

Abstract
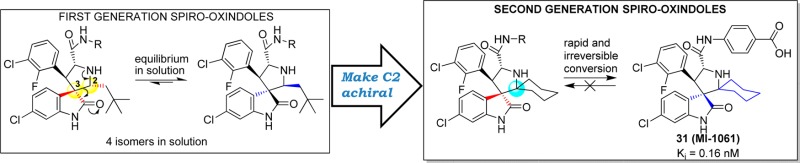
Inhibition of the MDM2–p53 protein–protein interaction is being actively pursued as a new anticancer therapeutic strategy, and spiro-oxindoles have been designed as a class of potent and efficacious small-molecule inhibitors of this interaction (MDM2 inhibitors). Our previous study showed that some of our first-generation spiro-oxindoles undergo a reversible ring-opening-cyclization reaction that, from a single compound in protic solution, results in an equilibrium mixture of four diastereoisomers. By exploiting the ring-opening-cyclization reaction mechanism, we have designed and synthesized a series of second-generation spiro-oxindoles with symmetrical pyrrolidine C2 substitution. These compounds undergo a rapid and irreversible conversion to a single, stable diastereoisomer. Our study has yielded compound 31 (MI-1061), which binds to MDM2 with Ki = 0.16 nM, shows excellent chemical stability, and achieves tumor regression in the SJSA-1 xenograft tumor model in mice.
Introduction
The powerful tumor suppressor p53 is a transcriptional factor and plays a key role in preventing tumor development. It is therefore not surprising that p53 function is compromised in most if not all human cancers. In approximately 50% of human cancers, the gene encoding p53 protein is mutated or deleted, which results in inactivation of its transcriptional activity and tumor suppressor function.1 In the other half of human cancers, p53 retains its wild-type status, but its function is inhibited by a variety of mechanisms.2 One major inhibitory mechanism is through the direct interaction between p53 and the human murine double-minute 2 (MDM2) protein.3−8 Overexpression of MDM2 by either gene amplification or other mechanisms has been observed in different types of human cancers.9 Furthermore, it has been observed that MDM2 gene amplification and p53 mutation are mutually exclusive in human cancers, highlighting a prominent role of MDM2 in suppressing p53 function.10,11
MDM2 inhibits wild-type p53 function by several mechanisms, which are distinct, but all are mediated through their direct binding.6 Upon binding, MDM2 ubiquitinates p53 by functioning as an E3 ligase and promotes proteasomal degradation of p53. Additionally, the interaction between MDM2 and p53 blocks the binding of p53 to targeted DNAs and transports p53 from the nucleus to the cytoplasm, rendering p53 ineffective as a transcriptional factor. Consequently, blocking the MDM2–p53 interaction with small-molecule inhibitors can reactivate the tumor suppressor function of wild-type p53, and this approach is being pursued as a new cancer therapeutic strategy.12−17
Using a structure-based approach, our laboratory has designed and synthesized a spiro-oxindole (1, Figure 1) as an inhibitor of the MDM2–p53 interaction (MDM2 inhibitor).18 Subsequently, potent and efficacious MDM2 inhibitors in this family were obtained through extensive optimization,19−22 and one such compound (SAR405838/MI-77301)23 has been advanced into clinical development.
Figure 1.
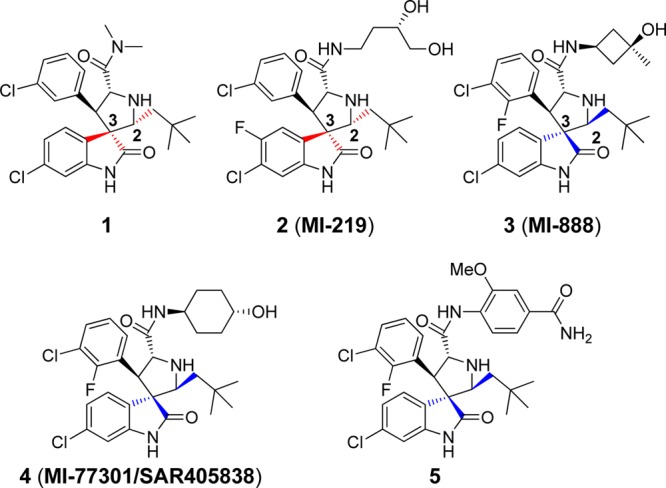
Previously reported spiro-oxindoles as inhibitors of MDM2–p53 interaction.
In the course of our research, it was discovered that, in protic solutions, some of the spiro-oxindoles are converted spontaneously into four diastereoisomers (Figure 2) which exist in equilibrium with one another.24 We recently reported a study of this phenomenon with compound 3 and its analogues (Figure 1),22,24 and the Roche group, using a different synthetic strategy, also observed the same isomerization in their preparation of compound 5 (Figure 1).25 Furthermore, it is likely that this isomerization accounts for the reported observation of other spiro-oxindole diastereoisomers in co-crystal structures with MDM2.26−28
Figure 2.
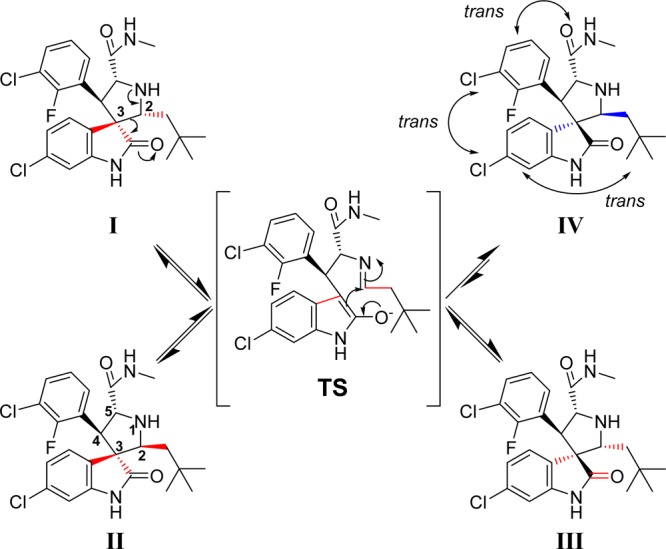
Proposed isomerization mechanism of spiro-oxindoles.
The proposed mechanism for the isomerization (Figure 2) involves a ring-opening retro-Mannich reaction between C2 and C3 of the pyrrolidine ring, generating the transition intermediate TS.22,25 Reconfiguration of the C2 and C3 pyrrolidine substituents and a subsequent Mannich reaction cyclization can generate any of the four diastereoisomers (I–IV, Figure 2), which then remain at equilibrium in solution. After equilibration, the major diastereoisomer was determined to have configuration IV, in which all the large substituents on the pyrrolidine ring are trans to one another (Figure 2). This diastereoisomer IV was isolated and shown to be the most stable and most biologically active of the diastereoisomers as MDM2 inhibitors.24
In this paper we report the design, synthesis, and evaluation of a series of new spiro-oxindoles that exploit the ring-opening-cyclization mechanism to obtain potent and chemically stable MDM2 inhibitors. Our study led to the discovery of 31 (MI-1061), which has excellent stability in solution and displays a high binding affinity (Ki = 0.16 nM) to MDM2. Significantly, 31 is orally bioavailable and achieves tumor regression in an SJSA-1 xenograft model in mice.
Design and Chemistry
Our study started with the analysis of the proposed mechanism in Figure 2, in which C2 and C3 of the pyrrolidine ring are directly involved in and affected by the ring-opening-cyclization reactions that underlie the isomerization. Because C2 and C3 are both chiral, the mechanism dictates the formation of four diastereoisomers.16 We initially rationalized that the presence of two identical substituents at C2 would reduce the number of diastereoisomers from four to two (Figure 3). We further predicted that making C2 symmetrical in this way would cause compounds with cis-configuration to reconfigure themselves through the retro-Mannich reaction, forming compounds with trans-configuration (Figure 3). Because compounds with trans-configuration experience less steric repulsion between the two bulky phenyl rings, they would be more stable than the corresponding compounds with cis-configuration and, judging by our previous binding data, may have superior potency for binding to MDM2 (Figure 3).
Figure 3.
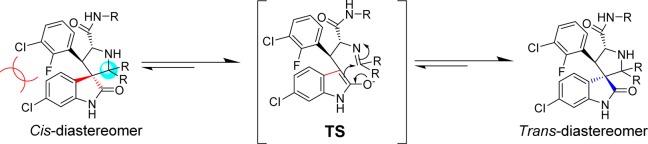
Design of second-generation spiro-oxindoles with symmetrical substituents at the C2 position.
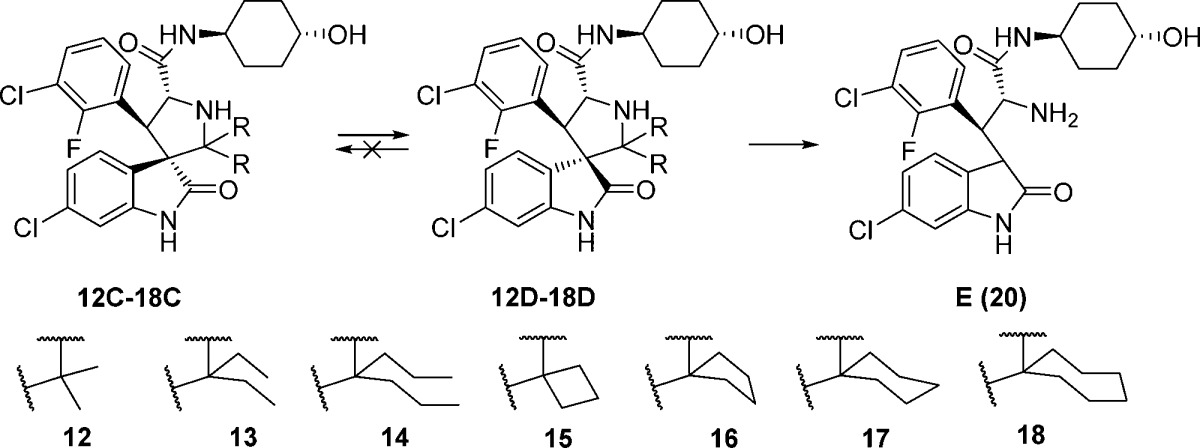
Accordingly, we designed a series of spiro-oxindoles with two identical substituents at C2 of the pyrrolidine. These included 2,2-dialkyl substituents of various sizes and 2-spirocycloalkyl substituents with different sized cycloalkyl groups. To synthesize these new compounds, we applied the same strategy used to obtain our first-generation spiro-oxindoles.18,22 The key step, building the spiro-oxindole scaffold, was the asymmetric 1,3-dipolar cycloaddition reaction illustrated in Scheme 1. It was discovered that the dimethyl ketal derivatives of acetone, 3-pentanone, or 4-heptanone (8–10) were necessary for the cycloaddition reactions that produced scaffolds 12A–14A. Unlike these acyclic ketones, the cyclic ketones undergo the cycloaddition reaction without prior conversion to their corresponding dimethyl ketals. Scaffolds 15A–18A were produced in this way. However, synthesis of scaffold 19A using either cyclo-octanone or its dimethyl ketal derivative 11 as the substrate of the cycloaddition reaction was unsuccessful, presumably due to the steric hindrance associated with the larger cyclo-octyl ring. The ring-opening of 12A–18A with trans-4-aminocyclohexanol and oxidative removal of the chiral auxiliary (1,2-diphenylethanol) on the pyrrolidine nitrogen from the resulting products (12B–18B) produced the target molecules (Scheme 2).
Scheme 1. Asymmetric 1,3-Dipolar Cycloaddition Reaction.

Scheme 2. Synthesis of Second-Generation Spiro-oxindoles.

Results and Discussion
In the first-generation spiro-oxindoles,22 four diastereoisomers could be detected after the oxidative removal of the chiral auxiliary, but in these second-generation spiro-oxindoles, only two diastereoisomers are possible because only one of the carbons (C3) involved in the ring-opening-cyclization reaction remains chiral. Analysis with ultra-performance liquid chromatography (UPLC) of the crude products of the removal of the chiral auxiliary from compounds 12B–18B (Scheme 2) revealed a fascinating trend. In the case of the acyclic pyrrolidine-2,2-dialkyl-substituted spiro-oxindoles (Table 1, entries 1–3), only one major compound was detected, and it was determined by NMR comparison (Supporting Information (SI)) to be the stable diastereoisomer, 12D–14D.
Table 1. Ratio of Diastereoisomers after Removal of the Chiral Auxiliary.
