

Abstract
Accurate and timely diagnosis of inherited bone marrow failure and inherited myelodysplastic syndromes is essential to guide clinical management. Distinguishing inherited from acquired bone marrow failure/myelodysplastic syndrome poses a significant clinical challenge. At present, diagnostic genetic testing for inherited bone marrow failure/myelodysplastic syndrome is performed gene-by-gene, guided by clinical and laboratory evaluation. We hypothesized that standard clinically-directed genetic testing misses patients with cryptic or atypical presentations of inherited bone marrow failure/myelodysplastic syndrome. In order to screen simultaneously for mutations of all classes in bone marrow failure/myelodysplastic syndrome genes, we developed and validated a panel of 85 genes for targeted capture and multiplexed massively parallel sequencing. In patients with clinical diagnoses of Fanconi anemia, genomic analysis resolved subtype assignment, including those of patients with inconclusive complementation test results. Eight out of 71 patients with idiopathic bone marrow failure or myelodysplastic syndrome were found to harbor damaging germline mutations in GATA2, RUNX1, DKC1, or LIG4. All 8 of these patients lacked classical clinical stigmata or laboratory findings of these syndromes and only 4 had a family history suggestive of inherited disease. These results reflect the extensive genetic heterogeneity and phenotypic complexity of bone marrow failure/myelodysplastic syndrome phenotypes. This study supports the integration of broad unbiased genetic screening into the diagnostic workup of children and young adults with bone marrow failure and myelodysplastic syndromes.
Introduction
Timely and accurate diagnosis of inherited bone marrow failure (BMF) and inherited myelodysplastic syndromes (MDS) is essential to ensure appropriate medical management and treatment.1,2 Early diagnosis of an underlying inherited BMF/MDS allows clinical monitoring for signs of clonal evolution in order to initiate hematopoietic stem cell transplantation prior to leukemia development. Secondary leukemias arising in these clinical contexts carry particularly poor prognoses. Many of these syndromes require reduced intensity transplantation regimens to avoid excessive toxicities. Furthermore, the recognition of an inherited disorder informs hematopoietic stem cell donor selection as it allows unambiguous identification of affected siblings and guides appropriate genetic counseling of family members.
Evidence-based guidelines for genetic screening for inherited BMF/MDS are scarce. A family history of MDS or leukemia is a clue to an underlying inherited cause. Physicians also rely on clinical stigmata of inherited BMF/MDS to guide diagnosis. Substantial phenotypic overlap between different syndromes, as well as absent, subtle, or previously unreported clinical findings of inherited BMF/MDS, render diagnosis challenging.3 The clinical benefit of applying next generation sequencing approaches for comprehensive genetic screening of seemingly idiopathic BMF/MDS remains a critical question.
To investigate the clinical utility of broad genomic analysis for cryptic or atypical presentations of inherited BMF/MDS, we developed a targeted capture gene panel coupled with high-throughput, multiplexed, massively parallel sequencing. This panel, referred to here as MarrowSeq, includes 85 genes responsible for inherited and acquired marrow failure syndromes and MDS. Using this assay, we queried patients deemed to have idiopathic disease for an underlying genetic cause of their BMF and MDS.
Methods
Subjects
The study was conducted in accordance with a protocol approved by the Institutional Review Board of Seattle Children’s Hospital and the Declaration of Helsinki. Informed consent was obtained for all study subjects. Samples were obtained from pediatric and adult patients with idiopathic BMF or MDS treated between 2000 and 2013 at Seattle Children’s Hospital, Seattle Cancer Care Alliance, University of Washington Medical Center, and Boston Children’s Hospital. Patients had been previously tested for mutations in various individual inherited marrow failure syndrome genes based on clinical history and physical findings but remained unclassified after genetic workup. Pediatric inclusion criteria were presentation to a pediatric hematology clinic with idiopathic marrow failure (hypoproductive cytopenias including any of the following: absolute neutrophil count less than 1.5×109/L, hemoglobin low for age, platelet count <150×109/L, hypocellular marrow for age) or MDS defined by WHO criteria.4 Adult inclusion criteria were presentation to an adult hematology clinic with marrow failure or MDS (defined as above) and either young age (<40 years) at presentation, or family history suggestive of inherited BMF/MDS regardless of age. Genomic DNA was isolated from peripheral blood, marrow mononuclear cells, and/or fibroblasts using the Allprep DNA/RNA kit (Qiagen) or as previously described.5
Genomics
Oligonucleotide probes were designed using the eArray website (Agilent Technologies). Probes of 120 basepairs (bp) tiled each region with 3x depth and covered coding regions, 20 bp of intronic sequence flanking each exon, as well as the promoter of DKC16 and the 5′UTR of ANKRD26.7 Pathogenic variants in an intronic region of GATA2 were described subsequently8 so all samples were screened for these mutations by Sanger sequencing. Mutations in RTEL1 and ERCC4 leading to dyskeratosis congenita9 and Fanconi anemia,10 respectively, were reported after completion of the initial screens and have been incorporated into subsequent gene capture pools.
