

Abstract
Antagonists of the CB1 receptor can be useful in the treatment of several diseases including obesity, diabetes, and liver disease. However, to date, the only clinically approved CB1 receptor antagonist, rimonabant, was withdrawn due to adverse CNS related side effects such as depression and suicidal ideation. Since rimonabant’s withdrawal, several groups have begun pursuing peripherally selective CB1 antagonists. These compounds are expected to be devoid of undesirable CNS related effects but maintain efficacy through antagonism of peripherally expressed CB1 receptors within target tissues. Reported here are our latest results toward development of a peripherally selective analog of the diphenyl purine CB1 antagonist otenabant 1. Compound 9 (N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}pentanamide) is a potent, orally absorbed antagonist of the CB1 receptor that is >50-fold selective for CB1 over CB2, highly selective for the periphery in a rodent model, and without efficacy in a series of in vivo assays designed to evaluate its ability to mitigate the central effects of Δ9-THC through the CB1 receptor.
Keywords: CB1, peripheral, antagonist, cannabinoid, topological polar surface area
INTRODUCTION
The cannabinoid receptors CB1 and CB2 are G-protein-coupled receptors (GPCRs) that primarily function as activators of inhibitory G proteins (Gi/o).1–4 CB1 receptors are expressed widely throughout the body; however, they are most heavily expressed in the central nervous system (CNS). CB2 receptors are found most abundantly in the periphery. Both receptors belong to an endocannabinoid system (ECS) that regulates many physiological systems and can be targeted to treat several serious disorders. Currently, various components of the ECS are under evaluation for the treatment of pain, inflammation, obesity, liver disease, and diabetes.2, 4
For example, antagonism of CB1 has been investigated for the treatment of metabolic disorders, such as diabetes and obesity. Past translational research in this regard led to the clinical approval of rimonabant (SR141716A) for weight loss in Europe. Rimonabant is a potent and selective CB1 receptor inverse agonist/antagonist. Regrettably, rimonabant produced serious side effects such as depression, anxiety, and suicidal ideation in a subset of patients. These CNS-related side effects led to the withdrawal of rimonabant from European markets and the drug was not approved for use in the United States.5, 6 Several other CB1 antagonists, such as otenabant (1) (Figure 1), taranabant, and ibipinabant, were pulled from clinical development upon discovery of rimonabant’s CNS-related side effects.7
Figure 1.
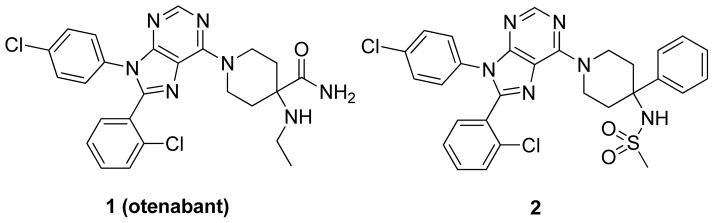
Otenabant and a peripherally selective analogue
A possible strategy to avoid CNS-related side effects is to generate CB1 antagonists that do not cross the blood-brain barrier (BBB). This strategy has been pursued by our group and others, and a small group of CB1 antagonists that are peripherally selective have been reported and are in various stages of development.8 Our group has focused on 1 as a starting point for further optimization.9 Despite being non-tissue selective, 1 has several properties that could be optimized to produce peripherally selective compounds, including a formula weight >500, a topological polar surface area (TPSA) of 102, and 3 H-bond donors.10 However, intramolecular H-bonding effectively lowers the polarity of 1, allowing for penetration into the CNS.10 In our initial publication, we reported several diphenyl purines with formula weights and TPSA similar to 1 but lacking the ability to form intramolecular H-bonds. To that end, we were able to discover 2, an analogue of 1 that has a formula weight >500 and a TPSA 101. However, unlike 1, 2 is a peripherally selective compound. Oral dosing of 2 at 10 mg/kg in Sprague-Dawley (SD) rats showed good oral absorption (Cmax = 1653 ng/g) and limited penetration into the CNS (brain to plasma ratios of 0.05–0.11 were observed).11
Based upon these promising results with 2, we investigated a series of analogues with substitutions at the 4-amino position of 3 (Figure 2) in an attempt to further limit CNS-penetration. While the 4-amino-4-phenyl piperidine portion of 2 was the most potent amino piperidine to be tested in our previous studies, these efforts focused on the 4-amino piperidine moiety of structure 3 due to its lower formula weight; 3 allows us to add steric bulk while maintaining the formula weight <600.
Figure 2.
