

Abstract
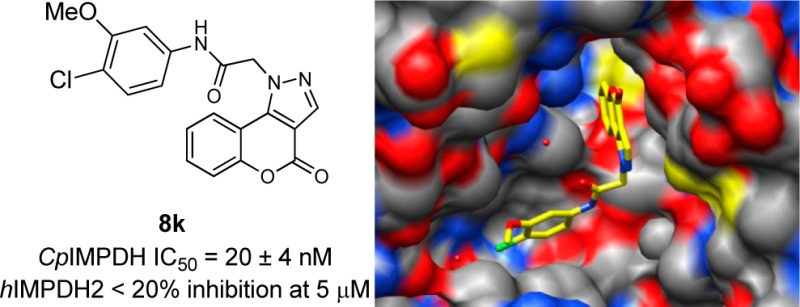
Cryptosporidium inosine 5′-monophosphate dehydrogenase (CpIMPDH) has emerged as a therapeutic target for treating Cryptosporidium parasites because it catalyzes a critical step in guanine nucleotide biosynthesis. A 4-oxo-[1]benzopyrano[4,3-c]pyrazole derivative was identified as a moderately potent (IC50 = 1.5 μM) inhibitor of CpIMPDH. We report a SAR study for this compound series resulting in 8k (IC50 = 20 ± 4 nM). In addition, an X-ray crystal structure of CpIMPDH·IMP·8k is also presented.
Introduction
Cryptosporidium parvum and Cryptosporidium hominis are intracellular protozoan parasites that invade the brush border epithelial cells of the small intestine. Cryptosporidiosis is prevalent in the developing world where it results in life-threatening diarrhea and severe malnutrition in children.1
Cryptosporidium oocysts are water-transmitted and highly resistant to water purification methods, also leading to significant disease burden in the developed world.2 Infections resolve in immunocompetent hosts but can be chronic and fatal in immunocompromised patients. Furthermore, because oocysts can readily be obtained and water supplies are relatively easily accessed, these organisms represent a credible bioterrorism threat.3 Currently, vaccine therapies against C. parvum and C. hominis are not available and the only approved drug, nitazoxanide, has an ill-defined mechanism of action and is not particularly effective.4 Thus, new chemotherapeutic agents are needed for the treatment of cryptosporidiosis.
One emerging molecular target for the treatment of cryptosporidiosis is the oxidoreductase inosine 5′-monophosphate dehydrogenase (IMPDH), which catalyzes the conversion of inosine-5′-monophosphate (IMP) into xanthosine-5′-monophosphate (XMP) as the rate-determining step in guanine nucleotide biosynthesis.5 Genomic analysis revealed that Cryptosporidium cannot synthesize purine nucleotides de novo.6−8 Instead, the parasite converts adenosine salvaged from the host into guanine nucleotides via a linear pathway dependent on IMPDH activity. Interestingly, these parasites appear to have obtained their IMPDH gene by lateral gene transfer from bacteria. Consequently, CpIMPDH is structurally distinct from mammalian IMPDH enzymes9 and is poorly inhibited by the prototypical human IMPDH inhibitor mycophenolic acid (CpIMPDH IC50 ∼ 10 μM; hIMPDH1 Ki = 33 nM; hIMPDH2 Ki ∼ 7 nM).10,11 These structural and mechanistic differences also provide an opportunity to design selective CpIMPDH inhibitors as therapeutic agents for treating cryptosporidiosis.12CpIMPDH inhibitors may also be effective against bacterial infections.13,14
Previously, we have reported the optimization of several structurally distinct compound series, including C64 and Q21,15−18 as well as the first demonstration of in vivo efficacy of a CpIMPDH inhibitor (e.g., P131) in a mouse model of cryptosporidiosis (Figure 1).19 This later study also revealed several additional hurdles required in the development of efficacious compounds, including preferential compound distribution to gastrointestinal enterocytes (as opposed to systemic distribution) and minimizing the impact of IMPDH inhibition on gut microbiome populations. The study reported herein is a continuation of our effort to identify and optimize structurally distinct CpIMPDH inhibitors and to develop a common pharmacophore as a guide for the future design of additional CpIMPDH inhibitors.
Figure 1.

Structures of previously described inhibitors C64 and Q21 that have been cocrystallized with CpIMPDH, P131 that demonstrated in vivo efficacy in a cryptosporidiosis animal model, and a new inhibitor 8a identified by HTS.
Our current structure–activity relationship (SAR) study was initiated based on 4-oxo-N-(3-methoxyphenyl)-[1]benzopyrano[4,3-c]pyrazole-1(4H)-acetamide (8a, Figure 1), identified by high throughput screening, as a moderately potent CpIMPDH inhibitor (IC50 = 1.5 ± 0.2 μM).
Results and Discussion
Chemistry
4-Oxo-[1]benzopyrano[4,3-c]pyrazole analogues (8a–n and 13a–f) were prepared using four general synthetic methods. The synthesis of analogues 8a–k used the methodology shown in Scheme 1 (method A). Anilines 2a–k were treated with bromoacetyl chloride, 3, in CH2Cl2 in the presence of K2CO3 to afford aryl amides 4a–k, which were treated with t-butyl carbazate in aqueous KHCO3 to provide the N-Boc-protected hydrazines 5a–k via an SN2 reaction. In the next step, trifluoroacetic acid was used to remove the t-butyl carbamate protecting group in 5a–k to give 6a–k, which were used without purification. The hydrazines 6a–k were refluxed in ethanol with 4-chloro-3-formylcoumarin (7a) in the presence of a catalytic amount of acetic acid to provide analogues 8a–k. The presence of the acid proved crucial for these reactions.20 The regioisomeric [1]benzopyrano[4,3-c]pyrazol-4(2H)-one derivative 9c was prepared using the methodology outlined in Scheme 2 (method B). 4-Hydroxycoumarin (9a) was treated with POCl3 and DMF, similar to standard Vilsmeier–Haack conditions, but at room temperature. The reaction was terminated by the addition of aqueous Na2CO3, which generated product 9b. Upon reaction with 6a in ethanol in the presence of DIPEA, the regioisomeric pyrazole 9c was obtained. Presumably, the terminal NH2 of hydrazine 6a condensed with the carbonyl of the vinylogous amide of 9b, which was followed by cyclization via an addition–elimination reaction to generate the isolated product.21
Scheme 1. Synthesis of 4-Oxo-[1]benzopyrano[4,3-c]pyrazole Derivatives 8a–k (Method A).

Reagents and conditions: (a) bromoacetyl chloride (3), K2CO3, CH2Cl2, 0 °C to rt; (b) t-butyl carbazate, KHCO3, EtOAc/H2O (1:2), 85 °C, 5 h; (c) TFA in CH2Cl2 (1:4), 2 h; (d) 4-chloro-3-formylcoumarin (7a), AcOH (cat), EtOH, 105 °C, 20 min.