Targeted gene capture and sequencing were performed as previously described.11,12 Reads were aligned to the human reference genome (hg19) using the Burrows-Wheeler aligner.13 Single nucleotide and small insertion-deletion (indel) variants were called by three independent bioinformatics pipelines, as previously described12,14,15 and results jointly analyzed. Alignment to the whole genome facilitated exclusion of variants that fell in pseudogenes. Copy number variants (CNVs) were identified as previously described.16
Variants were classified by predicted effect on protein function, as previously described.11,12 Variants previously reported as either germline or somatic pathogenic alleles for BMF/MDS were specifically noted. Loss-of-function or likely damaging mutations in genes not previously reported in inherited BMF/MDS were considered variants of unknown clinical significance. Variants of unknown clinical significance were evaluated as follows: 1) the likely impact of haploinsufficiency for truncating mutations was assessed based on published data regarding the function for the host gene and on modes of inheritance of phenotypes associated with the gene; and 2) in silico structural modeling for missense variants at highly conserved sites in critical domains.
Potentially damaging variants were validated by Sanger sequencing. Variants were confirmed as germline by sequencing fibroblast DNA. This analysis focused on identification of constitutional mutations in BMF/MDS genes.
Additional details on the design and methods of this study are provided in the Online Supplementary Methods.
Results
Validation of gene capture assay
Genes known to contribute to inherited bone marrow failure and myelodysplastic syndromes were selected for capture and sequencing (Table 1). Since acquired mutations that cause MDS may also be pathogenic when present as an inherited mutation, and vice versa, a broad gene panel was designed. Identified mutations were evaluated based on variant allele read fraction and by analysis of DNA sequences from a non-hematologic tissue to determine whether a mutation was constitutional. For all samples evaluated, median coverage across the 383kb targeted region was 549X, with 97.8% of bases having over 50X coverage and 98.2% of bases having over 10X coverage. This depth of coverage enabled identification of all classes of mutations, including point mutations, small indels, copy number variants, and genomic rearrangements. The assay was validated by blinded analysis of genomic DNA from 14 patients with known mutations in nine genes representing a variety of mutation classes (Online Supplementary Table S1). All mutations, including copy number variants (Figure 1A), were correctly identified.
Table 1.
Inherited bone marrow failure and myelodysplastic syndrome genes.
Figure 1.
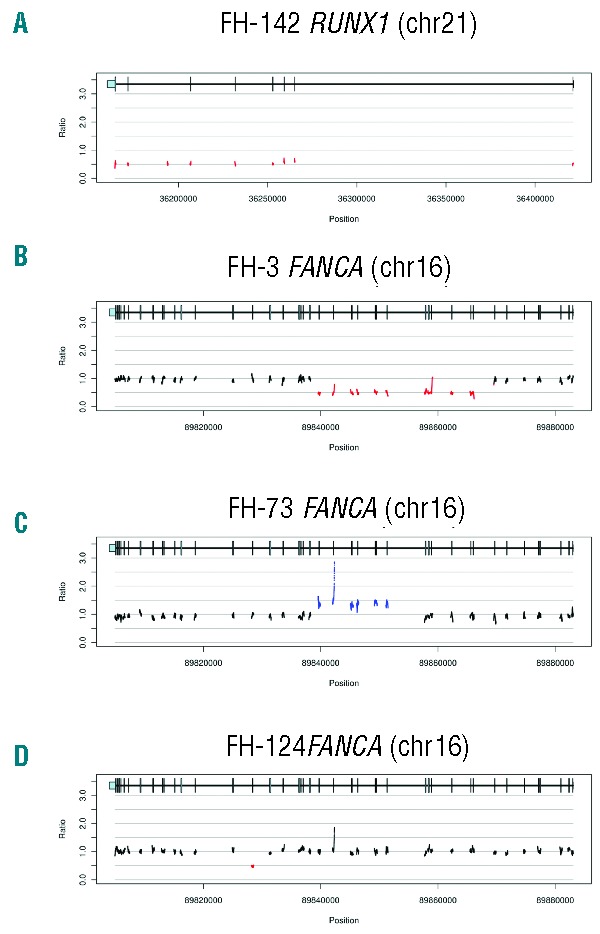
Detection of genomic copy number variants. Ratios of sample to median corrected depth of coverage within a flow cell lane are plotted across targeted genomic regions of the indicated gene. Diploid bases are shown in black. Deletions and duplications are shown in red and blue, respectively. Genomic positions of exons (vertical bars) and untranslated regions (light blue rectangles) are shown above ratio plots. (A) Whole gene deletion of RUNX1. No diploid bases were present in this region. (B) Deletion of FANCA exons 9–22. (C) Amplification of FANCA exons 15–22. (D) Deletion of FANCA exon 29.
Genetic determination of Fanconi anemia subtype
Simultaneous targeted capture and sequencing of all Fanconi anemia (FA) genes was applied to 6 patients who had been clinically diagnosed with Fanconi anemia. By prior complementation studies, 4 patients had been reported as subtype A (FA-A), and 2 patients remained unclassified after clinical complementation testing for A, C, and G subtypes. The critical mutations were unknown for all 6 patients. We identified biallelic deleterious mutations in FANCA in FH-9, FH-73, FH-124, and FH-241 (Figure 2A), confirming the results of their complementation tests. Three of the eight FANCA mutations were copy number variants (Figure 1B–D).