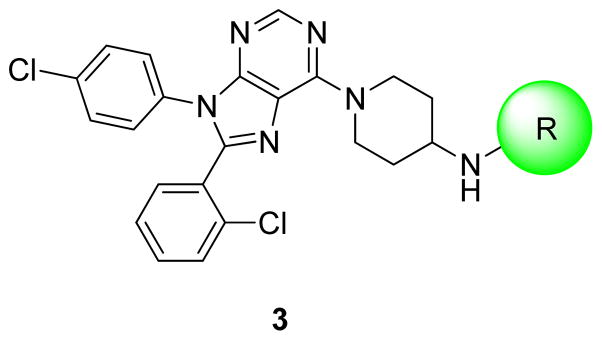
General strategy for compound development
RESULTS
Ligand Design and Synthesis
Of the compounds evaluated in our earlier studies that lacked additional substitution on the piperidine ring, the 4-amino piperidine compounds (represented by structure 3) were the most potent analogues.9 However, only a limited number of R groups were tested. In this study, a number of different R group substitutions were targeted to augment understanding of the existing structure activity relationship (SAR). Carbamates, amides, and sulfonamides were chosen for further examination because they were previously found to be favored over ureas.9
Synthesis of compounds was accomplished as described in Scheme 1. Carbamates were made by reacting the previous described amine 49 and the appropriate chloroformate. Amides 7-16 were synthesized from 4 using benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP), triethylamine and the appropriate amine or acid. Sulfamide 17 was synthesized by heating 4 with sulfamide in dioxane. Sulfonamide 18 was prepared by reacting 4, triethylamine, and trifluoromethanesulfonic anhydride in dichloromethane.
Scheme 1. Synthesis of compoundsa.

aReagents and conditions: (a) appropriate chloroformate, triethylamine (TEA), tetrahydrofuran (THF); (b) appropriate carboxylic acid, benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP), TEA, THF; (c) appropriate amino acid, BOP, TEA, THF; (d) sulfamide, dioxane, 80° C; (e) trifluoromethanesulfonic anhydride, TEA, dichloromethane.
Pharmacological Characterization
Compounds were tested in vitro for antagonist activity at CB1 using a calcium mobilization assay. This CB1 antagonist activity is expressed as an apparent dissociation affinity constant (Ke).12 Compounds that had Ke of <10 nM were assessed for selective affinity for CB1 versus CB2 receptors, as determined using a [3H]CP55940 displacement assay. For each receptor, values were expressed as the equilibrium dissociation constant (Ki).9 Selectivity was expressed as a ratio of these constants.
In general, excellent activity and potency were observed with these compounds (Table 1). Ten of the fourteen compounds had Ke < 10 nM and Ki < 20 nM at the CB1 receptor. All compounds tested were found to be selective for the CB1 versus CB2 receptor. Selectivity ranged from ~22-fold for 11 to >4000-fold for 7.
Table 1.
Pharmacological Assessment of Compounds
| Compound | R | Ke CB1 (nM) | Ki CB1a (nM) | Ki CB2a (nM) | Selectivity (CB1/CB2) |
|---|---|---|---|---|---|
| 5 | Me | 17 | |||
| 6 | Et | 2.3 | 3.57 | 426 | 119 |
| 7 | Ph | 4 | 2.55 | 10649 | 4176 |
| 8 | Cyclohexyl | 3.3 | 3.35 | 835 | 249 |
| 9 | n-Bu | 4.9 | 19.47 | 1046 | 54 |
| 10 | Cyclohexylmethyl | 2.1 | 1.93 | 2360 | 1223 |
| 11 | i-Bu | 5.4 | 4.01 | 89.5 | 22 |
| 12 | Cyclopentyl | 0.89 | 1.54 | 60.2 | 39 |
| 13 | 3-Methylbutyl | 7.4 | 2.67 | 215 | 81 |
| 14 | Cyclopentylmethyl | 2.9 | 2.02 | 569 | 282 |
| 15 | (Piperidin-1-yl)ethyl | 789 | |||
| 16 | Me2NCH2 | 140 | |||
| 17 | NH2 | 2768 | |||
| 18 | CF3 | 1.05 | 6.1 | 4501 | 738 |
Displacement was measured using [3H]CP55940
Pharmacokinetic studies of select compounds
Pharmacokinetic (PK) studies were performed with compounds 6 and 9. These compounds were chosen for several reasons. First, testing compounds with different functional groups could maximize the chances of obtaining a compound with a desirable PK profile. Therefore, one amide and one carbamate were chosen. Second, of the compounds with Ke < 10 nM and selectivity of >50 fold in each series, 6 and 9 had the lowest molecular weight and clog P value in their respective series. Compound 18 was excluded due to its relatively high formula weight. Finally, the shielding effect of bulk on the R group on polarity of the amide or carbamate functionality was taken into consideration. Excessive steric bulk adjacent to the amide or carbamate could effectively minimize the interaction of polar groups of the molecule with their surroundings, thereby increasing penetration across the BBB.
Both 6 and 9 were initially screened for CNS penetration by oral dosing at 10 mg/kg in Sprague-Dawley (SD) rats. Un-perfused brain and plasma samples were collected at 1, 2, 4, 8, and 24 hours post dose and samples were analyzed by LC/MS (data not shown). Brain to plasma ratios of 6 ranged from 0.14–0.46, representing significant brain penetration (data not shown). Minimal to no penetration into the CNS was observed with 9 (Table 2), with brain to plasma ratios ranging from 0.01–0.07. The blood volume of an un-perfused rodent brain is ~3–6%. 13, 14 Therefore, these PK data indicate that 9 has little to no penetration into the CNS and is peripherally selective.
Table 2.
