expression of various proteins was determined by the Western blot analysis. Equal protein loading was verified by actin. The numbers under each blot are intensity of the blot relative to that of the control (scramble siRNA). (G) MCF7 cells were treated with DMSO (-ve control), YM155 alone, embelin alone or YM155-embelin combinations for 48 h. Expression of LC3B was determined by the Western blot analysis. Equal protein loading was verified by actin.
As XIAP has been shown to be capable of inhibiting autophagy in colon cancer cells (Huang et al., 2013b), siRNA was applied to down-regulate XIAP to determine whether XIAP down-regulations could induce autophagy in breast cancer cells. Targeting XIAP by siRNA clearly induced the conversion of LC3B-II and AVOs accumulation in MCF7 and MDA-MB-231 cells (Figure 5D and 5E). Moreover, the expression of survivin was slightly decreased in the treated cells (Figure 5D). In consistent with Huang et al.'s findings, targeting XIAP by siRNA also increased the expression of beclin-1 and p62/SQSTM1 in MCF7 cells (Figure 5F). Western blot analysis also revealed that co-incubation with a XIAP pharmacological inhibitor, embelin, further increased LC3B-II conversion in the YM155-treated MCF7 cells (Figure 5G). Taken together, these results indicate that YM155 induces survivin and XIAP down-regulation and suggest that YM155 may induce autophagy in breast cancer cells through a survivin-XIAP-dependent mechanism.
Targeting survivin by YM155 induces autophagy-dependent DNA damage
A few studies suggested that YM155 is a survivin-independent DNA damaging agent. Interestingly, our previous study revealed that autophagy induction can induce DNA damage in cancer cells. Therefore, we hypothesized that YM155 could induce DNA damage through autophagy alterations in breast cancer cells. Western blot analysis revealed that YM155 increased the expression of γ-H2AX, a marker of DNA damage, in MCF7, TamR7, TamC3 and MDA-MB-231 breast cancer cells in a time-dependent manner (Figure 6A). To determine whether the YM155-induced γ-H2AX expression was caused by survivin down-regulation instead of off-target effects in breast cancer cells, survivin-specific siRNA was again applied. In consistent with the earlier results, targeting survivin by siRNA also increased the expression of γ-H2AX in the tested breast cancer cell lines (Figure 6B). Importantly, ectopic expression of survivin attenuated YM155's effect on the expression of γ-H2AX in MDA-MB-231 cells, indicating that YM155 induces DNA damage through a survivin-dependent mechanism (Figure 6C).
Figure 6.

YM155 induces autophagy-dependent DNA damage. (A) Breast cancer cells were treated with 2×IC50 YM155 for the indicated durations. Expression of survivin and γH2AX was examined by Western blotting. Equal protein loading was verified by actin. The numbers under each blot are intensity of the blot relative to that of the control (0 h post-treatment). (B) MCF7 and MDA-MB-231 breast cancer cells were treated with transfection reagent alone (vehicle control) or transfected with either scrambled siRNA or survivin siRNA for 72 h. Cell lysates were then prepared and subjected to Western blot analysis with antibodies to the indicated proteins. The numbers under each blot are intensity of the blot relative to that of the control (vehicle control). (C) YM155-treated MDA-MB-231 cells were transfected with either the control plasmid (pCMV6 empty vector) or pCMV6-survivin that overexpresses survivin (O/E survivin) for 72 h. Expression of γH2AX and survivin was examined by Western blotting. Equal protein loading was verified by actin. (D) Breast cancer cells were treated with either DMSO (control) or 2×IC50 YM155 with or without 3MA (8 mM) for 48 h. Expression of γH2AX was examined by Western blotting. Equal protein loading was verified by actin. The numbers under each blot are intensity of the blot relative to that of the cells treated with YM155 alone. (E) Cells were treated with either DMSO (control) or 2xIC50 YM155 with or without 3MA (8 mM) for 48 h. DNA damage was detected using comet assay. A statistically significant difference in the relative tail moment of cells treated with YM155 versus YM155 + 3MA is denoted by an asterisk; *P < 0.05. (F) Cells were treated with DMSO (control) or CQ for 48 h. Expression of LC3B-II was examined by Western blotting. Equal protein loading was verified by actin. DNA damage was detected using comet assay. (G) Breast cancer cells were treated with either DMSO (control) or 2×IC50 YM155 with or without BAF for 48 h. Expression of survivin and γH2AX was examined by Western blotting. Equal protein loading was verified by actin. (H) MDA-MB-231 cells were treated with either scramble siRNA or LC3B siRNA for 72 h or pretransfected with scrambled siRNA, LC3B siRNA for 24 h and subsequently co-treated with 2×IC50 YM155 for 48 h. Cell lysates were then prepared and subjected to Western blot analysis with antibodies to the indicated proteins. Equal protein loading was verified by actin.
