

Abstract
The ene reaction is a pericyclic process in which an alkene having an allylic hydrogen atom (the ene donor) reacts with a second unsaturated species (the enophile) to form a new product with a transposed π-bond. The aromatic ene reaction, in which the alkene component is embedded in an aromatic ring, has only been reported in a few (four) instances and has proceeded in low yield (≤6%). Here we show efficient aromatic ene reactions in which a thermally generated aryne engages a pendant m-alkylarene substituent to produce a dearomatized isotoluene, itself another versatile but rare reactive intermediate. Our experiments were guided by computational studies that revealed structural features conducive to the aromatic ene process. We proceeded to identify a cascade comprising three reactions: (i) hexadehydro-Diels-Alder (for aryne generation), (ii) intramolecular aromatic ene, and (iii) bimolecular Alder ene. The power of this cascade is evident from the structural complexity of the final products, the considerable scope, and the overall efficiency of these multi-stage, reagent- and byproduct-free, single-pot transformations.
Ene reactions1, which involve the attack of an alkene (the ene donor) by an unsaturated enophile (A=B or A≡B), are well known in organic chemistry2. In the process an allylic hydrogen atom is transferred to the enophile and the alkene π-bond is transposed (i.e., C1=C2–C3–H + A=B to H–B–A–C1–C2=C3). Aromatic ene reactions–those in which an arene bearing a benzylic C–H bond serves as the ene donor (cf. 1 to 2, Fig. 1a)–are extremely rare. The only (four) reported examples involve o-benzyne (3) itself as the enophile. Each proceeds in low yield (≤6%) and is presumed to follow the pathway shown in Fig. 1b (i.e., 1 to 5 via 4)3,4; the competing formation of bimolecular [4+2] Diels–Alder reaction products between o-benzyne (3, now acting as a dienophile) and toluene (the diene) perhaps has contributed to the fact that this fundamentally intriguing but rare type of ene reaction has since remained unexplored and unexploited.
Figure 1. Structural delineation and previous example of an aromatic ene reaction.

a, The minimal structural elements for an aromatic ene reaction: a potent enophile and an arene bearing a benzylic C–H bond as an ene donor. This transformation is rare because it requires the energetically demanding formation of a dearomatized isotoluene species (cf. 2). b, The only reported aromatic ene reactions involve the use of the highly energetic o-benzyne (3) in the role of enophile, which engages one of the alkylbenzenes toluene (1), ethylbenzene, cumene, or mesitylene as the ene donor. The intermediate isotoluene 4 was invoked to account for formation of product 5. Yields are marginal in part because products arising from [4+2] cycloaddition between 3 and the π-system in arene 1 are formed competitively.
Arynes are known to function as enophiles5. The earliest examples were sporadic and isolated in their nature, but recent studies have established synthetically useful ene reaction protocols involving arynes as the enophile (Fig. 2a–b). Lautens et al. has reported the wide scope of intramolecular ene reactions of (classically generated) benzyne derivatives like 7 (from 6) in which pendant alkenes cyclize efficiently to provide annelated products like 86,7. We recently described the intramolecular ene reaction of the aryne 10 to give 118. Cheng and coworkers have demonstrated the generality of a bimolecular propargylic ene reaction between o-benzyne (3, generated from 12 by the method of Kobayashi9) and an alkyne (cf. 13) to give an allene (cf. 14)10. Lee et al. have recently reported the considerable scope of reactions like 10 to 11 as well as an intramolecular propargylic ene reaction (15 to 17 via 16)11. Arynes are also known to participate in hetera-ene reactions with a phenol8,12 or aniline13 derivative serving as the ene donor. Here we show the first examples of efficient aromatic ene reactions. They were developed in the context of arynes like 19, which bear suitably disposed aromatic substituents bearing a m-alkyl substituent, and pass through isotoluene species like 20. These are reactive intermediates in their own right for which we also report here some unprecedented transformations.
Figure 2. Arynes as enophiles reacting with various types of ene donor.

a, An alkene (an Alder ene reaction)6,7,8. b, An alkyne (a propargylic ene reaction)10,11. c, An arene donor bearing a suitably disposed benzylic hydrogen atom (an aromatic ene reaction, this work).
