

Abstract
Juvenile myelomonocytic leukemia is a rare myeloproliferative disease in young children. While hematopoietic stem cell transplantation remains the only curative therapeutic option for most patients, children with juvenile myelomonocytic leukemia increasingly receive novel agents in phase I–II clinical trials as pre-transplant therapy or therapy for relapse after transplantation. However, response criteria or definitions of outcome for standardized evaluation of treatment effect in patients with juvenile myelomonocytic leukemia are currently lacking. Here we propose criteria to evaluate the response to the non-transplant therapy and definitions of remission status after hematopoietic stem cell transplantation. For the evaluation of non-transplant therapy, we defined 6 clinical variables (white blood cell count, platelet count, hematopoietic precursors and blasts in peripheral blood, bone marrow blast percentage, spleen size and extramedullary disease) and 3 genetic variables (cytogenetic, molecular and chimerism response) which serve to describe the heterogeneous picture of response to therapy in each individual case. It is hoped that these criteria will facilitate the comparison of results between clinical trials in juvenile myelomonocytic leukemia.
Introduction
Juvenile myelomonocytic leukemia (JMML) is a clonal disease in young children.1,2 Patients with JMML present with leukocytosis, monocytosis and splenomegaly, features similar to those observed in the myeloproliferative subtype of chronic myelomonocytic leukemia (CMML) in adults.3 Other clinical signs of JMML include thrombocytopenia, leukemic skin infiltration, elevation of fetal hemoglobin (HbF), and hypersensitivity of hematopoietic progenitors to granulocyte-macrophage colony-stimulating factor (GM-CSF).4 Approximately 90% of patients with JMML harbor largely mutually exclusive mutations in PTPN11, NF1, NRAS, KRAS, or CBL in their leukemic cells resulting in hyperactivation of the RAS-MAPK pathway.5–12
Hematopoietic stem cell transplantation (HSCT) is still the only curative therapy for the vast majority of JMML patients.13–15 However, with advances in understanding the underlying molecular mechanisms in JMML, the potential for the introduction of novel therapeutic agents has been recognized for some time. Several molecules, such as isotretinoin, zoledronic acid, and farnesyl transferase inhibitor R115777 have been evaluated in pre-HSCT windows or compassionately used.16–18 Recently, azacitidine, a DNA-hypomethylating agent, was reported to induce hematologic and molecular remissions in some children with JMML,19,20 and is currently being tested in clinical trials in Europe. Additional efforts are underway to employ therapeutic inhibition of the MEK/ERK and PI3K pathways.21–23
In order to evaluate the efficacy of any conventional or novel interventions for JMML, standardized criteria to define responses and relapse are urgently required. With the goal of defining widely accepted criteria of response to therapy, international experts from the European Working Groups of Myelodysplastic Syndromes in Childhood (EWOG-MDS), the Children’s Oncology Group (COG) from the US, and from the Japanese Society of Pediatric Hematology/Oncology met to find an agreement at the JMML International Symposium, New Orleans, USA, (6 December 2013). The agreed criteria are presented in this manuscript.
Proposal of response criteria in clinical trials of therapy in JMML
Previous efforts for standardized response criteria for non-HSCT therapy in JMML
In adults, standardized response criteria proposed by an International Working Group of MDS have been widely used in clinical trials for MDS as well as CMML.24,25 These criteria are, however, not applicable to JMML and myeloproliferative CMML because they focus on reduction of blast percentage and improvement of blood counts, and are not designed to evaluate myeloproliferative features such as organomegaly.
Bergstraesser et al. defined response criteria in JMML considering white blood cell (WBC) count, platelet count, as well as liver and spleen size.26 These authors retrospectively evaluated the efficacy of 129 treatment courses other than HSCT administered to 63 children with JMML and reported a significant correlation between WBC count or spleen size and the efficacy of non-HSCT therapy. This finding was later applied to response criteria proposed by Chan et al. describing complete response (WBC <20×109/L and normalization of spleen size) and partial response (<50% of initial WBC but total still greater than 20×109/L and 25% decrease in spleen size from initial size) based solely on these two criteria, WBC count and spleen size.27 Due to the rarity of JMML there is no published prospective clinical trial that has actually applied these criteria, and there are evident limitations when only these two variables are used. The definitions are only applicable in patients with leukocytosis (WBC ≥20×109/L) and splenomegaly (≥2 cm below the costal margin). However, among 497 JMML patients currently registered in the EWOG-MDS studies, 30% had a WBC count less than 20×109/L, and 12% a spleen size of less than 2 cm below the costal margin at diagnosis (EWOG-MDS, unpublished data, 2014). In addition, the criteria are not applicable to patients relapsing post HSCT who have reappearance of cytogenetic or molecular abnormalities after HSCT but who do not yet show the full clinical picture of relapse. Response criteria thus need to be applicable in many different clinical situations for a broad range of patients. Therefore, clinical variables other than WBC count and spleen size are needed, and cytogenetic and molecular variables are also necessary to describe disease status.