Next, we sought to determine whether the increase in γ-H2AX expression was a consequence of autophagy induction. Western blot analysis revealed that co-incubation with 3MA partially attenuated (≥50%) the YM155-induced γ-H2AX expression in MCF7, TamR7, TamC3 and MDA-MB-231 cells (Figure 6D). Comet assay was used as a second tool to visualize possible induction of DNA damage in the YM155-treated breast cancer cells. It was also used to determine whether the induced DNA damage was a consequence of autophagy alterations. Cells with increased DNA damage display increased migration of chromosomal DNA from the nucleus towards the anode, which resembles the shape of a comet (Speit and Hartmann, 2006). As shown in Figure 6E and 6F, YM155-treated cells showed increased amount of DNA in the comet tail, while this phenomenon was partially reversed (50–80%) by the co-incubation of autophagy inhibitors, 3MA and CQ in breast cancer cells. Importantly, co-incubation with another autophagy inhibitor, BAF and co-treatment with LC3B siRNA, also partially attenuated the effect of YM155 on the expression of γ-H2AX in MCF7 and MDA-MB-231 cells (Figure 6G, 6H and Supporting Information Fig. S4). These findings indicate that targeting survivin by YM155 induces DNA damage partially in an autophagy-dependent manner.
Because we found that YM155 induces XIAP down-regulation and XIAP down-regulation induces autophagy in breast cancer cells, we suspected that XIAP might also play a role in the YM155-modulated autophagy-induced DNA damage in cells. Western blot analysis revealed that targeting XIAP by either pharmacological inhibitor (embelin) or siRNA also increased the expression of γ-H2AX in MDA-MB-231 cells (Figure 7A and 7B). Moreover, co-treatment with the XIAP inhibitor, embelin, further increased the expression of γ-H2AX in the YM155-treated MCF7 cells (Figure 7C). These results indicate that YM155 may induce autophagy-dependent DNA damage through a survivin-XIAP-dependent mechanism.
Figure 7.
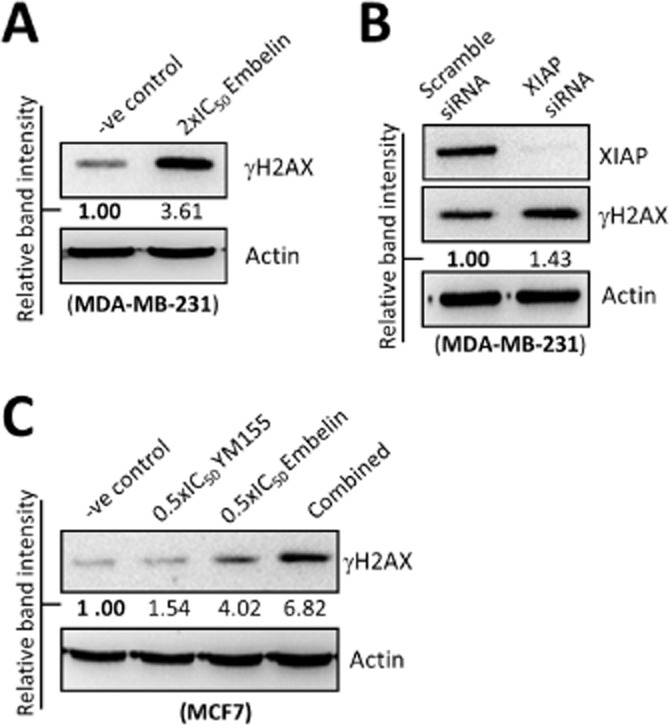
Targeting XIAP induces DNA damage in breast cancer cells. (A) MDA-MB-231 cells were treated with DMSO (-ve control) or 2×IC50 embelin for 48 h. Expression of γH2AX was examined by Western blotting. Equal protein loading was verified by actin. (B) MDA-MB-231 cells were transfected with either scramble siRNA or XIAP siRNA for 72 h. Expression of XIAP and γH2AX was examined by Western blotting. Equal protein loading was verified by actin. (C) MCF7 cells were treated with DMSO (-ve control), embelin or YM155 with or without embelin for 48 h. Expression of γH2AX was examined by Western blotting. Equal protein loading was verified by actin.
Targeting survivin by YM155 induces autophagic cell death in breast cancer cells
Autophagic dysfunction is associated with cancer, neurodegeneration, microbial infection and ageing. Paradoxically, although autophagy is primarily a protective process for the cell, it can also play a role in cell death (Mizushima et al., 2008). Interestingly, YM155 induces autophagy-dependent DNA damage, suggesting that YM155-modulated autophagy may promote cell death instead of cell survival in breast cancer cells. To determine whether the YM155-induced autophagy promotes cell death or cell survival, MTT assay was used to detect the viability of cells treated with YM155 alone or in combination with either 3MA, CQ or Z-DEVD-FMK. It is widely demonstrated that cells can induce prosurvival autophagy in response to serum starvations (Korkmaz et al., 2013). Here, Western blot analysis revealed that serum starvations induced LC3B-II conversion in MDA-MB-231 cells as expected (Figure 8A). In addition, co-treatment with either 3MA (autophagosome formation inhibitor) or CQ (autolysosome maturation inhibitor) significantly decreased the viability of MDA-MB-231 cells under serum starvations, indirectly reconfirming that both 3MA and CQ functioned as autophagy inhibitors not only at the molecular level, but also at the cellular level at our selected concentrations and treatment durations (Figure 8B). The MTT cell viability assay revealed that YM155 reduced cell viability of MCF7, TamR7, TamC3 and MDA-MB-231 cells in a concentration-dependent manner as expected (Figure 8C). Co-incubation with either 3MA or CQ was effective in inhibiting the YM155-induced loss of cell viability in MCF7, TamR7, TamC3 and MDA-MB-231 breast cancer cells (Figure 8C and 8D). In contrast, co-incubation with the autophagy inducer, rapamycin, potentiated the YM155-induced viability reduction in the tested breast cancer cells (Figure 8E). MTT cell viability assay also revealed that co-incubation with the pan-caspase inhibitor Z-DEVD-FMK partially attenuated the reduction of cell viability induced by YM155 only in TamC3 cells but not in MCF7, TamR7 and MDA-MB-231 cells (Figure 8C). These results complement with the above-mentioned findings that YM155 induces caspase-7 activation only in TamC3 cells and do not activate caspase-3 in breast cancer cells including the caspase-3-expressing MDA-MB-231 cells.