Scheme 2. Synthesis of Regioisomers 9c (Method B).

Reagents and conditions: (a) POCl3, DMF, 1,2-dichloroethane, rt, 12 h, then saturated aqueous Na2CO3; (b) t-butyl carbazate, KHCO3, ethyl acetate, 85 °C, 5 h, then TFA in CH2Cl2 (1:4), 2 h, rt; (c) DIPEA, EtOH, rt, 12 h.
The preparation of 8l–n, as analogues of 8k with additional substituents on the acetamide and [1]benzopyrano[4,3-c]pyrazole, is outlined in Scheme 3 (method C). Anilines 2l or 2k were treated with 3a or 3b to afford aryl amides 4l or 4m. A stronger base (e.g., K2CO3), organic solvent (e.g., acetone), and the presence of potassium iodide were required to displace the primary chloride of 4l to furnish 5l. In the case of 5m, DIPEA in toluene proved effective. The required intermediate 7b was synthesized from the corresponding 4-hydroxycoumarin following typical Vilsmeier–Haack conditions (see Experimental Section). Analogues 8l–n were synthesized from acid-catalyzed cyclization of hydrazines (5l–m des-Boc intermediates) and 7a or 7b using the same method described in Scheme 1.
Scheme 3. Synthesis of 4-Oxo-[1]benzopyrano[4,3-c]pyrazole Analogue 8l–n (Method C).

Reagents and conditions: (a) 21 (4-Cl-3-OMePhNHMe) or 2k (4-Cl-3-OMePhNH2), K2CO3, CH2Cl2, 0 °C to rt; (b) for 4l, t-butyl carbazate, K2CO3, KI, acetone, 65 °C, 18 h or for 4m, t-butyl carbazate, DIPEA, toluene 105 °C, 16 h; (c) TFA in CH2Cl2 (1:4), 2 h, then 7a (or 4-chloro-3-formyl-7-methylcoumarin, 7b), AcOH (cat.), EtOH, 105 °C, 20 min.
Initial attempts to synthesize analogues 13a–f following the methodology outlined in Scheme 1 (method A) proved problematic. Thus, an alternate method was developed that is shown in Scheme 4 (method D). Aldehydes 7a and 10a–b were refluxed in the presence of ethyl hydrazinoacetate hydrochloride and a catalytic amount of acetic acid in ethanol to afford pyrazoles 11a–c. Hydrolysis of the ester using 2 M aqueous LiOH in THF afforded acids 12b–c. In the case of 12a, the lactone ring also opened during this step and required relactonization using EDC and TEA in DMF. The acids 12a–c were treated with either 4-chloro-3-methoxyaniline or heterocyclic anilines in the presence of HBTU and DIPEA in DMF to afford analogues 13a–f. Intermediates 10a–b were again prepared using Vilsmeier–Haack reactions (see Experimental Section).
Scheme 4. Synthesis of Derivatives 13a–f (Method D).

Reagents and conditions: (a) ethyl hydrazinoacetate hydrochloride (14), AcOH (cat), EtOH, 105 °C, 0.5–2 h; (b) 12a, LiOH (aq), THF, rt, 12 h then EDC, TEA, DMF, 12b–c, aqueous LiOH, THF, rt, 12 h; (c) NH2R, HBTU, DIPEA, DMF, rt, 12 h.
Predicted Binding Mode of Inhibitor 8a with CpIMPDH·IMP
Inhibitor 8a was docked into the binding site observed in one of our previously reported crystal structures of the catalytic domain of CpIMPDH (PDB code: 4IXH)15 using AutoDock Tools 1.5.6. The top 10 binding conformations were examined, and the two best conformations (binding energies of −7.86 and −7.76 kcal/mol, respectively) were selected based on similarity of their binding modes with Q21,15 including π-interactions between the 4-oxo-[1]benzopyrano[4,3-c]pyrazole with the hypoxanthine of IMP and the 3-methoxyphenyl with Y358′ (where prime denotes a residue from the adjacent subunit). However, the hydrogen atom of the amide for these two conformations formed ionic–dipole interactions with two different oxygen atoms in the side chain of E329. Therefore, the conformation that formed an interaction similar to Q21 was selected as the predicted binding mode for the N-series and is shown in Figure 2.
Figure 2.
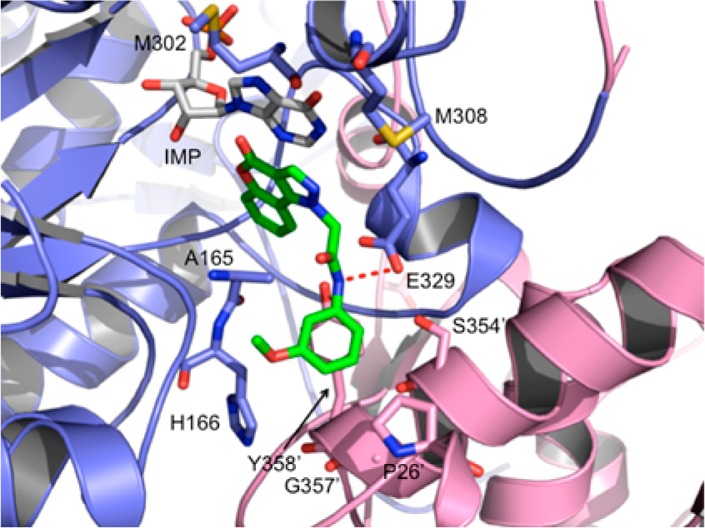
Predicted binding mode of 8a (green) in complex with CpIMPDH·IMP. The red dotted line indicates an ionic–dipole interaction between the amide of 8a and the side chain of E329. The phenyl ring of 8a is stacked above Y358′.
Evaluation of CpIMPDH Inhibition
Biological characterization of the 4-oxo-[1]benzopyrano[4,3-c]pyrazole derivatives was performed following our published procedures.15,16CpIMPDH was expressed and purified as previously reported.22−24 Enzymatic activity was monitored by NADH production.12 IC50 values were determined by averaging the results of three independent experiments unless otherwise noted.