Figure 2.
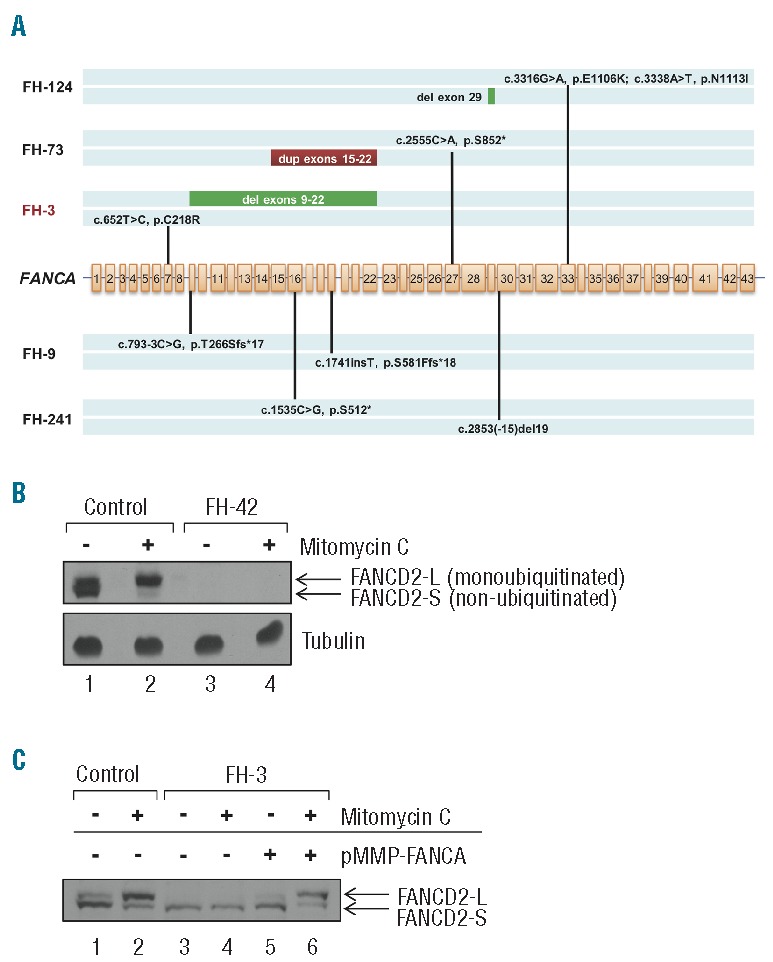
Targeted gene capture correction of Fanconi anemia subtype assignment. (A) Biallelic FANCA mutations identified by MarrowSeq in 5 patients. FA patient FH-3 (highlighted in red) was non-ACG subtype by clinical complementation testing. (B) Protein extracts of bone marrow fibroblasts isolated from healthy controls or Fanconi anemia patient FH-42 (FANCD2, p.[Leu683Pro];[Glu906Ilefs*4]) were immunoblotted for FANCD2 with or without 24-h mitomycin C treatment. Fibroblasts from FH-42 exhibit low FANCD2 protein expression (lanes 3 and 4) in comparison to cells from controls (lanes 1 and 2). α-tubulin was used to ascertain equivalent protein loading. (C) Functional validation of Fanconi anemia subtype A in FA patient FH-3 (FANCA p.[Cys218Arg];[Val265Leufs*8]). Protein extracts of bone marrow fibroblasts isolated from healthy control or FH-3 were immunoblotted for FANCD2 with or without 24-h mitomycin C treatment. Fibroblasts from healthy control show both non-ubiquitinated (FANCD2-S) and monoubiquitinated (FANCD2-L) FANCD2 forms (lane 1), with an increased ratio of monoubiquitinated FANCD2 relative to non-ubiquitinated FANCD2 upon mitomycin C treatment (lane 2). Fibroblasts from FH-3 show only the non-ubiquitinated FANCD2-S form with and without mitomycin C (lanes 3 and 4). FANCD2 monoubiquitination is restored upon infection with a pMMP retroviral vector encoding the wild-type FANCA cDNA (lanes 5 and 6).
Patient FH-42, without a previous subtype assignment, carried two damaging mutations in FANCD2: c.2715+1G>A (p.Glu906Ilefs*4) and c.2048T>C (p.Leu683Pro) (Online Supplementary Table S2). Sequencing of subcloned cDNA from this patient indicated that the two mutations were in trans and indicated the insertion of 27 base pairs of intron 28 in the FANCD2 transcript, consistent with aberrant splicing. Immunoblotting of protein from patient-derived fibroblast lysates showed nearly absent levels of FANCD2 protein (Figure 2B), confirming subtype D2 for this patient.