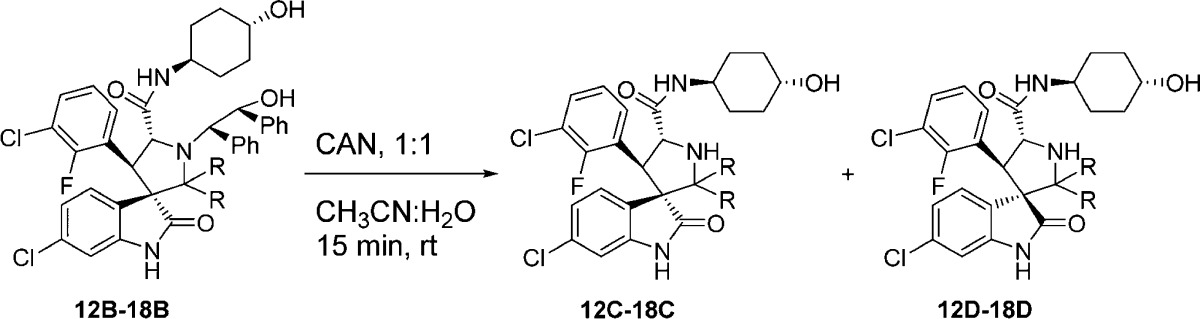
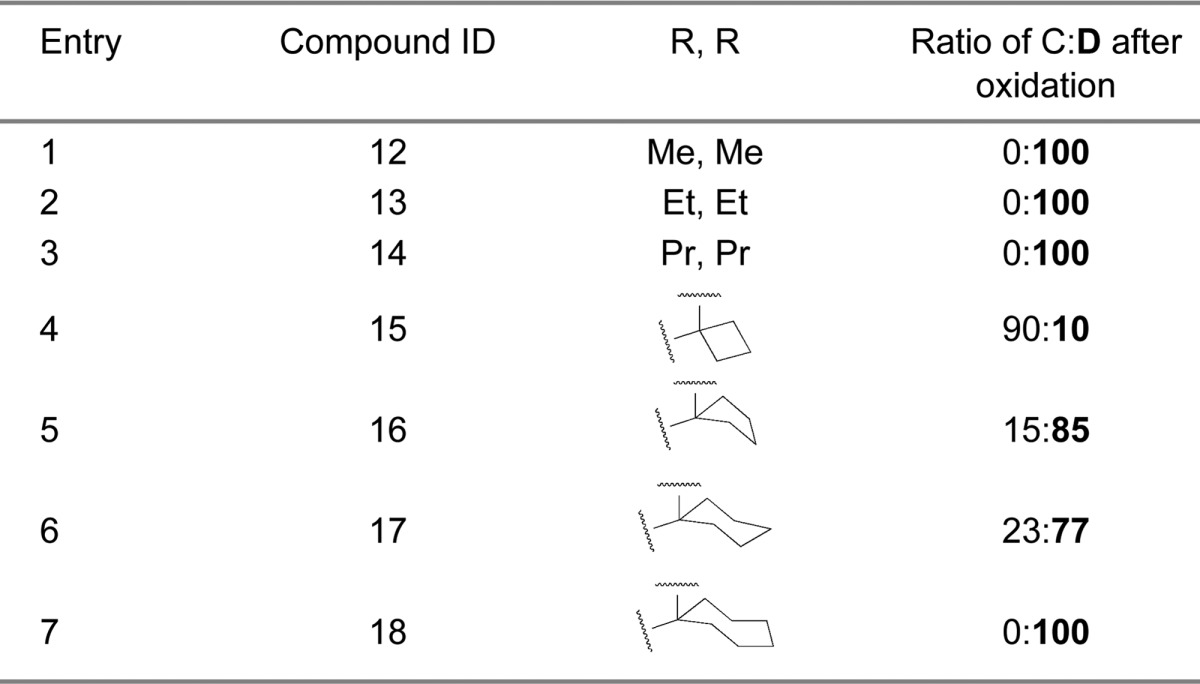
In the case of the pyrrolidine-2-spirocycloalkyl-substituted spiro-oxindoles (Table 1, entries 4–7), both C- and D-diastereoisomers were detected. As the ring size increased, the detected amount of the unstable C-diastereoisomers decreased from 90% in the case of the cyclobutyl compound 15C to 15%, 23%, and 0% in the cyclopentyl 16C, cyclohexyl 17C, and cycloheptyl 18C compounds, respectively.
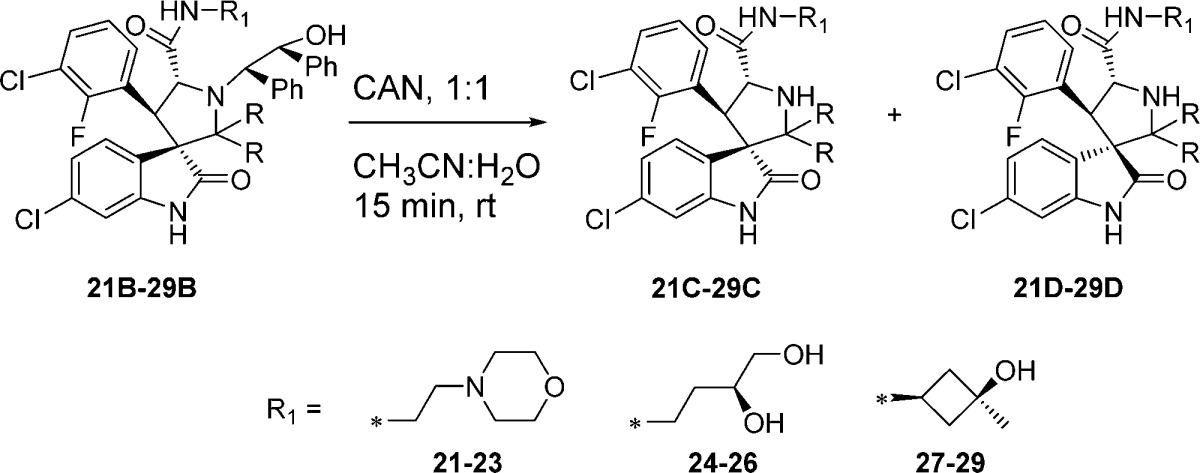
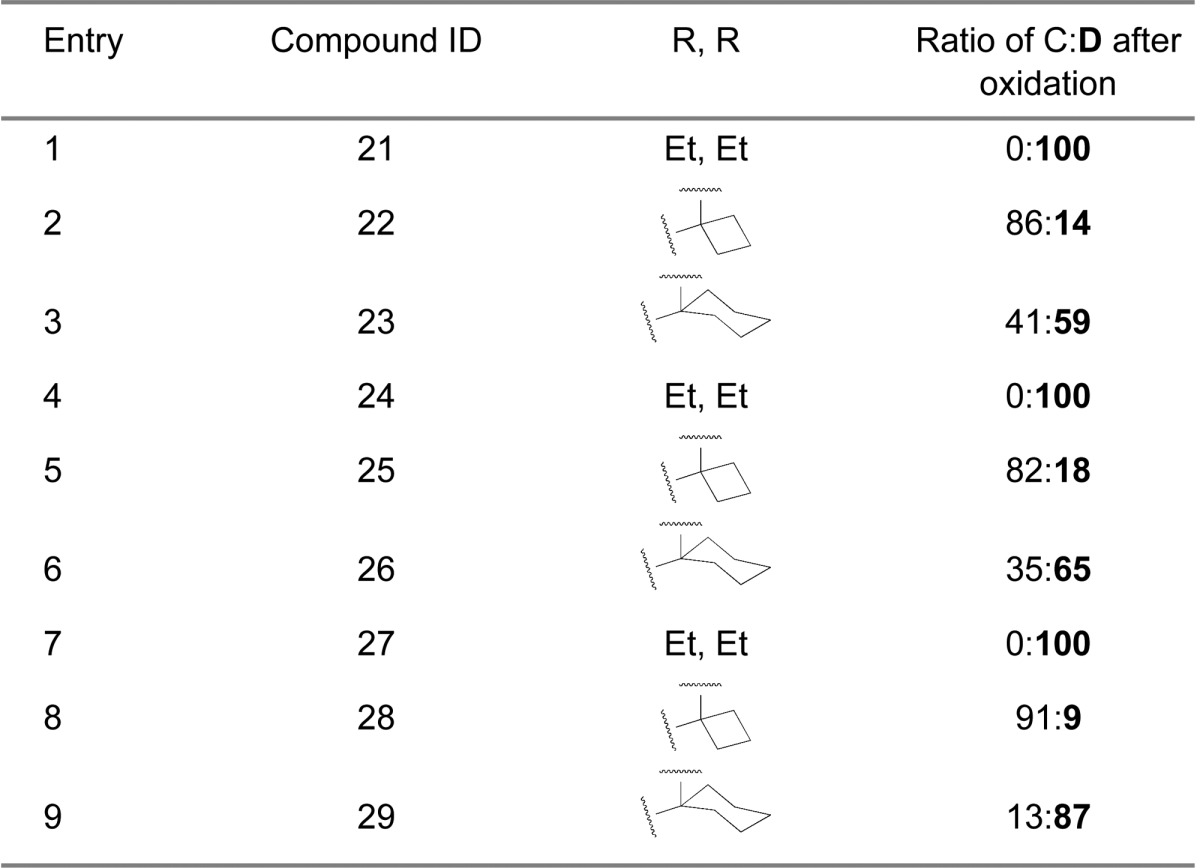
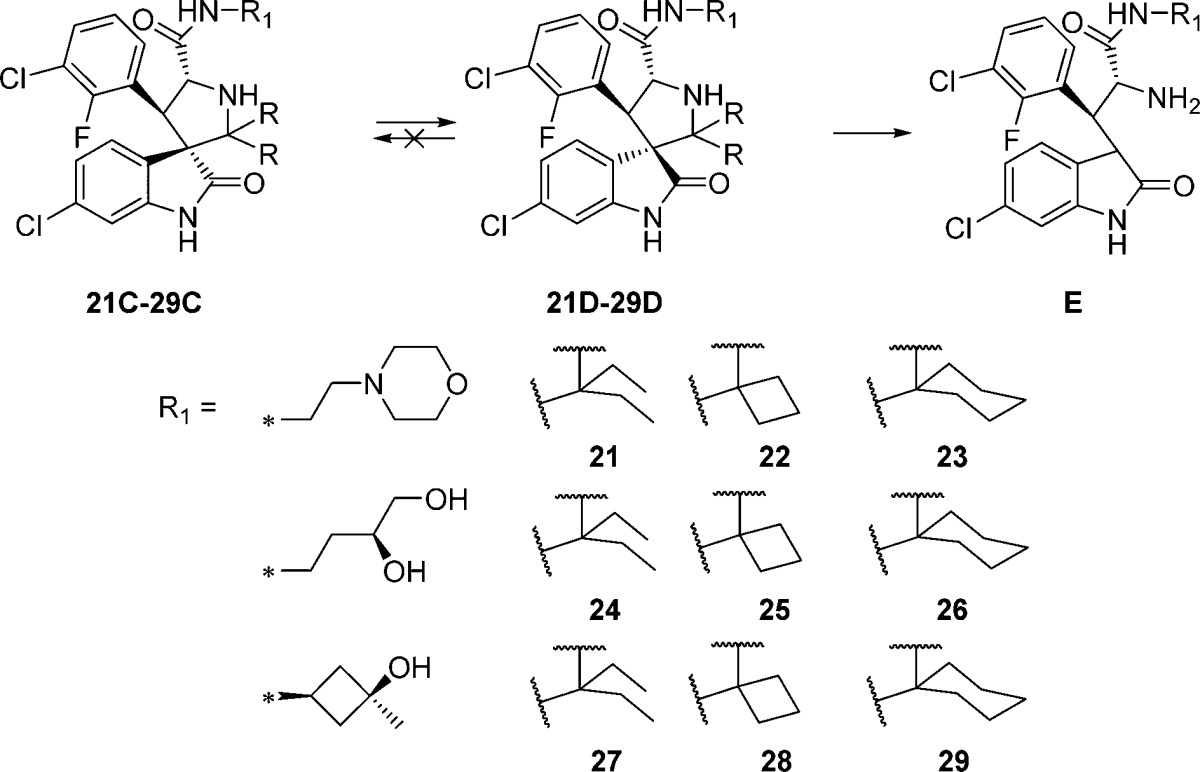
Next we examined the effect of varying the carboxamide substituent. The pyrrolidine-2,2-diethyl-, pyrrolidine-2-spirocyclobutyl-, and pyrrolidine-2-spirocyclohexylspiro-oxindole scaffolds were selected to further investigate the stability profiles of these second-generation spiro-oxindoles. The N-(2-morpholinoethyl)carboxamide, (S)-N-(3,4-dihydroxybutyl)carboxamide, and N-((1S,3S)-3-hydroxy-3-methylcyclobutyl)carboxamide substituents were selected because they were used in previously published first-generation spiro-oxindole MDM2 inhibitors.18−22,24 Combination of the second-generation scaffolds and the representative carboxamide substituents produced three series of compounds (21–23, 24–26, and 27–29), shown in Table 2. After removal of the chiral auxiliary from 21B–29B, the same trend was observed in each series. The acyclic pyrrolidine-2,2-diethyl spiro-oxindoles (Table 2, entries 1, 4, and 7) showed only the stable D-diastereoisomers in the crude products; the pyrrolidine-2-cyclobutyl compounds (Table 2, entries 2, 5, and 8) contained the unstable C-diastereoisomer as the major component in the crude reaction product, while the pyrrolidine-2-cyclohexyl compounds (Table 2, entries 3, 6, and 9) had the stable D-diastereoisomer as the major component in the crude product. This trend is consistent with the results shown in Table 1 for compounds 13, 15, and 17.
Table 2. Ratio of Diastereoisomers after Removal of the Chiral Auxiliary for Compounds with Varying Carboxamide Substituents.
To evaluate the stability of the individual products, their composition was analyzed at different time points after preparative HPLC purification. The composition relative to that observed immediately after purification was recorded, and the data are shown in Tables 3 and 4.