The regioisomeric derivative 9c did not inhibit CpIMPDH, indicating that the relative orientation of the anilide on the fused pyrazole was crucial for inhibitory activity (Table 1). Next, the SAR study focused on the monosubstituted aniline moiety of 8a. Analogues with a 2-chloro substituent (8c) showed no inhibitory activity. Replacement of the 3-methoxy with a chlorine (8b) likewise led to loss of inhibition. However, the 4-fluoro and 4-trifluoromethyl derivatives (8d and 8e) displayed a 3-fold increase in activity, indicating that monosubstitution on the amide phenyl moiety could provide only moderate increases in potency, similar to our observation with other inhibitor series.15,16,18 Therefore, disubstituted and fused anilines were examined. The 2,4-dichloro substituted analogue 8g showed no inhibition activity (Table 2). In light of this finding, combined with the result of 8c, it appeared that an ortho-chloro was not well tolerated in the binding pocket, possibly due to a clash with Y358′. However, the 3,4-dichloro analogue 8f displayed significantly improved potency. A further increase in potency was achieved by replacing the 3-chloro substituent with a methoxy (8k, CpIMPDH IC50 = 20 ± 4 nM). However, replacing the remaining chloro of 8k with another methoxy (8i) resulted in the loss of enzyme inhibition. Tethering the ethers into a dioxane ring (8h) also resulted in a significantly lower IC50 value compared to 8k. The loss of activity for these two derivatives containing electron donating groups in the para-position of the anilide is potentially due to weakening of the H-bond donating ability of the amide NH, which is a critical interaction with E329 observed in cocrystal structures of other CpIMPDH inhibitors.15,25 However, the naphthyl substituted analogue 8j demonstrated an IC50 value of 67 nM.
Table 1. Monosubstituted N-Phenyl 4-Oxo-[1]benzo Pyrano[4,3-c]pyrazole-1(4H)-acetamide Derivatives for CpIMPDH Inhibition.
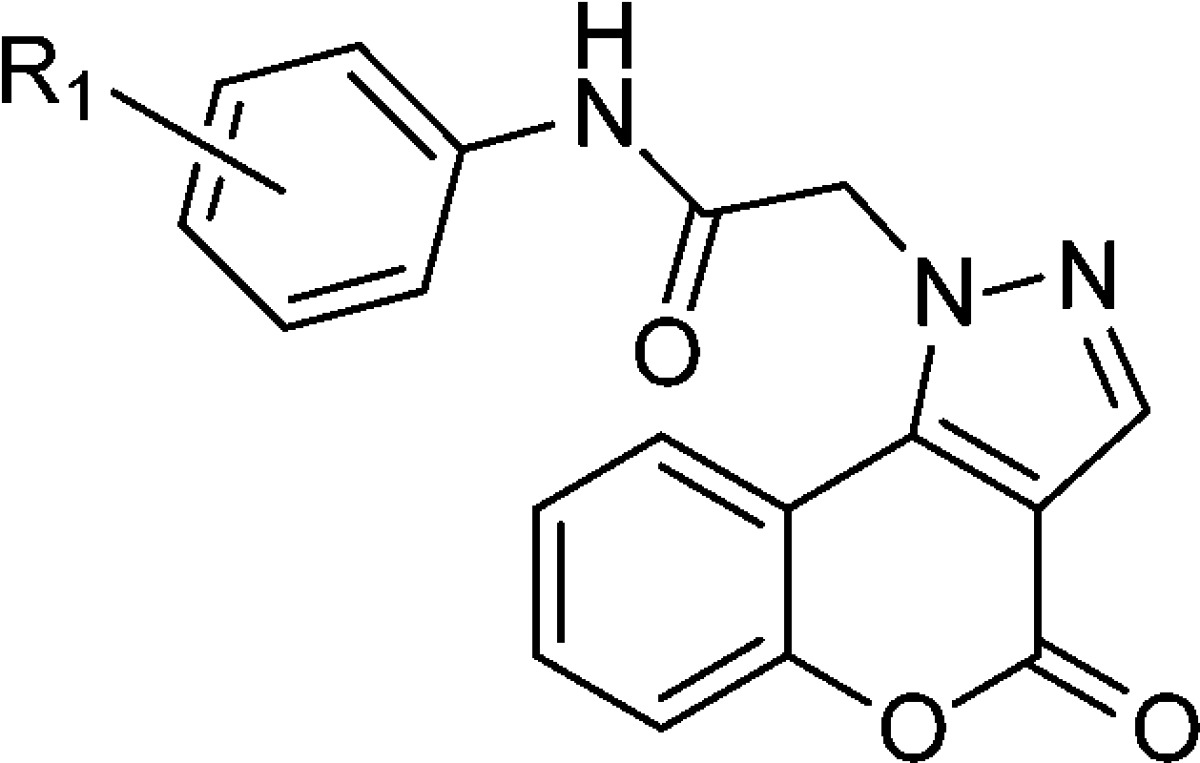
| ID | method | R1 | IC50 (nM) |
|---|---|---|---|
| 9c | B | >5000 | |
| 8a | A | 3-OMe | 1500 ± 200 |
| 8b | A | 3-Cl | >5000 |
| 8c | A | 2-Cl | >5000 |
| 8d | A | 4-F | 460 ± 95 |
| 8e | A | 4-CF3 | 580 ± 130 |
Table 2. Disubstituted N-Phenyl and N-Heteroaryl 4-Oxo-[1]benzopyrano[4,3-c]pyrazole-1(4H)-acetamide Derivatives for CpIMPDH Inhibition.