In vivo pharmacokinetic evaluation of compound 9a
| Time (hours) | Plasma conc. (ng/mL) | Brain conc. (ng/mL) | Brain/Plasma ratio |
|---|---|---|---|
| 1 | 730 | 24.3 | 0.03 |
| 2 | 1750 | 71.5 | 0.04 |
| 4 | 1565 | 103 | 0.07 |
| 8 | 1965 | 62.5 | 0.03 |
| 24 | 438 | 5.35 | 0.01 |
9 was dosed orally at 10 mg/kg in 3 SD rats per time-point
In vivo evaluation of central CB1 antagonism
Compound 9 was also evaluated for its ability to attenuate the central effects of a CB1 agonist in a series of in vivo tests. In this battery of tests, known as the “tetrad” assay, a classical CB1 agonist such as Δ9-THC produces suppression of spontaneous activity, antinociception, hypothermia, and catalepsy. These effects are believed to be mediated by CB1 receptors in the brain and spinal cord15 and are blocked by co-administration of rimonabant, a potent and CNS penetrant CB1 antagonist.15, 16
The effects of 9 on the tetrad were measured in adult ICR mice that were orally dosed with either vehicle or 10 mg/kg of 9 followed 30 min later by an i.v. injection of 30 mg/kg of Δ9-THC or vehicle. Spontaneous activity was measured in an automated locomotor activity chamber where beam breaks recorded horizontal movement. Total amulatory counts for this test are shown in Figure 3A. Antinociception was measured by the latency of mice to withdraw their tails from a warm water bath (55 °C) Results for this assay are expressed as percent of maximum possible effect (%MPE) and are shown in Figure 3B. Hypothermia was measured by comparing the differences in rectal temperature prior to dosing and post Δ9-THC dosing; these results are reported as change of temperature (Δ°C) and are shown in Figure 3C. Finally, catalepsy was measured by placing mice on an elevated ring apparatus and measuring the amount of time each mouse remained motionless during a 5 minute period following agonist administration. These data are reported as % ring immobility and are shown in Figure 3D. The 30 mg/kg dose of Δ9-THC significantly suppressed locomotor activity (panel A) and produced antinociception (panel B), hypothermia (panel C), and catalepsy (panel D), regardless of whether or not 9 was also administered (i.e., p < 0.05, main effect of Δ9-THC dose for each measure). Hence, 9 did not reverse any of the cannabimimetic effects of Δ9-THC. It also did not produce any significant effects in these tests by itself when tested in combination with vehicle.
Figure 3.

Effects of vehicle and Δ9-THC (30 mg/kg) tested in combination with vehicle (unfilled bars) or compound 9 (filled bars) on spontaneous activity (panel A), antinociception (panel B), rectal temperature (panel C), and catalepsy (panel D). Values represent the mean (± SEM) of 5–6 mice per group. Pound signs (#) indicate a significant main effect (p < 0.05) of Δ9-THC (right side of panels) compared to vehicle (left side of panels).
DISCUSSION
The goal of these studies was to produce a peripherally selective CB1 antagonist based on 1 as an improvement over our previously reported compound 2 that demonstrated ~10% brain penetration. To achieve this goal, the SAR around 1 was expanded. Several potent (Ke CB1 <10 nM) and selective (CB2/CB1 >50) amides were identified. Amides 8-14 with a variety of alkyl groups appeared to have significant activity at CB1. However, the presence of basic groups such as piperidine and dimethyl amino was detrimental to activity at CB1 as demonstrated by 15 and 16. Interestingly, affinity at the CB2 receptor could be modulated by changing the size and shape of the alkyl substituent as demonstrated by compounds 9-14. In general, amides with cyclic substituents, such as 8, 10, and 14 had excellent selectivity for CB1 over CB2. However, the cyclopentyl amide 12 was an exception to that trend. The reason/s behind this aberrant behavior is unclear at this time and will require additional studies in the future.
Carbamates 5-7 all had significant activity at the CB1 receptor. However, an order of magnitude decrease in activity was observed with the addition of a single methylene to 5 (forming 6). Aromatic carbamate 7 and ethyl carbamate 6 possessed both good affinity and selectivity. However, phenyl carbamate 7 was an order of magnitude more selective than ethyl carbamate 6 for CB1 over CB2. This result is consistent with the general trend observed for amides with cyclic substituents discussed above.
Sulfamide 17 did not have significant activity in the calcium mobilization assay. Sulfonamide 18 was very active at CB1, and it was very selective for CB1 over CB2. The results for 18 augment our previously reported data for the methanesulfonamide (Ke CB1 = 71 nM; CB2/CB1 Ki = 37) and the benzenesulfonamide (Ke CB1 = 0.3 nM; CB2/CB1 Ki = 4888) analogues of 3.9 Data for all three sulfonamides of 3 indicated that increased lipophilicity, and to a lesser extent mass, were advantageous for both activity at CB1 and selectivity for CB1 over CB2.
Of the two compounds submitted for PK analyses, only 9 was found to have selectivity for the periphery with brain to plasma levels ranging from 0.01–0.07. At only one time point, did the brain to plasma ratio exceed 0.04. A brain to plasma ratio of ≤0.06 in an un-perfused rodent brain indicates no discernible penetration into the CNS.13, 14 Thus, 9 is highly selective for the periphery and this lack of CNS penetration was also confirmed in the tetrad assay wherein the centrally mediated effects of Δ9-THC could not be reversed by 9.
