

Abstract
Naturally occurring chemoreceptors almost invariably employ structure-switching mechanisms, an observation that has inspired the use of biomolecular switches in a wide range of artificial technologies in the areas of diagnostics, imaging, and synthetic biology. In one mechanism for generating such behavior, clamp-based switching, binding occurs via the clamplike embrace of two recognition elements onto a single target molecule. In addition to coupling recognition with a large conformational change, this mechanism offers a second advantage: it improves both affinity and specificity simultaneously. To explore the physics of such switches we have dissected here the thermodynamics of a clamp-switch that recognizes a target DNA sequence through both Watson-Crick base pairing and triplex-forming Hoogsteen interactions. When compared to the equivalent linear DNA probe (which relies solely on Watson-Crick interactions), the extra Hoogsteen interactions in the DNA clamp-switch increase the probe's affinity for its target by ∼ 0.29 ± 0.02 kcal/mol/base. The Hoogsteen interactions of the clamp-switch likewise provide, however, an additional specificity check that increases the discrimination efficiency towards a single-base mismatch by 1.2 ± 0.2 kcal/mol. This, in turn, leads to a 10-fold improvement in the width of the “specificity window” of this probe relative to that of the equivalent linear probe. Given these attributes, clamp-switches should be of utility not only for sensing applications but also, in the specific field of DNA nanotechnology, for applications calling for a better control over the building of nanostructures and nanomachines.
Keywords: Clamp-Mechanism, Triplex, DNA Nanomachines, Biomolecular Switch, Molecular Beacons, Specificity, Ligand-Induced Fit
The use of binding-induced conformational changes to transduce binding events into useful outputs is ubiquitous throughout the cell. Protein- and RNA-based biomolecular switches, for example, undergo binding-induced changes in conformation or oligomerization and use this information to transduce chemical information into specific biochemical outputs. Motivated by the impressive performance of these naturally occurring biomolecular switches significant effort has gone into the design of similar molecular switches for use in artificial biotechnologies, including molecular diagnostics, synthetic biology, and imaging.1-11
Two general strategies are typically employed for engineering binding-induced molecular switches (Figure 1). The first strategy consists in re-engineering a recognition element so that it adopts a distorted conformation incapable of binding the target (the non-binding “off” state; Figure 1, top). This is typically achieved by stabilizing an alternative, non-binding state via the addition of non-native interactions. In the presence of a target ligand this non-binding “off” state, which is in equilibrium with the binding-competent “on” state, shifts toward the latter state via a population-shift mechanism.2, 12-16 The observed affinity of such switch is thus decreased as the stability of the non-native interactions increases. The second strategy used for designing binding-induced molecular switches consists in engineering a clamp-like mechanism, which employs two recognition elements that embrace a single copy of the target (Figure 1, bottom)5,8,17-18, thus leading to enhanced affinity (due to the larger recognition interface).17-21 Moreover, because clamp-switches recognize a single region of their target using multiple recognition elements, this improvement in affinity generally comes with an improvement in the gap between the affinity of the proper target and that of mismatched targets, thus potentially enhancing specificity.
Figure 1.
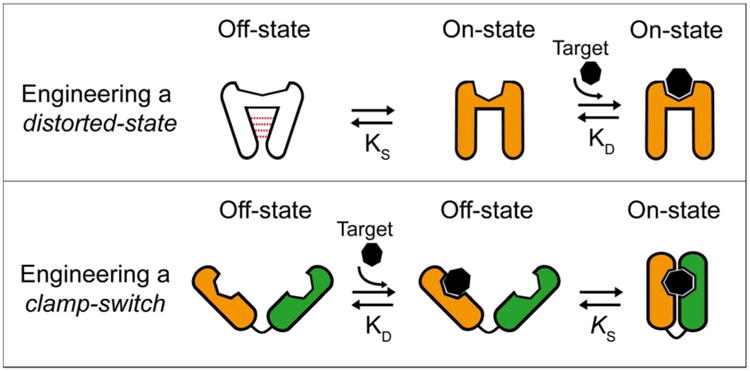
Two general strategies used to design binding-induced molecular switches. Top: A recognition element can be re-engineered into a switch by introducing non-native interactions (red dotted lines) that stabilize a distorted, non-binding “off” state. Upon target binding, the equilibrium between this distorted state and the binding-competent “on” state, KS, is switched to the latter state via a population-shift mechanism.22 The observed affinity (KD_obs) of such switch is thus arbitrarily decreased as KS is reduced (i.e., as the stability of the non-native interactions increases) via the following relationship: KD_obs = KD((1+KS)/KS), where KD is the affinity of the binding-competent state for the target. Bottom: Alternatively, clamp-like switches can be built by fusing together two recognition elements that embrace a single copy of the target in a complementary manner.5,8 In this mechanism the affinity of the switch for its target increases proportionally with KS (i.e., with the stability of the additional interactions) via the following relationship: KD_obs = KD/(1+KS). We recently described the thermodynamic basis for the design and optimization of biomolecular switches of the first type,22 providing a rational path to control and tune the sensitivity of many DNA-based probes.23-26 Here we similarly explore the thermodynamic basis for building biomolecular switches of the second type.
While biomolecular switches of the first type,22 have been thoroughly characterized, providing a rational path towards their design and optimization,22-26 the clamp-like mechanism, despite its promising properties, has seen much less investigation efforts. Thus motivated we explore here the thermodynamic basis for the design and optimization of clamp-like biomolecular switches. We do so using a model DNA-based nanodevice that recognizes a target oligonucleotide via both Watson-Crick base pairing and triplex-forming Hoogsteen interactions (Figure 2).