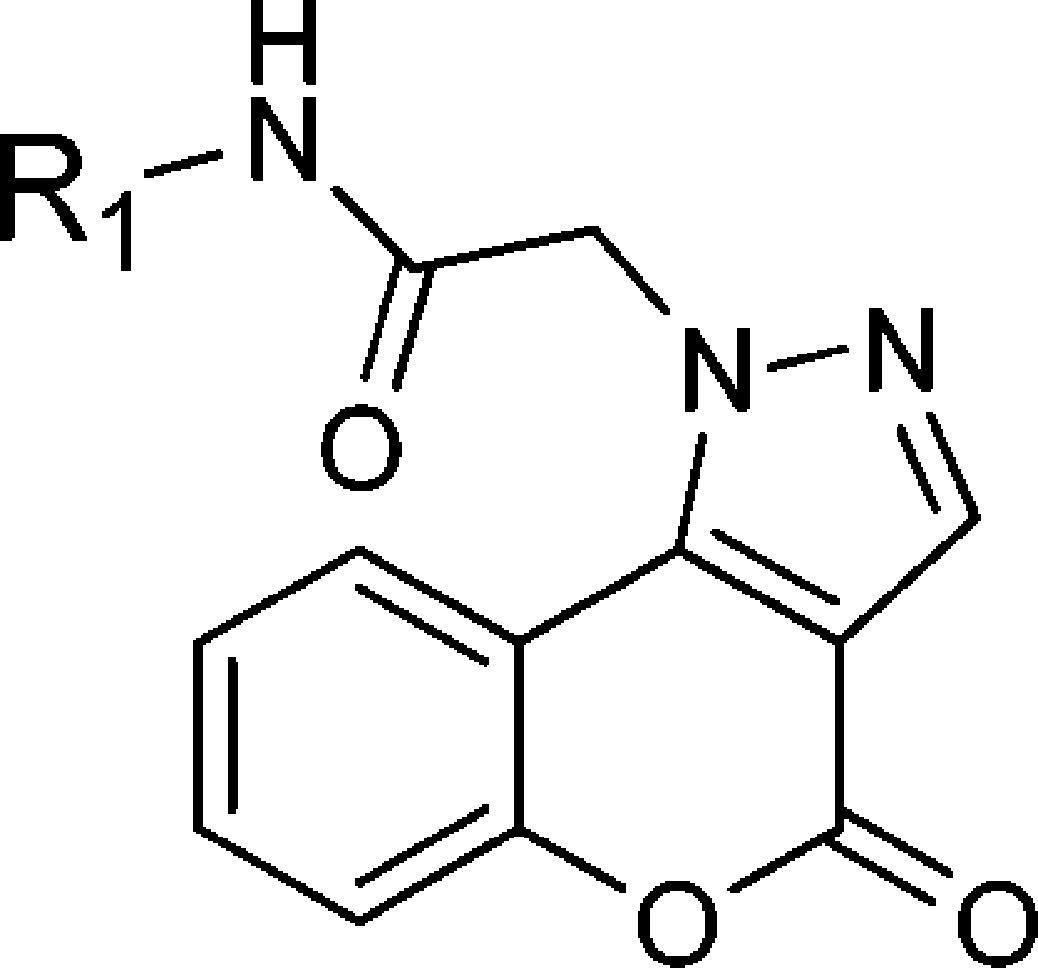
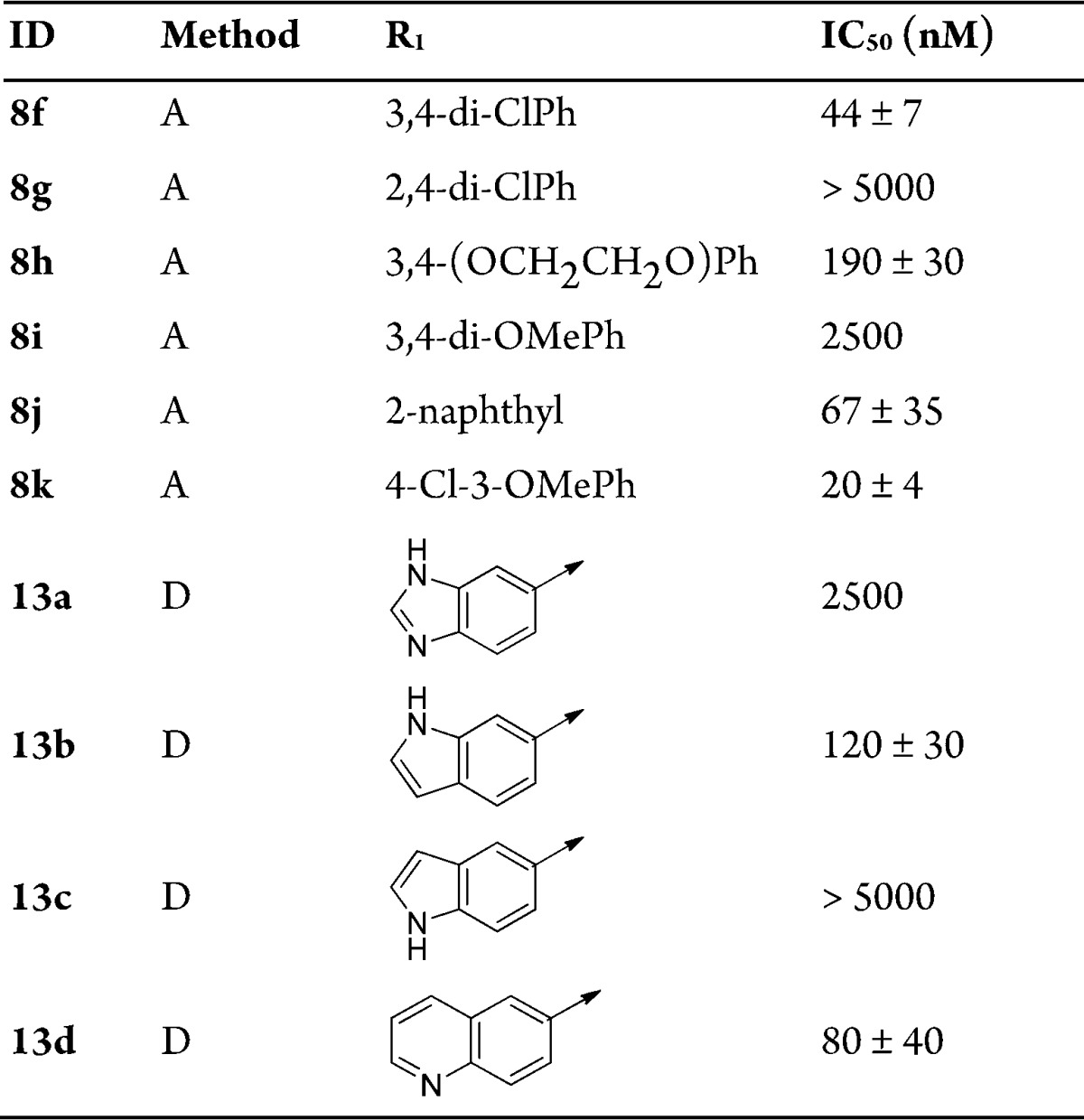
On the basis of these results, bioisosteres of the naphthyl were investigated. Benzo[d]imidazole 13a and 1H-indol-5-yl 13c had only weak or no inhibitory activity. However, the (1H)-indole 13b and quinolin-6-yl 13d showed moderate inhibition (CpIMPDH IC50 = 120 ± 30 and 80 ± 40 nM, respectively), albeit less than 8j.
Further modifications were performed on 8k that retained the 3-methoxy-4-chloro phenyl moiety (Table 3). Addition of a methyl group on the amide nitrogen (8l) resulted in loss of activity, again indicating the importance of the H-bond donor function of the amide NH. Addition of a methyl group on the methylene position (8m) resulted in a 3-fold loss of potency, likely due to a steric clash with the side chain of E329. The addition of a methyl to the 7-position of the 4-oxo-[1]benzopyrano[4,3-c]pyrazole ring system (8n) was also detrimental, revealing the steric limitation of this region of the molecule, likely the result of clashes with either M302 or the ribose of IMP. Replacing the lactone ring with an ether bridge (13e) resulted in a 3-fold loss of inhibition, indicating the carbonyl contributes to binding. Replacement of the lactone with an ethylene bridge (13f) resulted in a further decrease of potency. Therefore, the lactone appears essential for CpIMPDH inhibition, although the rationale for this observation was not obvious from the docking model.