Results and Discussion
Realization of the aromatic ene reaction has heretofore been elusive presumably for two fundamental reasons. First, the energetics of dearomatization associated with the primary ene event (cf. 1 to 2) require that the enophile be of inherently high reactivity. Second, o-benzyne, the only known enophile capable of overcoming this barrier, displays competitive [4+2] (Diels–Alder) reactivity toward the dienic character that is necessarily present in the requisite aromatic ring of the ene donor3,4,8. We felt that both of these challenges–the issues of inherent reactivity and selectivity–might be surmountable by use of properly designed aryne intermediates accessible by the hexadehydro-Diels–Alder (HDDA) reaction.
The HDDA reaction generates bicyclic benzyne derivatives merely by heating suitable triyne precursors [cf. 9 to 10 (Fig. 2a), 15 to 16 (Fig. 2b), and 18 to 19 (Fig. 2c)] and comprises a general and powerful strategy for accessing these reactive intermediates8,14. Importantly, the arynes are produced in the absence of external reagents. This feature affords them a longer lifetime and an accordingly greater opportunity for entering into transformations having relatively high activation energies. Thus, HDDA-derived arynes also comprise a valuable platform upon which to exploit relatively rare, if not unprecedented, types of reactivity [e.g., Si–O bond insertion8, phenol ene reaction8, benzenoid Diels–Alder cycloaddition (26a to 28a,8 Fig. 3c), C–H bond insertion15, aryne fluorination/trifluoromethylation16, and dihydrogen transfer from alkanes17].
Figure 3. Competition between aromatic ene and aromatic Diels-Alder pathways.

a, Computed (DFT) energetics of possibilities for the bimolecular reaction between toluene (1) and o-benzyne (3) leading to the aromatic ene product 4 (an isotoluene) vs. the Diels-Alder adducts 21a and 21b. b, Computed energetics of the analogous competitions for the tethered substrates 22 leading to the intramolecular ene product 23 vs. the [4+2] adduct 24; the potential energy surface is portrayed for the case where n = 2 and the comparative values of the ΔGTS are tabulated in the inset. c, Experimental results for the series of related HDDA substrates 26a-c; the first and last give, exclusively, the Diels–Alder products 28a and 28c whereas 26b gives only the aromatic ene product 27b.
Substrate design: the Diels-Alder vs. aromatic ene dichotomy
In the course of considering potential aromatic ene substrates like 19 (Fig. 2c), we reasoned that it should be possible to bias the reaction outcome away from an intramolecular Diels–Alder and towards the aromatic ene event (i.e., 19 to 20) by proper design of the tethered substrate. We have used computational methods to investigate the inherent energetic differences of reactions between o-benzyne (3) and toluene (1) (Fig. 3a). In those studies two competing modes of reactivity were examined–namely, aromatic ene (to 4, Figs. 1b and 3a) vs. bimolecular Diels-Alder [4+2] cycloaddition (to 21a and 21b, Fig. 3a). Density functional theory [DFT, M062X18/6-31G(d)] was used first to evaluate the relative stability of the primary products (4 and 21a/b) vis-à-vis the o-benzyne/toluene reactant pair. Both the aromatic ene (to 4) and Diels-Alder (to 21a/b) reactions were found to be quite exothermic (and to a similar degree), even though the toluene moiety is dearomatized in each of these three primary products. This reflects, largely, the loss of the high strain energy in an aryne (ca. 50 kcal•mol−1 19,20) and the net gain from the exchange of π- for σ-bonds that accompanies both the ene and Diels–Alder modes of reaction of the arene ring. We then located transition structures (TSs) for each of these competing reactions. A notable finding is that the activation energies (ΔG‡s) of the intermolecular aromatic ene vs. cycloaddition processes are very similar to one another (Fig. 3a). This is well in line with the reported ratios among the isolated products 5, 21a, and 21b (6:2:5)3,4. These results validated the merit of DFT analysis in this setting. We then used the same DFT method to study three benzyne derivatives 22 (n = 1, 2, and 3), each differing only in the length of the methylene chain joining the tolyl to benzyne moieties (Fig. 3b). We have described the reaction of substrate 26a (Fig. 3c) to give the Diels–Alder product 28a (85% isolated yield) to the exclusion of any observable amount of ene product 27a8. In view of that previous observation, it was reassuring to see that the computed relative TS energies for 22 (n = 3), having the same tether length as 26a, favors the intramolecular Diels–Alder pathway. Encouragingly, computation of either of the other substrates 22 (n = 1 or 2) showed a decided preference for the ene rather than the Diels–Alder event. The computed activation barrier for the aromatic ene reaction was considerably lower for the case where n = 2 than for n = 1. In the meantime, we synthesized substrate 26b, which contains a two-atom link, to learn if the aromatic ene pathway would be preferred for this structural motif. Indeed, heating triyne 26b produced only the aromatic ene-type product 27b (88% yield). We presumed that the initial intramolecular aromatic ene adduct cleanly rearomatized (cf. 23 to 25 in Fig. 3b and later discussion) under the reaction conditions. The complementary substrate 26c, which lacks methyl substitution on the pendant aryl ring and is, therefore, an impotent aromatic ene substrate, was then used to establish that a Diels–Alder cycloaddition is indeed feasible in this framework; product 28c was isolated in 83% yield. It is satisfying that computational study is capable of identifying the key structural attribute in the substrate—a two-atom linker between the aryne and trapping arene rings—that permits it to undergo efficient aromatic ene reaction.
