

Abstract
A more complete understanding of the mechanisms that regulate the angiogenic switch, which contributes to the conversion of small dormant tumors to actively growing malignancies, is important for the development of more effective anti-angiogenic strategies for cancer therapy. While significant progress has been made in understanding the complex mechanisms by which integrin αvβ3 expressed in endothelial cells governs angiogenesis, less is known concerning the ability of αvβ3 expressed within the tumor cell compartment to modulate the angiogenic output of a tumor. Here we provide evidence that αvβ3 expressed in melanoma cells may contribute to the suppression of IGFBP-4, an important negative regulator of IGF-1 signaling. Given the multiple context-dependent roles for αvβ3 in angiogenesis and tumor progression, our novel findings provide additional molecular insight into how αvβ3 may govern the angiogenic switch by a mechanism associated with a p38 MAPK and matrix metalloproteinases-dependent regulation of the endogenous angiogenesis inhibitor IGFBP-4.
Keywords: Angiogenesis, Integrin, MMPs, Melanoma, p38 MAPK, Insulin-like growth factor, IGFBP-4
Introduction
Angiogenesis, the process by which new blood vessels arise from preexisting vessels, is thought to play important roles in many normal and pathological events. Tumor angiogenesis may facilitate metastatic disease and in many cases has been correlated with a poor clinical prognosis [1–4]. Evidence indicates that small tumor lesions may remain dormant and fail to expand beyond a minimal size without a functional vascular network [5–10]. These studies and others suggest that in order for small tumors to expand, a shift in the expression of angiogenic regulatory molecules may occur, resulting in what has been termed the angiogenic switch [5–10].
Recent work has confirmed the utility of anti-angiogenic strategies in treating malignant tumors as indicated by the approval of anti-angiogenic drugs that target growth factor signaling [11–13]. However, several new studies have demonstrated the complexity involved in targeting secreted pro-angiogenic factors as a therapeutic modality. For example, studies have shown that while inhibition of growth factor signaling pathways or certain matrix metalloproteinases (MMPs) may lead to a reduction in tumor angiogenesis, malignant lesions may circumvent these initial inhibitory activities, contributing to increased tumor cell invasion and poor drug diffusion, ultimately resulting in limited durable responses [14–18]. These studies along with others are driving the need for a more comprehensive understanding of the mechanisms that regulate pathological angiogenesis in order to fully realize the potential of anti-angiogenic therapy.
Evidence suggests that the local tumor microenvironment plays an important role in controlling the pro- and anti-angiogenic balance within tumors [6, 19–24]. Several studies have provided evidence that alterations in the balance of pro- angiogenic growth factors such as VEGF, FGF-2 and PDGF, proteolytic enzymes such as MMPs and endogenous inhibitors such as TSP-1 and fragments of ECM proteins may govern whether a tumor remains dormant or expands [24–26]. Integrins are a family of heterodimeric cell surface proteins composed of non-covalently associated α and β chains that play important roles in establishing communication links between the extracellular microenvironment and cells [6, 27]. Integrins play multiple roles in angiogenesis and may act as functional hubs that integrate streams of molecular information between diverse cellular and non-cellular compartments [6, 27]. While it is well known that integrins can regulate multiple signaling cascades required for angiogenesis, the molecular details by which they coordinate these events are still not completely understood. To this end, pharmacological blockade of integrin αvβ3 inhibits angiogenesis and tumor growth [27–31]. However, lack of αvβ3 in transgenic mouse models enhanced angiogenesis and tumor growth [32, 33]. In other studies, mice expressing signaling deficient β3 integrin exhibited defective pathological vascular development and smaller tumors, which depended on reduced recruitment of bone marrow-derived cells [34]. These findings indicate that the roles of αvβ3 in governing pathological angiogenesis and tumor progression are complex, and further studies are needed to unravel the functions of this integrin.
Our prior studies have indicated that changes in structure of the ECM may facilitate the creation of a proangiogenic and tumor permissive environment in part by exposing cryptic integrin binding sites for tumor and stromal cells [19, 35, 36]. Given these findings, it is possible that the ECM may contribute to the control of the angiogenic switch. Previous studies suggest that endothelial cell interaction with denatured collagen suppresses expression of IGFBP-4 [37], an important regulator of IGF-1 signaling and angiogenesis [38–40]. However, the mechanisms by which cellular interactions with structurally altered collagen regulates IGFBP-4 is not known. Here we provide evidence that αvβ3 expressed in melanoma cells may contribute to the reduction of IGFBP-4 in part by a p38 MAPK-dependent mechanism that involves elevated levels of MMPs and enhanced degradation of IGFBP-4. Collectively, our novel finding provides new insight into the complex mechanisms by which αvβ3 may regulate angiogenesis and early tumor development by modulating the angiogenic output of melanoma tumors.
Experimental procedures
Reagents, kits, chemicals and antibodies
Bovine serum albumin (BSA), methanol, ethanol, acetone, and OCT embedding compound were all obtained from Sigma (St Louis, MO). Monoclonal antibodies (Mab) LM609 (anti-αvβ3) and anti-β3 integrin were obtained from Millipore (Bedford, MA). Anti-IGFBP-7 antibody was obtained from Abcam (Cambridge, MA). Anti-IGFBP-4, anti-MMP-2 antibody (K-20) and anti-tubulin antibodies were from Santa Cruz BioTechnology (Santa Cruz, CA). Antibodies to phosphorylated p38 MAPK and p38 isoforms were from Cell Signaling (Danvers, MA). Normal mouse IgG was obtained from Pierce (Rockford, IL). Anti-CD-31 antibody was obtained from BD Pharmingen (San Jose, CA). HRP-labeled goat anti-mouse and goat anti-rabbit antibodies were from BioSourse (Camarillo, CA). Alexa 488 and 594-conjugated secondary antibodies were obtained from Invitrogen (Carlsbad, CA). MTT proliferation assay kit was obtained from Millipore (Bedford, MA). BrdU cell proliferation assay kit was obtained from Calbiochem (San Diego, CA). P38 MAPK inhibitor (SB202190) was obtained from EMD chemicals (Gibbston, NJ). MMP inhibitor GM6001 was obtained from Millipore (Bedford, MA), and the selective MMP-2 [41] inhibitor ARP 100 was obtained from Cayman Chemical (Ann Arbor, MI). Mab D93 directed to denatured forms of collagen [42] was obtained from TRACON (San Diego, CA). Vector M.O.M Immunodetection Kit was obtained from Vector Laboratories (Burlingame, CA).