In conclusion, reported here are our latest efforts towards development of a peripherally selective CB1 antagonist for the treatment of a wide range of clinical indications. To date, two peripherally selective analogues of 1 have been identified (2 and 9), with 9 representing a significant improvement over 2 in the areas plasma concentration and BBB penetration. Although both compounds are slowly absorbed, plasma levels for 9 are greater than plasma levels of 2 at the Cmax (1965 ng/mL vs. 1653 ng/mL) and at the 8 h time point (1965 ng/mL vs. 914 ng/mL).9 While 2 has brain to plasma ratios that range from 0.05–0.11,9 9 has brain to plasma ratios ranging from 0.01–0.07. These data, along with the results from the tetrad, make a compelling case for the peripheral selectivity of 9. Efficacy studies are currently underway to test the utility of 9 in models of metabolic syndrome and liver disease.
EXPERIMENTAL SECTION
Chemistry General
Purity and characterization of compounds were established by a combination of HPLC, TLC, and NMR analytical techniques described below. 1H and 13CNMR spectra were recorded on a Bruker Avance DPX-300 (300 MHz) spectrometer and were determined in CHCl3-d or MeOH-d4 with tetramethylsilane (TMS) (0.00 ppm) or solvent peaks as the internal reference unless otherwise noted. Chemical shifts are reported in ppm relative to the solvent signal, and coupling constant (J) values are reported in hertz (Hz). Thin-layer chromatography (TLC) was performed on EMD pre-coated silica gel 60 F254 plates, and spots were visualized with UV light or I2 detection. Low-resolution mass spectra were obtained using a Waters Alliance HT/Micromass ZQ system (ESI). All test compounds were at least 95% pure as determined by HPLC on an Agilent 1100 system using an Agilent Zorbax SB-Phenyl, 2.1×150 mm, 5 μm column with gradient elution using the mobile phases (A) H2O containing 0.05% CF3COOH and (B) Methanol. A flow rate of 1.0 mL/min was used.
General procedure for making carbamates from 1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-amine (4)
To a solution of 1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-amine (4) (12.5 mg, 0.028 mmol, 1 eq.) in 2 mL of THF was added triethylamine (0.02 mL, 0.143 mmol, 5 eq.) and the appropriate chloroformate (2 eq). The reaction is stirred for 16h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexane to yield pure compound.
Methyl N-{1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}carbamate (5)
Reaction proceeded in 79% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.44 – 1.55 (m, 2 H) 2.13 (d, J=11.49 Hz, 2 H) 3.34 (t, J=12.24 Hz, 2 H) 3.68 (br. s., 3 H) 3.86 (br. s., 1 H) 4.60 (br. s., 1 H) 5.40 (br. s., 2 H) 7.13 – 7.45 (m, 7 H) 7.51 (d, J=6.88 Hz, 1 H) 8.38 (s, 1 H), [M + H]+ 497.8.
Ethyl N-{1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}carbamate (6)
Reaction proceeded in 55% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.25 (t, J=6.92 Hz, 3 H) 1.43 – 1.55 (m, 2 H) 2.13 (d, J=11.21 Hz, 2 H) 3.34 (t, J=12.01 Hz, 2 H) 3.86 (br. s., 1 H) 4.06 – 4.22 (m, 2 H) 4.58 (br. s., 1 H) 5.39 (br. s., 2 H) 7.14 – 7.43 (m, 7 H) 7.51 (d, J=6.69 Hz, 1 H) 8.38 (s, 1 H), [M + H]+ 511.3.
Phenyl N-{1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}carbamate (7)
Reaction proceeded in 50% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.59 – 1.72 (m, 2 H) 2.22 (d, J=11.40 Hz, 2 H) 3.37 (t, J=12.10 Hz, 2 H) 3.84 – 4.02 (m, 1 H) 4.99 (d, J=7.72 Hz, 1 H) 5.45 (br. s., 2 H) 7.02 – 7.45 (m, 12 H) 7.52 (d, J=6.88 Hz, 1 H) 8.40 (s, 1 H), [M + H]+ 559.8.
General procedure for making amides from 1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-amine (4) using carboxylic acids
To a solution of 1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-amine (4) (22 mg, 0.05 mmol, 1 eq.) in 2 mL of THF was added triethylamine (0.02 mL, 0.143 mmol, 3 eq.), (benzotria-zol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) (22 mg, 0.05 mmol, 1 eq.), and the appropriate carboxylic acid (1 eq). The reaction is stirred for 16h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexane to yield compound. The product was purified further by dissolving in ethyl acetate and precipitating with hexane. The solid, pure compound, was collected.
N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}cyclohexanecarboxamide (8)
Reaction proceeded in >99% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.15 – 2.17 (m, 14 H) 2.23 – 2.40 (m, 1 H) 3.31 (t, J=12.20 Hz, 2 H) 4.05 – 4.25 (m, 1 H) 5.22 – 5.61 (m, 3 H) 7.14 – 7.42 (m, 7 H) 7.51 (d, J=6.69 Hz, 1 H) 8.38 (s, 1 H), [M + H]+ 549.5.