The concept behind the proposed response criteria
As outlined above, response measurements in JMML need to be applicable to the highly heterogeneous clinical features at presentation and to individual response patterns to different therapeutic agents. Therefore, 6 clinical variables and 3 genetic variables were selected (Table 1). For each of these variables (v), complete response (vCR), partial response (vPR) and progressive disease (vPD) are defined. This makes the evaluation of heterogeneous effects of each intervention possible. Because we recognize that each patient can present with different clinical, cytogenetic and molecular features, the number of evaluable variables at the start of therapy differs among patients. Based on the cumulative response of these 9 variables, the clinical and genetic remission status can be described (Tables 2).
Table 1.
Variables for evaluation of response to therapy in JMML.
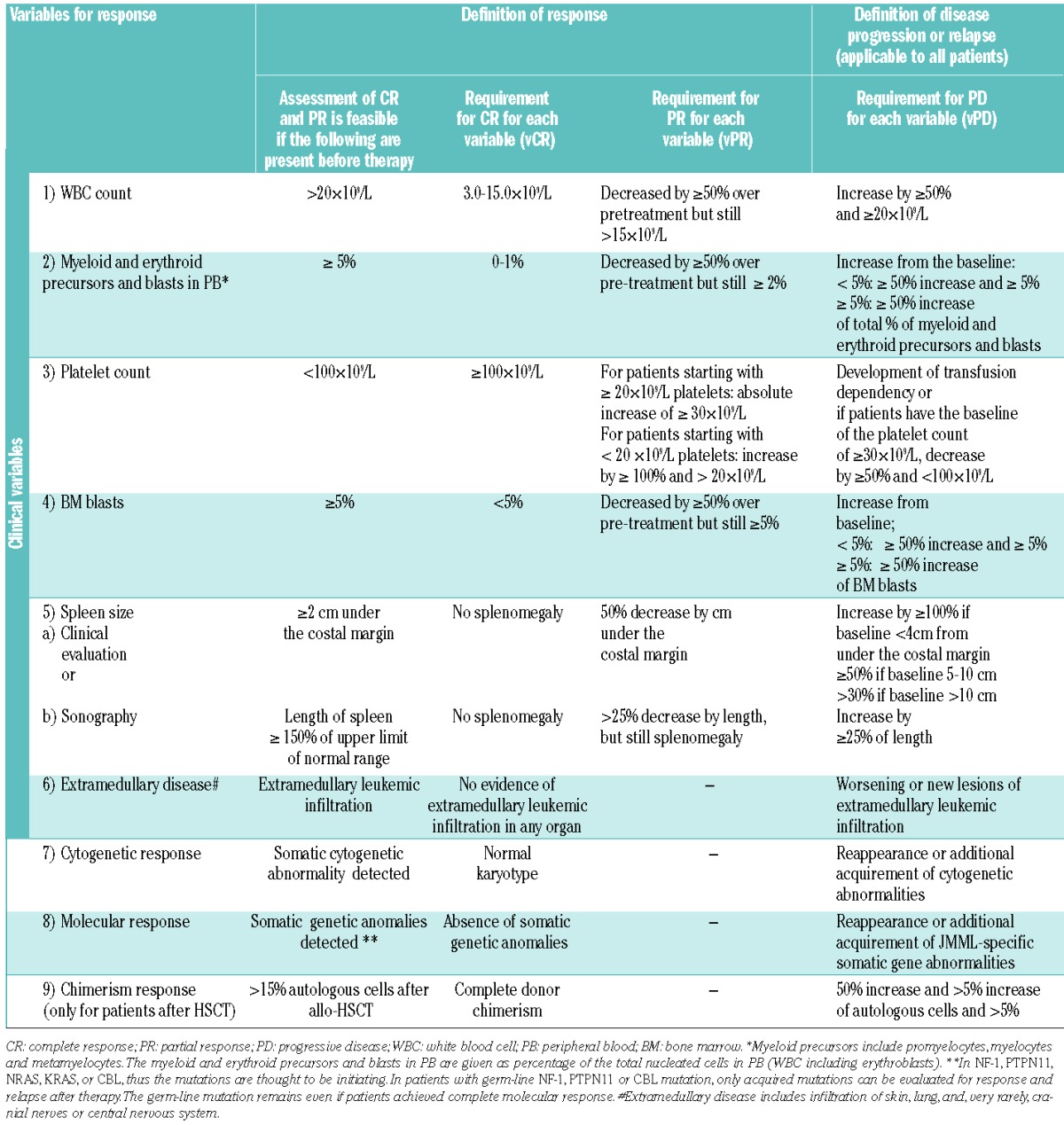
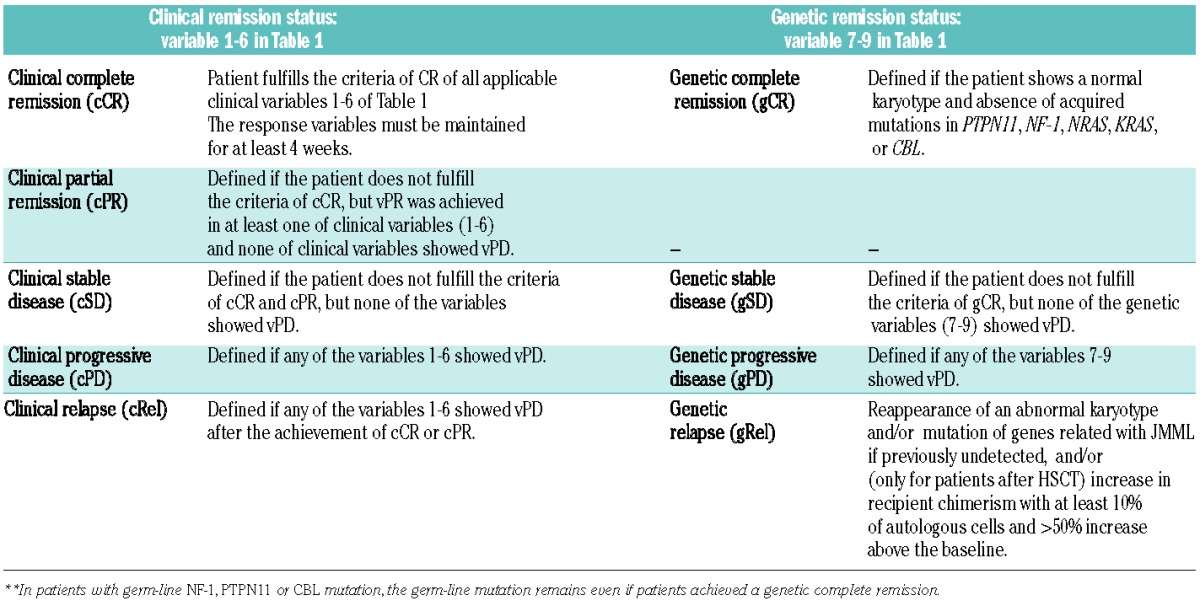
Table 2.
Definition of response following therapy other than HSCT in JMML.
Variables to evaluate response
In addition to WBC count and spleen size, clinical variables include presence of myeloid/erythroid precursors and blasts in peripheral blood, platelet count, percentage of blasts in bone marrow, and presence of extramedullary disease (Table 1).4 Two additional hallmarks of JMML, the level of hemoglobin F (HbF) corrected for age and the presence of monocytosis, have intentionally been excluded as response criteria for various reasons. High HbF levels at diagnosis are known to predict outcome but are dependent on karyotype.4 Moreover, so far there has been no report on serial HbF levels during the natural course of JMML or following HSCT. Therefore, further studies to evaluate HbF during the clinical course of JMML patients are necessary. Monocytosis more than 1.0×109/L is one of the diagnostic criteria of JMML,4 but the absolute monocyte count generally correlates with the WBC. Since the usefulness of the WBC count has been previously confirmed, we chose the WBC count, but not the monocyte count, as a variable to be evaluated.26 Moreover, monocytosis can be non-specific since it is observed in various conditions, such as infections or an early sign of bone marrow recovery after HSCT, and leukocytosis in JMML is manifested not only by monocytosis but also by circulating immature myeloid cells in the peripheral blood.