Cells and cell culture
Human M21 (αvβ3-positive) and M21L (αvβ3-negative) melanoma and parental ECV (αvβ3-positive) carcinoma cell lines were gifts from Dr. David Cheresh (UCSD, San Diego CA). M21β3-KD cell variant was generated by transfection with β3 integrin specific (ITGB3-shRNA) obtained from Origene Technologies (Rockville, MD). M21BP4 overexpressing tumor cell variant was generated by transfection of the IGFBP-4 full-length expression construct pCMV6-neo vector obtained from Origene Technologies (Rockville, MD). All control cell lines were generated by transfection of cloning vectors without cDNA or target shRNA inserts. Tumor cell variant ECVL (αvβ3 negative) was generated by 4 rounds of negative FAC sorting with Mab LM609. Tumor cells were maintained in RPMI (Gibco, Grand Island, NY), high glucose supplemented with 10 % fetal bovine serum (FBS), glutamine and penicillin/streptomycin at 37 °C with 5 % CO2. All cells were maintained as sub-confluent cultures and split 1:4 prior to use. Human umbilical vein endothelial cells (HUVEC) were obtained from Lonza (Walkersville, MD) and cultured in EBM-2 medium in the presence of 2 % FBS and supplements (Lonza). HUVECS were maintained as sub-confluent cultures and used between passages 4–6.
Immunohistochemical and immunofluorescence analysis
Briefly, frozen sections (4 lm) from tumors from each experimental condition were fixed with 50 % methanol and 50 % acetone at −20 °C and blocked with 2.5 % BSA in PBS. Tumor sections were subjected to standard hematoxylin and eosin (H&E) staining methods. For analysis of αvβ3 expression, tumor sections prepared as described above were blocked with 2.5 % BSA in PBS. Next, tissue sections were incubated with mouse on mouse Ig blocking reagent (Vector Laboratories) according to manufactures instruction and then incubated with anti-αvβ3 Mab LM609 (10 μg/ml). Tissues were washed and incubated with Alexa 594-conjugated anti-mouse secondary antibody (1:500). Photographs were taken at a magnification 200X.
Real-time quantitative RT-PCR
Real-time quantitative RT-PCR was carried out essentially as described [35]. Briefly, equal numbers of tumor cells (1.0 × 106) were plated and harvested. Total RNA was isolated using Qiagen RNeasy plus kit, (Valencia, CA). Real-time RT-PCR reactions were run in duplicate on a BioRad iQ5 system. Human IGFBP-4 primer sets included 5’-GAGCTGGGTGACACTGCTTG-3’and 5’-CCCACGAGGACCTCTACATCA-3’. β2-macroglobulin was used for normalization for all experiments, and primer sets for b2-macroglobulin included 5’’-GCCTGGAGGCTATCCAGCGTACT-3’ and 5’-ACATGGTTCACACGGCAGGCA-3’. Experiments were completed at least 3 times.
Proliferation assays
Equal numbers (1 × 103/well) of sub-confluent melanoma (M21 and M21L) or carcinoma (ECV and ECVL) tumor cell variants were resuspended in proliferation buffer (RPMI plus 1.0 % serum) and added to 96-well assay plates and incubated at 37 °C over a 3-day time course. Cell proliferation was quantified using the MTT proliferation assay kit measuring the relative levels of mitochondrial dehydrogenase according to the manufacturer's instructions. For HUVEC proliferation studies, sub-confluent cells (2,000/well) were washed and resuspended in HUVEC growth medium with 2.0 % FBS in the presence or absence 10 μl of 10× concentrated serum-free conditioned medium derived from 48-h cultures of equal numbers (5.0 × 106) of either M21 or M21L melanoma cells. Cell proliferation was measured 24 h later using the BrdU proliferation assay kit according to the manufacture's instructions. All growth assays were preformed at least 3 times.
Murine tumor growth assays
Briefly, sub-confluent cultures of human M21, M21L, M21β3-KD, M21Cont melanoma cells or ECV and ECVL carcinoma cells were harvested, washed and resuspended in sterile PBS. Human M21 melanoma cell variants (0.5 or 5.0 × 106) or ECV carcinoma cell variants (5.0 × 106) were injected subcutaneously into female nude mice in a total volume of 100 μl. For initial primary tumor growth analysis, tumor cells were allowed to grow from 4 to 19 days. At the end of the incubation period, tumor volume was estimated using the formula V = L2 × W/2, where V = volume, L = length and W = width [25, 29]. All experiments were preformed at least twice with 5–8 animals per condition.
Tumor angiogenesis assays
To quantify tumor-associated angiogenesis, microvascular density counts were performed as previously described [19, 36]. Briefly, tumors (N = 3–7) from each experimental condition were dissected, washed and embedded in OTC and snap-frozen. Frozen sections (5–6-μm) were fixed by incubation for 10 min in a solution of cold 50 % methanol and 50 % acetone. Tissues were blocked with 2.5 % BSA. Tissues were incubated with anti-CD31 antibody in 2.5 % BSA in PBS for 2 h at 37 °C. Tissues were washed and incubated with Alexa568-conjugated secondary (1:500 dilution in 1.0 % BSA in PBS). The number of CD31-positive tumor vessels was counted in either ten, 200× microscopic fields or from five, 100× microscopic fields from three individual tumors from each experimental condition. To assess the impact of IGFBP-4 on tumor angiogenesis, M21 cells overexpressing IGFBP-4 (M21BP4) and control-transfected (empty vector) cells (M21Con) were injected subcutaneously into nude mice and early tumor development and angiogenesis was examined 7 days later. At the end of the 7-day assay, mice were killed and tumors were harvested leaving the surrounding mouse skin intact. The number of vessels infiltrating the tumor from the surrounding mouse skin from each experimental condition was quantified using a dissecting scope. Experiments were completed twice with 5–6 tumors from each condition. To examine the impact of blocking tumor-associated αvβ3 has on tumor angiogenesis, M21 melanoma cells (1.0 or 5.0 × 106) were injected subcutaneously into nude mice. Three days later, mice were either not treated or injected (i.p) either daily (100 μg) or on days 3 and 5 (50 μg) with human-specific anti-αvβ3 Mab LM606 or a non-specific normal mouse control antibody. At the end of the 7-day assay, mice were killed and the subcutaneously growing tumors were harvested, and angiogenesis quantified as described above. Experiments were performed twice with 5–6 tumors from each condition.
MicroCT analysis of tumor vascular volume
MicroCT analysis was carried out essentially as described with some modifications [43]. Mice were anesthetized, and the thoracic cavity was opened exposing the heart. The right atrium was opened to serve as a drain vent. The animals were perfused with microfil medium mixture (Microfil MV-122, Flow Tech; Carver, MA) through the left ventricle at 3 ml/min. The animals were killed and placed at 4 °C overnight, to allow polymerization of microfil. After perfusion and solidification of the microfil contrast medium, tumors were dissected and scanned using a MicroCT unit (Scanco VivaCT-40, Scanco Medical, Basserdorf, Switzerland). Tumors were scanned at 10.5-μm resolution, with a voltage of 55 kVp and a current of 145 lA. A resolution was set to create a 2,048 × 2,048 pixel image matrix. The tomograms were globally thresholded based on X-ray attenuation and used to render binarized 3-D images of the tumors.