Table 3. Stability of Diastereoisomers of 12–18 in Solution in MeOH/H2O with 0.1% TFA.
| ratio
after HPLC C:D:E |
||||
|---|---|---|---|---|
| entry | compound | 0 h | 1 days | 2 days |
| 1 | 12D | 0:100:0 | 0:100:0 | 0:100:0 |
| 2 | 13D | 0:99:1 | 0:97:3 | 0:94:6 |
| 3 | 14D | 0:99.4:0.6 | 0:90:10 | 0:77:23 |
| 4 | 15C | 99.7:0.3:0 | 96:4:0 | 93:7:0 |
| 5 | 15D | 2:98:0 | 3:97:0 | 2:98:0 |
| 6 | 16C+16D | 73:27:0 | 0:100:0 | 0:100:0 |
| 7 | 16D | 0:100:0 | 0:100:0 | 0:100:0 |
| 8 | 17C+17D | 31:69:0 | 0:100:0 | 0:100:0 |
| 9 | 17D | 0:100:0 | 0:100:0 | 0:100:0 |
| 10 | 18D | 0:100:0 | 0:91:9 | 0:83:17 |
Table 4. Stability of Diastereoisomers 21–29 in MeOH/H2O Solution with 0.1% TFA.
| ratio
after HPLC C:D:E |
||||
|---|---|---|---|---|
| entry | compound | 0 h | 1 days | 2 days |
| 1 | 21D | 0:98:0.3 | 0:93:5 | 0:89:9 |
| 2 | 22C | 96:4:0 | 92:8:0 | 89:11:0 |
| 3 | 22D | 0:100:0 | 0:100:0 | 0:100:0 |
| 4 | 23C+23D | 53:47:0 | 0:99.3:0.7 | 0:99.3:0.7 |
| 5 | 23D | 0:100:0 | 0:100:0 | 0:99.8:0.2 |
| 6 | 24D | 0:100:0 | 0:98:2 | 0:97:3 |
| 7 | 25C | 100:0:0 | 98:2:0 | 97:3:0 |
| 8 | 25D | 0:100:0 | 0:100:0 | 0:100:0 |
| 9 | 26C+26D | 35:65:0 | 0:100:0 | 0:100:0 |
| 10 | 26D | 0:100:0 | 0:100:0 | 0:100:0 |
| 11 | 27D | 0:100:0 | 0:98:2 | 0:96:4 |
| 12 | 28C | 99.7:0.3:0 | 98:2:0 | 89:11:0 |
| 13 | 28D | 0:100:0 | 0:100:0 | 0:100:0 |
| 14 | 29C+29D | 32:68:0 | 0:100:0 | 0:100:0 |
| 15 | 29D | 0:100:0 | 0:100:0 | 0:100:0 |
Consistent with the data shown in Tables 1 and 2 obtained from the crude products, only the D-diastereoisomers were obtained for the acyclic compounds. However, as the size of the alkyl substituents increased from dimethyl to diethyl and di-n-propyl, a second compound (0%, 1%, and 0.6%, respectively) was detected (Table 3, 0 h, entries 1–3). This did not represent the unstable C-diastereoisomers but rather was found to be, after isolation and characterization (SI), the ring-opened primary amine E (20) formed as shown in Table 3. After 2 days in solution the difference in stability became more evident; the composition of the dimethyl compound 12D remained unchanged, while 6% and 23% of the ring-opened-elimination product 20 was observed in the solution of the diethyl (13D) and di-n-propyl (14D) compounds, respectively (Table 3). Similar ring-opened-elimination products were observed for the diethyl compounds (21D, 24D, and 27D) with different carboxamide substituents (Table 4). This indicates that the rate of decomposition to the ring-opened-elimination product E is proportional to the size of the alkyl substituent, indicating that large C2 dialkyl substituents introduce steric hindrance that slows the recyclization and allows for hydrolysis of the pyrrolidine-ring-opened imine intermediate (TS in Figure 3) to yield E.
As indicated above, both diastereoisomers were observed in the crude reaction product from the C2-spirocycloalkyl compounds (Table 1, entries 4–7; Table 2, entries 2-3, 5-6, and 8-9), and we presumed they could be isolated in pure form. However, pure C-diastereoisomers 15C, 22C, 25C, and 28C, could be isolated only in the case of the cyclobutyl compounds (Table 3, entry 4; Table 4, entries 2, 7, and 12). Isolation of the C-diastereoisomers of the cyclopentyl 16C and cyclohexyl 17C, 23C, 26C, and 29C compounds was unsuccessful. Fractions collected for the peaks corresponding to the diastereoisomers 16C, 17C, 23C, 26C, and 29C were immediately analyzed by UPLC and revealed to be mixtures which already showed 27%, 69% (Table 3, entries 6 and 8), 47%, 65%, and 68% (Table 4, entries 4, 9, and 14) conversion to their corresponding 16D, 17D, 23D, 26D, and 29D diastereoisomers. Isolation of the D-diastereoisomers with >98% purity (15D–18D, 22D–23D, 25D–26D, 28D–29D) was achieved for all the spirocycloalkyl compounds (Table 3, entries 5, 7, 9, and 10; Table 4, entries 3, 5, 8, 10, 13, and 15).
To assess the possibility of equilibrium between the C- and D-diastereoisomers, we analyzed the HPLC fractions over 2 days. After 1 day in solution, the cyclopentyl 16C and cyclohexyl 17C, 23C, 26C, and 29C diastereoisomer fractions (Table 3, entries 6 and 8; Table 4, entries 4, 9, and 14) had been completely converted to their corresponding diastereoisomers 16D, 17D, 23D, 26D, and 29D. The 15C, 22C, 25C, and 28C diastereoisomers of the cyclobutyl compounds, however, proved to be quite stable, showing only 7%, 11%, 3%, and 11% conversion to their corresponding 15D, 22D, 25D, and 28D diastereoisomers after 2 days (Table 3, entry 4; Table 4, entries 2, 7, and 12). None of the pure D-diastereoisomers of the cyclic compounds (15D–17D, Table 3, entries 5, 7, and 9; 22D–23D, 25D–26D, 28D–29D, Table 4, entries 3, 5, 8, 10, 13, and 15) revealed any detectable conversion to the corresponding C-diastereoisomers, and they maintained >98% purity after 2 days in solution, with the exception of the cycloheptyl compound 18D (Table 3, entry 10). This compound proved to be unstable, showing 9% and 17% decomposition, respectively, to compound 20 after 1 and 2 days in solution. These results show that no equilibrium exists between the C- and D-diastereoisomers for these second-generation spiro-oxindoles and that, if the carbon-2 substituents are too bulky, the ring-closure reaction simply becomes slower, resulting in hydrolysis of the iminium intermediate that leads to compound 20.
To support the proposed cis (C-) and trans (D-)configurations, we determined the crystal structures of 17B and 30. The intermediate 17B contains the cis-configuration, while 30 contains the trans-configuration, and both produce 17D with the trans-configuration. Equivalent NMR data were obtained for 17D prepared from 17B/C or 30 (Figure 4).
Figure 4.

Crystal structure of intermediate with starting cis-configuration and product having the more stable trans-configuration.
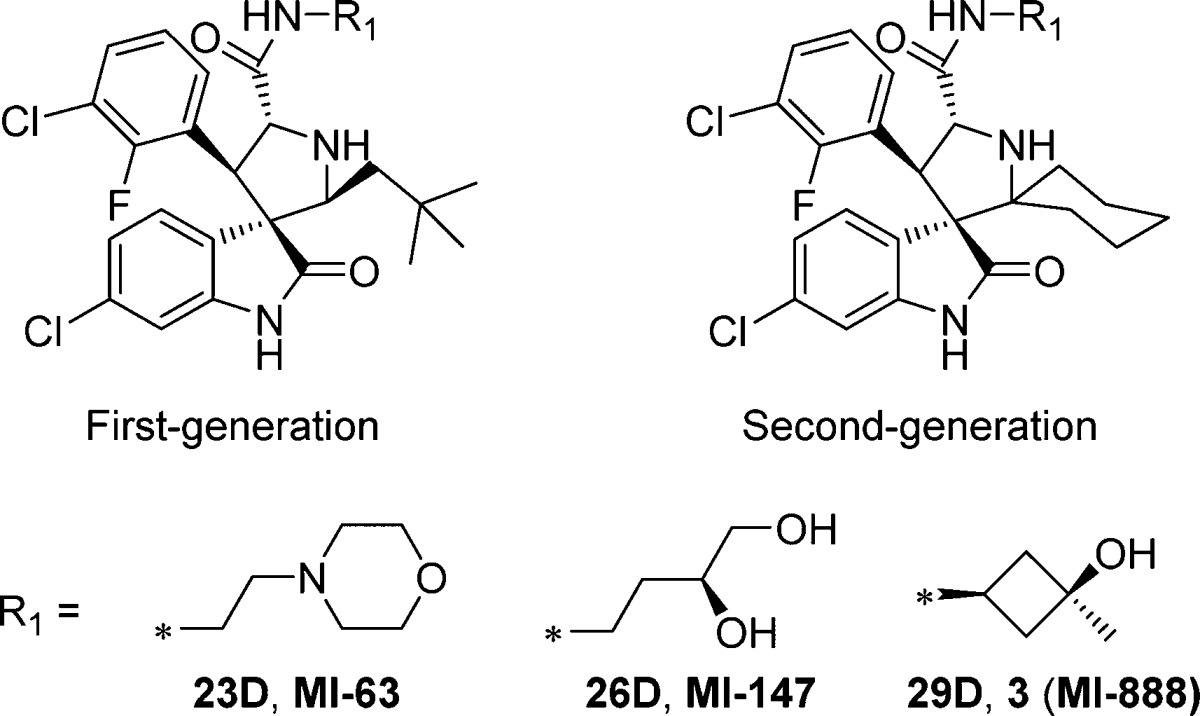
We next directly compared the stability, in solution over 6 days, of representative first-generation and second-generation spiro-oxindoles (MI-6324 vs 23D, MI-147(24) vs 26D, and 3 (MI-888)24 vs 26D) (Table 5). The first-generation spiro-oxindoles 3, MI-147, and MI-63 displayed 2.3%, 8.6%, and 17.3% isomerization, respectively, after 6 days in acetonitrile/water solution. In direct comparison, the second-generation compounds 23D, 26D, and 29D showed no detectable change in composition after 6 days in the same solution. Therefore, it can be concluded that the second-generation spiro-oxindoles with the D-configuration possess superior stability to the first-generation spiro-oxindoles.
Table 5. Stability Comparison of First-Generation and Second-Generation Spiro-oxindoles.
| % composition of
compound in 1:1 CH3CN/H2O |
|||||
|---|---|---|---|---|---|
| entry | compound | 0 h | 1 days | 3 days | 6 days |
| 1 | 23D | 100 | 100 | 99.6 | 99.2 |
| 2 | 26D | 100 | 100 | 100 | 100 |
| 3 | 29D | 100 | 100 | 100 | 100 |
| 4 | MI-63a | 98.09 | 90.79 | 86.9 | 82.7 |
| 5 | MI-147a | 97.45 | 95.80 | 95.7 | 91.4 |
| 6 | 3 (MI-888)a | 100 | 99.56 | 98.6 | 97.7 |
Reference (24).
Hence, our data showed that a symmetrical C2 in the pyrrolidine in these second-generation spiro-oxindoles results in destabilization of the C-diastereoisomer which, taking advantage of the rapid ring-opening-cyclization reaction, is quickly and irreversibly converted to the D-diastereoisomer.
Next the new spiro-oxindoles were evaluated for their binding affinities to MDM2. Only the compounds that could be obtained in pure form, i.e., 15C, 22C, 25C, 28C, 12D–18D, and 21D–29D, were tested for their binding to MDM2 and their inhibition of cell growth in the SJSA-1 cancer cell line. The results are shown in Table 6. Consistent with the data from the first-generation spiro-oxindoles,22,2415C, with the unstable C-configuration, displays a binding affinity (Ki = 234 nM) inferior to that of its corresponding stable diastereoisomer 15D, which has Ki = 19 nM. This difference in binding affinities between the diastereoisomers was maintained after changing the carboxamide substituents. Thus, 22D, 25D, and 28D have better binding affinities than their corresponding C-diastereoisomers 22C, 25C, and 28C.
Table 6. Structure–Activity Relationships of Second-Generation Spiro-oxindolesa.
| binding
affinity |
SJSA1 cell line | |||
|---|---|---|---|---|
| entry | compound | IC50 (nM) | Ki (nM) | IC50 (μM) |
| 1 | 12D | 151 ± 15 | 19 ± 2 | 2.9 ± 1.5 |
| 2 | 13D | 33 ± 4 | 3.4 ± 0.5 | 5.4 ± 2.1 |
| 3 | 14D | 775 ± 77 | 105 ± 10 | 6.9 ± 1.4 |
| 4 | 15C | 1719 ± 75 | 234 ± 10 | 2.0 ± 0.4 |
| 5 | 15D | 149 ± 32 | 19 ± 4 | 1.6 ± 0.4 |
| 6 | 16D | 35 ± 1 | 3.7 ± 0.1 | 0.65 ± 0.2 |
| 7 | 17D | 17.2 ± 4.6 | 2.9 ± 0.8 | 0.19 ± 0.04 |
| 8 | 18D | 15.6 ± 2.2 | 2.8 ± 0.3 | 2.1 ± 1.9 |
| 9 | 20 | >10 000 | NTb | |
| 10 | 21D | 167 ± 12 | 22 ± 2 | >10 |
| 11 | 22C | 9516 ± 888 | 1301 ± 121 | 1.65 ± 0.27 |
| 12 | 22D | 589 ± 84 | 80 ± 12 | 2.3 ± 0.28 |
| 13 | 23D | 21 ± 5 | 1.7 ± 0.6 | 0.30 ± 0.07 |
| 14 | 24D | 175 ± 32 | 23 ± 4.4 | >10 |
| 15 | 25C | 9058 ± 863 | 1238 ± 118 | 1.67 ± 0.35 |
| 16 | 25D | 494 ± 45 | 67 ± 6 | 2.08 ± 0.46 |
| 17 | 26D | 30 ± 6 | 3.0 ± 0.8 | 0.20 ± 0.18 |
| 18 | 27D | 118 ± 33 | 15 ± 5 | >10 |
| 19 | 28C | 7505 ± 928 | 1026 ± 127 | 1.46 ± 0.24 |
| 20 | 28D | 396 ± 49 | 53 ± 7 | 1.68 ± 0.14 |
| 21 | 29D | 20 ± 4 | 1.7 ± 0.6 | 0.16 ± 0.02 |
| 22 | 31 | 4.4 ± 1.1 | 0.16 ± 0.1 | 0.10 ± 0.03 |
Mean and standard deviation of at least three independent experiments.