Figure 8.

YM155 induces autophagic cell death in breast cancer cells. (A) MDA-MB-231 cells were cultured in RPMI containing either 10 or 1% FBS for 48 h. Expression of LC3B was examined by Western blotting. Equal protein loading was verified by actin. (B) MDA-MB-231 cells were cultured in RPMI containing 1% FBS and treated with either 3MA or CQ for 72 h. Cell viability was determined by the MTT assay. A statistically significant difference in the viability of cells treated with 1% FBS versus 1%FBS + 3MA/CQ is denoted by an asterisk; *P < 0.05. (C) Breast cancer cells were treated with the indicated concentrations of YM155 with or without 3MA or Z-DEVD-FMK for 72 h. Cell viability was determined by the MTT assay. (D) Breast cancer cells were treated with either DMSO (control) or 2×IC50 YM155 with or without CQ for 72 h. Cell viability was assessed by MTT assay. A statistically significant difference in the viability of cells treated with YM155 versus YM155 + CQ is denoted, *P < 0.05. (E) MDA-MB-231 cells were treated with DMSO (control), rapamycin or YM155 with or without rapamycin for 72 h. Cell viability was assessed by MTT assay. A statistically significant difference in the viability of cells treated with YM155 + rapamycin versus YM155 or rapamycin alone is denoted, *P < 0.05. (F) MDA-MB-231 cells were treated with either DMSO (-ve control) or 2×IC50 YM155 with or without CQ or BAF. Cytotoxicity was determined by LDH-cytotoxicity assay.
Besides cell viability as measured by the MTT assay, LDH-cytotoxicity assay was also used to determine whether inhibiting autophagy could actually block the breast cancer cell death induced by YM155. LDH-cytotoxicity assay is widely used to detect the release of LDH into the surrounding medium upon cell damage or lysis. The triple-negative caspase-3-expressing MDA-MB-231 cells were used as a model. Here, YM155 induced cell death in MDA-MB-231 breast cancer cells as expected. Co-incubation with the autophagy inhibitor, CQ or BAF, significantly attenuated the release of LDH from the YM155-treated cells, further supporting that YM155-modulated autophagy plays an important role in promoting cell death in breast cancer cells (Figure 8F).
Targeting survivin by YM155 induces autophagy-dependent caspase-7 cleavage
Because both 3MA and Z-DEVD-FMK could attenuate the effect (∼80%) of YM155 (15 nM, approx. 2×IC50) on cell viability in TamC3 cells, we suspected that caspase-7 might be partially activated by YM155 in an autophagy-dependent manner. Western blot analysis revealed that co-incubation with autophagy inhibitors, 3MA, CQ and BAF, inhibited YM155-induced caspase-7 cleavage (Figure 9A). Importantly, co-treatment with LC3B siRNA also attenuated YM155-induced caspase-7 cleavage in TamC3 cells (Figure 9B). In contrast, co-incubation with the autophagy inducer, rapamycin, further increased caspase-7 cleavage in the YM155-treated cells (Figure 9C). Taken together, these results indicate that YM155-modulated autophagy induces caspase-7 cleavage and the activation of caspase-7 may play an important role in reducing the cell viability of TamC3 cells.
Figure 9.
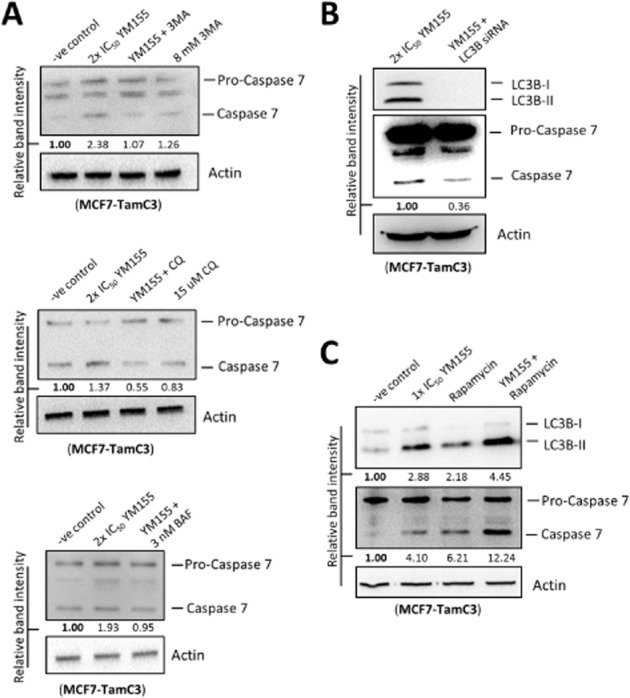
YM155 induces autophagy-dependent caspase-7 activation in MCF7-TamC3 cells. (A) MCF7-TamC3 cells were treated with either DMSO (-ve control) or 2×IC50 YM155 with or without 3MA, CQ, BAF or rapamycin for 48 h. Expression of caspase-7 was examined by Western blotting. Equal protein loading was verified by actin. The numbers under each blot are intensity of the blot relative to that of the control (-ve control). (B) MCF7-TamC3 cells were treated with YM155 with or without LC3B siRNA co-transfection. Expression of caspase-7 was examined by Western blotting. Equal protein loading was verified by actin. The numbers under each blot are intensity of the blot relative to that of the control (YM155 alone). (C) MCF7-TamC3 cells were treated with either DMSO (-ve control), 1×IC50 YM155, rapamycin, or YM155 with rapamycin for 48 h. Expression of LC3B and caspase-7 was examined by Western blotting. Equal protein loading was verified by actin. The numbers under each blot are intensity of blot relative to that of the control (-ve control).