Western blot analysis
Equal numbers of tumor cells from each experimental condition were harvested, and whole cell lysates and conditioned medium (CM) were collected after 24 or 48 h and concentrated 10X. For collection of CM following treatment with inhibitors, equal numbers of M21 cells were incubated with MMP inhibitor GM6001 (0–10.0 μM), the MMP-2 selective ARP 100 inhibitor (41) or DMSO for 48 h. For cell lysates, equal numbers of tumor cells from each experimental condition were washed and lysed in RIPA buffer (Santa Cruz) supplemented with protease inhibitor cocktail. Equal amounts (15–40 μg/lane) of cell lysates or CM were separated by SDS PAGE and transferred to PVDF membranes. Membranes were probed with antibodies directed to IGFBP-4, IGFBP-7, collagen, P38 MAPK, MMPs or tubulin. Western blots were visualized by chemiluminescence detection. Western blots were scanned, and fold change in band intensity was determined using Image J software (NIH).
Statistical analysis
Statistical analysis was performed using the InStat statistical program. Data were analyzed for statistical significance using Student T test. P values <0.05 were considered significant.
Results
Expression of αvβ3 integrin has minimal effects on tumor cell growth in vitro
Studies indicate that αvβ3 regulates tumor and endothelial cell behavior through multiple mechanisms [6, 13, 27]. While αvβ3 expressed in blood vessels plays a role in endothelial cell survival and angiogenesis, its expression in melanoma has been correlated with increased invasion and metastasis [6, 13, 27]. Previously, αvβ3 expressing M21 melanoma cells were shown to form larger tumors in mice as compared to M21L cells lacking αv, yet these cell variants exhibited minimal difference in growth rates in vitro [44]. To confirm these findings, we examined the growth of M21 cells expressing (M21 αvβ3+) or lacking (M21L αvβ3-) αvβ3 integrin in vitro. Consistent with previous studies [44], little if any change in growth was observed between these cell variants (Fig. 1a). These results are consistent with the possibility that melanoma cell-associated αvβ3 may alter the growth properties of these tumors by mechanisms that depend in part, on elements found within the tumor micro-environment. In this regard, we sought to determine whether the differential effects of αvβ3 on tumor cell growth in vivo were limited to M21 cells. To this end, ECV carcinoma cells were FAC sorted with anti-αvβ3-specific antibody LM609 to obtain a subpopulation (ECVL) lacking cell surface expression of αvβ3. As shown in Fig. 1b, while ECV cells readily expressed integrin αvβ3, 4 rounds of negative sorting resulted in isolation of ECVL cells that expressed little in any cell surface-αvβ3. To confirm our previous results and to assess the effects of αvβ3 on carcinoma cells, we examined the growth of these variants in vitro. As shown in Fig. 1c, little change in growth was observed between these ECV carcinoma cell variants.
Fig. 1.

Effects of αvβ3 integrin expression on tumor cell growth in vitro. The growth of tumor cells expressing (M21 melanoma and ECV carcinoma) or lacking (M21L melanoma and ECVL carcinoma) αvβ3 integrin was examined in vitro over a 3-day time course. a Quantification of M21 cell variant growth. b Flow cytometry analysis of the relative expression of αvβ3 integrin expressed as the mean fluorescence intensity (MFI) of ECV carcinoma cell variants. ECV-Cont, MFI of parental ECV cells stained with control secondary antibody only. ECV-αvβ3, MFI of parental ECV cells stained with Mab LM609. ECVL-Cont, MFI of ECVL cells derived from four rounds of αvβ3 negative selection and stained with control secondary antibody only. ECVL-αvβ3, MFI of ECVL cells derived from four rounds of αvβ3-negative selection and stained with Mab LM609. c Quantification of ECV cell variant growth. Data bars represent the mean optical density ±SE from triplicate wells. Experiments were completed three times with similar results
Expression of αvβ3 integrin differentially alters tumor growth in vivo
To determine whether αvβ3 alters tumor growth in vivo, nude mice were injected subcutaneously with M21 and M21L cells and tumors were allowed to grow over a time course. As shown in Fig. 2a, the growth of tumors expressing αvβ3 (M21) were accelerated resulting in significantly (P < 0.05) larger tumors as compared to tumors lacking αvβ3. In similar studies, the effects of αvβ3 on formation of small early-stage tumors resulting from injection of a lower number (0.5 × 106) of tumor cells were carried out. Consistent with our previous results, M21 tumors expressing αvβ3 were significantly (P < 0.05) larger as compared to M21L tumors lacking αvβ3 (Fig. 2b). To confirm that the small M21L tumors that formed remained αvβ3-negative, tumor sections were stained with Mab LM609 specific for human, but not mouse integrin αvβ3.
Fig. 2.

Differential impact of αvβ3 integrins on tumor growth and tumor angiogenesis in vivo. Nude mice were injected with tumor cells, and early-stage tumor growth and angiogenesis were examined. a M21 cell variants (5.0 × 106) were injected subcutaneously, and tumor growth monitored over a 19-day time course. Data bars represent mean tumor volumes ± SE from 8 to 9 mice per condition. b M21 cell variants (0.5 × 106/mouse) were injected subcutaneously, and tumor growth examined on day 7. c Representative examples of tumor tissues analyzed by H&E staining (Top) and for expression of αvβ3 (bottom) from each M21 tumor variant. Red color (bottom panels) indicates expression of αvβ3. Photographs were taken at ×200 magnification. d, e ECV cell variants (5.0 × 106/mouse) were injected subcutaneously into nude mice, and tumor growth examined on days 4 (d) and 14 (e). f Representative examples of tumor tissues analyzed by H&E staining (Top) and for expression of αvβ3 (bottom) from each ECV tumor variant. Red color (bottom panels) indicates expression of αvβ3. Photographs were taken at ×200 magnification. g, h Analysis of tumor-associated angiogenesis. Top panels Representative examples of CD31 expressing tumor vessels (Red) from each tumor variant. Photographs were taken at ×200 magnification. Bottom panels Quantification of tumor-associated CD31 expressing blood vessels. Data bars represent mean vessel counts ± SE per ×200 fields (n = 10) for each of three independent tumors from each condition. i The effects of serum-free ×10 conditioned medium (CM) from M21 or M21L cells on human umbilical vein endothelial cell (HUVEC) proliferation were examined. Data bars represent mean cell proliferation following incubation with CM from each cell variant, expressed as percent of control ± SE from four independent experiments. *Indicates P values <0.05
As shown in Fig. 2c, while viable tumors arising from M21 cells readily expressed αvβ3, viable M21L tumors lacked detectable expression of αvβ3. In similar studies, we examined the growth of ECV carcinoma cell variants in vivo. Consistent with our previous observations with M21 cells, ECVL carcinoma cells selected for lack of surface expression of αvβ3 also formed small viable tumors in vivo that were significantly (P < 0.05) reduced in size as compared to ECV cells (Fig. 2d, e). Importantly, these ECVL tumors also exhibited reduced levels of αvβ3 expression as compared to the parental ECV tumors (Fig. 2f). These data are in good agreement with previous studies and suggest that the differential impact of αvβ3 on tumor growth in vivo is not restricted to M21 melanoma cells.