Scope of the HDDA//aromatic ene reaction
To recapitulate, this unique HDDA//aromatic ene process (Table 1, top) converts a triyne substrate like 29 [bearing a (meta-alkyl)aryl substituent, connected through a two atom linker] via an aryne like 30 to an isotoluene intermediate like 31, which aromatizes to a final tricyclic product like 32. As with all HDDA-initiated reactions,8 this transformation is simply thermally induced, can be performed in a variety of non-participating solvents, and does not require particularly stringent reaction conditions (e.g., oxygen- or water-free). The broad scope of the process is demonstrated by the results summarized in Table 1. Highlights include: (i) Arynes having a variety of different electronic properties, as governed by the functional groups within the triyne tether (red), are effective enophiles (cf. 27b and 32a-e and j); (ii) the linker (blue) between the aryne enophile and arene ene donor can incorporate carbon, nitrogen, or oxygen atoms and can bear substitution (32h); (iii) substituents on the arene donor [e.g., alkoxy (32k), silyloxy (32f), or halide (32g)] are well-tolerated; (iv) on the other hand, minimally substituted arenes are also effective (cf. 32i); (v) the arene donor is not restricted to benzenoid derivatives; efficient formation of the pyridine-containing product 32j bodes well for the potential ene trapping by other heteroaromatics; (vi) secondary benzylic C–H bonds will participate in the ene transfer event, although at a slightly slower rate than that of a primary (cf. 1:2 ratio of products 32n:32o); and (vii) the ability to generate the reactive aryne intermediate in the absence of other reagents (i.e., by thermal activation only) was key to the uncovering of this aromatic ene reaction. This final point was reinforced by the outcome of our attempt to use the Lautens strategy (cf. 6 to 7 in Fig. 2a) to effect the aromatic ene trapping of the aryne 34 generated by base-induced elimination of HBr from bromoarene 33 (Fig. 4a). The biaryl 35 was formed, but in only ca. 2% yield. Products resulting from reduction (36)17 or amine trapping (37) of the intermediate aryne were formed predominantly.
Table 1.