N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}pentanamide (9)
Reaction proceeded in >99% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 0.84 – 0.98 (m, 3 H) 1.27 – 1.75 (m, 5 H) 2.05 – 2.23 (m, 3 H) 2.26 – 2.44 (m, 1 H) 3.31 (t, J=12.24 Hz, 2 H) 4.02 – 4.30 (m, 1 H) 5.22 – 5.63 (m, 3 H) 7.13 – 7.44 (m, 7 H) 7.51 (d, J=6.78 Hz, 1 H) 8.38 (s, 1 H), [M + H]+ 523.5.
N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}-2-cyclohexylacetamide (10)
Reaction proceeded in >99% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 0.86 – 1.00 (m, 3 H) 1.05 – 1.87 (m, 10 H) 2.02 – 2.18 (m, 4 H) 3.31 (t, J=12.10 Hz, 2 H) 4.10 – 4.28 (m, 1 H) 5.22 – 5.62 (m, 3 H) 7.14 – 7.43 (m, 7 H) 7.51 (d, J=6.78 Hz, 1 H) 8.38 (s, 1 H), [M + H]+ 562.2.
N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}-3-methylbutanamide (11)
Reaction proceeded in >99% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 0.97 (dd, J=9.94, 6.45 Hz, 6 H) 1.50 (qd, J=11.85, 3.81 Hz, 1 H) 2.01 – 2.17 (m, 4 H) 2.19 – 2.27 (m, 2 H) 3.31 (t, J=12.24 Hz, 2 H) 4.10 – 4.27 (m, 1 H) 5.39 (d, J=8.01 Hz, 3 H) 7.14 – 7.42 (m, 7 H) 7.51 (d, J=6.78 Hz, 1 H) 8.38 (s, 1 H), [M + H]+ 523.3.
N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}cyclopentanecarboxamide (12)
Reaction proceeded in 86% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.39 – 1.99 (m, 9 H) 2.06 – 2.21 (m, 3 H) 2.41 – 2.58 (m, 1 H) 3.31 (t, J=12.24 Hz, 2 H) 4.08 – 4.27 (m, 1 H) 5.40 (d, J=7.91 Hz, 3 H) 7.12 – 7.42 (m, 7 H) 7.51 (d, J=6.78 Hz, 1 H) 8.38 (s, 1 H), [M + H]+ 535.5.
N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}-4-methylpentanamide (13)
Reaction proceeded in 82% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 0.85 – 0.95 (m, 6 H) 1.40 – 1.62 (m, 5 H) 2.03 – 2.23 (m, 3 H) 2.34 (t, J=7.54 Hz, 1 H) 3.31 (t, J=12.24 Hz, 2 H) 4.08 – 4.29 (m, 1 H) 5.42 (d, J=7.91 Hz, 3 H) 7.14 – 7.43 (m, 7 H) 7.51 (d, J=7.16 Hz, 1 H) 8.38 (s, 1 H), [M + H]+ 537.5.
N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}-2-cyclopentylacetamide (14)
Reaction proceeded in 98% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.06 – 1.23 (m, 3 H) 1.44 – 1.69 (m, 5 H) 1.74 – 1.93 (m, 3 H) 2.06 – 2.28 (m, 3 H) 2.30 – 2.41 (m, 1 H) 3.32 (t, J=12.20 Hz, 2 H) 4.08 – 4.29 (m, 1 H) 5.41 (d, J=7.91 Hz, 3 H) 7.14 – 7.42 (m, 7 H) 7.51 (d, J=6.78 Hz, 1 H) 8.38 (s, 1 H), [M + H]+ 549.5.
General procedure for making amides from 1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-amine (4) using amino acids
To a solution of 1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-amine (4) (22 mg, 0.05 mmol, 1 eq.) in 2 mL of THF was added triethylamine (0.02 mL, 0.143 mmol, 3 eq.), (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) (22 mg, 0.05 mmol, 1 eq.), and the appropriate carboxylic acid (1 eq). The reaction is stirred for 16h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% CMA 80/dichloromethane to yield compound. The product was purified further by dissolving in ethyl acetate, washing with water, and precipitating with hexane. The solid, pure compound, was collected.
N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}-3-(piperidin-1-yl)propanamide (15)
Reaction proceeded in 41% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.52 – 1.60 (m, 4 H) 1.80 (br. s., 3 H) 1.98 – 2.14 (m, 3 H) 2.76 (t, J=5.93 Hz, 2 H) 2.93 (br. s., 2 H) 3.30 (br. s., 2 H) 3.40 (br. s., 2 H) 3.56 (br. s., 2 H) 3.97 – 4.15 (m, 1 H) 5.36 – 5.59 (m, 2 H) 6.46 – 6.65 (m, 1 H) 7.15 – 7.42 (m, 7 H) 7.54 (d, J=7.63 Hz, 1 H) 8.37 (s, 1 H), [M + H]+ 580.6.
N-{1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}-2-(dimethylamino)acetamide (16)
Reaction proceeded in 11% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.50 – 1.66 (m, 2 H) 2.01 – 2.13 (m, 2 H) 2.48 (s, 7 H) 2.64 (d, J=9.32 Hz, 1 H) 3.36 (t, J=11.82 Hz, 2 H) 4.05 – 4.26 (m, 1 H) 5.41 (br. s., 2 H) 7.08 (d, J=8.19 Hz, 1 H) 7.14 – 7.42 (m, 7 H) 7.51 (d, J=6.78 Hz, 1 H) 8.37 (s, 1 H), [M + H]+ 524.7.