While a number of mechanisms may contribute to the early growth advantage of αvβ3 expressing tumors in vivo, a possibility that has not been studied in detail involves the ability of αvβ3 to alter the angiogenic output of these tumors. Thus, we examined the relative levels of angio-genesis in tumors expressing or lacking αvβ3. As shown in Fig. 2g, h, αvβ3-expressing tumors had a significant (P < 0.05) 2.0–2.5-fold increase in CD31-expressing blood vessels within equivalent 200× microscopic fields as compared to those tumors lacking αvβ3. Finally, to examine whether soluble factors generated from the tumor cell variants may alter endothelial cell behavior, we examined the effects of conditioned medium (CM) from either M21 or M21L cells on the growth of human umbilical vein endothelial cells (HUVEC). As shown in Fig. 2i, addition of CM from M21L cells to HUVECs resulted in significantly (P < 0.05) reduced growth as compared to CM prepared from M21 cells. These data are consistent with differential expression of angiogenic regulatory molecules from these tumor variants.
Enhanced expression of IGFBP-4 in melanoma cells lacking αvβ3
Previous studies have suggested that small early-stage tumors may remain dormant and fail to expand without an adequate blood supply [5, 7–10]. While a number of mechanisms might explain the enhanced angiogenesis associated with αvβ3 expressing tumors in vivo, it is possible that αvβ3 may regulate vascularization and subsequent tumor growth in part by modulating expression of endogenous angiogenesis regulatory molecules. Indeed, we previously demonstrated that endothelial cell interaction with denatured collagen, an important αvβ3 ligand expressed selectively within the tumor microenvironment of melanomas, suppresses the expression of IGFBP-4 [37]. IGFBP-4 has been suggested to regulate tumor growth and angiogenesis [37–40, 45]. However, the mechanism by which binding to denatured collagen regulates IGFBP-4 expression is not known. Given the ability of αvβ3 to bind denatured collagen, we first examined IGFBP-4 mRNA levels in M21 and M21L cells. As shown in Fig. 3a, the levels of IGFBP-4 mRNA in M21L cells were enhanced as compared to M21 cells. To confirm the differential expression of IGFBP-4 between αvβ3 expressing cell variants, conditioned medium (CM) and cell lysates (LY) were examined. As shown in Fig. 3b, an approximately fourfold increase in the relative levels of IGFBP-4 was detected in CM and a twofold increase was observed in whole cell lysates of M21L cells lacking αvβ3 as compared to M21 cells expressing αvβ3. These findings are consistent with a possible role for αvβ3 in regulating expression of IGFBP-4.
Fig. 3.

Differential expression of IGFBP-4 in M21 melanoma cell variants. M21 tumor cell variants that express (M21) or lack (M21L) expression of αvβ3 were harvested from sub-confluent cultures. a The relative level of IGFBP-4 mRNA was analyzed by real-time RT-PCR. Data bars represent mean fold change ± SE from four independent experiments. Relative levels of IGFBP-4 in M21 cells were set to 1.0 for each experiment. b Analysis of IGFBP-4 protein in conditioned medium (CM) and whole cell lysates (LY) from M21 cell variants by Western blot. * Indicates P values <0.05
Knock down ofβ3 integrin enhances expression of IGFBP-4 in M21 cells and reduces tumor growth and angiogenesis in vivo
The lack of αvβ3 in M21L cells results from the lack of the av integrin chain, and thus, it is possible that additional av containing integrins might also contribute the altered expression of IGFBP-4 in our studies. To further examine the role of αvβ3 in regulating IGFBP-4, we knocked down expression of the β integrin in M21 cells (M21β-KD) by shRNA. As shown in Fig. 4a, the expression of β was reduced by greater than 85 % in M21β3-KD cells as compared to control-transfected M21 cells (M21Cont). To confirm the involvement of β3 integrin in the regulation of IGFBP-4 expression, we examined the relative levels of IGFBP-4 mRNA in these cell variants. As shown in Fig. 4b, an approximate fourfold increase in mRNA expression of IGFBP-4 was detected in M21β3-KD as compared to control-transfected M21 cells (M21Cont). To study these β3 integrin knocked down M21 cells in more detail, M21β3-KD cells were injected subcutaneously in mice and tumor growth was examined. As shown in Fig. 4c, M21β3-KD cells exhibited similar growth properties in vivo as was observed with M21L cells lacking αvβ3 and formed significantly smaller (P < 0.05) tumors as compared to M21Cont cells. While M21β3-KD lesions were smaller than M21Cont, these tumors contained viable cells (Fig. 4c bottom panel). Reduced tumor vascular density was also observed in M21β3-KD tumors as compared to control tumors (Fig. 4d). Finally, to confirm changes in tumor-associated angiogenesis among these tumor variants, we quantified total vascular volumes using μCT scanning [43]. As shown in Fig. 4e, a significant (P < 0.05) approximately 50 % reduction in total vessel volume was observed in M21β3-KD tumors as compared to M21Cont tumors following normalization for tumor size.
Fig. 4.

Reduction of integrin β3 enhances IGFBP-4 and reduces tumor growth and angiogenesis in vivo. Expression of β3 integrin was reduced in M21 cells by transfection with a β3-specific shRNA. a Western blot analysis of β3 protein in M21Cont and M21β3-KD cells. b Quantification of IGFBP-4 mRNA in M21Cont and M21β3-KD cells by real-time RT-PCR. c M21 tumor cell variants were injected in nude mice and allowed to grow for 17 days. Top panel Quantification of growth of M21Cont and M21β3-KD cells in nude mice. Data bars represent mean tumor volumes ± SE from 10 mice per condition. Bottom panel Example of tumor tissues from each tumor variant stained by H&E. Experiments were repeated twice with similar results. d. Quantification of the mean tumor vessels from each tumor variant. Data bars represent mean vessel counts ± SE per ×100 microscope fields (n = 5) for each of three independent tumors from each condition. e Total vascular volume was quantified from M21 tumor variants using lCT scanning of tumors injected with microfil. Top panels example of vascular networks from each tumor type. Bottom panel quantification of mean vascular volume ± SE from entire three-dimensional tumor mass from six tumors from each variant corrected for tumor size. *Indicates P values <0.05
Over-expression of IGFBP-4 in M21 cells reduces melanoma-associated angiogenesis in vivo
Previous studies have suggested that IGFBP-4 may inhibit endothelial tube formation in vitro and angiogenesis in vivo [38–40]. Moreover, our previous studies suggested that human biopsies of metastatic melanomas, which are often associated with high levels of angiogenesis, exhibited lower levels of IGFBP-4 as compared to primary melanomas [46]. To assess whether the elevated levels of angio-genesis observed within M21 tumors may be associated in part with altered levels of IGFBP-4, we overexpressed IGFBP-4 in M21 cells. As shown in Fig. 5a, M21 cells transfected with an IGFBP-4 expression construct had high levels of IGFBP-4 in conditioned medium (CM) and in whole cell lysates (LY), while little IGFBP-4 was detected in cells transfected with a control construct. To examine these cell variants in more detailed, IGFBP-4 over-expressing and control-transfected cells were injected into mice and allowed to form small early-stage tumor for 7 days and angiogenesis was assessed. As shown in Fig. 5b (top panel), M21Con tumors were associated with numerous blood vessels infiltrating the tumors from the surrounding skin, while IGFBP-4 overexpressing (M21BP4) tumors showed reduced blood vessels. While M21BP4 tumors were smaller than control M21Con tumors, these lesions still contained viable tumor cells (middle panel).