Since the spleen size is one of the variables evaluating treatment efficacy in JMML,26 reliable methods to measure the size of this variably shaped organ are required. Size determination by palpation with measurement of the length between the spleen tip and the left costal margin is clinically appropriate26,28–30 but may not be sufficient for clinical trials since its results are not verifiable. Measurements by computed tomography are to be avoided in children with JMML because of radiation issues, and magnetic resonance imaging generally requires deep sedation or anesthesia. For these reasons, we currently recommend evaluation of spleen size by ultrasound with the linear measurement of the splenic length, defined as the maximum distance between the dome and the tip of spleen in the right lateral decubitas position.31
The three genetic variables selected for evaluating response were cytogenetics, molecular alterations, and chimerism. Cytogenetic and molecular data must have been collected at diagnosis while chimerism is only applicable after HSCT. Abnormal karyotypes are observed in approximately 35% of JMML patients, with monosomy 7 being the most common aberration (25%).4 Oncogenic molecular alterations in PTPN11, NF-1, NRAS, KRAS or CBL, noted in approximately 90% of patients are increasingly important tools for diagnosis and follow up of JMML.5–12 We anticipate that additional somatic mutations will be discovered in JMML. Indeed, recently SETBP1 and JAK3 mutations were reported as a result of an exome sequencing project.32 However, until these markers are further validated, we would suggest that these mutations, which might indicate sub-clones, fall under the category of “acquired molecular abnormalities”. Analysis of donor chimerism has been a standard measurement to follow JMML patients given allogeneic HSCT. While discussion of the various methods used to determine donor chimerism is beyond the scope of this consensus report, there was broad agreement that unsorted cell donor chimerism should be a common measurement in clinical trials. Most JMML patients with persistent mixed chimerism experience clinical relapse of JMML,33 and are thus candidates for early intervention with innovative therapies prior to development of a full clinical relapse.
Definitions of response to therapy other than HSCT in JMML
Based on the response of each applicable variable listed in Table 1, the clinical and genetic remission status can be defined for each patient (Table 2). In patients who achieve genetic complete remission (gCR), JMML cells are considered to have been eradicated, irrespective of clinical remission status. Patients with gCR may have persistent splenomegaly or leukocytosis from causes other than JMML, such as infections. However, it is unlikely that such a patient has clinical signs of progressive disease of JMML in the presence of gCR. In such a patient, any new clonal abnormalities, other possible errors of genetic examinations, or other disorders which give rise to JMML-like clinical features, should be excluded.
Response criteria for clinical trials of HSCT
There is a consensus that criteria for remission after HSCT are somewhat different from those stated above for remission after non-HSCT. The remission criteria in HSCT recipients include the results of chimerism analyses (Table 3). Appraisal of methodological consideration of chimerism studies will change over time and is beyond the scope of this consensus document. Patients undergoing HSCT who achieve neutrophil engraftment and complete donor chimerism with disappearance of acquired cytogenetic and molecular abnormalities are considered to have a complete remission of JMML. In these individuals, complete remission is defined irrespective of the spleen size or WBC counts, since post HSCT, patients often have persistent splenomegaly and leukocytosis without active JMML due to infections, graft-versus-host disease or other hepatic complications. For the very few cases with mixed chimerism after HSCT and no diagnostic cytogenetic or molecular marker, definition of complete remission requires the resolution of all clinical features indicative of JMML (Table 3).
Table 3.
Definition of complete remission and relapse after hematopoietic stem cell transplantation in children with JMML.

Conclusion
In this paper we propose response and relapse criteria for patients who are diagnosed and treated for JMML, recognizing the complexities of disease presentation and therapeutic interventions. The usefulness and suitability of these criteria need to be proven in prospective clinical trials. It is likely that the proposed response criteria will require modifications in the future based on the accumulated experiences and advances of molecular biology in JMML. Because the goal of therapy of children with JMML is a cure, it is also important to be aware that responses need to be translated into an increase in long-term survival in well-controlled clinical trials.