NT = not tested.
We performed computational docking studies to investigate the binding models of the D-diastereoisomer 15D and C-diastereoisomer 15C in complex with MDM2 (Figure 5). Superposition of the modeled complex structure for 15D with the co-crystal structure of 4 complexed with MDM223 shows that the cyclobutyl in 15D and neopentyl in 4 occupy the Phe19 pocket, the same oxindole aryl group in both compounds projects into the Trp23 pocket, and the same 3-chloro-2-fluorophenyl group in both compounds occupies the Leu26 pocket, respectively (Figure 5A). Interestingly but consistent with our previous modeling prediction for MI-219,20 the predicted binding model for 15C shows that its cyclobutyl and 3-chloro-2-fluorophenyl substituents are reversed, as compared to the binding model for 15D, projecting into the Leu26 and Phe19 pockets, respectively. In the binding models for 15D and 4, these compounds benefit from several interactions with MDM2 protein: (1) their 3-chloro-2-fluoro-phenyl substituent in the Leu26 pocket has π–π stacking with His96; (2) the carbonyl group of the pyrrolidine carboxamide forms a hydrogen bond with His96 and; and (3) refolding of the N-terminal of MDM2 provides additional hydrophobic interactions with the 3-chloro-2-fluorophenyl substituent in the Leu26 pocket.23 These interactions are not present in the binding model for 15C and likely contribute to the superior MDM2 binding affinities for the D-diastereoisomers over their C-diastereoisomers.
Figure 5.
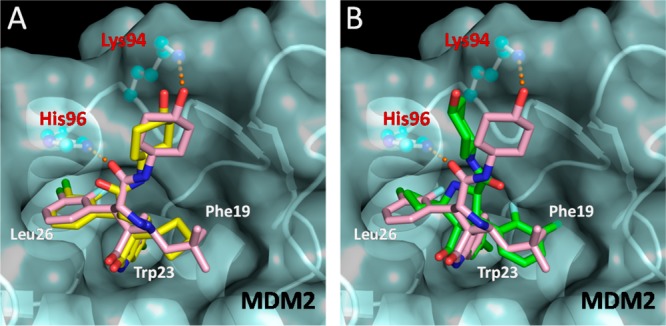
(A) Modeled binding mode of 15D (yellow) and (B) modeled binding mode of 15C (green), superimposed upon the co-crystal structure of 4 (pink) in MDM2 protein. Amino acid residues of the MDM2 protein are labeled in red, and the binding pockets are labeled in white. Docked models were obtained with GOLD, and figures were generated using Pymol.
Compounds 12D–18D, which possess the stable D-configuration, all display potent binding affinities with Ki values ranging from 2.8 to 105 nM (Table 6). In the acyclic modifications, the binding affinities are improved by 5-fold, from 19 to 3.4 nM, by increasing the substituent size from dimethyl in 12D to diethyl in 13D (Table 6, entries 1 and 2), but further increase in substituent size, to di-n-propyl in 14D, results in a 30-fold decrease in binding. The cyclic compounds 15D to 18D tolerate a substituent as large as a cycloheptyl group (18D). The spirocyclohexyl- and spirocycloheptyl-containing compounds 17D and 18D display the best Ki values (2.9 and 2.8 nM, respectively) in this series.
As expected, varying the carboxamide substituent did not change the trend. Independent of the carboxamide substituents, an improvement in binding affinities was observed going from the spirocyclobutyl (22D, 25D, and 28D) to the 2,2-diethyl (21D, 24D, and 27D) to the spirocyclohexyl (23D, 26D, 29D) compounds, respectively. However, in all cases, compounds 13D, 15D, and 17D, with the trans-4-hydroxycyclohexyl carboxamide substituent, showed higher binding affinities than their analogues having other carboxamide substituents.
We evaluated these new compounds for their cell growth inhibitory activity in the SJSA-1 cell line, which has wild-type p53 and overexpression of MDM2 protein due to MDM2 gene amplification. In general, all compounds with high binding affinities (Ki values in the low single digit nanomolar to sub-nanomolar) have sub-micromolar IC50 values in inhibition of cell growth (e.g., 16D, 17D, 23D, 26D, and 29D), with the exception of compounds 13D and 18D, which have IC50 = 5.4 and 2.1 μM, respectively. One factor that may contribute to their modest cellular potencies is that 13D and 18D are less stable in solution (Table 3, entries 2 and 10) showing decomposition to compound 20, which does not bind to MDM2. Other compounds with Ki > 10 nM to MDM2, such as 12D, 14D, 15C, 15D, 22C, 22D, 25C, 25D, and 28C, all have IC50 values in the low micromolar range, highlighting that very high affinities to MDM2 are necessary to achieve potent cell growth inhibition activity in the SJSA-1 cell line.
Among these new spiro-oxindoles, compound 17D possessed the most potent binding affinity for MDM2 and growth inhibition of SJSA-1 cell line and was therefore selected for further evaluation.
We further evaluated the stability of 17D, together with 3, in several solvents and found that 17D shows no detectable isomerization over a period of 7 days in solution, whereas 3 has 2–3% isomerization in CH3CN/H2O (Figure 6A) and MeOH/H2O (Figure 6B) but more than 10% isomerization in cell culture media (Figure 6C) over the same period. We concluded that 17D has excellent chemical stability in different solutions and has a superior stability compared to 3.
Figure 6.
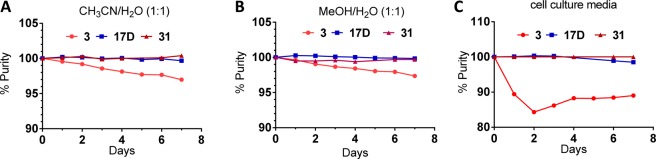
Stability comparison of 3, 17D, and 31 in (A) 1:1 CH3CN/H2O, (B) 1:1 MeOH/H2O, and (C) cell culture media.
For a potent and specific MDM2 inhibitor, its cellular activity should be dependent on the activation of wild-type p53. Hence, 17D was assessed for its cell activity and specificity in the HCT-116 p53+/+ colon cancer cell line and its p53 knockout isogenic HCT-116 p53–/– cell line (Table 7). In a cell growth assay, 17D has IC50 = 0.32 μM in the HCT-116 p53+/+ cell line but shows an IC50 > 10 μM in the isogenic HCT-116 p53–/– cell line, thus displaying high selectivity.
Table 7. Cell Growth Inhibition in HCT-116 p53+/+ and HCT-116 p53–/– Cell Linesa.
| cell growth
inhibition, IC50 (μM) |
|||
|---|---|---|---|
| entry | compound | HCT116 p53+/+ cell line | HCT116 p53–/– cell line |
| 1 | 17D | 0.32 ± 0.09 | >10 |
| 2 | 31 | 0.25 ± 0.05 | >10 |
Mean and standard deviation of at least three runs.
Based on its high binding affinity to MDM2, potent cell growth inhibitory activity, and excellent chemical stability, 17D was evaluated in pharmacodynamic (PD) and efficacy experiments in vivo in SCID mice bearing SJSA-1 osteosarcoma xenografts. A single oral 100 mg/kg dose of 17D effectively activates p53, as evidenced by robust accumulation of p53 protein itself, and MDM2 and p21 proteins, two p53-targeted gene products (Figure 7A). However, at 100 mg/kg 17D induces a minimal amount of PARP cleavage, indicative of modest apoptosis induction in the SJSA-1 tumor tissue. Consistent with the PD data, while 17D effectively inhibited tumor growth at 200 mg/kg with daily, oral administration for 2 weeks, it failed to achieve tumor regression (Figure 7B). Our in vitro and in vivo data indicated that while 17D represents a promising lead compound, it needs to be further optimized for potency and efficacy for therapeutic applications.
Figure 7.

(A) In vivo pharmacodynamics comparison using a single dose of 100 mg/kg of 17D or 31 in SCID mice bearing SJSA-1 tumor xenograft. (B) Efficacy of 17D in a SJSA-1 xenograft model. (C) Efficacy of 31 in an SJSA-1 xenograft model.
To further improve 17D as an MDM2 inhibitor, we focused our modifications on its 4-hydroxycyclohexyl group (Figure 8) since our previous studies have shown that this site plays a key role in modulation of binding affinity to MDM2, in vitro cellular potency, and in vivo efficacy.19,21,22 In the co-crystal structure of compound 4 complexed with MDM2, the 4-hydroxycyclohexyl group forms a hydrogen bonding interaction with a lsyine residue in MDM2.23 Therefore, we replaced the 4-hydroxycyclohexyl group with a benzoic acid group to enhance the interactions with the lysine residue, which yielded 31 (Figure 8). Of note, the benzoic acid group at this site was also employed by the Roche group for their design of MDM2 inhibitors.25,29,30 Compound 31 binds to MDM2 with Ki = 0.16 nM. In a cell growth assay, 31 achieves IC50 = 100 and 250 nM in the SJSA-1 and HCT-116 p53+/+ cell lines, respectively, and has IC50 > 10 000 nM in the p53 knockout cell line HCT-116 p53–/– cell line. Thus, 31 has a higher binding affinity to MDM2 and better cell growth inhibitory activity than 17D. Chemical stability testing further showed that compound 31 has excellent stability in three different solutions (Figure 6A–C).
Figure 8.
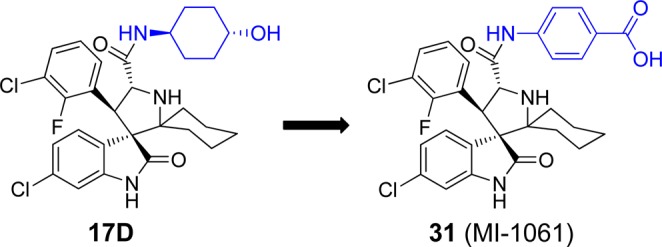
Design of a new analogue of 17D to further improve binding to MDM2.
We next evaluated 17D and 31 together for their ability to activate p53 in a PD experiment in the SJSA-1 tumor tissue harvested from mice treated with a single, oral dose of each compound at 100 mg/kg (Figure 7A). Both compounds effectively activated p53 in the SJSA-1 tumor tissue, leading to accumulation of p53, MDM2, and p21 proteins. Compound 31 effectively induced robust cleavage of PARP in the tumor, indicative of strong apoptosis induction, but 17D had a minimal effect on PARP cleavage. Encouraged by the strong p53 activation and apoptosis induction by 31 in the SJSA-1 tumor tissue, we next tested 31 for its efficacy in the SJSA-1 xenograft model. Consistent with the strong p53 activation and apoptosis induction in the tumor tissue, 31 demonstrated strong antitumor activity and achieved significant tumor regression when administered orally daily for 14 days at 100 mg/kg. At the end of treatment (day 25), the average tumor volume for the eight mice treated with 31 was reduced to 12 mm3 from 94 mm3 at the start of treatment, an 88% regression, and three of the mice had no detectable tumor. In comparison, 17D only inhibited tumor growth but did not induce tumor regression when dosed at 200 mg/kg, daily for 14 days when compared to the control group. All the mice treated with 17D or 31 suffered no weight loss and did not show any signs of toxicity during or after the treatment (SI).
Conclusion
In summary, we have designed and synthesized a new generation of spiro-oxindoles as MDM2 inhibitors, which exploit the ring-opening-cyclization reaction mechanism of this class of compounds. These second-generation compounds have a symmetrical pyrrolidine C2 position. Our data demonstrated that these second-generation spiro-oxindoles can be converted, rapidly and irreversibly, to the configuration preferred for binding to MDM2. To determine the optimal C2 substituents, a series of compounds was prepared with symmetrical acyclic and cyclic substituents at the C2 position of the pyrrolidine. It was found that increasing the size of the substituents in these compounds increases the rate of conversion to the trans-configured, stable D-diastereoisomers. Among these new spiro-oxindoles, compound 31 binds to MDM2 with a high affinity (Ki = 0.16 nM), potently activates p53, and induces apoptosis in the SJSA-1 xenograft tumor tissue in mice. Compound 31 is capable of achieving tumor regression in the SJSA-1 xenograft tumor model in mice with oral administration. Importantly, compound 31 has excellent chemical stability in solution, overcoming a limitation observed for the first-generation spiro-oxindole MDM2 inhibitors.