Fig. 5.

Reduced angiogenesis associated with M21 tumors over-expressing IGFBP-4. M21 cells were transfected with control construct (M21Con) or IGFBP-4 expression construct (M21BP4). a Western blot analysis of conditioned medium (CM) and whole cell lysates (LY) for IGFBP-4 expression. b M21 cells (0.5 × 106) overexpressing IGFBP-4 (M21BP4) or control-transfected cells (M21Con) were injected into mice, and tumors were allowed to form for 7 days. Tumor-associated angiogenesis was assessed by quantifying surface vessels infiltrating the tumors from the surrounding tissue. Top panel Examples of vessels infiltrating tumors from each condition. Middle panel Example of tissues from each tumor variant stained by H&E. Bottom panel Quantification of vessel infiltration of M21 melanomas from each condition. Data bars indicate mean number of surface vessels infiltrating the tumors ±SE from 3–4 tumors from each condition. c Tumor cell variants (5 × 106) were injected and tumors allowed to form for 7 days. Top panel Quantification of tumor size from each cell variant. Data bars represent mean tumor volumes ± SE from 7 to 8 tumors per condition. Bottom panel Example of tissues from each tumor variant stained by H&E. d Quantification of vessel infiltration of M21 melanomas from each condition. Data bars indicate mean number of surface vessels infiltrating the tumors ±SE from 7 to 8 tumors from each condition. *Indicates P values <0.05
Quantification (Fig. 5b bottom panel) of vessels infiltrating these tumors indicated significantly (P < 0.05) reduced levels associated with M21BP4 tumors as compared to controls. Given the small size of these tumors, we carried out additional experiments by injecting mice with larger numbers of tumor cells to allow quantification of tumor size. As shown in Fig. 5c (top panel), while early-stage M21BP4 tumors were slightly smaller than control M21 tumors, this small change in tumor size did not meet statistical significance (P > 0.05). However, results from quantification of M21BP4 tumor blood vessels indicated a significant reduction as compared to control tumors (Fig. 5d).
Reduction of IGFBP-4 in M21 cells expressing αvβ3 involves altered levels of p38 MAPK
Integrins are known to regulate multiple signaling cascades including the MAP kinase signaling pathways. Previous studies have implicated αvβ3 in the regulation of P38 MAPK signaling in melanoma and endothelial cells [47, 48]. Therefore, to begin to assess possible mechanisms to account for the altered IGFBP-4 in M21 cell variants, we first examined whether changes in αvβ3 expression in M21 cells were associated with altered MAP kinase levels. Melanoma cell variants were examined for levels of phosphorylated Erk and p38 MAPK. As shown in Fig. 6a, little change was observed between the levels of phosphorylated Erk in M21 and M21L cells. Interestingly, M21L cells exhibited an approximately 75 % reduction in the levels of phosphorylated p38 MAPK. Surprisingly, reduced levels of total p38 MAPK were also observed, suggesting a distinct change in the p38 MAPK levels in these melanoma cells. Given that multiple isoforms of p38 MAPK exist, we examined whether the reduction in p38 MAPK expression was associated with changes in specific isoforms. As shown in Fig. 6b, while a small reduction in the levels of p38c was observed in M21L cells as compared to M21 cells, no change in the levels of p38b was observed. In contrast, an approximately 70 % reduction in the relative levels of p38a was observed in M21L cells as compared to M21 cells. These findings suggest that the reduction of total p38 was at least in part due to a reduction of p38α.
Fig. 6.
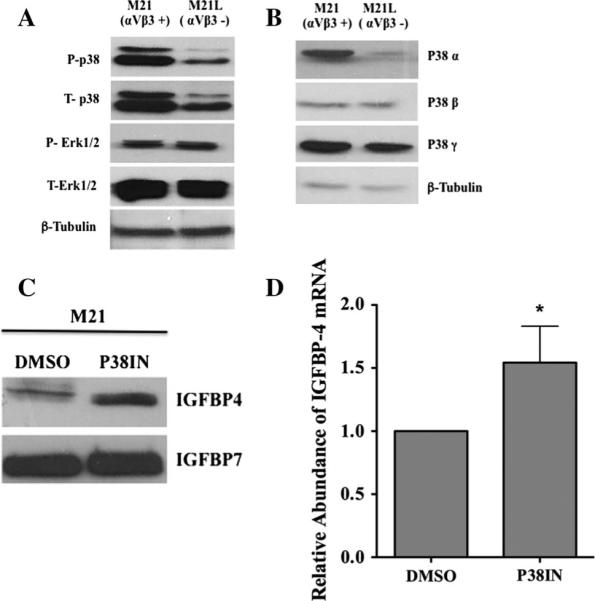
Regulation of IGFBP-4 expression in M21 cells involves altered p38 MAPK signaling. M21 melanoma cells expressing (M21) or lacking (M21L) αvβ3 were examined for the expression of total and activated p38 MAPK and Erk. a Western blots of the levels of total and phosphorylated p38 MAPK and Erk in M21 cell variant lysates. b Western blots of the levels of p38 MAPK isoforms in M21 cell variant lysates. c, d M21 cells were incubated with p38 inhibitor (P38IN) or control DMSO. The levels of IGFBP-4 were assessed by Western blot (c) or real-time RT-PCR (d). Experiments were completed 2–3 times with similar results. *Indicates P values <0.05
To examine whether the αvβ3-associated reduction of IGFBP-4 may be dependent in part on p38 MAPK, we treated αvβ3 expressing M21 cells with a p38 MAPK inhibitor and examined the relative levels of IGFBP-4. As shown in Fig. 6c, inhibition of p38 MAPK in M21 cells resulted in an approximately 2.6-fold increase in IGFBP-4 protein. Interesting, while inhibiting p38 enhanced the levels of IGFBP-4 protein, only a minimal change in the levels of mRNA was observed (Fig. 6d). These findings are consistent with a possible role for p38 MAPK in regulating the levels of IGFBP-4 protein by mechanisms associated in part with post-transcriptional events.