Clinical complementation testing of Patient FH-3 was negative for FA subtypes A, C, and G. However, genetic analysis revealed two mutations in FANCA for FH-3 (Figure 2A). No other pathogenic mutations were found in any other FA genes. By immunoblot analysis, fibroblasts of FH-3 were deficient in FANCD2 monoubiquitination, which was restored by the introduction of wild-type FANCA (Figure 2C), confirming the FA-A subtype for this patient.
Broad genetic screening identifies cryptic presentations of inherited BMF/MDS
We next tested for BMF/MDS gene mutations in 71 patients deemed to have idiopathic disease after clinical, laboratory, and clinically-directed genetic evaluation (Table 2 and Online Supplementary Table S3). Marrow failure in these patients remained unclassified after directed testing of candidate genes, chromosomal breakage testing for possible Fanconi anemia,18 telomere length analysis for possible dyskeratosis congenita,17 and tests of pancreatic enzyme levels for possible Shwachman-Diamond syndrome.19 Of the 71 subjects, 58 patients were age 18 years or younger and drawn from the pediatric clinic, and 13 patients were older than 18 years and drawn from the adult clinic. Thirty-two patients, including 6 of the 13 adults, had a positive family history. Eight patients carried damaging germline mutations in one of the BMF/MDS genes (Table 3). Five patients were heterozygous for damaging mutations in the hematopoietic transcription factor GATA2, despite the absence of clinical features of MonoMac20–23 or Emberger syndrome.24 GATA2 contains two Cys4 zinc finger domains, the first of which is responsible for interaction with co-regulator FOG1 (Friend of GATA1) and the second for mediating DNA binding. Two patients with idiopathic MDS carried nonsense mutations that truncate the protein in the first zinc finger domain (GATA2 c.988C>T [p.Arg330*]) or in the second zinc finger domain (GATA2 c.1084C>T [p.Arg362*]). GATA2 p.Arg330* was previously reported in an individual with MonoMac characteristics;23 our patient presented with familial leukemia/myelodysplasia. GATA2 p.Arg362* has not been previously reported. Two other patients carried GATA2 missense mutations altering highly-conserved residues in the second zinc finger domain: GATA2 c.1078T>A [p.Trp360Arg] and GATA2 c.1082G>A [p.Arg361His]. These missense mutations have not been previously reported, but GATA2 p.Arg361Leu and p.Arg361Cys have been reported in Emberger24 and MonoMac25 syndromes, respectively, and GATA2 p.Arg361His has been reported as an acquired mutation in acute myelogenous leukemia (AML).26 Trp360 resides in a LWRR motif that is conserved across N- and C-terminal zinc fingers in all human GATA proteins (Online Supplementary Figure S1A). Mutation of the buried hydrophobic Trp360 to a basic arginine residue would be predicted to disrupt the zinc-finger domain folding (Online Supplementary Figure S1B). These data support recent reports of cryptic presentations of GATA2 mutations without features of MonoMac syndrome, Emberger syndrome, or familial MDS in patients with BMF/MDS.25,27
Table 2.
Characteristics of patients with idiopathic BMF/MDS.
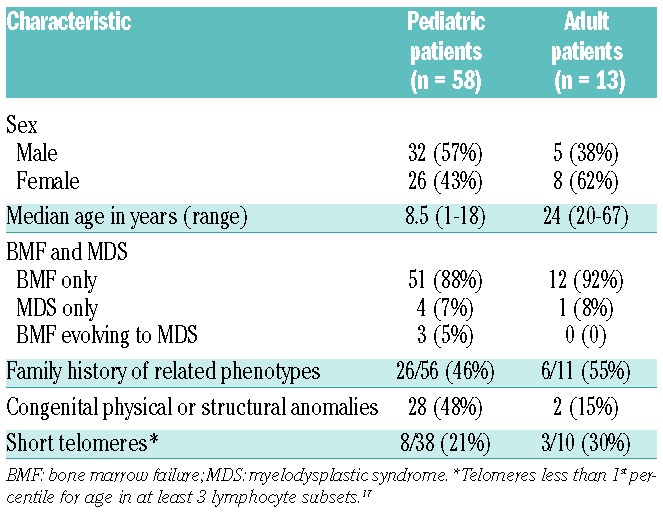
Table 3.
Clinical features and genetic diagnoses of patients with previously unclassified BMF/MDS.