Examples of the HDDA-initiated aromatic ene reaction.a,b
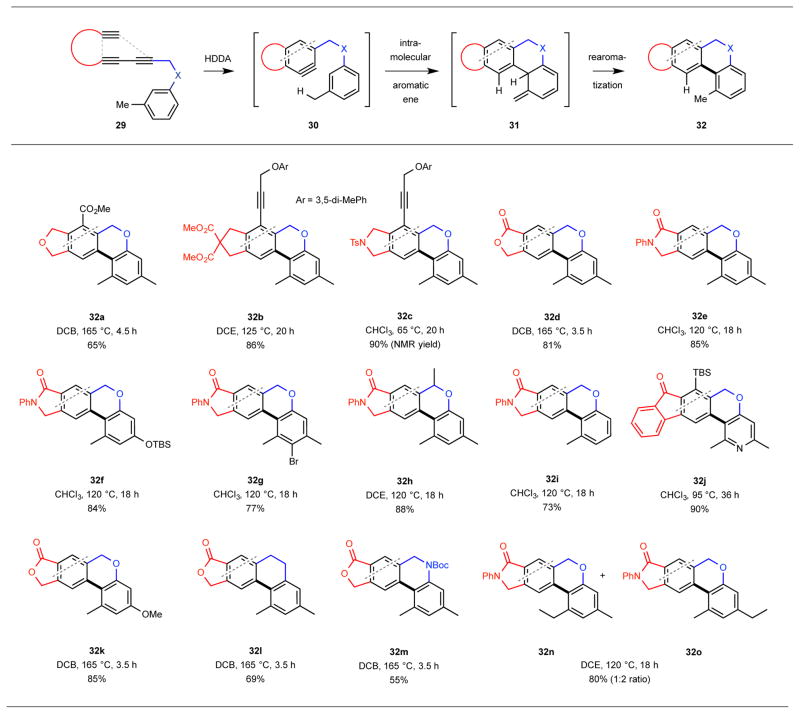
|
The dotted line in each structure shows the disconnection made possible by the HDDA reaction.
The bold bond in each indicates the C–C bond formed via the aromatic ene reaction.
Figure 4. Studies giving insight to a) the importance of reaction conditions and b/c) the mechanism of the HDDA//aromatic ene reactions.

a, Attempted aromatic ene reaction of the aryne 34 generated by elimination of HBr from the aryl bromide 33 shows an advantage of use of the reagent-free conditions of the HDDA reaction as the method for aryne formation. b, A pair of complementary deuterium-labeling experiments strongly implicate the intermediacy of the isotoluene 39. c, The cis-addition of carbon and deuterium across the maleic anhydride π-bond (see Supplementary Information for a discussion of the 1H NMR-based assignment of the structure 41-d3) provides strong mechanistic evidence for the concerted nature of the bimolecular (Alder) ene reaction.
Mechanistic investigations of the aromatic ene reaction
We have performed studies that bear on the mechanism of the final rearomatization event [cf. 23 to 25 (Fig. 3b) and 31 to 32 (Table 1, top)]. Thermal, suprafacial, unimolecular reorganization of isotoluene to toluene is symmetry-forbidden, and the antarafacial variant of this 1,3-sigmatropic rearrangement is geometrically unfavorable21, if not untenable. Thus, the 1,3-hydrogen atom migration event typically requires catalysis22. We speculated that adventitious water might be acting as a proton shuttle to promote conversion of 31 to 32 in a fashion like that known for keto-enol tautomerization23. To test this idea, we heated triyne 38 in the presence of D2O (specifically, D2O-saturated chloroform). The dominant product, 32e-d1 (Fig. 4b), was monodeuterated (ca. 93% deuterium incorporation, see Supplementary Information for analysis). A complementary experiment was performed in which the trideuterated substrate 38-d3 was heated, this time in the presence of H2O. In this case, the dominant product was 32e-d2 (Fig. 4b and Supplementary Information), in which one of the three aromatic deuterium atoms in triyne 38-d3 had been lost. These results are best explained by a rearomatization process like that portrayed in the isotoluene intermediate 39.
We next reasoned that because the isotoluene intermediate 39 formed via the initial aromatic ene reaction was sufficiently long-lived to encounter water, it might be possible to trap this species in an even more productive manner by an added external enophile. Indeed, when a 1,2-dichloroethane solution of the trideuterated substrate 38-d3 was heated in the presence of maleic anhydride24 (Fig. 4c), the adduct 41-d3 was formed. Moreover, the net cis-addition of carbon and deuterium across the enophile π-bond was stereospecific (coupling constant and chemical shift analysis of the 1H NMR spectrum, see Supplementary Information), providing evidence for a concerted ene reaction between maleic anhydride and 39-d3.