Discussion and conclusions
YM155 is the most advanced survivin small molecule inhibitor developed for cancer treatment and its target specificity and efficacy have been demonstrated and evaluated in various preclinical studies and phase I/II clinical trials respectively (Nakahara et al., 2007; Lewis et al., 2011; Cheson et al., 2012; Tolcher et al., 2012). However, the molecular mechanism of action of YM155 still remains controversial. In addition, it is unclear on whether this drug is applicable for patients with ER+/tamoxifen-resistant breast cancer. In this study, we found that YM155 is equally potent towards the parental ER+/caspase-3-deficient/tamoxifen-sensitive MCF7 breast cancer cells and its tamoxifen-resistant sublines in vitro. In addition, the ER−/HER2+ SK-BR-3 breast cancer cells and the triple-negative/caspase-3-expressing metastatic aggressive MDA-MB-231 breast cancer cells are also susceptible to YM155, suggesting that YM155 is a promising anticancer compound that has potential for the management of various breast cancer subtypes regardless of the expression of ER, HER2 and caspase-3. Moreover, we found that YM155 modulates autophagy instead of caspase-3-dependent apoptosis and induces autophagy-dependent DNA damage in breast cancer cells (Figure 10). Importantly, targeting survivin by siRNA induces similar cellular and molecular changes as compared with the YM155 treatment in breast cancer cells; further supporting that survivin depletion plays an important role in YM155-induced breast cancer cell death.
Figure 10.
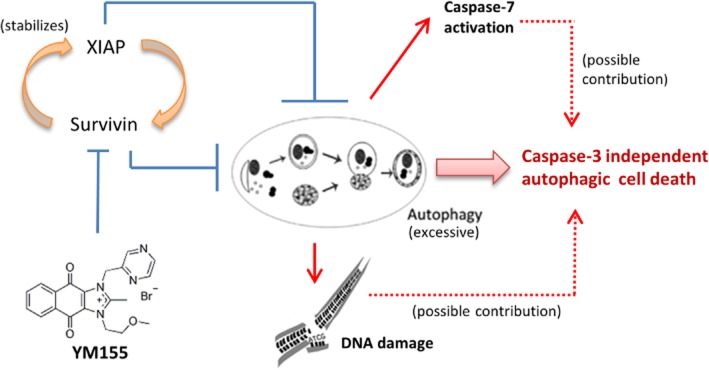
Proposed mechanism of action of YM155 in human breast cancer cells.
Because survivin inhibits apoptosis through both direct and indirect inhibitions of caspase-3, most studies on YM155 were carried out to determine whether YM155 is capable of down-regulating survivin expression and inducing caspase-9/-3 activation in cancer cells (Nakahara et al., 2007; Tao et al., 2012). However, our study reveals that YM155 modulates autophagy and induces autophagy-dependent but caspase-3-independent cell death in breast cancer cells. Autophagy is a double-edged sword. It has been shown that autophagy promotes cell survival in cells under genotoxic stress, metabolic stress and energy starvation (Ogata et al., 2006; Qiang et al., 2013). Alternatively, excessive autophagy may reduce cell viability by promoting autophagic cell death (Baehrecke, 2005). In this study, targeting survivin by YM155 increased the conversion of LC3B-II and the amount of autophagic vesicles present in cells. Moreover, co-incubation with different autophagy inhibitors, 3MA, CQ and BAF, attenuated the effect of YM155 on cell viability in MCF7, TamR7, TamC3 and MDA-MB-231 cells, whereas co-incubation with the well-known autophagy inducer, rapamycin, potentiated the effect of YM155, suggesting that YM155 induces autophagy and promotes autophagy-related cell death in breast cancer cells. Indeed, a recent study by Wang et al. shows that YM155 concurrently increases caspase-3 activity and induces autophagy-dependent apoptosis in prostate cancer cells (Wang et al., 2011), supporting that autophagy alteration plays an important role in YM155-induced cancer cell death. However, caspase-3 activation was undetected in the caspase-3-expressing MDA-MB-231 cells treated with YM155 in our study. While co-incubation of the pan-caspase inhibitor, Z-DEVD-FMK (60 uM), was effective in attenuating UV-induced (a pro-apoptotic stimulus) cell death in MDA-MB-231 breast cancer cells (Supporting Information Fig. S5), the same treatment was ineffective in inhibiting YM155-induced cell death in MCF7 (caspase-3-deficient), TamR7 (caspase-3-deficient) and MDA-MB-231 (caspase-3-expressing) cells, suggesting that caspase-3 activation is not a definite prerequisite event for YM155-induced breast cancer cell death. Caspase-7 is seldom demonstrated to be activated by autophagy in the past. In our study, the activation of caspase-7 in YM155-treated TamC3 cells seems to be autophagy-dependent, further supporting the importance of autophagy alteration in YM155-induced breast cancer cell death. However, while co-incubation of the pan-caspase inhibitor, Z-DEVD-FMK, partially attenuates the effect of YM155 on the cell viability of TamC3 cells, further investigations are required to determine whether caspase-7 activation actually plays an important role in YM155-induced TamC3 cell death.