Figure 2.
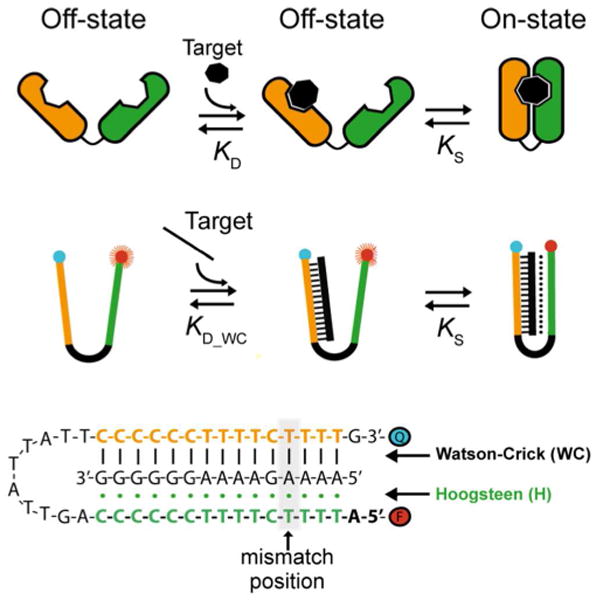
Here we used a model DNA-based nanoswitch to understand the thermodynamic basis of the enhanced affinity and specificity of clamp-switches. This DNA-switch is composed of two recognition domains separated by an unstructured 10-base loop. The first recognition domain (orange sequence) recognizes the target via Watson-Crick base pairing. Upon target binding, the double-stranded DNA is then recognized by the second recognition domain (a Triplex Forming Oligonucleotide, TFO, shown in green) through Hoogsteen base pairs, leading to the formation of a triplex DNA structure. To generate a measurable output, the switch is modified with a fluorophore/quencher pair that are brought into proximity upon formation of the triplex structure (signal-off). The affinity and specificity of such clamp-switch have been compared with those of a simple, linear DNA sequence that recognizes its target solely via Watson-Crick base pairing (non-switching probe, Figure S3) and that share the same common recognition element (orange strand). Such linear non-switching probe was also labeled with a fluorophore and a quencher at the two ends to observe a measurable output upon target binding.
Results and Discussion
As our test bed we have employed a simple, DNA-based clamp-switch composed of two recognition elements separated by an unstructured, 10-base loop (for other, similar examples see.refs27-31). The first recognition element, a 15-base polypyrimidine sequence (Figure 2, in orange), binds the target, a polypurine sequence, via Watson-Crick base pairing. The second recognition element, a polypyrimidine sequence (Figure 2, in green), then binds the so-formed duplex via sequence-specific Hoogsteen base pairing.32-33 The formation of this triplex conformation occurs through a structure-switching mechanism that leads to the switch's closure.27-31,34-36 In support of this proposed mechanism we note that, in the absence of complementary base pairing between the two recognition elements, we observe switch's closure only in the presence of the target (Figure S1). The switch's affinity towards a specific target is also strongly decreased at high pH or in the absence of Mg+2, conditions known to disrupt Hoogsteen interactions27-28 (Figure S2).
The affinity of the clamp-switch for its target (KD_clamp) depends on Watson-Crick (KD_WC) and Hoogsteen base pair interactions (this latter determining the switching equilibrium constant, KS), via the following equation:
| Eq. 1 |
To dissect the thermodynamics of this clamp-switch we have compared its affinity and specificity to those of a simple, linear DNA sequence that recognizes its target solely via Watson-Crick base pairing and that does not undergo any (energetically significant) conformational change (non-switching probe, Figure S3). For ease of comparison both probes share a common recognition element (orange strand in Figure 2). Because the linear probe does not undergo a structural switch and only form Watson-Crick base pairing, it can be used to determine KD_WC (see Figure 2). Together with the affinity of the clamp-switch probe (KD_clamp), this value provides a route to evaluating the contribution of the switching mechanism (KS) of the clamp-switch using Eq. 1.
Clamp-switch probes bind to their targets with greater affinity than do the equivalent linear DNA probes (Figure 3, S4).27-31,34-36 Indeed, the improvement in affinity is so great that we cannot directly measure the difference in binding energies for any single target. That is, because the affinity of the probes we tested can only be quantitatively measured over a specific concentration window, which is comprised between the concentration of the switch/probe (i.e., 2 nM) and the highest concentration of target that can be reasonably added to the working solution (here 0.1 mM), there is no single target for which both probes produce measurable dissociation constants. For example, while the clamp-switch exhibits micromolar affinity with a target as short as 8 bases (KD_clamp (8-base) = 1.4 μM; Figure 3, right), the linear non-switching probe does not exhibit any detectable binding with this same target at even the highest concentrations we have tested (100 μM). We have thus instead used extrapolations from data we collected to estimate the difference in the free energy with which each probe would bind a specific, 10-base target. We have taken two approaches to this end. The first is based on the observation that the affinity of the clamp-switch for a 10-base target matches the affinity with which the linear non-switching probe binds a longer, 12-base target (KD = 20 nM; Figure 3). A nearest-neighbor model37-39 predicts that these two extra G-C Watson-Crick base pairs should provide an additional 3.3 kcal/mol in binding energy, suggesting that this represents the extra stabilization provided by the Hoogsteen base pairing between the clamp-switch and the 10-base target. As a second means of estimating the Hoogsteen base-pairing contribution we predict the free energy with which the linear probe binds a 10-base target by extrapolation of the experimental data obtained with the same probe for longer targets (Figure 3, bottom). The difference between this extrapolated value (7.7 kcal/mol) and the experimental value for the clamp-switch binding to this same 10-base target (10.9 kcal/mol) is, at 3.2 kcal/mol, in good agreement with the value achieved with the first approach.