The p38 MAPK-associated reduction of IGFBP-4 depends on altered MMP levels
An important post-translational mechanism known to regulate IGFBP-4 is proteolytic degradation [49, 50]. Given previous studies implicating P38 MAPK in regulating matrix metalloproteinases (MMPs) and the ability MMPs to degrade IGFBP-4, we examined the relative levels of MMPs in cells expressing or lacking αvβ3. While minimal levels of MMP-9 were detected in M21 cell variants, MMP-2 was readily detected and exhibited differential expression between M21 cell variants. As shown in Fig. 7a, M21L cells exhibited greater than 50 % reduced levels of MMP-2 as compared to M21 cells as indicated by scanning image analysis of multiple experiments. Similar results were observed in conditioned medium when comparing M21β3-KD cells lacking αvβ3 to control-transfected M21Con-KD cells (Fig. 7b). While enhanced level of IGFBP-4 was detected in M21β3-KD cells (Fig. 4), IG FBP-7 levels appeared reduced in cells in which β3 inte-grin was reduced (Fig. 7b). However, the levels of collagen in these condition medium samples were not changed. Given that little if any change in IGFBP-7 expression was detected between M21 and M21L cells, it is not clear why knocking down β3 integrin chain was associated with reduced levels of IGFBP-7. Consistent with previous studies [51, 52] indicating the ability of p38 MAPK to regulate MMP expression, incubation of M21 cells with the p38 MAPK inhibitor reduced levels of MMP-2 by approximately 40 % as compared to control (Fig. 7c). While it is known that several proteases including multiple members of the MMP family [49, 50] can degrade IGFBP-4, given the differential expression of MMP-2 observed, we examined whether MMPs might contribute to the loss of IGFBP-4 in our studies. In this regard, we examined the effect of adding a broad spectrum MMP inhibitor (GM6001) on the level of IGFBP-4 in conditioned medium of M21 melanoma cells in which little IGFBP-4 is normally detected. As shown in Fig. 7d, incubation of cells with the MMP inhibitor increased detectable levels IGFBP-4. In a similar study, we examined the effects of the MMP-2 selective inhibitor ARP 100, which has been shown to exhibit minimal inhibitory activity toward other MMPs [41] on the levels of IGFBP-4. As shown in Fig. 7e, incubation of ARP 100 with M21 cells also enhanced detection of IGFBP-4. Importantly, while we cannot completely rule out that other MMPs in addition to MMP-2 contribute to the degradation of IGFBP-4 in M21 cells, our findings are consistent with the ability of p38 MAPK to regulate the levels of IGFBP-4, in part by an MMP-associated mechanism.
Fig. 7.

Reduction of IGFBP-4 depends in part on altered MMP expression. The relative level of MMP-2 and IGFBP-4 in conditioned medium (CM) from M21 cell variants was examined. a Western blot of CM from M21 and M21L cells for MMP-2 and IGFBP-7 protein. b Western blot of CM from M21Cont and M21β3-KD for MMP-2, IGFBP-7 and collagen. c Western blots of CM from M21 cells treated with the p38 inhibitor or DMSO for MMP-2 and IGFBP-7. d Western blot of CM from M21 cells treated with the MMP inhibitor GM6001 or DMSO for IGFBP-4 and IGFBP-7. e Western blot of CM from M21 cells treated with the MMP-2 selective inhibitor ARP 100 or DMSO for IGFBP-4 and IGFBP-7. Experiments were completed at least 2–3 times with similar results
Specific blockade of tumor cell-associated αvβ3 inhibits pathological angiogenesis in vivo
Numerous studies have provided evidence for a role for αvβ3 expressed in endothelial cells in regulating angio-genesis [27–31]. Interestingly, the role of αvβ3 in governing blood vessel development is complex as studies have suggested that αvβ3 may regulate vessel formation in both positive and negative fashion depending the cell types expressing the integrin and the tissue microenvironment in which angiogenesis is occurring. While the majority of studies have shown that pharmacological blockade of αvβ3 expressed within the endothelial cell compartment can inhibit endothelial cell proliferation and induce apoptosis, little evidence is available for the possibility that αvβ3 expressed within the tumor cell compartment may actively modulate angiogenesis.
To examine the effects of blocking αvβ3 expressed specifically within the tumor cell compartment, we examined the effects of anti-human αvβ3 Mab LM609 on tumor angiogenesis in mice. Importantly, it is well established that Mab LM609 does not bind or inhibit αvβ3 expressed on mouse blood vessels, yet readily binds and inhibits αvβ3 expressed on human M21 cells [29, 44]. This important differential binding characteristic allows us the ability to examine whether selective inhibition of αvβ3 expressed within melanoma cells rather than the murine stromal cell may impact tumor angiogenesis. To test this, mice were injected subcutaneously with αvβ3 expressing M21 cells. Following a 3-day incubation period, mice were either untreated or treated daily with anti-αvβ3 Mab LM609 or control antibody for 4 days. The resulting tumors were dissected and analyzed on day 7. Interestingly, even though Mab LM609 does not have the ability to bind and inhibit αvβ3 expressed in murine endothelial cells, reduced levels of blood vessels were observed infiltrating the tumors from mice injected with Mab LM609 as compared to controls (Fig. 8a top panel). While the M21 tumor from mice treated with Mab LM609 appeared smaller, these lesions contained viable tumor cells (Fig. 8a bottom panel). Given the small size of the tumors, we next carried out similar experiments and injected mice with increased numbers (5.0 × 106) of tumor cells to allow the formation of larger tumors within the 7-day incubation period. Mice were again untreated or treated with Mab LM609 or control, and tumor volumes and angiogenesis quantified on day 7. As shown in Fig. 8c, while tumors from mice treated with Mab LM609 were smaller than controls, this reduction did not meet statistical significance (P > 0.05). However, consistent with our previous results, tumors from mice treated with Mab LM609 exhibited significantly (P < 0.05) reduced levels of blood vessels as compared to controls (Fig. 8d). These data are consistent with the possibility that early blockade of αvβ3 expressed specifically within the tumor cells contributes to suppressing the angiogenic switch within these human melanoma cells.
Fig. 8.

Blocking tumor cell-associated αvβ3 inhibits pathological angiogenesis in vivo. M21 cells (1.0 × 106) were injected into mice and allowed to grow for 3 days. a Mice were untreated or treated daily with anti-human antibody LM609 on control normal mouse IgG (100 μg). Top panel Examples of blood vessels infiltrating tumors from each condition. Bottom panel Example of tumor tissue from each condition stained by H&E. b Quantification of blood vessels infiltrating tumors from the surrounding skin. Data bars indicate mean number of surface vessels infiltrating the tumors ±SE from 6 tumors from each condition. c Mice were injected with M21 cells (5 × 106) and treated on days 3 and 5 with Mab LM609 or normal mouse IgG (50 lg) and tumors analyzed on day 7. Data bars represent mean tumor volumes ± SE from 5 to 6 mice per condition. d Quantification of blood vessels infiltrating tumors from the surrounding skin. Data bars indicate mean number of surface vessels infiltrating the tumors ±SE from 5 to 6 tumors from each condition. *Indicates P values <0.05
Discussion
Recent evidence suggests that αvβ3 plays diverse roles in regulating cell behavior as targeting of this integrin in endothelial cells inhibits adhesion, migration and survival in vitro [25, 27–31]. Pharmacological inhibition of αvβ3 has also been shown to inhibit pathological angiogenesis in vivo using multiple models [27–31]. Interestingly, in other studies, lack of αvβ3 expression in mutant mice was associated with limited impact on normal vascular development [30, 31]. Surprisingly, studies have also shown that angio-genesis and tumor growth in mutant mice lacking β3 were enhanced, and this enhanced angiogenesis was associated with altered VEGFR signaling [53]. In one set of studies, enhanced but non-functional blood vessel formation was observed in mice expressing signaling deficient β3 integrin [54], while in a separate study investigators observed reduced pathological angiogenesis and smaller tumors in β3 signaling deficient mice, which was associated with reduced recruitment of bone marrow-derived stromal cells to the tumor and associated vasculature [34]. Interestingly, recent findings have also indicate that the role of αvβ3 integrin in angiogenesis may be time dependent as reduction of αvβ3 inhibited angiogenesis and subsequent tumor growth at an early time point but had little effect on later-stage tumors with pre-established vessels [55]. Given these findings, the divergent roles of αvβ3 may depend on a combination of distinct parameters such as the particular cell type expressing the integrin, the type and accessibility of αvβ3 ligands, the activation state of the integrin and the relative composition of secreted growth factors found locally within the tissue microenvironment in which angiogenesis is occurring [56]. In this regard, antagonists of αvβ3 inhibited FGF-2-induced angiogenesis while exhibiting minimal activity on angiogenesis induced by VEGF [57].