Under normal circumstances, increased autophagosome formation, which is characterized by increases in LC3B-II conversion, occurs during autophagy induction in cells. In addition, increased autophagosome formation increases autophagic flux, which is characterized by increases in rate of p62/SQSTM1 protein degradation. At the molecular level, p62/SQSTM1 acts as an adaptor between autophagic machinery and ubiquitinated proteins. Structurally, it contains an ubiquitin-associated domain (UBA), a LC3 interacting region (LIR) and two domains for oligomerization and polymerization (Puissant et al., 2012). It binds to ubiquitinated proteins and LC3B-II through its UBA domain and LIR region, respectively, and the interaction of p62/SQSTM1 with LC3B-II is required for the autophagy-mediated elimination of unfolded ubiquitinated long-life proteins. Following the fusion of lysosomes with autophagosomes to become autolysosomes, ubiquitinated proteins together with p62/SQSTM1 are degraded (Puissant et al., 2012). Thus, p62/SQSTM1 is a widely used marker of the autophagic flux. Surprisingly, YM155 induces biphasic p62/SQSTM1 expression (early phase p62/SQSTM1 degradation and late phase p62/SQSTM1 induction) in breast cancer cells. There are two possible explanations for this observation: (i) YM155 initially induces autophagy in breast cancer cells but inhibits the late stage of autophagy after prolonged treatment. The result is that autophagy continues in a futile cycle and the cell will not be able to recycle proteins. In this situation, p62/SQSTM1 and LC3B will continue to be made and will not turn over in the lysosome. Overproduction of autophagosomes can start to compromise cell physiology and lead to cell death (Yu et al., 2004). Thus, blocking the formation of autophagosome by either 3MA or CQ rescued the YM155-treated breast cancer cells. (ii) YM155 induces breast cancer cell autophagy. However, YM155 also induces p62/SQSTM1 gene transcription after prolonged treatment. Thus, p62/SQSTM1 degradation has not been observed in the YM155-induced autophagic cancer cells after 12 h. Interestingly, our study reveals that YM155 still promotes p62/SQSTM1 protein degradation but progressively up-regulates p62/SQSTM1 gene transcription after 24 h. A study by Jain et al. showed that the expression of p62/SQSTM1 can be up-regulated by NF-E2-related factor 2 in cells in response to oxidative stress (Jain et al., 2010). YM155-modulated autophagy may induce reactive oxygen species (ROS) production and create oxidative stress in cells, leading to the concurrent up-regulation of p62/SQSTM1 gene transcription and induction of DNA damage after prolonged treatment.
The autophagic function of survivin in cancer cells is unclear. Therefore, the molecular mechanism of YM155 on autophagy modulation or induction in cancer cells is seldom investigated in the past. Our study reveals that YM155 reduces XIAP expression in MCF7, TamR7, TamC3 and MDA-MB-231 breast cancer cells through a transcription-independent mechanism. In fact, YM155 also concurrently reduces XIAP expression and increases LC3B-II conversion in SK-BR-3 cells (Supporting Information Fig. S6). Noticeably, a recent study by Huang et al. shows that XIAP inhibits autophagy in colon cancer cells (Huang et al., 2013b). In consistent with the findings of Huang et al.'s study, down-regulation of XIAP by siRNA also increased LC3B-II conversion, beclin-1 and p62/SQSTM1 expressions, and autophagic vesicles formation in MCF7 and MDA-MB-231 breast cancer cells. Thus, YM155 may promote breast cancer autophagic cell death through survivin down-regulation and the subsequent reduction of XIAP.
It is also worth noting that apart from modulating autophagy, YM155 also induces DNA damage in breast cancer cells. Although it has been demonstrated that targeting survivin can induce DNA damage in certain cancer models such as non-small cell lung cancer and prostate cancer, the molecular mechanism of such DNA damage induction is seldom investigated (Iwasa et al., 2010; Glaros et al., 2012). Interestingly, a few studies suggested that YM155 is a direct DNA damaging agent and can induce DNA damage in cancer cells through a survivin-independent mechanism. However, in our study, Western blot analysis revealed that targeting survivin by siRNA induced DNA damage in MCF7 and MDA-MB-231 cells. In addition, ectopic expression of survivin was capable of inhibiting YM155-induced DNA damage in breast cancer cells, suggesting that survivin down-regulation plays an important role in YM155-induced DNA damage. Moreover, inhibition of autophagy by either pharmacological inhibitors (3MA, CQ and BAF) or LC3B siRNA was effective in inhibiting the DNA damage induced by YM155, indicating that such DNA damage is at least partially caused by an autophagy-dependent mechanism. Our previous study shows that inhibition of cathepsin S induces autophagy and autophagy-dependent ROS generation, leading to the induction of oxidative DNA damage and cell death (Huang et al., 2013a). Therefore, it will be interesting to determine whether YM155 induces DNA damage also through autophagy-dependent ROS generation in breast cancer cells.