Scope of the HDDA//aromatic ene//Alder ene cascade
The successful transformation of 38 to 41 via 39 (Fig. 4c) showed that it was possible to gang an additional (bimolecular) ene trapping reaction with the initial (unimolecular) aromatic ene reaction. This amounts to a HDDA//aromatic ene//Alder ene cascade (Table 2, top). This sequence is enabled because the isotoluene intermediate 39 is still endowed with a considerable portion of the potential energy embodied by the three alkyne units in the initial HDDA substrate 38. The scope of this three-stage cascade process can be seen from the examples shown in Table 2. Highlights include: (i) a variety of types of external enophiles can be used to trap the isotoluene intermediate; these include electron-deficient alkenes (maleic anhydride and maleimides, red), α-dicarbonyl compounds (pyruvates, glyoxalate, and N-methylisatin, blue), and an N-sulfonylimine (green); (ii) the overall cascade process is tolerant of a large number of inherently reactive functional groups including imide, anhydride, ketone, ester, halogen, alcohol, aldehyde, and sulfonamide that are present in the trapping enophiles and the newly formed products, demonstrating a considerable degree of chemoselectivity; (iii) the process is also compatible with a broad array of functionality in the aryne precursor–derivatives of isoindolone, phthalide, fluorenone, and indane can all be produced in high yields; (iv) the reaction can be conducted at relatively high substrate (triyne) concentration (0.2 to 0.3 M), which speaks to the inherently fast nature of the aromatic ene trapping step; (v) only 1.2 to 2 equivalents of external trapping enophiles were used, which suggests that the isotoluene intermediate has a relatively long lifetime and bodes well for application to convergent coupling synthesis strategies using complex triyne substrates and complex trapping enophiles; and (vi) various steps in the cascade are stereoselective, as indicated by the production of a 4:1 ratio of diastereomers 42i and its epimer from the corresponding (chiral) triyne. Although we do not know the exact origin of this selectivity, relative asymmetric induction in this case can occur at either (or both) of two stages. The aromatic ene substrate 44 can cyclize to either of the diastereotopic re and si faces at its (two equivalent) ortho-carbon atoms, and the approach of the Alder enophile to the isotoluene intermediate 43 can occur via either an endo or exo geometry.
Table 2.
Examples of the HDDA//aromatic ene//Alder ene cascade.a
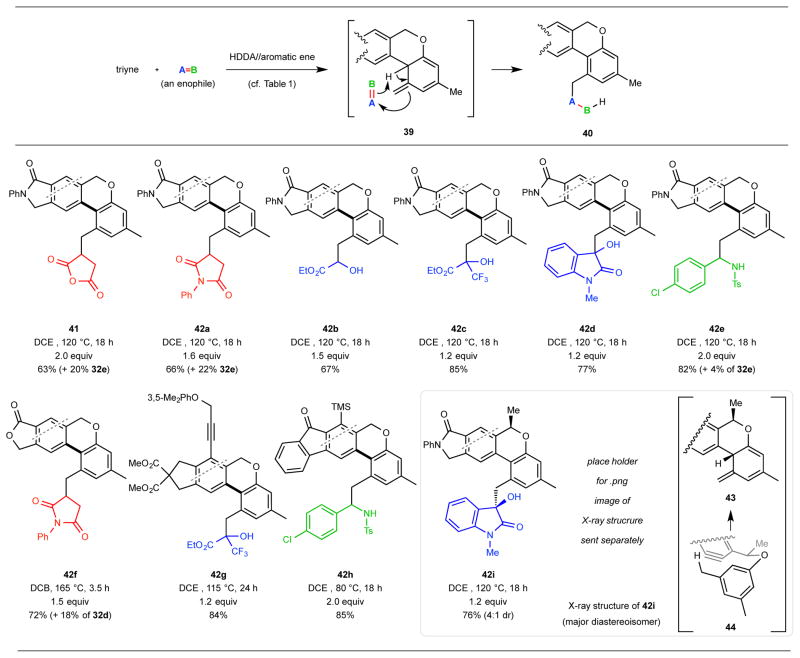
|
In the products 41 and 42a-i the moieties in red, blue and green are derived from trapping agents having
 ,
,
 and
and
 enophilic π-bonds, respectively. [or… C=C (red), C=O (blue) and C=N (green) enophilic π-bonds, respectively.]
enophilic π-bonds, respectively. [or… C=C (red), C=O (blue) and C=N (green) enophilic π-bonds, respectively.]