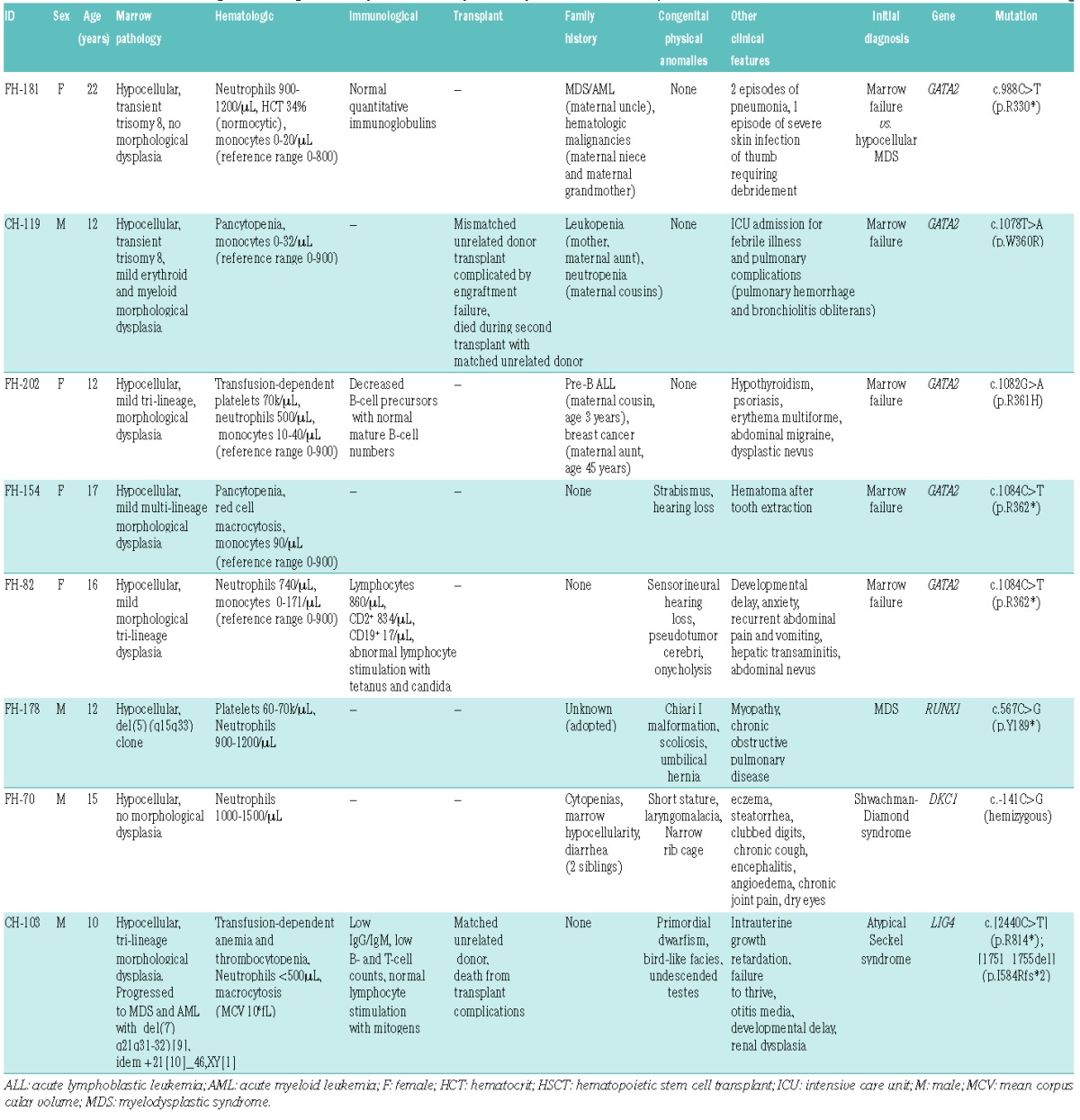
One patient (FH-178) presented with MDS. His antecedent medical history was significant for multiple congenital anomalies, neutropenia, and thrombocytopenia (Table 3). A heterozygous mutation (c.567C>G [p.Tyr189*]) in RUNX1 was identified. Heterozygous RUNX1 mutations cause familial platelet disorder with propensity to myeloid malignancy.28,29 RUNX1 p.Tyr189* truncates the protein C-terminal to the RUNT domain, with loss of the transactivation domain, and is predicted to function in a dominant negative fashion.30 One patient (FH-70) was originally diagnosed at an outside hospital with Shwachman-Diamond syndrome based on his clinical presentation of failure to thrive, neutropenia, enzyme-responsive steatorrhea, and low fecal elastase. He lacked mutations in SBDS and expressed normal levels of SBDS protein. Telomere lengths fell between the 1st and 10th percentiles for age in all 6 leukocyte subsets tested for FH-70, and hence did not raise prior clinical suspicion for dyskeratosis congenita using published criteria.18 We identified a hemizygous mutation in DKC1 (c.-141C>G) located in the promoter of the DKC1 gene. This promoter mutation was not found in normal control databases dbSNP138, the Exome Variant Server, nor 1000Genomes Project. Whole exome sequencing analysis of this family did not identify alternative candidate genes. This promoter mutation had been previously reported in 2 unrelated kindreds with X-linked dyskeratosis congenita and segregated with the dyskeratosis congenita (DC) phenotype.6,31 This mutation is located in a GC-rich element required for Sp1 transcription factor binding32 and leads to reduced transcript levels of DKC1.6 In DKC1 (c.-141C>G) patient-derived CD34+ cells, expression of DKC1WT, but not catalytically-inactive DKC1Asp125Ala, rescued hematopoietic colony formation.6 The absence of telomere lengths shorter than the first percentile was consistent with prior reports of other DC patients carrying this promoter mutation in DKC1.6 Thus, broad genetic screening identified a clinically unsuspected genetic syndrome presenting in an atypical fashion.