Table 3. Modifications of the Acetamide and 4-Oxo-[1]benzopyrano[4,3-c]pyrazole Regions of 8k for CpIMPDH Inhibition.
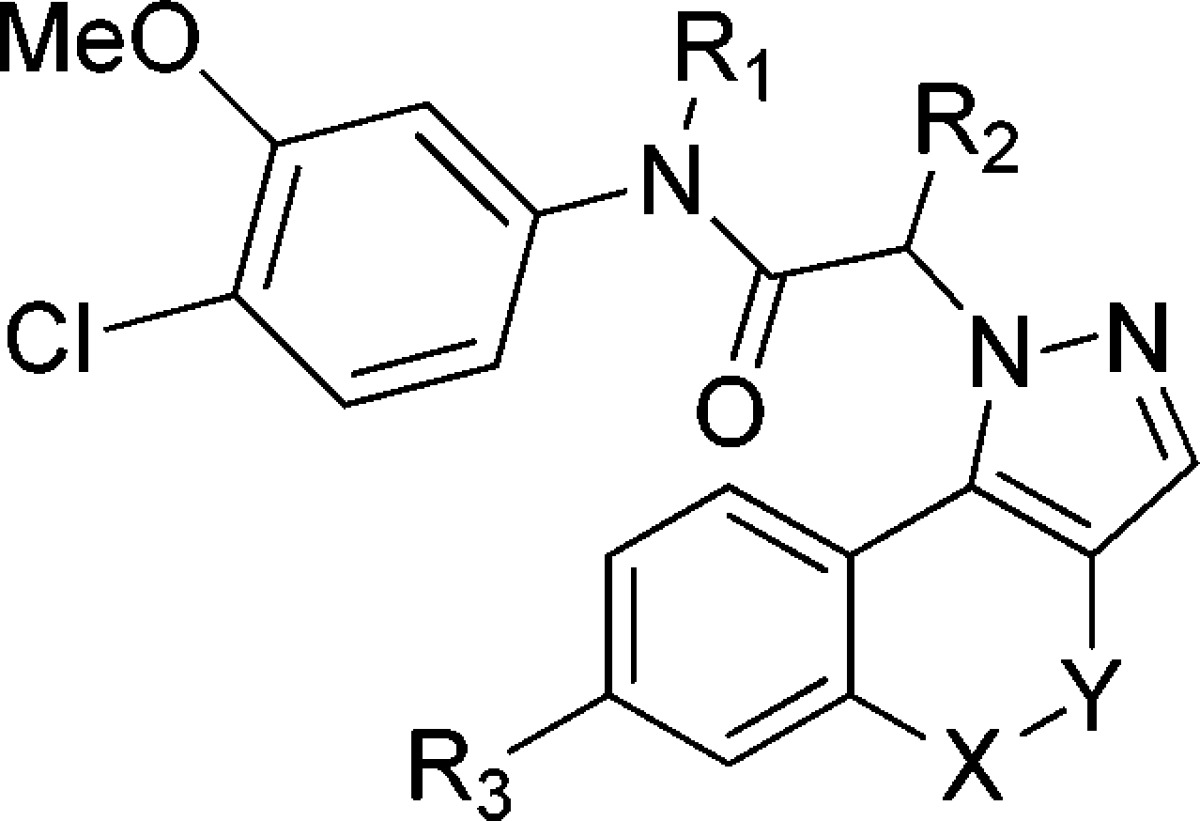
| ID | method | R1 | R2 | R3 | X | Y | IC50(nM) |
|---|---|---|---|---|---|---|---|
| 8k | A | H | H | H | O | C=O | 20 ± 4 |
| 8l | C | Me | H | H | O | C=O | >5000 |
| 8m | C | H | Me | H | O | C=O | 63 ± 11 |
| 8n | C | H | H | Me | O | C=O | 2000 |
| 13e | D | H | H | H | O | CH2 | 70 ± 27 |
| 13f | D | H | H | H | CH2 | CH2 | 100 ± 30 |
The original screening hit (8a) in addition to 11 other CpIMPDH inhibitors (e.g., 8d–f, 8h, 8j, 8k, 8m, 13b, and 13d–f) failed to inhibit hIMPDH2 (<20% inhibition at 5 μM), which also has high sequence identity (85%) to hIMPDH1. These results demonstrated that CpIMPDH inhibitory potency could be increased, while preserving selectivity against a human orthologue.
Crystal Structure of CpIMPDH·8k·IMP
The structure of a CpIMPDH complex with IMP and 8k was solved at 2.40 Å resolution using molecular replacement with the structure of apo CpIMPDH (PDB code: 3FFS)25 as the search model. Like the structures of CpIMPDH·IMP in complex with other inhibitors,15,25 the 4-oxo-[1]benzopyrano[4,3-c]pyrazole-based inhibitor interacts with residues from two adjacent subunits. One aromatic moiety of 8k, the 4-oxo[1]benzopyrano[4,3-c]pyrazole, π-stacks with the hydroxanthine of IMP (Figure 3A) in an interaction similar to that observed previously for two other CpIMPDH complexes and as predicted in the docking of 8a.15,26 The pyrazole portion interacts with the side chain of M308 and forms n−π* contacts between the carbonyl group of the 4-oxo[1]benzopyrano-[4,3-c]pyrazole moiety and the main chain carbonyl of M302.26 This latter interaction was not predicted in the docking model but provides an explanation for the importance of the lactone. The remaining portion of the inhibitor circumvents A165 and extends into the pocket formed at the subunit interface. The amide NH of 8k forms a H-bond with a side chain oxygen atom of E329 and is part of the extensive H-bonding network involving T221, S354′, and Y358′. Another common feature observed in all CpIMPDH inhibitor complexes is the interaction of the second aromatic moiety of the inhibitor with the side chain of Y358′. In the case of 8k, this interaction is observed for the 4-chloro-3-methoxyphenyl. In addition, this moiety is involved in contacts with H166 and P26′ via polar and van der Waals interactions. Inhibitor 8k does not extend as deep into the cavity formed at the subunit interface as does the 2-(4-pyridyl)benzoxazole derivative Q21 (Figure 3B).15 However, similarly to the bromo substituent of inhibitor C64,25 the chloro substituent of 8k is contacting the main chain carbonyl oxygen atom of G357′ (Figure 3C).
Figure 3.