N-{1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}aminosulfonamide (17)
To a solution of 1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-amine (4) (36.6 mg, 0.083 mmol, 1 eq.) in 2 mL of dioxane was added sulfamide (40 mg, 0.42 mmol, 5 eq.). The reaction was heated to 80° C for 16h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexane to yield 34 mg (79%) of desired compound. 1H NMR (300 MHz, METHANOL-d4) δ ppm 1.51 – 1.72 (m, 2 H) 2.15 (br. s., 2 H) 3.38 – 3.52 (m, 2 H) 3.54 – 3.65 (m, 1 H) 5.18 – 5.41 (m, 2 H) 7.24 – 7.50 (m, 7 H) 7.54 – 7.67 (m, 1 H) 8.23 (s, 1 H), [M + H]+ 518.5.
N-{1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl,}-1,1,1-trifluoromethanesulfonamide (18)
To a solution of 1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-amine (4) (27.9 mg, 0.064 mmol, 1 eq.) in 2 mL of dichloromethane was added triethylamine (0.027 mL, 0.191 mmol, 3 eq.) and the trifluoromethanesulfonic anhydride (0.01 mL, 0.069 mmol, 1 eq.). The reaction is stirred for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexane to yield 9 mg (25%) of desired compound. 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.60 – 1.76 (m, 2 H) 2.20 (d, J=12.62 Hz, 2 H) 3.30 (t, J=12.57 Hz, 2 H) 3.71 – 3.92 (m, 1 H) 4.95 (d, J=8.48 Hz, 1 H) 5.50 (d, J=12.34 Hz, 2 H) 7.14 – 7.43 (m, 7 H) 7.50 (d, J=6.97 Hz, 1 H) 8.39 (s, 1 H) [M + H]+ 571.7.
Calcium mobilization and radioligand displacement assays
Each compound was pharmacologically characterized using a functional fluorescent CB1 activated Gαq16-coupled intracellular calcium mobilization assay in CHO-K1 cells, as has been described in our previous publications and apparent affinity (Ke) values were determined.13 Briefly, CHO-K1 cells were engineered to co-express human CB1 and Gqα16. Activation of CB1 by an agonist then leads to generation of inositol phospahatase 3 (IP3) and activation of IP3 receptors, which leads to mobilization of intracellular calcium. Calcium flux was monitored in a 96-well format using the fluorescent dye Calcein-4 AM in an automated plate-reader (Flexstation, Molecular Devices). The antagonism of a test compound was measured by its ability to shift the concentration response curve of the synthetic CB1 agonist CP55940 rightwards using the equation:
where DR is the EC50 ratio of CP55940 in the presence or absence of a test agent. Standard errors were between 5–25% of mean in most cases and have been left out for clarity.
Further characterization of select compounds was performed using radioligand displacement of [3H]SR141716 and equilibrium dissociation constant (Ki) values were determined as described previously.13 Selectivity of these compounds at CB1 versus CB2 was also determined by obtaining Ki values at either receptor using displacement of [3H]CP55940 in membranes of CHO-K1 cells over-expressing either receptor. Data reported are average values from 3–6 measurements. The standard errors for most measurements were between 5–25% of mean and have been left out from the tables and figures for clarity.
Pharmacokinetic studies
Male Sprague Dawley (SD) rats aged 9 weeks at time of dosing were acquired from Charles River Laboratories and were dosed orally. Doses were formulated in corn oil and administered by gavage. Animals were sacrificed at various time-points and analyzed. Samples were prepared and analyzed as follows: Plasma (50 μL) was mixed with 300 μL of acetonitrile, vortexed, and centrifuged at 9000 g for 5 min. Supernatant was transferred to autosampler vials with low-volume inserts and injected without dilution. Brain was homogenized with 50:50 ethanol:water (3:1, v/v) using a Potter Elvehjem type homogenizer. Homogenate (50 μL) was mixed with 300 μL of acetonitrile, vortexed and centrifuged at 9000 g for 5 min. Supernatant was transferred to autosampler vials with low-volume inserts and injected without dilution. Standards were prepared as above for each compound in blank plasma, blank liver homogenate, and blank brain homogenate. Standards used were within 15% of nominal; except for 20% at LOQ. Compounds for LC-MS/MS analyses were supplied at 1 mg/mL in methanol. The stock solutions were further diluted to ~100 ng/mL. The 100 ng/mL solutions were used to optimize the mass spectrometer for MRM transitions and mass spectrometer parameters. Infusion and flow injection optimization were also performed. LC-MS/MS was conducted using an Applied Biosystems API 4000 coupled with an Agilent 1100 HPLC system. Chromatography was performed with a Phenomenex Luna C18 (50 × 2 mm, 5μm) column. Mobile phases were 0.1% formic acid and 10 mM ammonium formate in water (A), and 0.1% formic acid and 10 mM ammonium formate in methanol (B). Initial conditions were 10 % B and held for one minute, followed by a linear gradient to 90% B over 5 minutes. 90% B was held for 2 minutes before returning to initial conditions. Compound 9 was analyzed with multiple reactions monitoring in the positive mode with a transition of 522.922→368.0. The following parameters were used, DP=171, CE=51, and CXP=30.