In agreement with context-dependent functions of αvβ3, activated αvβ3 in tumor cells growing within the brain microenvironment resulted in enhanced pathological angiogenesis and elevated levels of VEGF, resulting from a mechanism associated with the inhibition of the translational repressor 4E-binding protein-1 (4E-BP1) [58]. These same tumor cells expressing activated αvβ3 growing within the mammary fat pad failed to exhibit a similar response [58]. These findings are consistent with the concept that the particular tissue microenvironment in which αvβ3 is expressed may impact its ability to alter the angiogenic output of a given tumor and thus the angiogenic switch. Consistent with this possibility, studies have suggested that αvβ3-dependent interactions with the ECM may regulate shear stress-induced release of FGF-2 from endothelial cells [59]. Here we provide evidence that M21 melanoma cells as well as ECV carcinoma cells expressing αvβ3 formed significantly larger tumors with elevated levels of angiogenesis as compared to corresponding tumor cells that lacked αvβ3. Given that αvβ3 expressing tumor cell variants exhibited similar growth properties in vitro, it is likely that the formation of these small early-stage tumors in our studies depends on additional mechanisms regulated by factors governed in part by the local microenvironment. To this end, we provide evidence that the expression of the endogenous angiogenesis inhibitor IGFBP-4 is controlled in part by αvβ3 in M21 cells. These findings are consistent with our previous report, indicating a reduction in tumor-associated IGFBP-4 in biopsies of metastatic human melanomas as compared to primary melanoma [46], given that metastatic melanoma have been shown to express high levels of αvβ3 as compared to primary melanoma. This αvβ3-mediated reduction of IGFBP-4 in melanoma cells depends in part on p38 MAPK since L21L cells lacking αvβ3 exhibited reduced levels of p38a and elevated levels of IGFBP-4. Moreover, blocking p38 activation in αvβ3 expressing M21 cells enhanced the levels of IGFBP-4. The ability of p38 MAPK to alter IGFBP-4 levels appears to depend on post-transcriptional events as minimal changes in IGFBP-4 mRNA were observed, yet the levels of IGFBP-4 protein were enhanced by 2.6 fold. In this regard, p38 MAPK has been suggested to regulate the expression of MMPs, and MMPs can degrade IGFBP-4 [49, 50]. Our studies indicate elevated expression of MMP-2 and little detectable IGFBP-4 in conditioned medium of M21 cells, while enhanced levels of IGFBP-4 were observed in conditioned medium from M21L cells that lacked αvβ3 and that expressed reduced levels of p38 MAPK and MMP-2. Importantly, blocking p38 MAPK in M21 cells reduced levels of MMP-2 and enhanced levels of IGFBP-4. Moreover, blocking MMP activity with either a broad spectrum MMP inhibitor or an MMP-2 selective inhibitor resulted in the enhanced accumulation of IGFBP-4 in M21 cells. These novel findings are consistent with a mechanism by which p38 MAPK contributes to the regulation of IGFBP-4 by altering MMP-mediated degradation. It would be interesting to speculate that given that αvβ3 expressing metastatic melanoma cells have been suggested to express high levels of MMPs, the reduction in the endogenous angiogenesis inhibitor IGFBP-4 expression observed in our previous studies [46] might be associated in part with enhanced MMP-mediated degradation.
Our studies suggest that the elevated levels of IGFBP-4 in melanoma cell variants lacking αvβ3 (M21L and M21β3-KD) may contribute to the reduction in the early onset of angiogenesis and the smaller tumor size observed with these melanomas. In support of this possibility, αvβ3 expressing M21 cells overexpressing IGFBP-4 exhibited reduced tumor growth and angiogenic response as compared to αvβ3-positive M21 cells expressing the control vector. While it is possible that additional secreted factors regulated by αvβ3 in these cells contribute to the differential angiogenic output, overexpression of IGFBP-4 in M21 cells inhibited melanoma-associated angiogenesis in vivo.
Interestingly, previous studies have indicated that av integrins expressed in melanoma cells as well as in endothelial cells can suppress p53 activity [60, 61]. P53 has been shown to play an important role in regulating the angiogenic switch in tumors by enhancing the expression of pro-angiogenic factors such as FGF-2, HIF1α, VEGF and PDGF and by reducing the expression of endogenous angiogenesis inhibitors such as TSP-1 and prolyl hydroxylase leading to reduced levels anti-angiogenic collagen fragments [62]. These findings are consistent with the possibility that the accelerated early tumor growth observed with melanoma cells expressing αvβ3 may be associated with αvβ3-dependent activation of a proangiogenic program resulting from altering the balance of pro-angiogenic factors and endogenous angiogenesis inhibitors. Moreover, the importance of αvβ3 expressed within the melanoma cell compartment in regulating the angiogenic switch is further supported by our observations that targeting tumor cell-associated αvβ3, but not other murine host cells, resulted in a reduction of early onset tumor angiogenesis. Taken together, our studies provide new insight into the context-dependent role of αvβ3 in controlling the balance of angiogenic regulatory factors, which may contribute to the overall control of early melanoma tumor growth.
Acknowledgments
This work was supported in part by National Institutes of Health grants CA91645 (to P.C.B), Grant HL65301 (to R.F), Grants HL083151 and P20RR15555 (Protein, Nucleic Acid Analysis) to (C.P.H.V.), and Grant P20 RR181789 (Bioinformatics Core) to D. M. Wojchowski. This work was also supported by NIH Center of Biomedical Research Excellence 5P30GM103392 (PI: R. Friesel), the Maine Cancer Foundation (MCF) grant to L. W. C., and by institutional support from the Maine Medical Center.
Footnotes
Conflict of interest The authors declare no conflict of interest.
Contributor Information
Liangru W. Contois, Maine Medical Center Research Institute, Center for Molecular Medicine, 81 Research Drive, Scarborough, ME 04074, USA
Abebe Akalu, Maine Medical Center Research Institute, Center for Molecular Medicine, 81 Research Drive, Scarborough, ME 04074, USA.
Jennifer M. Caron, Maine Medical Center Research Institute, Center for Molecular Medicine, 81 Research Drive, Scarborough, ME 04074, USA
Eric Tweedie, Maine Medical Center Research Institute, Center for Molecular Medicine, 81 Research Drive, Scarborough, ME 04074, USA.
Alexandra Cretu, University of Pennsylvania, 3451 Walnut Street, Philadelphia, PA 19104, USA.
Terry Henderson, Maine Medical Center Research Institute, Center for Molecular Medicine, 81 Research Drive, Scarborough, ME 04074, USA.