Experimental Section
General Information
Unless otherwise stated, all reactions were performed under a nitrogen atmosphere in dry solvents under anhydrous conditions, and all commercial reagents were used as supplied without further purification. NMR spectra were obtained on a Bruker 300 UltraShield spectrometer at a 1H frequency of 300 MHz and 13C frequency of 75 MHz. Chemical shifts (δ) are reported in parts per million (ppm) relative to an internal standard. The final products were purified on a preparative HPLC (Waters 2545, Quaternary Gradient Module) with a SunFire Prep C18 OBD 5 μm 50 × 100 mm reverse-phase column. The mobile phase was a gradient of solvent A (0.1% TFA in water) and solvent B (0.1% TFA in MeOH) at a flow rate of 40 mL/min and 1%/2 min increase of solvent B. CH3CN as solvent B failed to separate the diastereoisomers. All final compounds have purity ≥95% as determined by Waters ACQUITY UPLC using reverse-phase column (SunFire, C18, 5 μm, 4.6 × 150 mm) and a solvent gradient of A (0.1% of TFA in water) and solvent D (0.1% of TFA in MeOH).
Synthesis and characterization of compound 20, 22C, 25C, 28C, 21D–29D, and 30 are described in the SI. The procedure for monitoring the stability of 3, 17D, and 31 in cell culture media is the same as described previously.24
3,3-Dimethoxypentane (9)
p-Toluenesulfonic acid (PTSA) (29 mg, cat) was added to a solution of 3-pentanone (10 g, 116 mmol), trimethyl orthoformate (30.8 g, 290 mmol) in methanol (41 mL). After standing overnight, the reaction was poured into ice and extracted with Et2O. The combined ether extracts were washed with saturated sodium bicarbonate and brine, dried over sodium sulfate, and filtered, and then the solvent was removed by rotary evaporation using mild vacuum to produce 9 (11.2 g, 73% yield) as a colorless liquid. 1H NMR (300 MHz, CDCl3) δ ppm 3.15 (s, 6H), 1.59 (q, J = 7.5 Hz, 4H), 0.82 (t, J = 7.5 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ ppm 104.27, 47.73(2C), 24.43(2C), 7.98(2C).
4,4-Dimethoxyheptane (10)
Starting with 4-heptanone, compound 10 (10.52 g, 74% yield) was prepared according to the procedure described for the preparation of 9. 1H NMR (300 MHz, CDCl3) δ ppm 3.14 (s, 6H), 1.59–1.49 (m, 4H), 1.35–1.19 (m, 4H), 0.92 (t, J = 7.3 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ ppm 103.45, 47.79(2C), 35.03(2C), 17.23(2C), 14.56(2C).
1,1-Dimethoxycyclooctane (11)
Starting with cyclo-octanone, compound 11 (2.23 g, 82% yield) was prepared according to the procedure described for the preparation of 9. 1H NMR (300 MHz, CDCl3) δ ppm 3.14 (s, 6H), 1.82–1.73 (m, 4H), 1.56 (br. s, 10H); 13C NMR (75 MHz, CDCl3) δ ppm 103.95, 47.81(2C), 30.48(2C), 28.31(2C), 24.68, 21.44(2C).
(3S,3′S,4′R,8′S,8a′R)-6-Chloro-8′-(3-chloro-2-fluorophenyl)-6′,6′-dimethyl-3′,4′-diphenyl-3′,4′,8′,8a′-tetrahydrospiro[indoline-3,7′-pyrrolo[2,1-c][1,4]oxazine]-1′,2(6′H)-dione (12A)
In a sealed tube, (E)-6-chloro-3-(3-chloro-2-fluorobenzylidene)indolin-2-one (6, 500 mg, 1.62 mmol), (5R,6S)-5,6-diphenyl-2-morpholinone (7, 492 mg, 1.94 mmol), and 2,2-dimethoxypropane (506 mg, 4.86 mmol) were suspended in a mixture of toluene (7 mL) and THF (0.7 mL) and heated to 140 °C. After 1 h, the reaction was allowed to cool to room temperature, and the solvent was removed by rotary evaporation. The crude product was purified by column chromatography (the compound eluted with 100% dichloromethane) to give 133 mg (14% yield) of 12A as a light yellow solid. 1H NMR (300 MHz, CDCl3) δ ppm 7.63 (br s, 1H), 7.59–7.02 (m, 14H), 6.86 (t, J = 8.0 Hz, 1H), 6.70 (d, J = 1.6 Hz, 1H), 5.72 (d, J = 4.7 Hz, 1H), 5.08 (d, J = 8.8 Hz, 1H), 4.95–4.81 (m, 2H), 1.46 (s, 3H), 0.72 (s, 3H); 13C NMR (75 MHz, CDCl3) δ ppm 178.03, 172.51, 156.72 (d, JC–F = 251.38 Hz), 142.75, 137.97, 135.03, 135.00, 129.87, 129.55, 129.40, 128.91, 128.50, 128.29, 128.17, 127.87, 125.03 (d, JC–F = 12.89 Hz), 124.31 (d, JC–F = 4.69 Hz), 124.20, 122.36, 121.19 (d, JC–F = 19.41 Hz), 110.06, 84.35, 69.20, 66.34, 61.82, 60.37, 42.02, 24.75, 20.74; ESI-MS m/z 601.33 (M+H)+.
(3S,3′S,4′R,8′S,8a′R)-6-Chloro-8′-(3-chloro-2-fluorophenyl)-6′,6′-diethyl-3′,4′-diphenyl-3′,4′,8′,8a′-tetrahydrospiro[indoline-3,7′-pyrrolo[2,1-c][1,4]oxazine]-1′,2(6′H)-dione (13A)
Starting with 3,3-dimethoxypentane (9), compound 13A (166 mg, 16% yield) was prepared according to the procedure described for the preparation of 12A. 1H NMR (300 MHz, CDCl3) δ ppm 8.76 (s, 1H), 7.54 (t, J = 6.7 Hz, 1H), 7.29–7.02 (m, 10H), 6.98–6.80 (m, 4H), 6.62 (dd, J = 1.7, 8.2 Hz, 1H), 6.39 (d, J = 8.3 Hz, 1H), 5.22 (d, J = 10.8 Hz, 1H), 4.97 (d, J = 3.3 Hz, 1H), 4.68 (d, J = 10.9 Hz, 1H), 2.51–2.33 (m, 1H), 1.93–1.66 (m, 2H), 1.58–1.41 (m, 1H), 0.64 (t, J = 7.4 Hz, 3H), 0.58 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ ppm 182.54, 170.19, 156.84 (d, JC–F = 247.24 Hz), 142.44, 139.19, 136.48, 134.29, 130.01, 128.76, 128.70, 128.17, 128.03, 127.87, 127.22, 126.63, 126.44, 126.40, 125.91 (d, JC–F = 14.75 Hz), 124.56 (d, JC–F = 4.50 Hz), 121.38, 121.33 (d, JC–F = 18.72 Hz), 110.52, 81.36, 75.59, 66.97, 60.16, 58.29, 52.07, 27.61, 23.99, 8.95, 8.29; ESI-MS m/z 629.00 (M+H)+.
(3S,3′S,4′R,8′S,8a′R)-6-Chloro-8′-(3-chloro-2-fluorophenyl)-3′,4′-diphenyl-6′,6′-di-n-propyl-3′,4′,8′,8a′-tetrahydrospiro[indoline-3,7′-pyrrolo[2,1-c][1,4]oxazine]-1′,2(6′H)-dione (14A)
Starting with 4,4-dimethoxyheptane (10), compound 14A (266 mg, 25% yield) was prepared according to the procedure described for the preparation of 12A. 1H NMR (300 MHz, CDCl3) δ ppm 9.16 (s, 1H), 7.58 (t, J = 6.7 Hz, 1H), 7.28–7.05 (m, 10H), 6.97 (d, J = 3.1 Hz, 1H), 6.91–6.78 (m, 3H), 6.64 (dd, J = 1.9, 8.2 Hz, 1H), 6.39 (d, J = 8.3 Hz, 1H), 5.25 (d, J = 10.9 Hz, 1H), 4.97 (d, J = 3.4 Hz, 1H), 4.69 (d, J = 10.9 Hz, 1H), 2.27 (t, J = 12.1 Hz, 1H), 1.87–1.67 (m, 2H), 1.48–1.34 (m, 1H), 1.33–1.17 (m, 1H), 1.16–0.99 (m, 1H), 0.94–0.66 (m, 8H); 13C NMR (75 MHz, CDCl3) δ ppm 182.90, 170.18, 156.83 (d, JC–F = 247.50 Hz), 142.56, 139.13, 136.50, 134.31, 130.00, 128.66, 128.60, 128.16, 128.05, 127.85, 127.21, 126.53, 126.39, 126.27 (d, JC–F = 2.89 Hz), 125.94 (d, JC–F = 14.61 Hz), 124.61 (d, JC–F = 4.24 Hz), 121.42, 121.35 (d, JC–F = 18.59 Hz), 110.59, 81.21, 75.16, 67.20, 60.21, 58.29, 51.86, 38.18, 34.47, 17.92, 17.09, 15.29, 14.66; ESI-MS m/z 657.08 (M+H)+.
(3′S,4′R,7′S,8′S,8a′R)-6″-Chloro-8′-(3-chloro-2-fluorophenyl)-3′,4′-diphenyl-3′,4′,8′,8a′-tetrahydro-1′H-dispiro[cyclobutane-1,6′-pyrrolo[2,1-c][1,4]oxazine-7′,3″-indoline]-1′,2″-dione (15A)
Starting with cyclobutanone, compound 15A (684 mg, 69% yield) was prepared according to the procedure described for the preparation of 12A. 1H NMR (300 MHz, CDCl3) δ ppm 9.12 (s, 1H), 7.37–7.05 (m, 11H), 6.96–6.73 (m, 5H), 6.15 (d, J = 3.6 Hz, 1H), 4.96 (d, J = 3.7 Hz, 1H), 4.74 (d, J = 9.2 Hz, 1H), 4.59 (d, J = 9.2 Hz, 1H), 3.01–2.86 (m, 1H), 2.68 (q, J = 9.34 Hz, 1H), 2.12 (q, J = 9.7 Hz, 1H), 1.73–1.46 (m, 2H), 1.36–1.15 (m, 1H); 13C NMR (75 MHz, CDCl3) δ ppm 178.27, 169.84, 156.63 (d, JC–F = 249.92 Hz), 142.50, 137.66, 136.09, 134.88, 130.20, 128.74, 128.52, 128.49, 128.28, 127.80, 127.58, 127.40 (d, JC–F = 2.70 Hz), 126.30, 125.65, 124.48 (d, JC–F = 13.11 Hz), 124.26 (d, JC–F = 4.08 Hz), 122.51, 121.50 (d, JC–F = 19.15 Hz), 111.15, 81.99, 74.00, 62.67, 60.99, 58.25, 46.55, 32.72, 28.22, 14.49; ESI-MS m/z 613.17 (M+H)+.
(3′S,4′R,7′S,8′S,8a′R)-6″-Chloro-8′-(3-chloro-2-fluorophenyl)-3′,4′-diphenyl-3′,4′,8′,8a′-tetrahydro-1′H-dispiro[cyclopentane-1,6′-pyrrolo[2,1-c][1,4]oxazine-7′,3″-indoline]-1′,2″-dione (16A)
In a round-bottom flask, (E)-6-chloro-3-(3-chloro-2-fluorobenzylidene)indolin-2-one (6, 500 mg, 1.62 mmol), (5R,6S)-5,6-diphenyl-2-morpholinone (7, 492 mg, 1.94 mmol), and cyclopentanone (409 mg, 4.86 mmol) were suspended in toluene (10 mL) and heated at reflux at an oil bath temperature of 140 °C. After heating at reflux for 2 h, the reaction was allowed to cool to room temperature, and the solvent was removed by rotary evaportion. The crude product was purified by column chromatography (the compound was eluted with 100% CH2Cl2) to give 562 mg (55% yield) of 16A as a yellow solid. 1H NMR (300 MHz, CDCl3) δ ppm 8.98 (s, 1H), 7.28–7.01 (m, 12H), 6.96–6.77 (m, 4H), 6.44 (d, J = 4.2 Hz, 1H), 5.03 (d, J = 9.5 Hz, 1H), 4.87 (d, J = 4.4 Hz, 1H), 4.79 (d, J = 9.5 Hz, 1H), 2.41–2.26 (m, 1H), 2.22–2.07 (m, 1H), 1.66–1.12 (m, 6H); 13C NMR (75 MHz, CDCl3) δ ppm 180.08, 171.03, 156.64 (d, JC–F = 248.76 Hz), 142.37, 138.85, 136.05, 134.47, 130.02, 128.54, 128.48, 128.41, 128.26, 128.21, 127.68, 127.48 (d, JC–F = 3.21 Hz), 126.79, 126.27, 125.30 (d, JC–F = 13.47 Hz), 124.33 (d, JC–F = 4.40 Hz), 121.90, 121.53 (d, JC–F = 19.02 Hz), 110.76, 82.04, 81.26, 64.57, 60.33, 59.43, 49.27, 36.21, 28.99, 23.88, 23.69; ESI-MS m/z 627.16 (M+H)+.