Patient CH-103 was originally diagnosed with Seckel syndrome based on his presentation with short stature, microcephaly, bird-like facies, and mental retardation (Table 3). He subsequently developed marrow failure evolving to MDS and AML. Seckel syndrome with marrow failure is associated with mutations in the ATR gene, but no deleterious ATR mutations were identified for CH-103. Instead, CH-103 harbored biallelic truncating mutations LIG4 (c.2440C>T [p.Arg814*])33 and LIG4 (c.1751_1755delTAAGA [p.Ile584Argfs*2]). LIG4 encodes DNA ligase IV. LIG4 mutations are associated with an autosomal recessive syndrome that, like Seckel syndrome, is characterized by microcephaly, facial dysmorphism, growth retardation, developmental delay, and pancytopenia, but MDS and AML have not been previously reported for LIG4 syndrome.33 DNA ligase IV functions in DNA double-strand break repair via non-homologous end-joining and V(D)J recombination.34 Patients with LIG4 syndrome exhibit sensitivity to ionizing radiation, so the correct diagnosis informs choice of treatments and imaging modalities. Patients with LIG4 syndrome develop pancytopenia33,35 and lymphoid malignancies.36–38 However, MDS and AML were not previously associated with LIG4 syndrome and thus directed testing for LIG4 mutations had not been pursued for this patient. Therefore, broad multi-gene sequencing approach overcomes limitations posed by incomplete published knowledge of the range of clinical phenotypes for these syndromes.
Discussion
Currently, the selection of specific genetic tests for a given patient is driven by clinical suspicion based on history, physical examination and laboratory evaluation. Our results demonstrate that multi-gene screening identifies patients harboring mutations in known BMF/MDS genes that were clinically unsuspected and therefore were not diagnostically pursued. Our study subjects were selected after a diagnostic workup including prior screening for cryptic presentations of inherited marrow failure syndromes such as chromosomal breakage testing for Fanconi anemia,18 telomere length testing for dyskeratosis congenita,39,40 and pancreatic enzyme testing for Shwachman-Diamond syndrome.41 Our results, therefore, underestimate the frequency of genetic causes of BMF/MDS in children, young adults, and adults with suggestive family histories.
Of the patients for whom no mutations were found in any of the 85 BMF/MDS genes, 47 patients had family history and/or syndromic features suggestive of inherited, or at least constitutional, disease. Our results, therefore, suggest that additional as yet unidentified genes remain to be found for BMF and MDS. Further investigation with whole exome sequencing or whole genome sequencing may identify new genetic causes of inherited BMF/MDS. Conversely, of the 8 patients for whom constitutional damaging mutations were identified, only 4 had a suggestive family history. Of the genetic diagnoses in the absence of a family history, 2 most likely involve de novo mutation (of GATA2 in FH-82 and FH-154), one involved recessive inheritance (of LIG4 in CH-103), and one occurred in an adopted child with no available family history (RUNX1 in FH-178).
Substantial phenotypic overlap among inherited BMF/MDS syndromes, as well as pleiotropy and variable expressivity within syndromes, further complicate diagnosis based purely on clinical presentation. Identification of causal genetic lesions thus plays a critical role in the diagnostic workup. Our multiplexed genetic approach resolved the diagnosis of 2 patients, FH-70 and CH-103, by simultaneously revealing the absence of mutations in genes associated with their initial clinical diagnoses (SBDS and ATR, respectively) and identifying pathogenic mutations in clinically unsuspected genes (DKC1 and LIG4, respectively).
For the purposes of clinical diagnostic testing, custom targeted gene panels offer important advantages over whole exome sequencing. Clinical genetic testing requires sensitive and accurate mutation detection across a defined set of clinically actionable genes with reporting of results in a timely manner. Whole exome sequencing, while well suited for gene discovery, does not ensure deep coverage across all exons of clinical interest. In contrast, high depth of coverage across all genes of interest is achieved through targeted gene capture.42 The deep coverage afforded by this targeted sequencing approach detected CNVs with a single assay, in contrast to alternative approaches combining targeted capture, array comparative genomic hybridization (aCGH), and RNA sequencing.43 This is especially important for inherited BMF/MDS which are frequently caused by deletions in ribosomal protein genes,44,45 RUNX1,28,46 and FANCA.47,48 Finally, whole exome sequencing queries thousands of genes unrelated to BMF/MDS and thus may reveal incidental mutations not immediately relevant to the diagnosis that prompted the sequencing. Patients for whom a germline cause for his or her disease is not revealed by MarrowSeq may benefit from whole exome sequencing for gene discovery in a research setting. New BMF/MDS genes can be incorporated into probe sets for MarrowSeq as they are reported.
These results demonstrate the clinical utility of broad screening of apparently idiopathic cases of BMF and MDS to identify cryptic presentations of inherited disease. Genomic analysis revealed previously unreported phenotypes due to mutations in known genes for syndromic disease. The integration of genomic analysis with clinical and laboratory evaluation in the diagnosis of patients with BMF/MDS can guide clinical management. Multiplexed, clinically unbiased genetic screening provides a powerful approach to elucidate the genetic heterogeneity and phenotypic complexity of inherited syndromes.