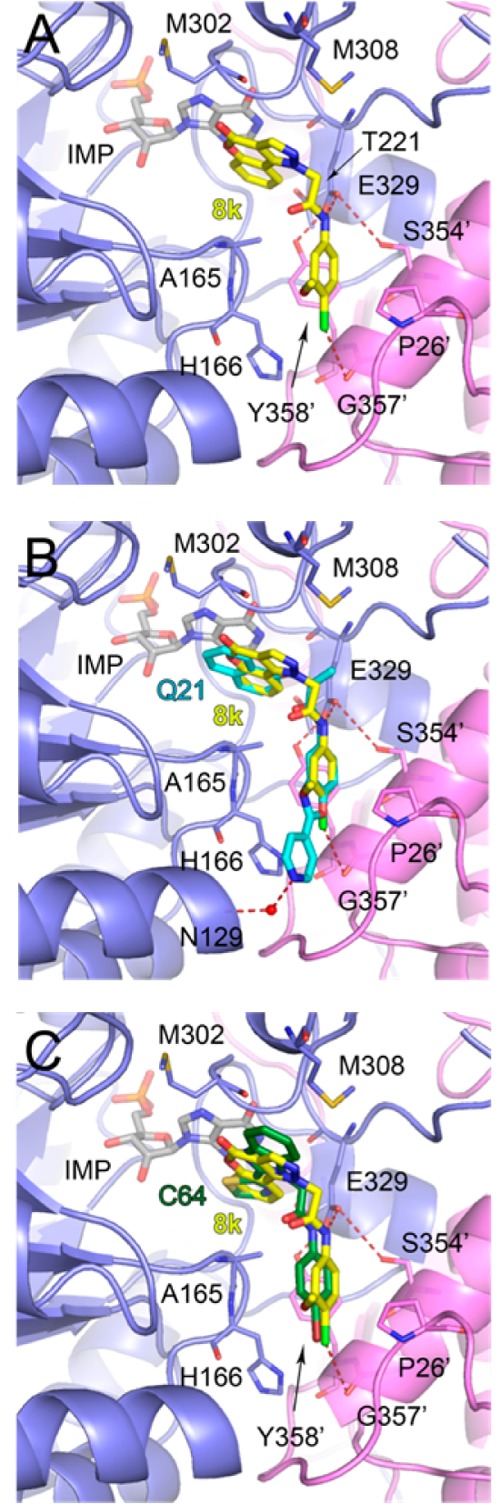
(A) Structure of CpIMPDH·IMP·8k. (B) Overlay of CpIMPDH structures with 8k and Q21. (C) Overlay of CpIMPDH structures with 8k and C64. In all panels, chains A (slate) and D (violet) are shown in cartoon representations. Residues involved in IMP and inhibitor binding are shown as sticks. IMP (light gray), 8k (yellow), C64 (green), and Q21 (teal) are shown as sticks. Hydrogen and halogen bonds are depicted as red dashed lines and a water molecule as a red sphere. Prime indicates residues from an adjacent monomer.
Conclusion
An SAR study of CpIMPDH inhibitor 8a was conducted with guidance from an in silico docking model based on a previously crystallized CpIMPDH inhibitor complex. The orientation of the anilide on the fused pyrazole was crucial, a 4-chloro-3-methoxy substitution on the anilide (e.g., 8k) achieved the greatest potency among this series of derivatives, and the secondary amide and the lactone of the 4-oxo-[1]benzopyrano[4,3-c]pyrazole were vital for binding. Overall, this study provides a structurally distinct inhibitor series that will further assist in the continuing development of CpIMPDH inhibitors for the treatment of cryptosporidiosis and possibly other infectious diseases.13,14 Finally, a crystal structure of CpIMPDH·IMP and 8k (e.g., N109) provides further support for a general binding mode of CpIMPDH inhibitors featuring three key interactions: (i) π-interaction between an aryl/heteroaryl moiety and the hydroxanthine of IMP, (ii) H-bond with E329, and (iii) extension of an aryl/herteroaryl group into an adjacent subunit forming interactions with Y358′. This pharmacophore provides a template that can be extended to the discovery of other structurally distinct chemical scaffolds of selective CpIMPDH inhibitors.
Experimental Section
Chemistry
All test compounds had a purity ≥95% as determined by HPLC analyses.
General Procedure for 4a–m
To a suspension of 2a–l (6.5 mmol) in 15 mL of CH2Cl2 at 0 °C, acyl halides (3 or 3a–b, 8.5 mmol) and K2CO3 (1.25 g, 9.1 mmol) were added. The reaction was maintained at 0 °C for 20 min and then allowed to warm to rt. Saturated aqueous NaHCO3 was added. The reaction mixture was extracted with CH2Cl2, and the organic layer was washed by brine, dried over anhydrous Na2SO4, filtered, and evaporated in vacuo. The product was purified by column chromatography (0–50% EtOAc in hexane) to afford 4a–m.
General Procedure for 5a–k
Compound 4a–k (5.6 mmol), t-butyl carbazate (11.2 mmol), and KHCO3 (16.8 mmol) were suspended in 30 mL of EtOAC and H2O (1:2). The mixture was refluxed at 85 °C for 5 h. The mixture was allowed to cool to rt, and the crude product was extracted with EtOAC, dried with anhydrous MgSO4, filtered, and concentrated. The product was purified by column chromatography (10–70% EtOAc in hexane).
t-Butyl 2-(2-((4-Chloro-3-methoxyphenyl)(methyl)amino)-2-oxoethyl)hydrazinecarboxylate (5l)
Compound 4l (1 mmol), t-butyl carbazate (2 mmol), K2CO3 (3 mmol), and KI (2 mmol) were suspended in 20 mL of acetone. The mixture was heated at 65 °C for 18 h, allowed to cool to rt, and then evaporated to dryness. The residue was diluted with aqueous NH4Cl and extracted with EtOAc (20 mL × 3). The organic layer was dried over anhydrous MgSO4, filtered, and concentrated. The product was purified by silica gel column chromatography (5% EtOAc in CH2Cl2) to give 5l as an oil (66% yield).
t-Butyl 2-(1-((4-Chloro-3-methoxyphenyl)amino)-1-oxopropan-2-yl)hydrazinecarboxylate (5m)
Compound 4m (1.7 mmol), t-butyl carbazate (3.3 mmol), and DIPEA (3.3 mmol) were suspended in 10 mL of toluene. The mixture was refluxed at 105 °C for 16 h, allowed to cool to rt, and then evaporated to dryness. The residue was diluted with aqueous NH4Cl and extracted with EtOAc (20 mL × 3). The organic layer was dried over anhydrous MgSO4, filtered, and concentrated. The crude product was purified by silica gel chromatography (0–50% EtOAc in hexane) to give 5m as a solid (83% yield).