(3′S,4′R,7′S,8′S,8a′R)-6″-Chloro-8′-(3-chloro-2-fluorophenyl)-3′,4′-diphenyl-3′,4′,8′,8a′-tetrahydro-1′H-dispiro[cyclohexane-1,6′-pyrrolo[2,1-c][1,4]oxazine-7′,3″-indoline]-1′,2″-dione (17A)
Starting with cyclohexanone, compound 17A (665 mg, 64% yield) was prepared according to the procedure described for the preparation of 16A. 1H NMR (300 MHz, CDCl3) δ ppm 9.24 (s, 1H), 7.79 (t, J = 6.4 Hz, 1H), 7.35–7.03 (m, 11H), 6.94 (d, J = 1.4 Hz, 1H), 6.80 (d, J = 6.7 Hz, 2H), 6.61 (dd, J = 1.4, 8.2 Hz, 1H), 6.29 (d, J = 8.2 Hz, 1H), 5.53 (d, J = 11.3 Hz, 1H), 4.95 (d, J = 1.4 Hz, 1H), 4.68 (d, J = 11.3 Hz, 1H), 2.50 (d, J = 12.6 Hz, 1H), 2.07 (d, J = 12.1 Hz, 1H), 1.53–1.01 (m, 7H), 0.93–0.70 (m, 1H); 13C NMR (75 MHz, CDCl3) δ ppm 183.39, 170.16, 156.79 (d, JC–F = 246.90 Hz), 142.81, 139.07, 136.64, 134.25, 129.95, 129.12, 128.69, 128.11, 127.50, 127.03, 126.58, 126.41, 126.21, 126.01, 125.58, 124.69 (d, JC–F = 4.04 Hz), 121.43, 121.21, 110.62, 80.82, 74.37, 68.50, 61.01, 59.25, 51.78, 36.88, 29.98, 25.30, 22.62, 22.36; ESI-MS m/z 641.08 (M+H)+.
(3′S,4′R,7′S,8′S,8a′R)-6″-Chloro-8′-(3-chloro-2-fluorophenyl)-3′,4′-diphenyl-3′,4′,8′,8a′-tetrahydro-1′H-dispiro[cycloheptane-1,6′-pyrrolo[2,1-c][1,4]oxazine-7′,3″-indoline]-1′,2″-dione (18A)
Starting with cycloheptanone, compound 18A (315 mg, 30% yield) was prepared according to the procedure described for the preparation of 16A. In this case the reaction was heated for just 30–60 min, a longer reaction time produces lower yields. 1H NMR (300 MHz, CDCl3) δ ppm 8.42 (s, 1H), 7.30–7.11 (m, 8H), 7.11–7.00 (m, 4H), 6.92 (t, J = 7.9 Hz, 1H), 6.83 (d, J = 1.3 Hz, 1H), 6.79–6.67 (m, 2H), 6.57 (d, J = 3.7 Hz, 1H), 5.01 (d, J = 10.0 Hz, 1H), 4.96 (d, J = 3.89 Hz, 1H), 4.73 (d, J = 9.9 Hz, 1H), 2.44–2.26 (m, 2H), 1.80–0.77 (m, 10H); 13C NMR (75 MHz, CDCl3) δ ppm 180.42, 171.02, 156.73 (d, JC–F = 248.72 Hz), 142.04, 138.73, 136.22, 134.33, 129.96, 129.11, 128.80, 128.33 (d, JC–F = 2.37 Hz), 128.25, 128.11, 127.58, 127.16 (d, JC–F = 2.77 Hz), 126.61, 125.94, 125.45 (d, JC–F = 13.66 Hz), 124.35 (d, JC–F = 4.48 Hz), 121.71, 121.49 (d, JC–F = 19.14 Hz), 110.60, 81.90, 77.38, 66.65, 59.72, 59.12, 48.96, 37.05, 32.44, 30.12, 29.77, 24.54, 22.68; ESI-MS m/z 655.25 (M+H)+.
(3R,4′S,5′R)-6-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1R,4R)-4-hydroxycyclohexyl)-2′,2′-dimethyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (12D, TFA Salt)
trans-4-Aminocyclohexanol (127 mg, 1.11 mmol) was added to solution of 12A (133 mg, 0.22 mmol) in THF (3 mL) and heated at reflux. After heating at reflux overnight, the reaction was cooled to room temperature, and the solvent was removed by rotary evaporation. The crude 12B was purified by column chromatography to produce 107 mg (68% yield) of 12B. Cerium ammonium nitrate (162 mg, 0.296 mmol) was added to a solution of the resulting 12B (107 mg, 0.148 mmol) in MeCN (3 mL) and stirred for 5 min at room temperature, and then H2O (3 mL) was added. After the reaction was stirred for an additional 10 min, it was quenched with saturated sodium bicarbonate, brine was added, and the solution was extracted with EtOAc. The EtOAc solution was dried over sodium sulfate and filtered through Celite, and the solvent was removed by rotary evaporation to produce crude 12D. The crude material was dissolved in a 3:1 mixture of MeOH:H2O that was acidified with TFA, and this solution was immediately analyzed by UPLC (data shown in Table 1) and purified by preparative HPLC (mobile phase was a gradient of solvent A (0.1% of TFA in H2O) and solvent B (0.1% of TFA in MeOH) at a flow rate of 40 mL/min and 1%/2 min increase of solvent B). The combined fractions of the pure compound were immediately analyzed by UPLC (data shown in Table 3, “0 h” column) and concentrated by rotary evaporation. The concentrate was redissolved in a minimum amount of MeCN, H2O was added, and the solution was frozen and lyophilized to produce the TFA salt of 12D (45 mg, 47% yield) as a white powder. 1H NMR (300 MHz, CD3OD) δ ppm 8.16 (d, J = 7.7 Hz, 1H), 7.67–7.59 (m, 1H), 7.51 (dd, J = 2.1, 8.2 Hz, 1H), 7.43–7.35 (m, 1H), 7.22–7.13 (m, 1H), 7.11 (dd, J = 1.9, 8.2 Hz, 1H), 6.80 (d, J = 1.9 Hz, 1H), 5.09 (d, J = 11.2 Hz, 1H), 4.83 (d, J = 11.3 Hz, 1H), 3.69–3.53 (m 1H), 3.50–3.35 (m, 1H), 2.08–1.84 (m, 5H), 1.84–1.73 (m, 1H), 1.65–1.51 (m, 1H), 1.41 (s, 3H), 1.39–1.06 (m, 3H), 1.04–0.87 (m, 1H); 13C NMR (75 MHz, CD3OD) δ ppm 178.50, 167.15, 157.75 (d, JC–F = 249.27 Hz), 145.27, 137.16, 132.41, 128.94 (d, JC–F = 2.15 Hz), 128.69, 126.51 (d, JC–F = 4.6 Hz), 123.57, 122.27 (d, JC–F = 19.05 Hz), 122.10, 121.95 (d, JC–F = 12.41 Hz), 111.86, 69.95, 69.91, 66.66, 61.69, 47.23, 34.32, 34.26, 30.92, 30.78, 29.84, 24.62, 22.36; ESI-MS m/z 520.67 (M+H)+.
(3R,4′S,5′R)-6-Chloro-4′-(3-chloro-2-fluorophenyl)-2′,2′-diethyl-N-((1R,4R)-4-hydroxycyclohexyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (13D, TFA Salt)
Starting with 13B (69 mg), compound 13D (23 mg, 38% yield) was prepared according to the procedure described for the preparation of 12D. 1H NMR (300 MHz, CD3OD) δ ppm 8.13 (d, J = 7.7 Hz, 1H), 7.67–7.58 (m, 1H), 7.54 (dd, J = 2.2, 8.3 Hz, 1H), 7.43–7.34 (m, 1H), 7.22–7.12 (m, 1H), 7.09 (dd, J = 1.9, 8.3 Hz, 1H), 6.75 (d, J = 1.9 Hz, 1H), 5.05 (d, J = 11.2 Hz, 1H), 4.81 (d, J = 11.3 Hz, 1H), 3.70–3.54 (m, 1H), 3.49–3.35 (m, 1H), 2.77–2.59 (m, 1H), 2.42–2.24 (m, 1H), 2.24–2.09 (m, 1H), 2.09–1.94 (m, 1H), 1.93–1.83 (m, 2H), 1.83–1.71 (m, 1H), 1.63–1.50 (m, 1H), 1.41–0.82 (m, 7H), 0.57 (t, J = 7.6 Hz, 3H); 13C NMR (75 MHz, CD3OD) δ ppm 178.36, 167.49, 157.79 (d, JC–F = 249.25 Hz), 144.68, 136.95, 132.44, 129.05, 128.87, 126.45 (d, JC–F = 4.77 Hz), 124.00, 123.50, 122.17 (d, JC–F = 19.39 Hz), 121.45 (d, JC–F = 12.15 Hz), 111.76, 77.69, 76.20, 69.88, 66.54, 61.71, 34.30, 34.22, 30.93, 30.70, 29.93, 26.00, 25.23, 7.82, 7.38; ESI-MS m/z 548.42 (M+H)+.
(3R,4′S,5′R)-6-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1r,4R)-4-hydroxycyclohexyl)-2-oxo-2′,2′-di-n-propylspiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (14D, TFA salt)
Starting with 14B (174 mg), compound 14D (71 mg, 46% yield) was prepared according to the procedure described for the preparation of 12D. 1H NMR (300 MHz, CD3OD) δ ppm 8.15 (d, J = 7.3 Hz, 1H), 7.61 (t, J = 6.7 Hz, 1H), 7.52 (dd, J = 1.6, 8.3 Hz, 1H), 7.38 (t, J = 7.2 Hz, 1H), 7.16 (t, J = 8.1 Hz, 1H), 7.10 (dd, J = 1.4, 8.1 Hz, 1H), 6.75 (s, 1H), 5.01 (d, J = 11.1 Hz, 1H), 4.81 (d, J = 11.2 Hz, 1H), 3.70–3.55 (m, 1H), 3.49–3.36 (m, 1H), 2.52 (t, J = 13.0 Hz, 1H), 2.18 (t, J = 13.0 Hz, 1H), 2.10–1.68 (m, 6H), 1.66–1.53 (m, 1H), 1.53–1.11 (m, 7H), 1.11–0.86 (m, 2H), 0.81 (t, J = 6.8 Hz, 3H), 0.75–0.57 (m, 1H); 13C NMR (75 MHz, CD3OD) δ ppm 178.45, 167.82, 157.78 (d, JC–F = 249.65 Hz), 144.71, 136.92, 132.38, 128.98, 128.91, 126.42 (d, JC–F = 4.81 Hz), 124.06, 123.50, 122.15 (d, JC–F = 19.28 Hz), 121.72 (d, JC–F = 11.61 Hz), 111.75, 75.43, 69.91, 66.76, 61.83, 36.07, 35.06, 34.47, 34.33, 34.26, 30.98, 30.77, 17.65, 17.16, 14.60, 14.39; ESI-MS m/z 576.50 (M+H)+.