Acknowledgments
We thank the JMML Foundation for their continuous efforts to nurture the discussion among clinicians and scientists working in the field of JMML and organizing an annual International JMML Symposium. The project described here was supported by award number R13CA132568 from the National Cancer Institute to the JMML Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. This project was also supported by a grant from the Parent Initiative for Children with Cancer, Freiburg (Förderverein für krebskranke Kinder e.V. Freiburg i. Br.) to the Coordinating Study Center of the EWOG-MDS in Freiburg, Germany.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Niemeyer CM, Kratz CP. Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia: molecular classification and treatment options. Br J Haematol. 2008;140(6):610–24. [DOI] [PubMed] [Google Scholar]
- 2.Loh ML. Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br J Haematol. 2011; 152(6):677–87. [DOI] [PubMed] [Google Scholar]
- 3.Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick H, et al. The chronic myeloid leukaemias: guidelines for distinguishing chronic granulocytic, atypical chronic myeloid, and chronic myelomonocytic leukaemia. Proposals by the French-American- British Cooperative Leukaemia Group. Br J Haematol. 1994; 87(4):746–54. [DOI] [PubMed] [Google Scholar]
- 4.Niemeyer CM, Arico M, Basso G, Biondi A, Cantu RA, Creutzig U, et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS). Blood. 1997;89(10):3534–43. [PubMed] [Google Scholar]
- 5.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic PTPN11 mutations in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003; 34(2):148–50. [DOI] [PubMed] [Google Scholar]
- 6.Kratz CP, Niemeyer CM, Castleberry RP, Cetin M, Bergsträsser E, Emanuel PD, et al. The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood. 2005;106(6):2183–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyauchi J, Asada M, Sasaki M, Tsunematsu Y, Kojima S, Mizutani S. Mutations of the N-ras gene in juvenile chronic myelogenous leukemia. Blood. 1994;83(8):2248–54. [PubMed] [Google Scholar]
- 8.Side LE, Emanuel PD, Taylor B, Franklin J, Thompson P, Castleberry RP, et al. Mutations of the NF1 gene in children with juvenile myelomonocytic leukemia without clinical evidence of neurofibromatosis, type 1. Blood. 1998;92(1):267–72. [PubMed] [Google Scholar]
- 9.Flotho C, Valcamonica S, Mach-Pascual S, Schmahl G, Corral L, Ritterbach J, et al. Ras mutations and clonality analysis in children with juvenile myelomonocytic leukemia (JMML). Leukemia. 1999;13(1):32–7. [DOI] [PubMed] [Google Scholar]
- 10.Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archambeault S, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. 2009; 114(9):1859–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, Bunin NJ, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet. 2010;42(9):794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Vries AC, Zwaan CM, van den Heuvel-Eibrink MM. Molecular basis of juvenile myelomonocytic leukemia. Haematologica. 2010; 95(2):179–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Locatelli F, Nöllke P, Zecca M, Korthof E, Lanino E, Peters C, et al. Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood. 2005;105(1):410–9. [DOI] [PubMed] [Google Scholar]
- 14.Yabe M, Sako M, Yabe H, Osugi Y, Kurosawa H, Nara T, et al. A conditioning regimen of busulfan, fludarabine, and melphalan for allogeneic stem cell transplantation in children with juvenile myelomonocytic leukemia. Pediatr Transplant. 2008; 12(8):862–7. [DOI] [PubMed] [Google Scholar]
- 15.Locatelli F, Crotta A, Ruggeri A, Eapen M, Wagner JE, Macmillan ML, et al. Analysis of risk factors influencing outcomes after cord blood transplantation in children with juvenile myelomonocytic leukemia: a EURO-CORD, EBMT, EWOG-MDS, CIBMTR study. Blood. 2013;122(12):2135–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castleberry RP, Emanuel PD, Zuckerman KS, Cohn S, Strauss L, Byrd RL, et al. A pilot study of isotretinoin in the treatment of juvenile chronic myelogenous leukemia. N Engl J Med. 1994;331(25):1680–4. [DOI] [PubMed] [Google Scholar]