Figure 3.
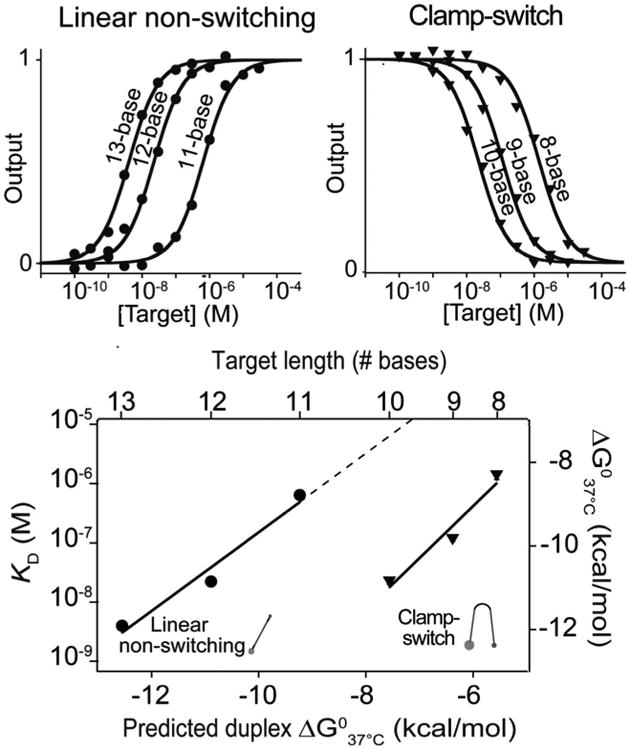
The affinity of the clamp-switch (top, right) is greater than that of the linear non-switching probe (top, left), enough so that a target must be ∼20% longer to bind the later as tightly. Bottom: Shown are the experimentally derived affinities (and the equivalent free energies) of a clamp-switch and a linear non-switching probe using the same Watson-Crick recognition element versus target length (and the nearest-neighbor model predicted binding energy). 37-39 The binding curves were obtained by adding increasing concentration of perfectly matched targets of different length to a 2 nM concentration of clamp-switch (top, right) or linear non-switching (top, left) probe in 100 mM Tris buffer, 10 mM MgCl2, pH 7.0 at 37°C.
Given that, at pH 7, the free energy of forming a CGC Hoogsteen base pairs is nearly equal to that of forming TAT Hoogsteen interaction40-41, the 3.3 kcal/mol additional energy provided by the extra 10 Hoogsteen base pairs in the clamp-switch suggests that each Hoogsteen base pair provides ∼0.33 kcal/mol/base in stabilization, which is in close agreement with previous reports.40,42 The 3.3 kcal/mol in additional binding energy provided by the 10 extra Hoogsteen base-pairs also corresponds to a clamp-switch equilibrium constant, KS, of 190. This, as expected (Eq. 1) improves the affinity of the clamp-switch by ∼200-fold relative to that of the linear probe.
The enhanced affinity of the clamp-switches is not found in other mechanisms of coupling binding to a large-scale conformational change. Specifically, the affinity of clamp-switches is greater than that of the equivalent (i.e., same recognition site) switch, which uses the other commonly employed switching mechanism: an engineered distorted state (Figure 1, top). To show this we have compared the clamp-switch with the equivalent molecular beacon, a commonly employed optical or electrochemical approach for the detection of specific DNA sequences.1,43-45 A molecular beacon is a fluorophore-and-quencher-modified DNA strand that forms a low-emissive stem-loop conformation due to hybridization of its complementary ends. This structure opens –thus producing enhanced fluorescence- when a target hybridizes to the loop, breaking the stem and segregating the fluorophore/quencher pair (Figure S3). In contrast to the nanomolar affinity that the clamp-switch shows for targets as short as 10 bases, the molecular beacon does not reach this affinity until targets of at least 15 bases are used (Figure S5). This occurs because molecular beacons employ an engineered distorted state (here a stem-loop structure), the stabilization energy of which competes with target binding (Figure S3). This effect is more readily apparent if we compare the affinity of the linear probe and the equivalent molecular beacon since we are able to compare directly the affinities of these probes for a single target of the same length (i.e. 13 bases). While the linear non-switching probe shows nanomolar affinity (KD = 4 nM) for a 13-base target, the affinity of the molecular beacon for this same target is some 40-fold poorer (KD= 162 nM) (Figure 3 and Figure S5).
In addition to improve binding affinity, the clamp-switch mechanism should, at least in principle, also enhance specificity.17-21 To explore this we have compared the affinities of our clamp-switch against a perfectly matched and a single-base mismatched target (see Figure 2 for mismatch location). In order to describe specificity quantitatively we use the discrimination factor, Q, which is the ratio of the output signal produced by the perfectly matched target (χPM) to that of the mismatched target (χMM) (Figure 4)46 and the specificity window, defined here as the range of target concentration at which we observe a value of Q equal or above 5 (thus representing a 20% interfering signal). The specificity window of the simple linear non-switching probe spans about an order of magnitude in target concentration (Figure 4, bottom). The specificity window of the clamp-switch, in contrast, is 10 times wider (Figure 4, top). Due to the experimental limitations described above, however, the specificity of the clamp-switch probe was determined using a shorter target (10-base) than that employed to test the specificity of the linear non-switching probe (13-base) which could, in theory, also lead to higher specificity. To rule this out we performed simulations using the nearest–neighbor model,37-39 which confirm that the small difference in target length does not account for the large difference in specificity we observe (Figure S6).