Conclusion
In conclusion, by capitalizing on the reagent-free, aryne-generating HDDA reaction and taking guidance from computational chemistry, we designed substrates that reveal the generality of a heretofore rare and elusive type of ene reaction in which an arene bearing a benzylic C–H bond functions as the ene donor. We have shown that the isotoluenes generated by this HDDA-enabled “aromatic ene” reaction can rearomatize either by net rearrangement or through interception by an external enophiles. Thus, two complementary overall transformations have emerged. In the first, the isotoluene rearomatizes to provide polycyclic products like 32a-o (Table 1) in a process explained by water-mediated proton shuttling. In the second, the reactive isotoluene is further engaged by an external enophile to give products of yet greater structural complexity (cf. 41 and 42a-i, Table 2). This ene-upon-ene cascade reaction involves the overall formation of four carbon-carbon bonds and three rings, requires no external reagents, and generates no byproducts. The discovery of this efficient aromatic ene reaction further attests to the importance of a key feature of the HDDA cycloisomerization—namely, its ability to deliver aryne intermediates in the absence of the potentially interfering reagents that typically accompany aryne formation by classical methods.25 Finally, this work presages additional choreographed cascades that can capitalize on the high potential energy innate to alkynes. Possibilities are under investigation here.
Methods
Full experimental procedures for preparation of and complete spectroscopic characterization data for all new compounds (aromatic ene substrates and products) and a description of the computational protocols and results can be found in the Supplementary Information.
General procedure for the HDDA//aromatic ene reaction
A solution of the triyne precursor in the indicated solvent (ca. 0.03 M) was placed in a vial closed with a Teflon-lined cap and heated at the indicated temperature (external bath). The product was purified by chromatography on silica gel after the indicated time.
General procedure for the HDDA//aromatic ene//Alder ene cascade
A solution of the triyne precursor in the indicated solvent (0.2–0.3 M) and the corresponding enophile was placed in a vial closed with a Teflon-lined cap and heated at the indicated temperature (external bath). Unless otherwise noted, the product was purified by chromatography on silica gel after the indicated time.
Supplementary Material
Acknowledgments
D.N. was partially supported by a University of Minnesota Graduate School Doctoral Dissertation Fellowship. Financial support was provided by the National Institutes of Health (CA76497 and GM65597).
Footnotes
Author Contributions D.N and T.R.H. designed the experiments, analyzed the data, and wrote the manuscript.
Supplementary information and chemical compound information are available in the online version of the paper.
Reprints and permissions information is available online at www.nature.com/reprints.
Competing financial interest The authors declare no competing financial interest.
References
- 1.Alder K, Pascher F, Schmitz A. Über die Anlagerung von Maleinsäure-anhydrid und Azodicarbonsäure-ester an einfach ungesättigte Koh an einfach ungesättigte Kohlenwasserstoffe. Zur Kenntnis von Substitutionsvorgängen in der Allyl-Stellung. Ber Dtsch Chem Ges. 1943;76:27. [Google Scholar]
- 2.Hoffman HMR. The ene reaction. Angew Chem Int Ed. 1969;8:556–577. [Google Scholar]
- 3.Brinkley YJ, Friedman L. Novel ene and insertion reactions of benzyne and alkylbenzenes. Tetrahedron Lett. 1972;13:4141–4142. [Google Scholar]
- 4.Tabushi I, Yamada H, Yoshida Z, Oda R. Reactions of benzyne with substituted benzenes. Bull Chem Soc Jpn. 1977;50:285–290. [Google Scholar]
- 5.Hoffmann RW. Dehydrobenzene and Cycloalkynes (Organic Chemistry, A Series of Monographs) Vol. 118. Academic; 1967. [Google Scholar]