Acknowledgments
The authors would like to thank Marshall Horwitz and Penny Jeggo for helpful discussion, Scott Coats and Sommer Castro for sample processing, Dana Matthews, Blythe Thomson, Barbara Small, Jessica Pollard, Paul Carpenter, Melissa Forouhar, Paul Hendrie, Bart Scott, Pam Becker, and Terry Gernsheimer for patient referrals, Kathleen McGregor for her assistance in the marrow failure/MDS clinic, and James Wacker, Celeste Oglesby, and Lauren DePue for clinical research assistance. Regulatory support to A.S. was obtained through the University of Washington Northwest Institute of Genetic Medicine from Washington State Life Sciences Discovery funds (grant 2065508).
Footnotes
Funding
This work was supported by NIH/NIDDK grants R24DK093425 and R24DK099808 (A.S., M.C.K., J.A.), R24DK094746 (M.D.F., D.A.W.), T32AG000057 (M.Y.Z.), and the Ghiglione Aplastic Anemia Fund and the Julian’s Dinosaur Guild at Seattle Children’s Hospital (A.S.). M.C.K. is an American Cancer Society professor.
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Shimamura A. Clinical approach to marrow failure. ASH Educ Program Book. 2009;2009(1):329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Churpek JE, Lorenz R, Nedumgottil S, Onel K, Olopade OI, Sorrell A, et al. Proposal for the clinical detection and management of patients and their family members with familial myelodysplastic syndrome/acute leukemia predisposition syndromes. Leuk Lymphoma. 2013;54(1):28–35. [DOI] [PubMed] [Google Scholar]
- 3.Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24(3):101–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–51. [DOI] [PubMed] [Google Scholar]
- 5.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellodi C, McMahon M, Contreras A, Juliano D, Kopmar N, Nakamura T, et al. H/ACA small RNA dysfunctions in disease reveal key roles for noncoding RNA modifications in hematopoietic stem cell differentiation. Cell Rep. 2013;3(5):1493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pippucci T, Savoia A, Perrotta S, Pujol-Moix N, Noris P, Castegnaro G, et al. Mutations in the 5 UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant form of inherited thrombocytopenia, THC2. Am J Hum Genet. 2011;88(1):115–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu AP, Johnson KD, Falcone EL, Sanalkumar R, Sanchez L, Hickstein DD, et al. GATA2 haploinsufficiency caused by mutations in a conserved intronic element leads to MonoMAC syndrome. Blood. 2013;121(19):3830–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet. 2013;92(3):448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet. 2013;92(5):800–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh T, Lee MK, Casadei S, Thornton AM, Stray SM, Pennil C, et al. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci USA. 2010;107(28):12629–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108(44):18032–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma Oxf Engl. 2009;25(14): 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pritchard CC, Salipante SJ, Koehler K, Smith C, Scroggins S, Wood B, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16(1):56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154(3):518–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nord AS, Lee M, King M-C, Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics. 2011;12(1):184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alter BP, Baerlocher GM, Savage SA, Chanock SJ, Weksler BB, Willner JP, et al. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood. 2007;110(5):1439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinto FO, Leblanc T, Chamousset D, Roux GL, Brethon B, Cassinat B, et al. Diagnosis of Fanconi anemia in patients with bone marrow failure. Haematologica. 2009;94(4):487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ip WF, Dupuis A, Ellis L, Beharry S, Morrison J, Stormon MO, et al. Serum pancreatic enzymes define the pancreatic phenotype in patients with Shwachman-Diamond syndrome. J Pediatr. 2002;141(2): 259–65. [DOI] [PubMed] [Google Scholar]
- 20.Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118(10):2653–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118(10):2656–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kazenwadel J, Secker GA, Liu YJ, Rosenfeld JA, Wildin RS, Cuellar-Rodriguez J, et al. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood. 2012;119(5): 1283–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pasquet M, Bellanné-Chantelot C, Tavitian S, Prade N, Beaupain B, LaRochelle O, et al. High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood. 2013;121(5):822–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, Woollard WJ, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet. 2011;43(10):929–31. [DOI] [PubMed] [Google Scholar]
- 25.Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123(6):809–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fasan A, Eder C, Haferlach C, Grossmann V, Kohlmann A, Dicker F, et al. GATA2 mutations are frequent in intermediate-risk karyotype AML with biallelic CEBPA mutations and are associated with favorable prognosis. Leukemia. 2013;27(2):482–5. [DOI] [PubMed] [Google Scholar]