Figure 4.
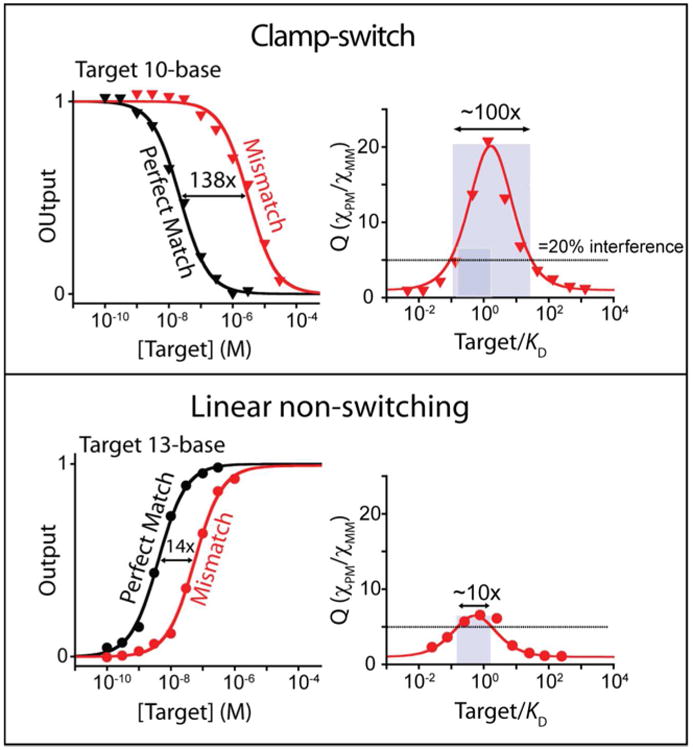
The additional Hoogsteen base pairs in our clamp-switch (top) increase the probe's specificity by 10-fold over that of the equivalent simple linear non-switching probe (bottom). To demonstrate this we challenged both probes against a perfectly matched target and a single-base mismatched target. Shown (right-hand column) is the discrimination factor, Q, the ratio between perfect match and mismatch outputs, as a function of target concentration. We have also highlighted the specificity window (gray rectangles), defined here as the range of target concentration at which we observe a value of Q equal or above 5 (thus representing a 20% interfering signal). This window is more than an order of magnitude broader for the clamp-switch. These binding curves were obtained by adding increasing concentration of perfectly matched and 1-base mismatched DNA targets to a 2 nM concentration of clamp-switch (top) or linear non-switching (bottom) probe in 100 mM Tris buffer, 10 mM MgCl2, pH 7.0 at 37°C.
The different specificity windows of the two probes provide an additional insight on how the clamp-switch mechanism leads to enhanced specificity. Specifically, the 138-fold difference in affinity between the perfectly matched and the single-based mismatched target for the clamp-switch suggests that the mismatch is 3.10 kcal/mol less stable than the perfectly matched target (Figure 4). For the linear probe, in contrast, the 14-fold difference in affinity between the perfectly matched and the mismatched target gives a mismatch destabilization of only 1.66 kcal/mol. The extra Hoogsteen interactions in the clamp mechanism thus improve the specificity of the clamp-switch by ca. 1.44 kcal/mol.
In contrast to the clamp-switch mechanism, the distorted recognition element strategy does not enhance specificity over that of the non-switching linear probe. The stem-loop distorted switch (i.e., the molecular beacon –see Figure S3), for example, exhibits a specificity window similar to the one of the linear non-switching probe (Figure S7). This is likely attributable to the fact that the non-native interactions introduced in this class of switches do not alter the binding interface between the switch and the target (Figure 1, top).25
To further explore the improved performance of clamp-switches we have also employed urea denaturation experiments, which provide a route towards determining their switching and binding thermodynamics22, 47-49 (Figure 5, and see SI for detailed information about this method). An advantage that such experiments have over the more traditional measurement of binding curves is that they provide a means of determining the free energy of association between two biomolecules in presence of saturating amount of one of them. The approach thus allows us to compare the free energy of association of the clamp-switch and linear probes when each is bound to the same target. Using this approach we find that the difference in free energy with which the clamp-switch binds mismatched and perfectly matched targets is, at 2.1 ± 0.2 kcal/mol, significantly higher than the 0.9 ± 0.1 kcal/mol difference observed for the linear non-switching probe (Figure 5). Of note, this extra 1.2 ± 0.2 kcal/mol in discrimination energy provided by the Hoogsteen interactions in the clamp-switch agrees well with the value determined above using binding curves (i.e., 1.44 kcal/mol). The 4.1 ± 0.2 kcal/mol (0.29 ± 0.02 kcal/mol/base) additional interaction energy provided by the 14 Hoogsteen interactions in the clamp-switch is likewise close to the ∼0.33 kcal/mol/base value estimated from the extrapolation procedure using the 10-base target (Figure 3).
Figure 5.