In conclusion, YM155 induces autophagy-dependent DNA damage and autophagic cell death in various breast cancer subtypes regardless of the expression of ER, HER2 and caspase-3. These key features distinguish this treatment from other previously developed anti-breast cancer therapies, such as hormone therapy and anti-HER2 therapy, that they can only be applied to treat ER+ breast cancer and HER2+ breast cancer respectively. Because YM155 is already undergoing a phase II clinical evaluation in patients with breast cancer (clinical trial identifier: NCT01038804), findings of this study will provide important information for clinicians in understanding the mechanism of action of this compound and to select possible drugs that can be used in combination with YM155 in the future.
Acknowledgments
We would like to thank Professor Daniel Klionsky and Professor Chia-Ching (Josh) Wu for their wonderful advice on this study. This work is kindly supported by the following grants: NSC101-2320-B-006-041 and NSC102-2320-B-006-038 (National Science Council, Taiwan, ROC), NHRI-EX102-10237SC (National Health Research Institutes, Taiwan, ROC), ‘Aim for the Top University Project for National Cheng Kung University’ (Ministry of Education, Taiwan, ROC) and CMNCKU10111 (Chi Mei Medical Center, Taiwan, ROC).
Glossary
- 3MA
3-methyladenine
- BAF
bafilomycin A1
- CQ
chloroquine
- ER (also known as NR3A)
oestrogen receptor
- HER2
human epidermal growth factor receptor 2
- IAPs
inhibitor-of-apoptosis proteins
- PR (also known as NR3C3)
progesterone receptor
- XIAP
X-linked inhibitor of apoptosis protein
- YM155
sepantronium bromide
- Z-DEVD-FMK
Z-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-fluoromethylketone
Author contributions
S. M. C., Y. C. C., C. Y. L., J. Y. C. and C. H. A. C conceived and designed the experiments. S. M. C., C. Y. L., Y. C. C., J. Y. C. L., H. H. C., C. W. K., K. Y. L., S. L. T., S. H. C., C. F. L. and C. C. H performed the experiments. S. M. C., Y. C. C., C. Y. L. and C. H. A. C analysed the data. J. Y. C. L., E. L., J. R. K. and C. H. A. C. wrote and proofread the paper.
Conflict of interest
We declare we have no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12935
Figure S1 YM155 increases the size of AVOs present in breast cancer cells. Breast cancer cells were treated with 2×IC50 YM155 for 48 h and were subsequently processed by MDC staining. The size of AVOs (green puncta) in cells was analysed by the ImageJ software.
Figure S2 Down-regulation of LC3B attenuates YM155-induced AVOs formation in breast cancer cells. MCF7 cells were pretransfected with either scamble siRNA or LC3B siRNA for 24 h and were subsequently co-treated with or without YM155 for 24 h. Cells were stained with MDC and formation of AVOs was determined using fluorescence microscopy.
Figure S3 Survivin regulates the expression of XIAP in breast cancer cells. MDA-MB-231 cells were transfected with either pCMV6-GFP, a plasmid that overexpresses GFP, or pCMV6-GFP-survivin, a plasmid that overexpresses the GFP-tagged survivin. Expression of various proteins was examined by Western blotting. Equal protein loading was verified by actin.
Figure S4 Inhibition of autophagy attenuates YM155-induced DNA damage in MCF7 cells. MCF7 cells were treated with either DMSO (control) or 2×IC50 YM155 with or with BAF for 48 h. Expression of γH2AX was examined by Western blotting. Equal protein loading was verified by actin.
Figure S5 Caspase-inhibition attenuates UV-induced cell death in breast cancer cells. The UV-treated (100 J·m−2) MDA-MB-231 cells were co-treated with or without Z-DEVD-FMK for 72 h. Percentage of cell death was determined by trypan blue exclusion assay. A statistically significant difference in the percentage of cell death of cells treated with UV versus UV + Z-DEVD-FMK is denoted by ‘*’. *P < 0.05.