Tetrad Test
Male adult ICR mice, obtained from Harlan (Indianapolis, IN) and housed singly in polycarbonate mouse cages, were used for assessment of locomotor suppression, antinociception, hypothermia, and catalepsy. Separate mice were used for testing each condition in this battery of procedures. All animals were maintained in a temperature-controlled (20–22°C) environment with a 12-hour light-dark cycle (lights on at 6 a.m.) and had free access to food and water when in their home cages. The in vivo studies reported in this manuscript were carried out in accordance with guidelines published in the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011). Δ9-THC, obtained from the National Institute on Drug Abuse (NIDA, Bethesda, MD) through the NIDA Drug Supply Program, was mixed in a vehicle of 7.8% Polysorbate 80 N.F. (VWR, Marietta, GA), and 92.2% sterile saline USP (Butler Schein, Dublin, OH) and was given by i.v. injection. Compound 9 was dissolved in peanut oil and administered via oral gavage at a volume of 5 ml/kg.
Measurement of spontaneous activity occurred in standard Plexiglas locomotor activity chambers (33 cm × 51 cm × 23 cm). Beam breaks were recorded by San Diego Instruments Photobeam Activity System software (model SDI: V-71215, San Diego, CA) on a computer located in the experimental room. The apparatus contained two 4-beam infrared arrays that measured horizontal movement. A glass beaker filled with water heated to 55°C was used for the warm water tail withdrawal procedure. A digital thermometer (Physitemp Instruments, Inc., Clifton, NJ) was used to measure rectal temperature. The ring immobility device consisted of an elevated metal ring (diameter = 5.5 cm, height = 16 cm) attached to a metal stand.
Each mouse was tested in a battery of four tests, in which cannabinoid agonists produced a characteristic profile of in vivo effects:17 suppression of locomotor activity, antinociception, decreased rectal temperature and ring immobility. Prior to injection, rectal temperature and baseline latency in the warm water tail withdrawal test were measured in the mice. The latter procedure involved immersing the mouse’s tail into a water bath maintained at 55°C and recording latency (in s) for tail removal. Control latencies were 1–3 s. A 10 s maximal latency was used in order to avoid damage to the mouse’s tail. After measurement of temperature and baseline tail withdrawal latency, mice were dosed with vehicle or with 10 mg/kg 9 via oral gavage. Thirty minutes later, mice were injected i.v. in the tail vein with vehicle or with 30 mg/kg Δ9-tetrahydrocannabinol (Δ9-THC). After 5 min, they were placed into individual activity chambers for 10 min. Spontaneous activity was measured as the total number of beam interruptions during the entire session. Tail-withdrawal latency was measured again at 20 min post-injection. Rectal temperature was measured again at 30 min after injection. At 40 min post-injection, the mice were placed on the elevated ring apparatus, and the amount of time the animals remained motionless during a 5 min period was recorded.
Rectal temperature values were expressed as the difference between control temperature (before injection) and temperature following drug administration (Δ°C). Spontaneous activity was measured as total number of photocell beam interruptions during the 10 min session. Antinociception was expressed as the percent maximum possible effect (MPE) using a 10 s maximum test latency as follows: [(test−control)/(10−control)]×100. During assessment for catalepsy, the total amount of time (in s) that the mouse remained motionless on the ring apparatus (except for breathing and whisker movement) was measured and was used as an indication of catalepsy-like behavior. This value was divided by 300 s and multiplied by 100 to obtain a percent immobility. Factorial ANOVAs (Δ9-THC dose X 9 dose) were used to analyze results of tetrad tests. Significant main effects and interactions were further analyzed with Tukey post hoc tests (Δ = 0.05) as necessary.
Acknowledgments
The authors wouxld like to thank Ann Gilliam for performing the binding assays. We express our gratitude to the NIDA drug supply program for providing radiolabeled probes and to Dr. Brian Thomas for supplying the CB1 cells. This research was funded by research grants AA019740-01 to R.M. and DA-003672 to J.W.