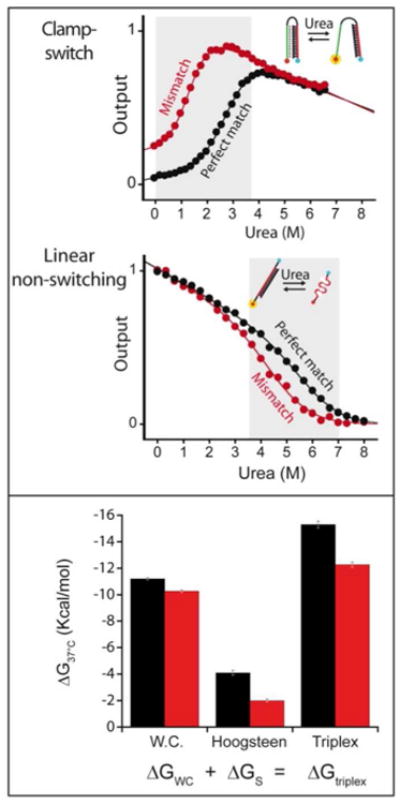
Urea denaturation curves (see SI for more details) provide a means by which to compare both the specificity and affinity of the clamp-switch probe with those of the equivalent, non-switching linear probe using a single 14-base perfect match/mismatch target pair (both at a saturating concentration of 10 μM). Note that the unfolding/dissociation of the clamp-switch occurs at much lower urea concentrations thus suggesting a sequential unfolding of the triplex probe through: 1) denaturation of the triplex Hoogsteen interactions followed by 2) denaturation of the remaining duplex. We determined ΔGWC, the free energy of association of the linear probe for both the perfect match (11.2 ± 0.1 kcal/mol) and the mismatch (10.3 ± 0.1 kcal/mol) (see SI, eq. 7). Using a two-state approximation (see SI, eq.13), we found that the clamp-switch free energy of opening (Hoogsteen interactions), ΔGS, is 4.1 ± 0.2 kcal/mol and 2.0 ± 0.1 kcal/mol for the perfect match and mismatch target respectively. We note that the 4.1 ± 0.2 kcal/mol is in excellent agreement with the results obtained using binding curve analysis (0.29 ± 0.02 kcal/mol/base ∼ 4.1 kcal/mol/14-bases; Figure 3). The difference in free energies for perfect match and mismatch targets obtained with the linear probe (11.2 (±0.1) - 10.3 (±0.1) = 0.9 ± 0.1 kcal/mol) and the clamp-switch (4.1 (±0.2) - 2.0 (±0.1) = 2.1 ± 0.2 kcal/mol) are also in good agreement with the results obtained above from extrapolations of the binding curve (Figure 4), and confirm the enhanced specificity of the clamp-switch relative to the linear, non-switching probe.
Conclusion
In this work we have explored the thermodynamics by which clamp-based molecular switches improve both the affinity and specificity of recognition.18-22, 27-31,34-36 Using a simple DNA model clamp-switch, the triplex-forming probe, we showed that clamp-switches recognize their specific target through two sequential binding events which sum up to provide a higher binding free energy and greater discrimination efficiency than those observed for either the equivalent, non-switching linear probe or a switch based on the engineering of a distorted state. These advantages likely explains why evolution employs clamp-like strategies so frequently when it builds its signaling mechanisms.8,50-52
The simplicity of the clamp-switch strategy may also inspire us towards engineering switches with improved affinity and specificity. For example, binding-activated molecular probes could be engineered to detect DNA sequences with much higher specificity than those based on simple Watson-Crick base pairing. Of note, triplex forming sequences are common enough that it is straightforward to select unique sites in human or pathogen genomes.52-53 In addition, the clamp-switch strategy dissected here could also be used to engineer highly specific structure-switching biosensors using more complex recognition elements that can include aptamers54 and proteins.17-21 Finally, with their ability to detect very short targets with high affinity and specificity, DNA clamp-switches should find many applications for building new DNA nanostructures, DNA nanomachines and DNA origami,55-61 where the ability to specifically and tightly bind short DNA sequences will lead to improved structural control.
Materials and Methods
HPLC purified oligonucleotides labeled with a FAM (5-carboxyfluorescein) at the 5′ end and a BHQ-1 (black hole quencher 1) at the 3′ end were purchased from Sigma-Genosys. We used a triplex-clamp switch, a linear probe and a molecular stem-loop beacon all of them bearing the same 15-base recognition element. The sequences of the probes were as follows.
Triplex clamp-switch: 5′-A-TTTTCTTTTCCCCCC-AGTTATTATT-CCCCCCTTTTCTTTT-G-3′
Linear non-switching probe: 5′-A-CCCCCCTTTTCTTTT-G-3′
Molecular beacon: 5′-A-CTCGC-CCCCCCTTTTCTTTT-GCGAG-G-3′
For all the sequences above the bases in bold represent the recognition element (red portion in Figure S3). In the molecular beacon sequence above the underlined bases represent the stem portion (black and green portion in Figure S3, top). In the clamp-switch the underlined bases represent the random loop sequence (black portion in Figure S3, middle) and the italic bases represent the triplex forming oligonucleotide sequence (green portion in Figure S3, middle). The sequences, sensing principles and the expected conformational change of the three probes used in this work are depicted in Figure S3.
Perfect match and mismatch targets were also purchased from Sigma-Genosys. The sequences of the probes were as follows.
Perfect match targets:
15-base: 5′-AAAAGAAAAGGGGGG-3′
14-base: 5′- AAAAGAAAAGGGGG-3′
13-base: 5′- AAAAGAAAAGGGG-3′
12-base: 5′- AAAAGAAAAGGG-3′
11-base: 5′- AAAAGAAAAGG-3′
10-base: 5′- AAAAGAAAAG-3′
9-base: 5′- AAAAGAAAA-3′
8-base: 5′- AAAAGAAA-3′
7-base: 5′- AAAAGAA-3′
Mismatch targets have the same sequence of the perfect match targets except for a mutated base in position 4 where the A base was substituted with a C base. For example for the 14-base mismatch target the sequence was as follows:
14-base: 5′- AAACGAAAAGGGGG -3′
Where the underlined base represents the mismatch position.