(3′S,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1R,4R)-4-hydroxycyclohexyl)-2″-oxodispiro[cyclobutane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (15C, TFA Salt)
Starting with 15B (493 mg), compound 15C was prepared according to the procedure described for the preparation of 12D. The crude sample contains 15C and 15D (Table 1, entry 4). One-third of the crude was purified by preparative HPLC, which separated the isomers 15C (81 mg, 55% yield) and 15D (9 mg, 6% yield), which were stable enough to be characterized. 15C: 1H NMR (300 MHz, CD3OD) δ ppm 8.28 (d, J = 7.7 Hz, 1H), 7.42–7.32 (m, 1H), 7.24–7.16 (m, 1H), 7.12–7.03 (m, 1H), 6.96–6.89 (m, 3H), 4.82 (d, J = 11.1 Hz, 1H), 4.44 (d, J = 11.2 Hz, 1H), 3.73–3.57 (m, 1H), 3.54–3.40 (m, 1H), 2.67–2.52 (m, 2H), 2.42–2.26 (m, 1H), 2.25–1.77 (m, 5H), 1.72–1.50 (m, 2H), 1.39–1.20 (m, 3H), 1.19–1.01 (m, 1H); 13C NMR (75 MHz, CD3OD) δ ppm 178.26, 166.49, 157.72 (d, JC–F = 248.78 Hz), 145.52, 136.79, 132.02, 128.60 (d, JC–F = 2.52 Hz), 128.53, 126.07 (d, JC–F = 4.82 Hz), 124.20, 123.88 (d, JC–F = 13.56 Hz), 123.02, 122.53 (d, JC–F = 18.31 Hz), 112.14, 73.33, 70.01, 63.09, 61.70, 34.42, 34.37, 31.00, 30.95, 30.14, 15.47; ESI-MS m/z 532.50 (M+H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1R,4R)-4-hydroxycyclohexyl)-2″-oxodispiro[cyclobutane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (15D, TFA Salt)
Isomerization of 15C to produce 15D was accomplished by refluxing a solution of 15C (20 mg) in a 3:1 mixture of MeOH:H2O with 0.1% TFA. After 2 days, the reaction was cooled, purified by preparative HPLC, concentrated, redisolved in water, frozen, and lyophilized to produce 15D as a white powder. 1H NMR (300 MHz, CD3OD) δ ppm 8.19 (d, J = 7.6 Hz, 1H), 7.64 (dd, J = 1.2, 8.2 Hz, 1H), 7.57–7.48 (m, 1H), 7.44–7.35 (m, 1H), 7.19–7.10 (m, 2H), 6.87 (d, J = 1.6 Hz, 1H), 4.95 (d, J = 10.9 Hz, 1H), 4.48 (d, J = 10.9 Hz, 1H), 3.71–3.55 (m, 1H), 3.50–3.36 (m, 1H), 3.10–2.82 (m, 2H), 2.63–2.46 (m, 1H), 2.11–1.72 (m, 5H), 1.69–1.54 (m, 1H), 1.49–1.12 (m, 4H), 1.06–0.85 (m, 1H); 13C NMR (75 MHz, CD3OD) δ ppm 178.06, 167.26, 157.67 (d, JC–F = 248.91 Hz), 145.33, 137.35, 132.44, 128.35, 127.90, 126.61 (d, JC–F = 4.92 Hz), 123.94, 122.36 (d, JC–F = 18.92 Hz), 122.19 (d, JC–F = 12.81 Hz), 121.97, 112.16, 72.28, 69.92, 64.97, 62.52, 47.33, 47.29, 34.34, 34.29, 31.14, 30.96, 30.84, 28.67, 14.94; ESI-MS m/z 532.42 (M+H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1R,4R)-4-hydroxycyclohexyl)-2″-oxodispiro[cyclopentane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (16D, TFA Salt)
Starting with 16B (274 mg), compound 16D was prepared according to the procedure described for the preparation of 12D. The crude sample contains 16C and 16D (Table 1, entry 5). Preparative HPLC separated the isomers; however, the fractions corresponding to 16C already revealed some conversion to 16D (Table 3, entry 6). Because of the rapid isomerization, isolation of pure 16C (mixture: 37 mg, 15% yield) was not possible, and only 16D (123 mg, 50% yield) was characterized. 16D: 1H NMR (300 MHz, CD3OD) δ ppm 8.24 (d, J = 7.7 Hz, 1H), 7.62–7.50 (m, 2H), 7.43–7.34 (m, 1H), 7.19–7.12 (m, 1H), 7.10 (dd, J = 2.0, 8.2 Hz, 1H), 6.81 (d, J = 1.9 Hz, 1H), 5.07 9d, J = 11.0 Hz, 1H), 4.69 (d, J = 11.1 Hz, 1H), 3.70–3.56 (m, 1H), 3.49–3.36 (m, 1H), 2.81–2.68 (m, 1H), 2.53–2.39 (m, 1H), 2.24–2.11 (m, 1H), 2.11–1.44 (m, 9H), 1.37–1.17 (m, 3H), 1.03–0.86 (m, 1H); 13C NMR (75 MHz, CD3OD) δ ppm 178.90, 167.28, 157.70 (d, JC–F = 249.06 Hz), 145.40, 137.21, 132.38, 128.82 (d, JC–F = 2.25 Hz), 128.39, 126.57 (d, JC–F = 4.90 Hz), 123.67, 122.33 (d, JC–F = 19.10 Hz), 122.20 (d, JC–F = 12.68 Hz), 122.06, 111.97, 80.49, 69.92, 65.76, 62.14, 49.80, 48.32, 34.88, 34.73, 34.34, 34.27, 30.97, 30.82, 24.78, 24.31; ESI-MS m/z 546.33 (M+H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1R,4R)-4-hydroxycyclohexyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (17D, TFA Salt)
Starting with 17B (224 mg), compound 17D was prepared according to the procedure described for the preparation of 12D. The crude sample contains 17C and 17D (Table 1, entry 6). Preparative HPLC separated the isomers; however, the fractions corresponding to 17C already displayed conversion to 17D (Table 3, entry 8). Because of the rapid isomerization, isolation of pure 17C (mixture: 49 mg, 25% yield) was not possible, and only 17D (67 mg, 34% yield) was characterized. 17D: 1H NMR (300 MHz, CD3OD) δ ppm 8.18 (d, J = 7.5 Hz, 1H), 7.69–7.59 (m, 1H), 7.48 (dd, J = 2.1, 8.2 Hz, 1H), 7.44–7.34 (m, 1H), 7.21–7.13 (m, 1H), 7.10 (dd, J = 1.8, 8.2 Hz, 1H), 6.79 (d, J = 1.8 Hz, 1H), 5.09 (d, J = 11.1 Hz, 1H), 4.78 (d, J = 11.2 Hz, 1H), 3.72–3.54 (m, 1H), 3.50–3.35 (m, 1H), 2.85 (d, J = 9.8 Hz, 1H), 2.19 (d, J = 12.5 Hz, 1H), 2.02–1.82 (m, 5H), 1.82–1.69 (m, 3H), 1.67–1.13 (m, 7H), 1.02–0.82 (m, 1H); 13C NMR (75 MHz, CD3OD) δ ppm 178.22, 167.68, 157.78 (d, JC–F = 249.08 Hz), 145.27, 137.11, 132.38, 129.50 (d, JC–F = 2.50 Hz), 128.89, 126.49 (d, JC–F = 4.89 Hz), 123.42, 122.42, 122.22 (d, JC–F = 19.24 Hz), 121.92 (d, JC–F = 12.12 Hz), 111.78, 73.30, 69.88, 67.72, 62.25, 46.75, 46.70, 34.31, 34.23, 32.10, 31.35, 30.98, 30.73, 25.34, 23.11, 21.69; ESI-MS m/z 560.58 (M+H)+.
(3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-N-((1R,4R)-4-hydroxycyclohexyl)-2″-oxodispiro[cycloheptane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamide (18D, TFA Salt)
Starting with 18B (146 mg), compound 18D (56 mg, 43% yield) was prepared according to the procedure described for the preparation of 12D. The crude sample contains only 18D (Table 1, entry 7). 1H NMR (300 MHz, CD3OD) δ ppm 8.22 (d, J = 7.6 Hz, 1H), 7.66–7.53 (m, 2H), 7.43–7.34 (m, 1H), 7.19–7.11 (m, 1H), 7.08 (dd, J = 1.9, 8.2 Hz, 1H), 6.79 (d, J = 1H), 5.10 (d, J = 11.3 Hz, 1H), 4.84 (d, J = 11.2 Hz, 1H), 3.69–3.53 (m, 1H), 3.49–3.35 (m, 1H), 3.13–2.99 (m, 1H), 2.41 (dd, J = 9.6, 15.6 Hz, 2H), 1.95–1.83 (m, 2H), 1.83–1.66 (m, 4H), 1.66–1.39 (m, 5H), 1.35–0.83 (m, 6H); 13C NMR (75 MHz, CD3OD) δ ppm 178.43, 167.16, 157.78 (d, JC–F = 249.13 Hz), 145.18, 137.17, 132.37, 129.09 (d, JC–F = 2.68 Hz), 128.83, 126.44 (d, JC–F = 4.87 Hz), 123.45, 122.25 (d, JC–F = 19.13 Hz), 121.84, 121.69 (d, JC–F = 12.44 Hz), 111.81, 77.13, 69.89, 67.35, 61.75, 47.46, 47.41, 37.17, 35.11, 34.30, 34.23, 32.33, 31.29, 30.92, 30.80, 25.18, 23.65; ESI-MS m/z 574.33 (M+H)+.
4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)benzoic Acid (31, TFA Salt)
CDI (525 mg, 3.24 mmol), DIEA (0.941 mL, 5.4 mmol), and DMAP (catalytic) were added to a solution of the carboxylic acid intermediate 30 (500 mg, 1.08 mmol) dissolved in 1,2-dichloroethane, and the resulting solution was heated to 40 °C. After 30 min, methyl 4-aminobenzoate (816 mg, 5.4 mmol) was added to the reaction, which was then heated at reflux. After heating at reflux overnight, the solvent was removed, and the crude product was purified by column chromatography to give 265 mg (41% yield) of intermediate 31-methylester as a white solid. The resulting methyl ester intermediate, 31-methylester (265 mg, 0.44 mmol), was dissolved in THF (10 mL) and MeOH (5 mL), and then LiOH·H2O (56 mg, 1.33 mmol) and NaOH (53 mg, 1.33 mmol) were added, followed by H2O (10 mL). After 2 h, TLC showed the reaction was complete, and then 3 mL of TFA was added, the mixture was stirred briefly, and the solvent was evaporated. The resulting oil was redissolved in 3:1 MeOH:H2O and then purified by preparative HPLC. The mobile phase was a gradient flow of solvent A (H2O with 0.1% TFA) and solvent B (MeOH with 0.1% TFA). The eluent was lyophilized to give 235 mg (31% yield, 2 steps) of the title compound 31 as a white solid. 1H NMR (300 MHz, CD3OD) δ ppm 7.98 (d, J = 8.7 Hz, 2H), 7.75–7.61 (m, 3H), 7.52 (dd, J = 2.4, 8.2 Hz, 1H), 7.39–7.29 (m, 1H), 7.16 (t, J = 8.0 Hz, 1H), 7.09 (dd, J = 1.8, 8.2 Hz, 1H), 6.78 (d, J = 7.8 Hz, 1H), 5.21 (d, J = 10.6 Hz, 1H), 4.94 (d, J = 10.6 Hz, 1H), 2.73 (d, J = 10.32 Hz, 1H), 2.11 (d, J = 13.6 Hz, 1H), 2.01–1.47 (m, 6H), 1.32–1.09 (m, 2H); 13C NMR (75 MHz, CD3OD) δ ppm 178.60, 169.24, 169.13, 157.71 (d, JC–F = 249.13 Hz), 145.23, 142.83, 136.68, 131.97, 131.91(2C), 129.54 (d, JC–F = 2.62 Hz), 129.03, 128.14, 126.23 (d, JC–F = 4.65 Hz), 123.17, 122.21 (d, JC–F = 19.19 Hz), 120.43(2C), 111.55, 72.58, 68.95, 63.25, 46.92, 32.45, 31.95, 25.77, 23.36, 22.18; ESI-MS m/z 582.17 (M+H)+.
Diastereomer Composition Analysis
The crude products from the removal of the chiral auxiliary from 12B–18B and 21B–29B, separately, were dissolved in 3:1 MeOH:H2O and 0.1% TFA. An aliquot (250 μL) was immediately analyzed by analytical reverse-phase UPLC (mobile phase gradient of MeOH:H2O), and their compositions are shown in Tables 1 and 2. The prepared crude solutions were immediately purified by reverse-phase preparative HPLC (mobile phase gradient of MeOH:H2O). Immediately upon collecting the fraction corresponding to the C- or D-diastereomers, an aliquot (250 μL) was taken and immediately analyzed by analytical reverse-phase UPLC. The same samples were then analyzed each day to determine their change in composition, and the data are shown in Tables 3 and 4.
Preparation of Samples for Analysis of Stability in Cell Culture Medium
3(22,24) or 17D or 31 (3.0 mg) was dissolved in cell growth medium (3 mL) containing 10% FBS, and the solution was incubated at 37 °C. At approximately the same time each day, 0.25 mL of the solution was taken into a 1.5 mL microcentrifuge tube, and MeCN was added to make a total volume of 1 mL. The sample was sonicated in a water bath for 2 min and then centrifuged at 14 000 rpm for 10 min. The supernatant aliquot (250 μL) was taken and mixed with 250 μL of water. The final sample was immediately analyzed by analytical reverse-phase UPLC.
Fluorescence Prolarization (FP)-Based Protein Binding Assay
The binding affinity of MDM2 inhibitors was determined by an optimized, sensitive, and quantitative FP-based binding assay, using a recombinant human His-tagged MDM2 protein (residues 1–118) and a FAM tagged p53-based peptide as the fluorescent probe.
The design of the fluorescent probe was based upon a previously reported high affinity p53-based peptidomimetic compound (5-FAM-βAla-βAla-Phe-Met-Aib-pTyr-(6-Cl-LTrp)-Glu-Ac3c-Leu-Asn-NH2).31 This tagged peptide was named as PMDM6-F. The equilibrium dissociation constant (Kd) of PMDM6-F to the MDM2 protein was determined to be 1.4 ± 0.3 nM by monitoring the total fluorescence polarization of mixtures composed with the fluorescent probe at a fixed concentration and the MDM2 protein with increasing concentrations up to full saturation. Fluorescence polarization values were measured using the Infinite M-1000 plate reader (Tecan U.S., Research Triangle Park, NC) in Microfluor 1 96-well, black, round-bottom plates (Thermo Scientific). In the saturation experiments, 1 nM of PMDM6-F and increasing concentrations of proteins were added to each well to a final volume of 125 μL in the assay buffer (100 mM potassium phosphate, pH 7.5, 100 μg/mL bovine γ-globulin, 0.02% sodium azide (Invitrogen), with 0.01% Triton X-100 and 4% DMSO). Plates were mixed and incubated at room temperature for 30 min with gentle shaking to ensure equilibrium. The polarization values in millipolarization units (mP) were measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm. The Kd value was then calculated by fitting the sigmoidal dose-dependent FP increases as a function of protein concentrations using Graphpad Prism 6.0 software (Graphpad Software, San Diego, CA).