- 17.Castleberry RP, Loh ML, Jayaprakash N, Peterson A, Casey V, Chang M, et al. Phase II window study of the farnesyltransferase inhibitor R115777 (Zarnestra®) in untreated juvenile myelomonocytic leukemia (JMML): A Children’s Oncology Group study [abstract]. Blood. 2005; 106:727a. [Google Scholar]
- 18.Shimada H, Shima H, Shimasaki N, Yoshihara H, Mori T, Takahashi T. Little response to zoledronic acid in a child of juvenile myelomonocytic leukemia (JMML) harboring the PTPN11 mutation. Ann Oncol. 2005;16(8):1400. [DOI] [PubMed] [Google Scholar]
- 19.Furlan I, Batz C, Flotho C, Mohr B, Lübbert M, Suttorp M, et al. Intriguing response to azacitidine in a patient with juvenile myelomonocytic leukemia and monosomy 7. Blood. 2009;113(12):2867–8. [DOI] [PubMed] [Google Scholar]
- 20.Cseh A, Niemeyer CM, Catalá A, Dworzak M, Hasle H, van den Heuvel-Eibrink MM, et al. Therapy with azacitidine in pediatric MDS and JMML A retrospective survey of the EWOG-MDS study. Haematologica. 2012;97:S3. [Google Scholar]
- 21.Lyubynska N, Gorman MF, Lauchle JO, Hong WX, Akutagawa JK, Shannon K, et al. A MEK inhibitor abrogates myeloproliferative disease in Kras mutant mice. Sci Transl Med. 2011;3:76ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang T, Krisman K, Theobald EH, Xu J, Akutagawa J, Lauchle JO, et al. Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J Clin Invest. 2013;123(1):335–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodwin CB, Li XJ, Mali RS, Chan G, Kang M, Liu Z, et al. PI3K p110delta uniquely promotes gain-of-function Shp2-induced GM-CSF hypersensitivity in a model of JMML. Blood. 2014;23(18):2838–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheson BD, Bennett JM, Kantarjian H, Pinto A, Schiffer CA, Nimer SD, et al. Report of an international working group to standardize response criteria for myelodysplastic syndromes. Blood. 2000;96(12):3671–4. [PubMed] [Google Scholar]
- 25.Cheson BD, Greenberg PL, Bennett JM, Lowenberg B, Wijermans PW, Nimer SD, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108(2):419–25. [DOI] [PubMed] [Google Scholar]
- 26.Bergstraesser E, Hasle H, Rogge T, Fischer A, Zimmermann M, Noellke P, et al. Non-hematopoietic stem cell transplantation treatment of juvenile myelomonocytic leukemia: a retrospective analysis and definition of response criteria. Pediatr Blood Cancer. 2007;49(5):629–33. [DOI] [PubMed] [Google Scholar]
- 27.Chan RJ, Cooper T, Kratz CP, Weiss B, Loh ML. Juvenile myelomonocytic leukemia: a report from the 2nd International JMML Symposium. Leuk Res. 2009;33(3):355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ades L, Sekeres MA, Wolfromm A, Teichman ML, Tiu RV, Itzykson R, et al. Predictive factors of response and survival among chronic myelomonocytic leukemia patients treated with azacitidine. Leuk Res. 2013;37(6):609–13. [DOI] [PubMed] [Google Scholar]
- 29.Aribi A, Borthakur G, Ravandi F, Shan J, Davisson J, Cortes J, et al. Activity of decitabine, a hypomethylating agent, in chronic myelomonocytic leukemia. Cancer. 2007;109(4):713–7. [DOI] [PubMed] [Google Scholar]
- 30.Wattel E, Guerci A, Hecquet B, Economopoulos T, Copplestone A, Mahé B, et al. A randomized trial of hydroxyurea versus VP16 in adult chronic myelomonocytic leukemia. Groupe Francais des Myelodysplasies and European CMML Group. Blood. 1996;88(7):2480–7. [PubMed] [Google Scholar]
- 31.Lamb PM, Lund A, Kanagasabay RR, Martin A, Webb JA, Reznek RH, et al. Spleen size: how well do linear ultrasound measurements correlate with three-dimensional CT volume assessments? Br J Radiol. 2002; 75(895):573–7. [DOI] [PubMed] [Google Scholar]
- 32.Sakaguchi H, Okuno Y, Muramatsu, Yoshida K, Shiraishi Y, Takahashi M, et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet. 2013;45(8):937–41. [DOI] [PubMed] [Google Scholar]
- 33.Yoshimi A, Niemeyer CM, Bohmer V, Duffner U, Strahm B, Kreyenberg H, et al. Chimaerism analyses and subsequent immunological intervention after stem cell transplantation in patients with juvenile myelomonocytic leukaemia. Br J Haematol. 2005;129(4):542–9. [DOI] [PubMed] [Google Scholar]