Figure S6 YM155 induces conversion of LC3B-II and expression of γH2AX in SK-BR-3 cells. SK-BR-3 cells were treated with either DMSO (-ve control) or 2×IC50 YM155 for 48 h. Expression of various proteins was examined by Western blotting. Equal protein loading was verified by actin.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Nuclear hormone receptors. Br J Pharmacol. 2013a;170:1652–1675. doi: 10.1111/bph.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catlytic receptors. Br J Pharmacol. 2013b;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013c;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–921. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- Cheng Q, Ling X, Haller A, Nakahara T, Yamanaka K, Kita A, et al. Suppression of survivin promoter activity by YM155 involves disruption of Sp1-DNA interaction in the survivin core promoter. Int J Biochem Mol Biol. 2012;3:179–197. [PMC free article] [PubMed] [Google Scholar]
- Cheson BD, Bartlett NL, Vose JM, Lopez-Hernandez A, Seiz AL, Keating AT, et al. A phase II study of the survivin suppressant YM155 in patients with refractory diffuse large B-cell lymphoma. Cancer. 2012;118:3128–3134. doi: 10.1002/cncr.26510. [DOI] [PubMed] [Google Scholar]
- Devarajan E, Sahin AA, Chen JS, Krishnamurthy RR, Aggarwal N, Brun AM, et al. Down-regulation of caspase 3 in breast cancer: a possible mechanism for chemoresistance. Oncogene. 2002;21:8843–8851. doi: 10.1038/sj.onc.1206044. [DOI] [PubMed] [Google Scholar]
- Dohi T, Okada K, Xia F, Wilford CE, Samuel T, Welsh K, et al. An IAP-IAP complex inhibits apoptosis. J Biol Chem. 2004;279:34087–34090. doi: 10.1074/jbc.C400236200. [DOI] [PubMed] [Google Scholar]
- Early Breast Cancer Trialists' Collaborative Group (EBCTCG) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- Encarnacion CA, Ciocca DR, McGuire WL, Clark GM, Fuqua SAW, Osborne CK. Measurement of steroid-hormone receptors in breast-cancer patients on tamoxifen. Breast Cancer Res Treat. 1993;26:237–246. doi: 10.1007/BF00665801. [DOI] [PubMed] [Google Scholar]
- Giaccone G, Zatloukal P, Roubec J, Floor K, Musil J, Kuta M, et al. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J Clin Oncol. 2009;27:4481–4486. doi: 10.1200/JCO.2008.21.1862. [DOI] [PubMed] [Google Scholar]
- Glaros T, Stockwin L, Mullendore M, Smith B, Morrison B, Newton D. The ‘survivin suppressants’ NSC 80467 and YM155 induce a DNA damage response. Cancer Chemother Pharmacol. 2012;70:207–212. doi: 10.1007/s00280-012-1868-0. [DOI] [PubMed] [Google Scholar]
- Huang C-C, Chen K-L, Cheung CHA, Chang J-Y. Autophagy induced by cathepsin S inhibition induces early ROS production, oxidative DNA damage, and cell death via xanthine oxidase. Free Radic Biol Med. 2013a;65:1473–1486. doi: 10.1016/j.freeradbiomed.2013.07.020. [DOI] [PubMed] [Google Scholar]
- Huang X, Wu Z, Mei Y, Wu M. XIAP inhibits autophagy via XIAP-Mdm2-p53 signalling. EMBO J. 2013b;32:2204–2216. doi: 10.1038/emboj.2013.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasa T, Okamoto I, Takezawa K, Yamanaka K, Nakahara T, Kita A, et al. Marked anti-tumour activity of the combination of YM155, a novel survivin suppressant, and platinum-based drugs. Br J Cancer. 2010;103:36–42. doi: 10.1038/sj.bjc.6605713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Lamark T, Sjøttem E, Bowitz Larsen K, Atesoh Awuh J, Øvervatn A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkmaz G, Tekirdag KA, Ozturk DG, Kosar A, Sezerman OU, Gozuacik D. MIR376A is a regulator of starvation-induced autophagy. PLoS ONE. 2013;8:e82556. doi: 10.1371/journal.pone.0082556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung E, Kannan N, Krissansen GW, Findlay MP, Baguley BC. MCF-7 breast cancer cells selected for tamoxifen resistance acquire new phenotypes differing in DNA content, phospho-HER2 and PAX2 expression, and rapamycin sensitivity. Cancer Biol Ther. 2010;9:717–724. doi: 10.4161/cbt.9.9.11432. [DOI] [PubMed] [Google Scholar]
- Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KD, Samlowski W, Ward J, Catlett J, Cranmer L, Kirkwood J, et al. A multi-center phase II evaluation of the small molecule survivin suppressor YM155 in patients with unresectable stage III or IV melanoma. Invest New Drugs. 2011;29:161–166. doi: 10.1007/s10637-009-9333-6. [DOI] [PubMed] [Google Scholar]
- Li XQ, Dang XG, Sun XB. Expression of survivin and VEGF-C in breast cancer tissue and its relation to lymphatic metastasis. Eur J Gynaecol Oncol. 2012;33:178–182. [PubMed] [Google Scholar]
- Minematsu T, Iwai M, Sugimoto K, Shirai N, Nakahara T, Usui T, et al. Carrier-mediated uptake of 1-(2-methoxyethyl)-2-methyl-4,9-dioxo-3-(pyrazin-2-ylmethyl)-4,9-dihydro-1H-naphtho[2,3-d]imidazolium bromide (YM155 monobromide), a novel small-molecule survivin suppressant, into human solid tumor and lymphoma cells. Drug Metab Dispos. 2009;37:619–628. doi: 10.1124/dmd.108.025254. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafo DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114(Pt 20):3619–3629. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, et al. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007;67:8014–8021. doi: 10.1158/0008-5472.CAN-07-1343. [DOI] [PubMed] [Google Scholar]
- Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, et al. Broad spectrum and potent antitumor activities of YM155, a novel small-molecule survivin suppressant, in a wide variety of human cancer cell lines and xenograft models. Cancer Sci. 2011a;102:614–621. doi: 10.1111/j.1349-7006.2010.01834.x. [DOI] [PubMed] [Google Scholar]
- Nakahara T, Yamanaka K, Hatakeyama S, Kita A, Takeuchi M, Kinoyama I, et al. YM155, a novel survivin suppressant, enhances taxane-induced apoptosis and tumor regression in a human Calu 6 lung cancer xenograft model. Anticancer Drugs. 2011b;22:454–462. doi: 10.1097/CAD.0b013e328344ac68. [DOI] [PubMed] [Google Scholar]
- Niemann A, Takatsuki A, Elsässer HP. The lysosomotropic agent monodansylcadaverine also acts as a solvent polarity probe. J Histochem Cytochem. 2000;48:251–258. doi: 10.1177/002215540004800210. [DOI] [PubMed] [Google Scholar]
- Ogata M, Hino S-I, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puissant A, Fenouille N, Auberger P. When autophagy meets cancer through p62/SQSTM1. Am J Cancer Res. 2012;2:397–413. [PMC free article] [PubMed] [Google Scholar]
- Qiang L, Wu C, Ming M, Viollet B, He YY. Autophagy controls p38 activation to promote cell survival under genotoxic stress. J Biol Chem. 2013;288:1603–1611. doi: 10.1074/jbc.M112.415224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocr Relat Cancer. 2004;11:643–658. doi: 10.1677/erc.1.00776. [DOI] [PubMed] [Google Scholar]
- Satoh T, Okamoto I, Miyazaki M, Morinaga R, Tsuya A, Hasegawa Y, et al. Phase I study of YM155, a novel survivin suppressant, in patients with advanced solid tumors. Clin Cancer Res. 2009;15:3872–3880. doi: 10.1158/1078-0432.CCR-08-1946. [DOI] [PubMed] [Google Scholar]
- Speit G, Hartmann A. The comet assay: a sensitive genotoxicity test for the detection of DNA damage and repair. Methods Mol Biol. 2006;314:275–286. doi: 10.1385/1-59259-973-7:275. [DOI] [PubMed] [Google Scholar]
- Tao YF, Lu J, Du XJ, Sun LC, Zhao X, Peng L, et al. Survivin selective inhibitor YM155 induce apoptosis in SK-NEP-1 Wilms tumor cells. BMC Cancer. 2012;12:619. doi: 10.1186/1471-2407-12-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolcher AW, Mita A, Lewis LD, Garrett CR, Till E, Daud AI, et al. Phase I and pharmacokinetic study of YM155, a small-molecule inhibitor of survivin. J Clin Oncol. 2008;26:5198–5203. doi: 10.1200/JCO.2008.17.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolcher AW, Quinn DI, Ferrari A, Ahmann F, Giaccone G, Drake T, et al. A phase II study of YM155, a novel small-molecule suppressor of survivin, in castration-resistant taxane-pretreated prostate cancer. Ann Oncol. 2012;23:968–973. doi: 10.1093/annonc/mdr353. [DOI] [PubMed] [Google Scholar]
- Wang Q, Chen Z, Diao X, Huang S. Induction of autophagy-dependent apoptosis by the survivin suppressant YM155 in prostate cancer cells. Cancer Lett. 2011;302:29–36. doi: 10.1016/j.canlet.2010.12.007. [DOI] [PubMed] [Google Scholar]
- Yamashita SI, Masuda Y, Kurizaki T, Haga Y, Murayama T, Ikei S, et al. Survivin expression predicts early recurrence in early-stage breast cancer. Anticancer Res. 2007;27(4C):2803–2808. [PubMed] [Google Scholar]
- Yu WH, Kumar A, Peterhoff C, Shapiro Kulnane L, Uchiyama Y, Lamb BT, et al. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for β-amyloid peptide over-production and localization in Alzheimer's disease. Int J Biochem Cell Biol. 2004;36:2531–2540. doi: 10.1016/j.biocel.2004.05.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 YM155 increases the size of AVOs present in breast cancer cells. Breast cancer cells were treated with 2×IC50 YM155 for 48 h and were subsequently processed by MDC staining. The size of AVOs (green puncta) in cells was analysed by the ImageJ software.
Figure S2 Down-regulation of LC3B attenuates YM155-induced AVOs formation in breast cancer cells. MCF7 cells were pretransfected with either scamble siRNA or LC3B siRNA for 24 h and were subsequently co-treated with or without YM155 for 24 h. Cells were stained with MDC and formation of AVOs was determined using fluorescence microscopy.
Figure S3 Survivin regulates the expression of XIAP in breast cancer cells. MDA-MB-231 cells were transfected with either pCMV6-GFP, a plasmid that overexpresses GFP, or pCMV6-GFP-survivin, a plasmid that overexpresses the GFP-tagged survivin. Expression of various proteins was examined by Western blotting. Equal protein loading was verified by actin.
Figure S4 Inhibition of autophagy attenuates YM155-induced DNA damage in MCF7 cells. MCF7 cells were treated with either DMSO (control) or 2×IC50 YM155 with or with BAF for 48 h. Expression of γH2AX was examined by Western blotting. Equal protein loading was verified by actin.
Figure S5 Caspase-inhibition attenuates UV-induced cell death in breast cancer cells. The UV-treated (100 J·m−2) MDA-MB-231 cells were co-treated with or without Z-DEVD-FMK for 72 h. Percentage of cell death was determined by trypan blue exclusion assay. A statistically significant difference in the percentage of cell death of cells treated with UV versus UV + Z-DEVD-FMK is denoted by ‘*’. *P < 0.05.
Figure S6 YM155 induces conversion of LC3B-II and expression of γH2AX in SK-BR-3 cells. SK-BR-3 cells were treated with either DMSO (-ve control) or 2×IC50 YM155 for 48 h. Expression of various proteins was examined by Western blotting. Equal protein loading was verified by actin.