General Procedures for 8a–n
Compounds 5a–m (0.8 mmol) were treated with 2.5 mL of TFA in CH2Cl2 (1:4) at rt for 2 h. The mixture was evaporated in vacuo to afford 6a–m as colorless oils. The intermediates 6a–m were dissolved in 2 mL of EtOH, which was then added to a suspension of 7a–b (0.8 mmol) in 2 mL of EtOH. Acetic acid (20 μL) was added. The mixture was refluxed at 105 °C for 20 min then allowed to cool to rt. The mixture was evaporated in vacuo, partitioned between aqueous NaHCO3 and EtOAc. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated. The products 8a–n was collected after silica gel chromatography using 20–85% EtOAc in hexane as eluent.
3-[(Dimethylamino)methylene]chroman-2,4-dione (9b)
4-Hydroxycoumarin (9a, 9.2 mmol) in 1.5 mL of DMF added to 10 mL of 1,2-dichloroethane. To this solution was added 1.1 mL of POCl3. The mixture was stirred at rt for 12 h. Next, saturated aqueous Na2CO3 was added. The reaction mixture was extracted with EtOAc, dried over anhydrous MgSO4, filtered, and concentrated. The crude product was purified by silica gel chromatography (0–25% EtOAc in CH2Cl2) to give 9b (48% yield) as a yellow solid.
N-(3-Methoxyphenyl)-[1]benzopyrano[4,3-c]pyrazol-4(2H)-one (9c)
Intermediate 6a (2.1 mmol, prepared from 5a following the procedure described above) was dissolved in 3 mL of EtOH then was added to 9b (2.1 mmol) suspended in 3 mL of EtOH. To this mixture was added DIPEA (8.4 mmol). The reaction mixture was stirred at rt for 12 h and then evaporated in vacuo. The crude product was partitioned between aqueous NH4Cl and EtOAc. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated. The material was purified by silica gel chromatography using 0–20% EtOAc in CH2Cl2 as eluent to give 9c as a white solid (56% yield).
General Procedure for 11a–c
Compounds 7a or 10a–b (8.6 mmol) were suspended in 15 mL of EtOH with ethyl hydrazinoacetate hydrochloride (9.5 mmol). Acetic acid (30 μL) was added. The mixture was refluxed at 105 °C for 30 min (for 11a) or 2 h (for 11b–c) and then allowed to cool to rt. The reaction mixture was evaporated in vacuo, partitioned between aqueous NaHCO3 and EtOAc. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated. The crude product was purified by silica gel chromatography using 20–75% EtOAc in hexane as eluent to give products 11a–c.
[1]Benzopyrano[4,3-c]pyrazole-1(4H)acetic Acid (12a)
Compound 11a (7.3 mmol) was dissolved in 30 mL of THF, and then 18 mL of aqueous LiOH (2M) was added. The mixture was stirred at rt for 12 h, and then 5% aqueous HCl was added until the pH = 2. The mixture was evaporated to dryness, and then the residue was dissolved in EtOAc, concentrated to dryness, and used without further purification. The intermediate was dissolved in 8 mL of DMF, and then 3.6 mL of TEA and 1.23 g of EDC were added in order to reform the lactone. The mixture was stirred at rt for 18 h, and then 5% aqueous HCl was added until the pH = 2. The mixture was evaporated in vacuo, partitioned between EtOAc and aqueous NH4Cl. The organic layer was evaporated in vacuo to give 12a, which was used without further purification (75% yield).
General Procedure for 12b–c
Compound 11b–c (4.6 mmol) was dissolved in 15 mL of THF, and 9 mL of aqueous LiOH (2M) was added. The mixture was stirred at rt for 12 h, then 5% aqueous HCl was added until pH = 2. The mixture was evaporated in vacuo. The material was partitioned between EtOAc and H2O. The organic layer was evaporated and 12b–c used without further purification.
General Procedure for 13a–f
Acids 12a–c (1.3 mmol), anilines (1.2 mmol), and DIPEA (5.2 mmol) were dissolved in 6 mL of DMF. To this solution was added HBTU (1.3 mmol). The mixture was stirred at rt for 12 h. Aqueous NH4Cl (20 mL) was added and mixture extracted with EtOAc (20 mL × 3). The organic extracts were combined, dried over anhydrous MgSO4, filtered, and concentrated to give material that was purified by silica gel chromatography using 0–100% EtOAc in hexane as eluent or recrystallization in 70% EtOAc in hexane to give 13a–f.
Acknowledgments
This work was supported by the National Institute of Allergy and Infectious Diseases (U01AI075466 and R01AI93459 to L.H., R56AI106743 to G.D.C. and Contracts HHSN272200700058C and HHSN272201200026C to the Center for Structural Genomics of Infectious Diseases, University of Chicago, IL) and the New England Regional Center of Excellence for Biodefense and Emerging Infectious Diseases. We also thank Minyi Gu for help with gene cloning. The use of the 19-ID beamline at the Structural Biology Center at the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357.