- 27.Dickinson RE, Milne P, Jardine L, Zandi S, Swierczek SI, McGovern N, et al. The evolution of cellular deficiency in GATA2 mutation. Blood. 2014;123(6):863–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song W-J, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23(2):166–75. [DOI] [PubMed] [Google Scholar]
- 29.Liew E, Owen C. Familial myelodysplastic syndromes: a review of the literature. Haematologica. 2011;96(10):1536–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mangan JK, Speck NA. RUNX1 mutations in clonal myeloid disorders: from conventional cytogenetics to next generation sequencing, a story 40 years in the making. Crit Rev Oncog. 2011;16(1–2):77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knight SW, Vulliamy TJ, Morgan B, Devriendt K, Mason PJ, Dokal I. Identification of novel DKC1 mutations in patients with dyskeratosis congenita: implications for pathophysiology and diagnosis. Hum Genet. 2001;108(4):299–303. [DOI] [PubMed] [Google Scholar]
- 32.Salowsky R, Heiss NS, Benner A, Wittig R, Poustka A. Basal transcription activity of the dyskeratosis congenita gene is mediated by Sp1 and Sp3 and a patient mutation in a Sp1 binding site is associated with decreased promoter activity. Gene. 2002;293(1–2):9–19. [DOI] [PubMed] [Google Scholar]
- 33.O’Driscoll M, Cerosaletti KM, Girard P-M, Dai Y, Stumm M, Kysela B, et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell. 2001;8(6): 1175–85. [DOI] [PubMed] [Google Scholar]
- 34.Frank KM, Sekiguchi JM, Seidl KJ, Swat W, Rathbun GA, Cheng H-L, et al. Late embryonic lethality and impaired V (D)J recombination in mice lacking DNA ligase IV. Nature. 1998;396(6707):173–7. [DOI] [PubMed] [Google Scholar]
- 35.Gruhn B, Seidel J, Zintl F, Varon R, Tönnies H, Neitzel H, et al. Successful bone marrow transplantation in a patient with DNA ligase IV deficiency and bone marrow failure. Orphanet J Rare Dis. 2007;2(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toita N, Hatano N, Ono S, Yamada M, Kobayashi R, Kobayashi I, et al. Epstein–Barr virus-associated B-cell lymphoma in a patient with DNA ligase IV (LIG4) syndrome. Am J Med Genet A. 2007; 143A(7):742–5. [DOI] [PubMed] [Google Scholar]
- 37.Bacon CM, Wilkinson SJ, Spickett GP, Barge D, Lucraft HH, Jackson G, et al. Epstein-Barr virus–independent diffuse large B-cell lymphoma in DNA ligase 4 deficiency. J Allergy Clin Immunol. 2013;131(4):1237–9.e1. [DOI] [PubMed] [Google Scholar]
- 38.Riballo E, Critchlow SE, Teo S-H, Doherty AJ, Priestley A, Broughton B, et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr Biol. 1999;9(13):699–S2. [DOI] [PubMed] [Google Scholar]
- 39.Yamaguchi H, Baerlocher GM, Lansdorp PM, Chanock SJ, Nunez O, Sloand E, et al. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood. 2003;102(3):916–8. [DOI] [PubMed] [Google Scholar]
- 40.Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005;352(14):1413–24. [DOI] [PubMed] [Google Scholar]
- 41.Myers KC, Bolyard AA, Otto B, Wong TE, Jones AT, Harris RE, et al. Variable clinical presentation of Shwachman-Diamond syndrome: update from the North American Shwachman-Diamond Syndrome Registry. J Pediatr. 2014;164(4):866–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puchalka J, Kohistani N, Klein C. Novel NGS-based platforms for molecular diagnosis of severe congenital neutropenia. Blood. 2013;122(21):1034–1034.23798711 [Google Scholar]
- 43.Chandrasekharappa SC, Lach FP, Kimble DC, Kamat A, Teer JK, Donovan FX, et al. Massively parallel sequencing, aCGH, and RNA-Seq technologies provide a comprehensive molecular diagnosis of Fanconi anemia. Blood. 2013;121(22):e138–e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farrar JE, Vlachos A, Atsidaftos E, Carlson-Donohoe H, Markello TC, Arceci RJ, et al. Ribosomal protein gene deletions in Diamond-Blackfan anemia. Blood. 2011;118(26):6943–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quarello P, Garelli E, Brusco A, Carando A, Mancini C, Pappi P, et al. High frequency of ribosomal protein gene deletions in Italian Diamond-Blackfan anemia patients detected by multiplex ligation-dependent probe amplification assay. Haematologica. 2012;97(12):1813–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Béri-Dexheimer M, Latger-Cannard V, Philippe C, Bonnet C, Chambon P, Roth V, et al. Clinical phenotype of germline RUNX1 haploinsufficiency: from point mutations to large genomic deletions. Eur J Hum Genet. 2008;16(8):1014–8. [DOI] [PubMed] [Google Scholar]
- 47.Morgan NV, Tipping AJ, Joenje H, Mathew CG. High frequency of large intragenic deletions in the Fanconi anemia group A gene. Am J Hum Genet. 1999;65(5):1330–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castella M, Pujol R, Callén E, Trujillo JP, Casado JA, Gille H, et al. Origin, functional role, and clinical impact of Fanconi anemia FANCA mutations. Blood. 2011;117(14): 3759–69. [DOI] [PMC free article] [PubMed] [Google Scholar]