- 6.Candito DA, Panteleev J, Lautens M. Intramolecular aryne–ene reaction: synthetic and mechanistic studies. J Am Chem Soc. 2011;133:14200–14203. doi: 10.1021/ja205405n. [DOI] [PubMed] [Google Scholar]
- 7.Candito DA, Dobrovolsky D, Lautens M. Development of an intramolecular aryne ene reaction and application to the formal synthesis of (±)-crinine. J Am Chem Soc. 2012;134:15572–15580. doi: 10.1021/ja306881u. [DOI] [PubMed] [Google Scholar]
- 8.Hoye TR, Baire B, Niu D, Willoughby PH, Woods BP. The hexadehydro-Diels–Alder reaction. Nature. 2012;490:208–212. doi: 10.1038/nature11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Himeshima Y, Sonoda T, Kobayashi H. Fluoride-induced 1,2-elimination of o-trimethylsilylphenyl triflate to benzyne under mild conditions. Chem Lett. 1983;12:1211–1214. [Google Scholar]
- 10.Jayanth TT, Jeganmohan M, Cheng M, Chu S, Cheng C. Ene reaction of arynes with alkynes. J Am Chem Soc. 2006;128:2232–2233. doi: 10.1021/ja058418q. [DOI] [PubMed] [Google Scholar]
- 11.Karmakar R, Mamidipalli P, Yun SY, Lee D. Alder-ene reactions of arynes. Org Lett. 2013;15:1938–1941. doi: 10.1021/ol4005905. [DOI] [PubMed] [Google Scholar]
- 12.Truong T, Daugulis O. Divergent reaction pathways for phenol arylation by arynes: Synthesis of helicenes and 2-arylphenols. Chem Sci. 2013;4:531–535. doi: 10.1039/c2sc21288a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pirali T, Zhang F, Miller AH, Head JL, McAusland D, Greaney MF. Transition-metal-free direct arylation of anilines. Angew Chem Int Ed. 2012;51:1006–1009. doi: 10.1002/anie.201106150. [DOI] [PubMed] [Google Scholar]
- 14.Baire B, Niu D, Willoughby PH, Woods BP, Hoye TR. Synthesis of complex benzenoids via the intermediate generation of o-benzynes through the hexadehydro-Diels–Alder reaction. Nature Protocols. 2013;8:501–508. doi: 10.1038/nprot.2013.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yun SY, Wang KP, Lee NK, Mamidipalli P, Lee D. Alkane C–H activation by aryne intermediates with a silver catalyst. J Am Chem Soc. 2013;135:4668–4671. doi: 10.1021/ja400477r. [DOI] [PubMed] [Google Scholar]
- 16.Wang KP, Yun SY, Mamidipalli P, Lee D. Unified approaches for fluorination, trifluoromethylation, and trifluoromethylthiolation of arynes. Chem Sci. 2013;4:3205–3211. [Google Scholar]
- 17.Niu D, Willoughby PH, Baire B, Woods BP, Hoye TR. Alkane desaturation via concerted double hydrogen atom transfer to benzyne. Nature. 2013;501:531–534. doi: 10.1038/nature12492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states,_and transition elements: two new functionals and systematic testing_of four M06-class functionals and 12 other functionals. Theor Chem Acc. 2008;120:215–241. [Google Scholar]
- 19.Ajaz A, Bradley AZ, Burrell RC, Li WHH, Daoust KJ, Bovee LB, DiRico KJ, Johnson RP. Concerted vs. stepwise mechanisms in dehydro-Diels Alder reactions. J Org Chem. 2011;76:9320–9328. doi: 10.1021/jo201567d. [DOI] [PubMed] [Google Scholar]
- 20.Jiao H, Schleyer PVR, Warmuth R, Houk KN, Beno BR. Theoretical studies of the structure, aromaticity, and magnetic properties of o-benzyne. Angew Chem Int Ed. 1997;36:2761–2764. [Google Scholar]
- 21.Rey M, Huber UA, Dreiding AS. Formation of homofulvenes. Tetrahedron Lett. 1968;32:3583–3588. [Google Scholar]
- 22.Kopecky KR, Lau M. Thermal reaction between 5-methylene-1,3-cyclohexadiene and styrene. J Org Chem. 1978;43:525–526. [Google Scholar]
- 23.Chiang Y, Kresge AJ, Santaballa JA, Wirz J. Ketonization of acetophenone enol in aqueous buffer solutions. Rate–equilibrium relations and mechanism of the “uncatalyzed” reaction. J Am Chem Soc. 1988;110:5506–5510. [Google Scholar]
- 24.Alder VK, Schmitz-Josten R. Über die Addition von Maleinsäure-anhydrid an Styrol. Liebigs Ann Chem. 1955;595:1–37. [Google Scholar]
- 25.Kitamura T. Synthetic methods for the generation and preparative application of benzyne. Aust J Chem. 2010;63:987–1001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.