Glossary
Abbreviations Used
- Cp
Cryptosporidium parvum
- DIPEA
N,N-diisopropylethylamine
- IMP
inosine 5′-monophosphate
- IMPDH
IMP dehydrogenase
- ND
not determined
- HBTU
N,N,N,N-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
- EDC
1-ethyl-3-(3-dimethylamino-propyl)carbodiimide
- rt
room temperature
- TEA
triethylamine
- TFA
trifluoroacetic acid
- XMP
xanthosine 5′-monophosphate
Supporting Information Available
Procedures for 2l, 7b, and 10a–b, compound characterization, IC50 determinations, gene cloning, protein expression, crystallization, and statistics for data collection/refinement of the X-ray crystal structure. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
PDB ID code: 4QJ1.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Striepen B. Parasitic infections: time to tackle cryptosporidiosis. Nature 2013, 503, 189–191. [DOI] [PubMed] [Google Scholar]; b Guerrant R. L.; Oria R. B.; Moore S. R.; Oria M. O.; Lima A. A. Malnutrition as an enteric infectious disease with long-term effects on child development. Nutr. Rev. 2008, 66, 487–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayer R. Cryptosporidium: a water-borne zoonotic parasite. Vet. Parasitol. 2004, 126, 37–56. [DOI] [PubMed] [Google Scholar]
- DuPont H. L.; Chappell C. L.; Sterling C. R.; Okhuysen P. C.; Rose J. B.; Jakubowski W. The infectivity of Cryptosporidium parvum in healthy volunteers. N. Engl. J. Med. 1995, 332, 855–859. [DOI] [PubMed] [Google Scholar]
- Mead J. R. Cryptosporidiosis and the challenges of chemotherapy. Drug Resist. Updates 2002, 5, 47–57. [DOI] [PubMed] [Google Scholar]
- Hedstrom L. IMP dehydrogenase: structure, mechanism, and inhibition. Chem. Rev. 2009, 109, 2903–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striepen B.; Pruijssers A. J.; Huang J.; Li C.; Gubbels M. J.; Umejiego N. N.; Hedstrom L.; Kissinger J. C. Gene transfer in the evolution of parasite nucleotide biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 3154–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P.; Widmer G.; Wang Y.; Ozaki L. S.; Alves J. M.; Serrano M. G.; Puiu D.; Manque P.; Akiyoshi D.; Mackey A. J.; Pearson W. R.; Dear P. H.; Bankier A. T.; Peterson D. L.; Abrahamsen M. S.; Kapur V.; Tzipori S.; Buck G. A. The genome of Cryptosporidium hominis. Nature 2004, 431, 1107–1112. [DOI] [PubMed] [Google Scholar]
- Abrahamsen M. S.; Templeton T. J.; Enomoto S.; Abrahante J. E.; Zhu G.; Lancto C. A.; Deng M.; Liu C.; Widmer G.; Tzipori S.; Buck G. A.; Xu P.; Bankier A. T.; Dear P. H.; Konfortov B. A.; Spriggs H. F.; Iyer L.; Anantharaman V.; Aravind L.; Kapur V. Complete genome sequence of the apicomplexan Cryptosporidium parvum. Science 2004, 304, 441–445. [DOI] [PubMed] [Google Scholar]
- Striepen B.; White M. W.; Li C.; Guerini M. N.; Malik S. B.; Logsdon J. M. Jr.; Liu C.; Abrahamsen M. S. Genetic complementation in apicomplexan parasites. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 6304–6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison A. C.; Kowalski W. J.; Muller C. D.; Eugui E. M. Mechanisms of action of mycophenolic acid. Ann. N. Y. Acad. Sci. 1993, 696, 63–87. [DOI] [PubMed] [Google Scholar]
- Allison A. C.; Eugui E. M. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 2000, 47, 85–118. [DOI] [PubMed] [Google Scholar]
- Umejiego N. N.; Gollapalli D.; Sharling L.; Volftsun A.; Lu J.; Benjamin N. N.; Stroupe A. H.; Riera T. V.; Striepen B.; Hedstrom L. Targeting a prokaryotic protein in a eukaryotic pathogen: identification of lead compounds against cryptosporidiosis. Chem. Biol. 2008, 15, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom L.; Liechti G.; Goldberg J. B.; Gollapalli D. R. The antibiotic potential of prokaryotic IMP dehydrogenase inhibitors. Curr. Med. Chem. 2011, 18, 1909–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandapati K.; Gorla S. K.; House A. L.; McKenney E. S.; Zhang M.; Rao S. N.; Gollapalli D. R.; Mann B. J.; Goldberg J. B.; Cuny G. D.; Glomski I. J.; Hedstrom L. Repurposing Cryptosporidium inosine 5′-monophosphate dehydrogenase inhibitors as potential antibacterial agents. ACS. Med. Chem. Lett. 2014, 5, 846–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorla S. K.; Kavitha M.; Zhang M.; Chin J. E.; Liu X.; Striepen B.; Makowska-Grzyska M.; Kim Y.; Joachimiak A.; Hedstrom L.; Cuny G. D. Optimization of benzoxazole-based inhibitors of Cryptosporidium parvum inosine 5′-mono phosphate dehydrogenase. J. Med. Chem. 2013, 56, 4028–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorla S. K.; Kavitha M.; Zhang M.; Liu X.; Sharling L.; Gollapalli D. R.; Striepen B.; Hedstrom L.; Cuny G. D. Selective and potent urea inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J. Med. Chem. 2012, 55, 7759–7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C. R.; Gorla S. K.; Kavitha M.; Zhang M.; Liu X.; Striepen B.; Mead J. R.; Cuny G. D.; Hedstrom L. Phthalazinone inhibitors of inosine-5′-monophosphate dehydrogenase from Cryptosporidium parvum. Bioorg. Med. Chem. Lett. 2013, 23, 1004–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirubakaran S.; Gorla S. K.; Sharling L.; Zhang M.; Liu X.; Ray S. S.; Macpherson I. S.; Striepen B.; Hedstrom L.; Cuny G. D. Structure–activity relationship study of selective benzimidazole-based inhibitors of Cryptosporidium parvum IMPDH. Bioorg. Med. Chem. Lett. 2012, 22, 1985–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorla S. K.; McNair N. N.; Yang G.; Gao S.; Hu M.; Jala V. R.; Haribabu B.; Striepen B.; Cuny G. D.; Mead J. R.; Hedstrom L. Validation of IMP dehydrogenase inhibitors in a mouse model of cryptosporidiosis. Antimicrob. Agents Chemother. 2014, 58, 1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leleti M. R.; Pennell A. M. K.; Thomas W. D.; Zhang P.. CXCR4 modulators. WO2007115231, 2007.
- Sosnovskikh V. Y.; Moshkin V. S.; Kodess M. I. On the reaction of 3-cyanochromones with phenyl- and methylhydrazines: Structural revision and a simple synthesis of chromeno[4,3-c]pyrazol-4-ones. J. Heterocycl. Chem. 2010, 47, 629–633. [Google Scholar]
- Farazi T.; Leichman J.; Harris T.; Cahoon M.; Hedstrom L. Isolation and characterization of mycophenolic acid-resistant mutants of inosine-5′-monophosphate dehydrogenase. J. Biol. Chem. 1997, 272, 961–965. [DOI] [PubMed] [Google Scholar]
- Mortimer S. E.; Hedstrom L. Autosomal dominant retinitis pigmentosa mutations in inosine 5′-monophosphate dehydrogenase type I disrupt nucleic acid binding. Biochem. J. 2005, 390, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umejiego N. N.; Li C.; Riera T.; Hedstrom L.; Striepen B. Cryptosporidium parvum IMP dehydrogenase: identification of functional, structural, and dynamic properties that can be exploited for drug design. J. Biol. Chem. 2004, 279, 40320–40327. [DOI] [PubMed] [Google Scholar]
- MacPherson I. S.; Kirubakaran S.; Gorla S. K.; Riera T. V.; D’Aquino J. A.; Zhang M.; Cuny G. D.; Hedstrom L. The structural basis of Cryptosporidium-specific IMP dehydrogenase inhibitor selectivity. J. Am. Chem. Soc. 2010, 132, 1230–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen F. H.; Baalham C. A.; Lommerse J. P. M.; Raithby P. R. Carbonyl–carbonyl interactions can be competitive with hydrogen bonds. Acta Crystallogr., Sect. B: Struct. Sci. 1998, B54, 320–329. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.