Abbreviations Used
- CB1
Cannabinoid Receptor 1
- CB2
Cannabinoid Receptor 2
- CMA 80
80% chloroform, 18% methanol, and 2% ammonium hydroxide
- CNS
Central Nervous System
- BBB
Blood-Brain Barrier
- TPSA
Topological Polar Surface Area
- ECS
Endocannabinoid System
- CBR
Cannabinoid Receptors
- Ke
apparent affinity constant
- MDCK-mdr1
Madin-Darby canine kidney cells transfected with the human MDR1 gene
- A
Apical
- B
basal
- BOP
Benzotriazole-1-yl-oxytris(dimethylamino)phosphonium hexafluorophosphate
- CHO-K1
Chinese Hamster Ovary Cells
- IP3
Inositol Phospahatase 3
- MRM
Multiple Reaction Monitoring
- LOQ
Below Limit of Quantitation
- NA
not applicable
Footnotes
Supporting Information
HPLC and melting point data of target compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bouaboula M, Bianchini L, McKenzie FR, Pouyssegur J, Casellas P. Cannabinoid receptor CB1 activates the Na+/H+ exchanger NHE-1 isoform via Gi-mediated mitogen activated protein kinase signaling transduction pathways. FEBS Lett. 1999;449:61–5. doi: 10.1016/s0014-5793(99)00395-6. [DOI] [PubMed] [Google Scholar]
- 3.Barth F, Rinaldi-Carmona M, Millan J, Derocq J-M, Bouaboula M, Casellas P, Congy C, Oustric D, Sarran M, Calandra B, Portier M, Shire D, Breliere J-C, Le Fur G. SR144528, A Potent and Selective Antagonist of the CB2 Receptor. Vol. 1997. Internation Cannabinoid Research Society; Stone Mountain, GA: ICRS; Stone Mountain, GA: 1997. p. 11. [Google Scholar]
- 4.Bouaboula M, Perrachon S, Milligan L, Canat X, Rinaldi-Carmona M, Portier M, Barth F, Calandra B, Pecceu F, Lupker J, Maffrand JP, Le Fur G, Casellas P. A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1. Evidence for a new model of receptor/ligand interactions. J Biol Chem. 1997;272:22330–9. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- 5.Janero DR, Makriyannis A. Cannabinoid receptor antagonists: pharmacological opportunities, clinical experience, and translational prognosis. Expert Opin Emerg Drugs. 2009;14:43–65. doi: 10.1517/14728210902736568. [DOI] [PubMed] [Google Scholar]
- 6.Di Marzo V. CB(1) receptor antagonism: biological basis for metabolic effects. Drug Discov Today. 2008;13:1026–41. doi: 10.1016/j.drudis.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 7.Lee HK, Choi EB, Pak CS. The current status and future perspectives of studies of cannabinoid receptor 1 antagonists as anti-obesity agents. Curr Top Med Chem. 2009;9:482–503. doi: 10.2174/156802609788897844. [DOI] [PubMed] [Google Scholar]
- 8.Kunos G, Tam J. The case for peripheral CB(1) receptor blockade in the treatment of visceral obesity and its cardiometabolic complications. Br J Pharmacol. 2011;163:1423–31. doi: 10.1111/j.1476-5381.2011.01352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fulp A, Bortoff K, Zhang Y, Seltzman H, Mathews J, Snyder R, Fennell T, Maitra R. Diphenyl purine derivatives as peripherally selective cannabinoid receptor 1 antagonists. J Med Chem. 2012;55:10022–32. doi: 10.1021/jm301181r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griffith DA, Hadcock JR, Black SC, Iredale PA, Carpino PA, DaSilva-Jardine P, Day R, DiBrino J, Dow RL, Landis MS, O’Connor RE, Scott DO. Discovery of 1-[9-(4-chlorophenyl)-8-(2-chlorophenyl)-9H-purin-6-yl]-4-ethylaminopiperidine-4-carboxylic acid amide hydrochloride (CP-945,598), a novel, potent, and selective cannabinoid type 1 receptor antagonist. J Med Chem. 2009;52:234–7. doi: 10.1021/jm8012932. [DOI] [PubMed] [Google Scholar]
- 11.Fulp A, Bortoff K, Seltzman H, Zhang Y, Mathews J, Snyder R, Fennell T, Maitra R. Design and synthesis of cannabinoid receptor 1 antagonists for peripheral selectivity. J Med Chem. 2012;55:2820–34. doi: 10.1021/jm201731z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Gilliam A, Maitra R, Damaj MI, Tajuba JM, Seltzman HH, Thomas BF. Synthesis and biological evaluation of bivalent ligands for the cannabinoid 1 receptor. J Med Chem. 2010;53:7048–60. doi: 10.1021/jm1006676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chugh BP, Lerch JP, Yu LX, Pienkowski M, Harrison RV, Henkelman RM, Sled JG. Measurement of cerebral blood volume in mouse brain regions using micro-computed tomography. Neuroimage. 2009;47:1312–8. doi: 10.1016/j.neuroimage.2009.03.083. [DOI] [PubMed] [Google Scholar]
- 14.Edvinsson L, Nielsen KC, Owman C. Circadian rhythm in cerebral blood volume of mouse. Experientia. 1973;29:432–3. doi: 10.1007/BF01926763. [DOI] [PubMed] [Google Scholar]
- 15.Wiley JL, Martin BR. Cannabinoid pharmacological properties common to other centrally acting drugs. Eur J Pharmacol. 2003;471:185–193. doi: 10.1016/s0014-2999(03)01856-9. [DOI] [PubMed] [Google Scholar]
- 16.Compton DR, Aceto MD, Lowe J, Martin BR. In vivo characterization of a specific cannabinoid receptor antagonist (SR141716A): inhibition of delta 9-tetrahydrocannabinol-induced responses and apparent agonist activity. J Pharmacol Exp Ther. 1996;277:586–94. [PubMed] [Google Scholar]
- 17.Martin BR, Compton DR, Thomas BF, Prescott WR, Little PJ, Razdan RK, Johnson MR, Melvin LS, Mechoulam R, Ward SJ. Behavioral, biochemical, and molecular modeling evaluations of cannabinoid analogs. Pharmacol Biochem Behav. 1991;40:471–478. doi: 10.1016/0091-3057(91)90349-7. [DOI] [PubMed] [Google Scholar]