All experiments were conducted in 100 mM Tris buffer, 10 mM MgCl2, pH 7.0 at 37°C unless otherwise stated. All fluorescence measurements were obtained using a Cary Eclipse Fluorimeter with excitation at 480 (± 5) nm and acquisition between 514 and 520 nm. Ultrapure urea was obtained from Sigma-Aldrich. Urea unfolding curves were obtained using 10 nM of the relevant probe (clamp-switch or linear probe) by sequentially increasing the urea concentration of a 0 M urea sample with 8 M urea containing the same concentration of probe. The fluorescence of the open state was set relative to 1.
Binding curves were obtained using 2 nM of the relevant probe (clamp-switch, linear or stem-loop probe) and were fitted to a single-site binding mechanism ([X] = target concentration; FB= fluorescence in the presence of saturating concentration of target; F[T]= fluorescence in the presence of different concentration of target; F0 = background fluorescence):
| Eq. 2 |
Supplementary Material
Acknowledgments
Funding Sources: This work was supported by Bill & Melinda Gates Foundation through the Grand Challenges Explorations (OPP1061203) (FR), by the International Research Staff Exchange Scheme (IRSES) grant under the Marie Curie Actions program (FR), by the NIH through grant R01EB007689 (KWP) and by the National Sciences and Engineering Research Council of Canada through grant 436381-2013 (AVB). FR is supported by a Marie Curie Outgoing Fellowship (IOF) (Proposal No. 298491 under FP7-PEOPLE-2011-IOF). AI is supported by the Canada-Italy innovation award.
Footnotes
Author Contributions: The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tyagi S, Kramer FR. Molecular Beacons: Probes That Fluoresce Upon Hybridization. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 2.Vallée-Bélisle A, Plaxco KW. Structure-Switching Biosensors: Inspired by Nature. Curr Opin Struct Biol. 2010;20:518–526. doi: 10.1016/j.sbi.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radley TL, Markowska AI, Bettinger BT, Ha JH, Loh SN. Allosteric Switching by Mutually Exclusive Folding of Protein Domains. J Mol Biol. 2003;332:529–536. doi: 10.1016/s0022-2836(03)00925-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stratton MM, Mitrea DM, Loh SN. A Ca2+-Sensing Molecular Switch Based on Alternate Frame Protein Folding. ACS Chem Biol. 2008;3:723–732. doi: 10.1021/cb800177f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stratton MM, Loh SN. Converting a Protein Into a Switch for Biosensing and Functional Regulation. Protein Sc. 2011;20:19–29. doi: 10.1002/pro.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wright CM, Heins RA, Ostermeier M. As Easy as Flipping a Switch? Curr Opin Struct Biol. 2007;11:342–346. doi: 10.1016/j.cbpa.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Ostermeier M. Designing Switchable Enzymes. Curr Opin Struct Biol. 2009;19:442–448. doi: 10.1016/j.sbi.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koide S. Generation Of New Protein Functions by Nonhomologous Combinations and Rearrangements of Domains and Modules. Curr Opin Biotech. 2009;20:398–404. doi: 10.1016/j.copbio.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The Fluorescent Toolbox for Assessing Protein Location and Function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 10.Isaacs FJ, Dwyer DJ, Collins JJ. RNA Synthetic Biology. Nat Biotechnol. 2006;24:545–554. doi: 10.1038/nbt1208. [DOI] [PubMed] [Google Scholar]
- 11.Sallee NA, Yeh BJ, Lim WA. Engineering Modular Protein Interaction Switches by Sequence Overlap. J Am Chem Soc. 2007;129:4606–4611. doi: 10.1021/ja0672728. [DOI] [PubMed] [Google Scholar]
- 12.Ma B, Kumar S, Tsai C, Nussinov R. Folding Funnels and Binding Mechanisms. Protein Eng. 1999;129:713–720. doi: 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- 13.Tsai CJ, Ma B, Nussinov R. Folding and Binding Cascades: Shifts in Energy Landscapes. Proc Natl Acad Sci U S A. 1999;96:9970–9972. doi: 10.1073/pnas.96.18.9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar S, Ma B, Tsai C, Sinha N, Nussinov R. Folding and Binding Cascades: Dynamic Landscapes and Population Shifts. Protein Sc. 2000;9:10–19. doi: 10.1110/ps.9.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma B, Shatsky M, Wolfson HJ, Nussinov R. Multiple Diverse Ligands Binding at a Single Protein Site: a Matter of Pre-Existing Populations. Protein Sc. 2002;11:184–197. doi: 10.1110/ps.21302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plaxco KW, Soh HT. Switch Based Biosensors: a New Approach Towards Real-Time in Vivo Molecular Detection. Trends Biotech. 2011;29:1–5. doi: 10.1016/j.tibtech.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herschlag D. The Role of Induced Fit and Conformational Changes of Enzymes in Specificity and Catalysis. Bioorg Chem. 1988;16:62–96. [Google Scholar]