IC50 and Ki values of tested compounds were determined in a dose-dependent competitive binding experiment. Mixtures of 5 μL of the tested compound with different concentrations in DMSO and 120 μL of pre-incubated protein/fluorescent probe complex with fixed concentrations in the assay buffer (100 mM potassium phosphate, pH 7.5, 100 μg/mL bovine γ-globulin, 0.02% sodium azide, with 0.01% Triton X-100) were added into assay plates and incubated at room temperature for 30 min with gentle shaking. Final concentrations of the protein and fluorescent probe in the competitive assays were 10 and 1 nM, respectively, and final DMSO concentration was 4%. Negative controls containing protein/fluorescent probe complex only (equivalent to 0% inhibition), and positive controls containing free fluorescent probe only (equivalent to 100% inhibition), were included in each assay plate. FP values were measured as described above. IC50 values were determined by nonlinear regression fitting of the sigmoidal dose-dependent FP decreases as a function of total compound concentrations using Graphpad Prism 6.0 software (Graphpad Software, San Diego, CA). Ki values of competitive inhibitors were obtained directly by nonlinear regression fitting as well, based upon the Kd values of the probe to different proteins and concentrations of the proteins and probes in the competitive assays.32,33
Cell Growth Inhibition Assay
The SJSA-1 osteosarcoma tumor cell line was purchased from the American Type Culture Collection (ATCC). Cells were seeded in 96-well flat bottom cell culture plates at a density of (3–4) × 103 cells/well, grown overnight, and then incubated with compounds at different concentrations. The rate of cell growth inhibition after treatment with different concentrations of a compound was determined by assaying with a lactate dehydrogenasebased WST-8 assay (WST-8; Dojindo Molecular Technologies Inc., Gaithersburg, MD). WST-8 solution was added to each well to a final concentration of 10%, and then the plates were incubated at 37 °C for 2–3 h. The absorbance of the samples was measured at 450 nm using a TECAN ULTRA reader. The concentration of a compound that inhibited cell growth by 50% (IC50) was calculated by comparing absorbance of the untreated cells with the treated cells.22
Pharmacodynamic (PD) Study in SJSA-1 Xenograft Model
Xenograft tumors were developed by subcutaneous injection of 5 × 106 of SJSA-1 cancer cells with 50% Matrigel on the dorsal side of SCID mice (from Charles River). When the tumors reached an average volume of ∼100 mm3, mice were treated with a single dose of vehicle control, 17D at 100 mg/kg, or 31 at 100 mg/kg, all via oral gavage. Mice were sacrificed at indicated time points and tumors harvested, which were then analyzed by Western blotting for p53 activation and apoptosis.
For Western blot analysis, tumor tissues were lysed in ice-cold RIPA buffer: 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate, and 1 μg/mL leupeptin. The expressions of indicated proteins in the whole cell lysates were detected by Western blot analysis using the following antibodies: anti-p53 (OP43, Calbiochem), anti-MDM2 (sc-965, Santa Cruz), anti-p21 (556431, BD Biosciences), anti-PARP (9542, Cell Signaling Technology), anti-caspase-3 (AAP-113, Stressgen Bioreagents), and HRP-conjugated anti-GAPDH (sc-5778, Santa Cruz).22
In Vivo Efficacy Study
Xenograft tumors (one tumor per mouse) were developed by subcutaneous injection of 5 × 106 of SJSA-1 cancer cells with 50% Matrigel on the dorsal side of SCID mice, purchased from Charles River. When tumors reached an average volume of ∼100 mm3, mice were randomized into different groups of eight and were treated with vehicle control, 17D at 200 mg/kg, 31 at 100 mg/kg daily for 14 days via oral gavage. Tumor sizes and animal weights were measured 3 times per week during treatment. Data are presented as mean tumor volumes ± SEM. Statistical analyses were performed using two-way ANOVA and unpaired two-tailed t test, using Prism (version 4.0, GraphPad, La Jolla, CA). P < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by grants from the NCI/NIH R01CA121279, University of Michigan Cancer Center grant P30CA046592, Sanofi, and Ascenta Therapeutics Inc. We thank Dr. G. W. A. Milne for editing of the manuscript.
Glossary
ABBREVIATIONS USED
- C2
carbon-2
- CAN
cerium ammonium nitrate
- CDI
carbonyldiimidazole
- DIEA
N,N-diisopropylethylamine
- DMAP
dimethylaminopyridine
- EDCI
1-ethyl-(3-(dimethylamino)propyl)carbodiimide
- EtOAc
ethyl acetate
- HOBt
hydroxybenzotriazole
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- UPLC
ultra-performance liquid chromatography
Supporting Information Available
Diastereoisomer assignment, detailed synthesis of compounds, animal weights in efficacy experiments, crystal structures of compounds 17B and 30, and a molecular string file (CSV) for compounds reported in this study and their associated biochemical binding data and cell growth inhibitory activity. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ A.A. and W.S. contributed equally.
The authors declare the following competing financial interest(s): Shaomeng Wang, Angelo Aguilar, Wei Sun, Liu Liu, Jianfeng Lu, and Donna McEachern are inventors on the MDM2 inhibitors described in this study, which have been licensed by Ascenta and Sanofi from the University of Michigan. Shaomeng Wang has also received research funding from Ascenta and Sanofi and owns stocks in Ascenta.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hainaut P.; Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv. Cancer Res. 2000, 77, 81–137. [DOI] [PubMed] [Google Scholar]
- Vousden K. H.; Lane D. P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [DOI] [PubMed] [Google Scholar]
- Momand J.; Zambetti G. P.; Olson D. C.; George D.; Levine A. J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [DOI] [PubMed] [Google Scholar]
- Freedman D. A.; Wu L.; Levine A. J. Functions of the MDM2 oncoprotein. Cell. Mol. Life Sci. 1999, 55, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond G. L.; Hu W. W.; Levine A. J. MDM2 is a central node in the p53 pathway: 12 years and counting. Curr. Cancer Drug Targets 2005, 5, 3–8. [DOI] [PubMed] [Google Scholar]
- Wu X.; Bayle J. H.; Olson D.; Levine A. J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [DOI] [PubMed] [Google Scholar]
- Juven-Gershon T.; Oren M. Mdm2: the ups and downs. Mol. Med. 1999, 5, 71–83. [PMC free article] [PubMed] [Google Scholar]
- Momand J.; Wu H. H.; Dasgupta G. MDM2 - master regulator of the p53 tumor suppressor protein. Gene 2000, 242, 15–29. [DOI] [PubMed] [Google Scholar]
- Momand J.; Jung D.; Wilczynski S.; Niland J. The MDM2 gene amplification database. Nucleic Acids Res. 1998, 26, 3453–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvarts A.; Steegenga W. T.; Riteco N.; van Laar T.; Dekker P.; Bazuine M.; van Ham R. C.; van der Houven van Oordt W.; Hateboer G.; van der Eb A. J.; Jochemsen A. G. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996, 15, 5349–5357. [PMC free article] [PubMed] [Google Scholar]
- Oliner J. D.; Kinzler K. W.; Meltzer P. S.; George D. L.; Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [DOI] [PubMed] [Google Scholar]
- Vassilev L. T.; Vu B. T.; Graves B.; Carvajal D.; Podlaski F.; Filipovic Z.; Kong N.; Kammlott U.; Lukacs C.; Klein C.; Fotouhi N.; Liu E. A. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [DOI] [PubMed] [Google Scholar]
- Vassilev L. T. p53 Activation by small molecules: application in oncology. J. Med. Chem. 2005, 48, 4491–4499. [DOI] [PubMed] [Google Scholar]
- Shangary S.; Wang S. Targeting the MDM2–p53 interaction for cancer therapy. Clin. Cancer Res. 2008, 14, 5318–5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Zhao Y.; Bernard D.; Aguilar A.; Kumar S. Targeting the MDM2–p53 Protein-Protein Interaction for New Cancer Therapeutics. Top. Med. Chem. 2012, 8, 57–80. [Google Scholar]
- Zhao Y.; Bernard D.; Wang S.. Small Molecule inhibitors of MDM2–p53 and MDMX–p53 interaction as new cancer therapeutics. BioDiscovery 2013, 8. [Google Scholar]
- Carry J. C.; Garcia-Echeverria C. Inhibitors of the p53/hdm2 protein-protein interaction-path to the clinic. Bioorg. Med. Chem. Lett. 2013, 23, 2480–2485. [DOI] [PubMed] [Google Scholar]
- Ding K.; Wang G. P.; Deschamps J. R.; Parrish D. A.; Wang S. M. Synthesis of spirooxindoles via asymmetric 1,3-dipolar cycloaddition. Tetrahedron Lett. 2005, 46, 5949–5951. [Google Scholar]
- Ding K.; Lu Y.; Nikolovska-Coleska Z.; Wang G.; Qiu S.; Shangary S.; Gao W.; Qin D.; Stuckey J.; Krajewski K.; Roller P. P.; Wang S. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2–p53 interaction. J. Med. Chem. 2006, 49, 3432–3435. [DOI] [PubMed] [Google Scholar]
- Shangary S.; Qin D.; McEachern D.; Liu M.; Miller R. S.; Qiu S.; Nikolovska-Coleska Z.; Ding K.; Wang G.; Chen J.; Bernard D.; Zhang J.; Lu Y.; Gu Q.; Shah R. B.; Pienta K. J.; Ling X.; Kang S.; Guo M.; Sun Y.; Yang D.; Wang S. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 3933–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S.; Qin D.; Shangary S.; Chen J.; Wang G.; Ding K.; McEachern D.; Qiu S.; Nikolovska-Coleska Z.; Miller R.; Kang S.; Yang D.; Wang S. Potent and orally active small-molecule inhibitors of the MDM2–p53 interaction. J. Med. Chem. 2009, 52, 7970–7973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Yu S.; Sun W.; Liu L.; Lu J.; McEachern D.; Shargary S.; Bernard D.; Li X.; Zhao T.; Zou P.; Sun D.; Wang S. A potent small-molecule inhibitor of the MDM2–p53 interaction (MI-888) achieved complete and durable tumor regression in mice. J. Med. Chem. 2013, 56, 5553–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Sun W.; Zhao Y.; McEachern D.; Meaux I.; Barriere C.; Stuckey J. A.; Meagher J. L.; Bai L.; Liu L.; Hoffman-Luca C. G.; Lu J.; Shangary S.; Yu S.; Bernard D.; Aguilar A.; Dos-Santos O.; Besret L.; Guerif S.; Pannier P.; Gorge-Bernat D.; Debussche L. SAR405838: An Optimized Inhibitor of MDM2–p53 Interaction That Induces Complete and Durable Tumor Regression. Cancer Res. 2014, 74, 5855–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Liu L.; Sun W.; Lu J.; McEachern D.; Li X.; Yu S.; Bernard D.; Ochsenbein P.; Ferey V.; Carry J. C.; Deschamps J. R.; Sun D.; Wang S. Diastereomeric spirooxindoles as highly potent and efficacious MDM2 inhibitors. J. Am. Chem. Soc. 2013, 135, 7223–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu L. H.; Li Z. Z.; Gu C.; Fishlock D. Synthesis of a Spiroindolinone Pyrrolidinecarboxamide MDM2 Antagonist. Org. Process Res. Dev. 2013, 17, 247–256. [Google Scholar]
- Popowicz G. M.; Czarna A.; Wolf S.; Wang K.; Wang W.; Domling A.; Holak T. A. Structures of low molecular weight inhibitors bound to MDMX and MDM2 reveal new approaches for p53-MDMX/MDM2 antagonist drug discovery. Cell Cycle 2010, 9, 1104–1111. [DOI] [PubMed] [Google Scholar]
- Popowicz G. M.; Domling A.; Holak T. A. The structure-based design of Mdm2/Mdmx–p53 inhibitors gets serious. Angew. Chem., Int. Ed. 2011, 50, 2680–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Lopez de Turiso F.; Sun D.; Rew Y.; Bartberger M. D.; Beck H. P.; Canon J.; Chen A.; Chow D.; Correll T. L.; Huang X.; Julian L. D.; Kayser F.; Lo M. C.; Long A. M.; McMinn D.; Oliner J. D.; Osgood T.; Powers J. P.; Saiki A. Y.; Schneider S.; Shaffer P.; Xiao S. H.; Yakowec P.; Yan X.; Ye Q.; Yu D.; Zhao X.; Zhou J.; Medina J. C.; Olson S. H. Rational design and binding mode duality of MDM2–p53 inhibitors. J. Med. Chem. 2013, 56, 4053–4070. [DOI] [PubMed] [Google Scholar]
- Ding Q.; Zhang Z.; Liu J. J.; Jiang N.; Zhang J.; Ross T. M.; Chu X. J.; Bartkovitz D.; Podlaski F.; Janson C.; Tovar C.; Filipovic Z. M.; Higgins B.; Glenn K.; Packman K.; Vassilev L. T.; Graves B. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Chu X. J.; Liu J. J.; Ding Q.; Zhang J.; Bartkovitz D.; Jiang N.; Karnachi P.; So S. S.; Tovar C.; Filipovic Z. M.; Higgins B.; Glenn K.; Packman K.; Vassilev L.; Graves B. Discovery of Potent and Orally Active p53-MDM2 Inhibitors RO5353 and RO2468 for Potential Clinical Development. ACS Med. Chem. Lett. 2014, 5, 124–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Echeverria C.; Chene P.; Blommers M. J.; Furet P. Discovery of potent antagonists of the interaction between human double minute 2 and tumor suppressor p53. J. Med. Chem. 2000, 43, 3205–3208. [DOI] [PubMed] [Google Scholar]
- Wang Z. X. An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule. FEBS Lett. 1995, 360, 111–114. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Mayhood T.; Lipari P.; Wang Y.; Durkin J.; Syto R.; Gesell J.; McNemar C.; Windsor W. Fluorescence polarization assay and inhibitor design for MDM2/p53 interaction. Anal. Biochem. 2004, 331, 138–146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.