- 18.Vinkenborg JL, Nicolson TJ, Bellomo EA, Koay MS, Rutter GA, Merkx M. Genetically Encoded FRET Sensors to Monitor Intracellular Zn2+ Homeostasis. Nat Methods. 2009;6:737–740. doi: 10.1038/nmeth.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson KA. Role of Induced Fit in Enzyme Specificity: A Molecular Forward/Reverse Switch. J Biol Chem. 2008;283:26297–26301. doi: 10.1074/jbc.R800034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang J, Koide A, Makabe K, Koide S. Design of Protein Function Leaps by Directed Domain Interface Evolution. Proc Natl Acad Sci U S A. 2008;105:6578–6583. doi: 10.1073/pnas.0801097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang J, Makabe K, Biancalana M, Koide A, Koide S. Structural Basis for Exquisite Specificity of Affinity Clamps, Synthetic Binding Proteins Generated Through Directed Domain-Interface Evolution. J Mol Biol. 2009;392:1221–1231. doi: 10.1016/j.jmb.2009.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vallée-Bélisle A, Ricci F, Plaxco KW. Thermodynamic Basis for the Optimization of Binding-Induced Biomolecular Switches and Structure-Switching Biosensors. Proc Natl Acad Sci U S A. 2009;106:13802–13807. doi: 10.1073/pnas.0904005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ricci F, Vallée-Bélisle A, Plaxco KW. High-Precision, in Vitro Validation of the Sequestration Mechanism for Generating Ultrasensitive Dose-Response Curves in Regulatory Networks. PLoS Comp Biol. 2011;7:e1002171. doi: 10.1371/journal.pcbi.1002171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ricci F, Vallée-Bélisle A, Porchetta A, Plaxco KW. Rational Design of Allosteric Inhibitors and Activators Using the Population-Shift Modelin Vitro Validation and Application to an Artificial Biosensor. J Am Chem Soc. 2012;134:15177–15180. doi: 10.1021/ja304672h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vallée-Bélisle A, Ricci F, Plaxco KW. Engineering Biosensors With Extended, Narrowed, or Arbitrarily Edited Dynamic Range. J Am Chem Soc. 2012;134:2876–2879. doi: 10.1021/ja209850j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porchetta A, Vallée-Bélisle A, Plaxco KW, Ricci F. Using Distal-Site Mutations and Allosteric Inhibition to Tune, Extend, and Narrow the Useful Dynamic Range of Aptamer-Based Sensors. J Am Chem Soc. 2012;134:20601–20604. doi: 10.1021/ja310585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kandimalla ER, Agrawal S. Single-Strand-Targeted Triplex Formation: Stability, Specificity and Rnase H Activation Properties. Gene. 1994;149:115–121. doi: 10.1016/0378-1119(94)90419-7. [DOI] [PubMed] [Google Scholar]
- 28.Kandimalla ER, Manning A, Agrawal S. Single Strand Targeted Triplex Formation: Physicochemical and Biochemical Properties of Foldback Triplexes. J Biomol Str Dyn. 1996;14:79–90. doi: 10.1080/07391102.1996.10508931. [DOI] [PubMed] [Google Scholar]
- 29.Kandimalla ER, Agrawal S. Single-Strand-Targeted Triplex Formation. Destabilization of Guanine Quadruplex Structures by Foldback Triplex-Forming Oligonucleotide. Nucleic Acids Research. 1995;23:1068–1074. doi: 10.1093/nar/23.6.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xodo LE, Manzini G, Quadrifoglio F. Spectroscopic and Calorimetric Investigation on the DNA Triplex Formed by D(CTCTTCTTTCTTTTCTTTCTTCTC) and D(GAGAAGAAAGA) at Acidic pH. Nucleic Acids Research. 1990;18:3557–3564. doi: 10.1093/nar/18.12.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee IB, Lee JY, Lee NK, Hong SC. Direct Observation of the Formation of DNA Triplexes by Single-Molecule FRET Measurement. Current Applied Physics. 2012;12:1027–1032. [Google Scholar]
- 32.Hoogsteen K. The Structure of Crystals Containing a Hydrogen Bonded Complex of 1-Methylthymine and 9-Methyladenine. Acta Crystallogr. 1959;12:822–823. [Google Scholar]
- 33.Hoogsteen K. The Crystal and Molecular Structure of a Hydrogen-Bonded Complex Between 1-Methylthymine and 9-Methyladenine. Acta Crystallogr. 1963;16:907–916. [Google Scholar]
- 34.Giovannangeli C, Thuong NT, Helene C. Oligonucleotide Clamps Arrest DNA Synthesis on a Single-Stranded DNA Target. Proc Natl Acad Sci U S A. 1993;90:10013–10017. doi: 10.1073/pnas.90.21.10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trkulja I, Biner SM, Langenegger SM, Häner R. A Molecular Probe for the Detection of Homopurine Sequences. ChemBioChem. 2007;8:25–27. doi: 10.1002/cbic.200600378. [DOI] [PubMed] [Google Scholar]
- 36.Fatthalla MI, Pedersen EB. Improved DNA Clamps by Stacking to Adjacent Nucleobases. Helvetica Chimica Acta. 2012;95:1538–1547. [Google Scholar]
- 37.Owczarzy R, Tataurov AV, Wu Y, Manthey JA, McQuisten KA, Almabrazi HG, Pedersen FK, Lin Y, Garretson J, McEntaggart NO, et al. IDT SciTools: A Suite for Analysis and Design of 572 Nucleic Acid Oligomers. Nucleic Acids Res. 2008;368:163–169. doi: 10.1093/nar/gkn198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santalucia J., Jr A Unified View of Polymer, Dumbbell, and Oligonucleotide DNA Nearest-Neighbor Thermodynamics. Proc Natl Acad Sci. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santalucia J, Jr, Hicks D. The Thermodynamics of DNA Structural Motifs. Annu Rev Byophys Biomol Struct. 2004;33:415–440. doi: 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- 40.Roberts R, Crothers D. Prediction of The Stability of DNA Triplexes. Proc Natl Acad Sci U S A. 1996;93:4320–4325. doi: 10.1073/pnas.93.9.4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Volker J, Klump HH. Electrostatic Effect in DNA Triple Helices. Biochemistry. 1994;33:13502–13508. doi: 10.1021/bi00249a039. [DOI] [PubMed] [Google Scholar]
- 42.Best GC, Dervan PB. Energetics of Formation of Sixteen Triple Helical Complexes Which Vary at a Single Position Within a Pyrimidine Motif. J Am Chem Soc. 1195;117:1187–1193. [Google Scholar]
- 43.Marras SAE, Tyagi S, Kramer FR. Real-Time Assays With Molecular Beacons and Other Fluorescent Nucleic Acid Hybridization Probes. Clin Chim Acta. 2006;363:48–60. doi: 10.1016/j.cccn.2005.04.037. [DOI] [PubMed] [Google Scholar]
- 44.Ricci F, Plaxco KW. E-DNA Sensors for Convenient, Label-Free Electrochemical Detection of Hybridization. Microch Acta. 2008;163:149–155. [Google Scholar]
- 45.Tyagi S, Bratu DP, Kramer FR. Multicolor molecular beacons for allele discrimination. Nat Biotechnol. 1998;16:49–53. doi: 10.1038/nbt0198-49. [DOI] [PubMed] [Google Scholar]
- 46.Zhang DY, Chen SX, Yin P. Optimizing the Specificity of Nucleic Acid Hybridization. Nature Chem. 2012;4:208–214. doi: 10.1038/nchem.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pace CN. Determination and Analysis of Urea and Guanidine Hydrochloride Denaturation Curves. Meth Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 48.Shelton VM, Sosnick TR, Pan T. Applicability of Urea in the Thermodynamic Analysis of Secondary and Tertiary RNA Folding. Biochemistry. 1999;38:16831–16839. doi: 10.1021/bi991699s. [DOI] [PubMed] [Google Scholar]
- 49.Santoro MM, Bolen DW. Unfolding Free Energy Changes Determined by the Linear Extrapolation Method. 1. Unfolding of Phenylmethanesulfonyl α-Chymotrypsin Using Different Denaturants. Biochemistry. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 50.Rossmann MG, Moras D, Olsen KW. Chemical and Biological Evolution of Nucleotide Binding Protein. Nature. 1974;250:194–199. doi: 10.1038/250194a0. [DOI] [PubMed] [Google Scholar]
- 51.Blake CCF. Do Genes-In-Pieces Imply Proteins-In-Pieces? Nature. 1978;273:267–268. [Google Scholar]
- 52.Pawson T, Nash P. Assembly of Cell Regulatory Systems Through Protein Interaction Domains. Science. 2003;300:445–452. doi: 10.1126/science.1083653. (52) Goñi, J. R.; de la Cruz, X.; Orozco, M. Triplex-Forming Oligonucleotide Target Sequences in the Human Genome. Nucleic Acids Res. 2004, 32, 354–360. [DOI] [PubMed] [Google Scholar]
- 53.Duca M, Vekhoff P, Oussedik K, Halby L, Arimondo PB. The Triple Helix: 50 Years Later, the Outcome. Nucleic Acids Res. 2008;36:5123–5138. doi: 10.1093/nar/gkn493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muller J, Wulffen B, Potzsch B, Mayer G. Multidomain Targeting Generates a High-Affinity Thrombin-Inhibiting Bivalent Aptamer. ChemBioChem. 2007;8:2223–2226. doi: 10.1002/cbic.200700535. [DOI] [PubMed] [Google Scholar]
- 55.Yurke B, Turberfield AJ, Mills AP, Jr, Simmel FC, Neumann JL. A DNA-Fuelled Molecular Machine Made of DNA. Nature. 2000;406:605–608. doi: 10.1038/35020524. [DOI] [PubMed] [Google Scholar]
- 56.Nimjee SM, Rusconi CP, Sullenger BA. Aptamers: an Emerging Class of Therapeutics. Annu Rev Med. 2005;56:555–583. doi: 10.1146/annurev.med.56.062904.144915. [DOI] [PubMed] [Google Scholar]
- 57.Seelig G, Soloveichik D, Zhang DY, Winfree E. Enzyme-Free Nucleic Acid Logic Circuits. Science. 2006;314:1585–1588. doi: 10.1126/science.1132493. [DOI] [PubMed] [Google Scholar]
- 58.Yin P, Choi HMT, Calvert CR, Pierce NA. Programming Biomolecular Self-Assembly Pathways. Nature. 2008;451:318–322. doi: 10.1038/nature06451. [DOI] [PubMed] [Google Scholar]
- 59.Andersen ES, Dong M, Nielsen MM, Jahn K, Subramani R, Mamdouh W, Golas MM, Sander B, Stark H, Oliveira CLP, et al. Self-Assembly of a Nanoscale DNA Box With a Controllable Lid. Nature. 2009;459:73–76. doi: 10.1038/nature07971. [DOI] [PubMed] [Google Scholar]
- 60.Keefe AD, Pai S, Ellington A. Aptamers as Therapeutics. Nature Reviews Drug Discovery. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krishnan Y, Simmel FC. Nucleic Acid Based Molecular Devices. Angew Chem Int Ed. 2011;50:3124–3156. doi: 10.1002/anie.200907223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.