Lucy Liaw, Maine Medical Center Research Institute, Center for Molecular Medicine, 81 Research Drive, Scarborough, ME 04074, USA.
Robert Friesel, Maine Medical Center Research Institute, Center for Molecular Medicine, 81 Research Drive, Scarborough, ME 04074, USA.
Calvin Vary, Maine Medical Center Research Institute, Center for Molecular Medicine, 81 Research Drive, Scarborough, ME 04074, USA.
Peter C. Brooks, Maine Medical Center Research Institute, Center for Molecular Medicine, 81 Research Drive, Scarborough, ME 04074, USA
References
- 1.Varney ML, Jonansson SL, Singh RK. Tumor-associated infiltration, neovascularization and aggressiveness in malignant melanoma: role of monocyte chemotactic protein-1 and vascular endothelial growth factor-A. Melanoma Res. 2005;15:417–425. doi: 10.1097/00008390-200510000-00010. [DOI] [PubMed] [Google Scholar]
- 2.Demirkesen C, Buyukpinarbasili N, Ramazangoglu R, Ollguz O, Mandel NM, et al. The correlation of angiogenesis with metastasis in primary cutaneous melanoma: a comparative analysis of microvessel density, expression of vascular endothelial growth factor and basic fibroblast growth factor. Pathology. 2006;38:132–137. doi: 10.1080/00313020600557565. [DOI] [PubMed] [Google Scholar]
- 3.Massi D, Franchi A, Borgognoni L, Paglierani M, Reali UM, et al. Tumor angiogenesis as a prognostic factor in thick cutaneous malignant melanoma. A quantitative morphological analysis. Virchow Arch. 2002;440:22–28. doi: 10.1007/s004280100480. [DOI] [PubMed] [Google Scholar]
- 4.Wu S, Singh S, Varney ML, Kindle S, Singh RK. Modulation of CXCL-8 expression in human melanoma cells regulates tumor growth, angiogenesis, invasion, and metastasis. Cancer Med. 2012;3:306–317. doi: 10.1002/cam4.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naumov GN, Bender E, Zurakowski D, Kang SY, Sampson D, et al. A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J Natl Cancer Inst. 2006;98:316–325. doi: 10.1093/jnci/djj068. [DOI] [PubMed] [Google Scholar]
- 6.Contois L, Akalu A, Brooks PC. Integrins as functional hubs in the regulation of pathological angiogenesis. Semin Cancer Biol. 2009;19:318–328. doi: 10.1016/j.semcancer.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Indraccolo S. Insights into the regulation of tumor dormancy by angiogenesis in experimental tumors. Adv Exp Med Biol. 2013;734:37–52. doi: 10.1007/978-1-4614-1445-2_3. [DOI] [PubMed] [Google Scholar]
- 8.Kang SY, Watnick RS. Regulation of tumor dormancy as a function of tumor-mediated paracrine regulation of stromal Tsp-1 and VEGF. APMIS. 2008;116:638–647. doi: 10.1111/j.1600-0463.2008.01138.x. [DOI] [PubMed] [Google Scholar]
- 9.Almog N, Ma L, Rachowdhury R, Schwager C, Erber R, et al. Transcriptional switch of dormant tumors to fast growing angiogenic phenotype. Cancer Res. 2009;69:836–944. doi: 10.1158/0008-5472.CAN-08-2590. [DOI] [PubMed] [Google Scholar]
- 10.Moserle L, Amadori A, Indraccolo S. The angiogenic switch: implications in the regulation of tumor dormancy. Curr Mol Med. 2009;8:935–941. doi: 10.2174/156652409789712800. [DOI] [PubMed] [Google Scholar]
- 11.Chung AS, Lee J, Ferrara N. Targeting the tumor vasculature: insight from physiological angiogenesis. Nat Rev Cancer. 2010;10:505–513. doi: 10.1038/nrc2868. [DOI] [PubMed] [Google Scholar]
- 12.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17:1359–1369. doi: 10.1038/nm.2537. [DOI] [PubMed] [Google Scholar]
- 14.Ebos JM, Lee CR, Kerbel RS. Tumor and host-mediated pathways of resistance and disease progression in response to anti-angiogenic therapy. Clin Cancer Res. 2009;16:5020–5025. doi: 10.1158/1078-0432.CCR-09-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebos JM, Lee CR, Cruz-Munzo W, Bjarmason GA, Christensen JG, et al. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, et al. Antiangiogenic therapy elicits malignant progression of tumors to increase local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hangai M, Kitaya N, Chan CK, Kim JJ, Werb Z, et al. Matrix metaaloproteinase-9-dependent exposure of a cryptic migratory control site in collagen is required before retinal angiogenesis. Am J Pathol. 2002;161:1429–1437. doi: 10.1016/S0002-9440(10)64418-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Q, Jin M, Yang F, Zhu J, Xiao Q, et al. Matrix metalloproteinases: inflammatory regulators of cell behavior in vascular formation and remodeling. Mediators Inflamm. 2013 doi: 10.1155/2013/928315. doi:10.1155/2013/928315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu X, Rodriguez D, Petitclerc E, Kim JJ, Hangai M, et al. Proteolytic exposure of a cryptic site within collagen type-IV is required for angiogenesis and tumor growth in vivo. J Cell Biol. 2001;154:1069–1079. doi: 10.1083/jcb.200103111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Assal Y, Mie M, Kobatake E. The promotion of angiogenesis by growth factors integrated with ECM proteins through coiled-coil structures. Biomaterials. 2013;34:3315–3323. doi: 10.1016/j.biomaterials.2013.01.067. [DOI] [PubMed] [Google Scholar]
- 22.Davis GE. Angiogenesis and proteinase: influence on vascular morphogenesis, stabilization and regression. Drug Discov Today Dis Models. 2011;8:13–20. doi: 10.1016/j.ddmod.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sweet DT, Chen Z, Wiley DM, Bautch VL, Tzima E. The adaptor protein Shc integrates growth factor and ECM signaling during postnatal angiogenesis. Blood. 2012;119:1946–1955. doi: 10.1182/blood-2011-10-384560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sacharidou A, Stratman AN, Davis GE. Molecular mechanisms controlling vascular lumen formation in three-dimensional extracellular matrices. Cells Tissues Organs. 2012;195:122–143. doi: 10.1159/000331410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petitclerc E, Boutaud A, Prestrayko A, Xu J, Sado Y, et al. New functions for non-collagenous domains of human collagen type-IV: novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J Biol Chem. 2000;275:8051–8061. doi: 10.1074/jbc.275.11.8051. [DOI] [PubMed] [Google Scholar]
- 26.Ren B, Yee KO, Lawer J, Khosrav-Far J. Regulation of tumor angiogenesis by thrombospondin-1. Biochim Biophys Acta. 2006;1765:178–188. doi: 10.1016/j.bbcan.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin αvβ3 for angiogenesis. Science. 1994;264:569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 29.Brooks PC, Montgomery AM, Rosenfeld M, Reisfeld RA, Hu T, et al. Integrin αvβ3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 1994;79:1157–1164. doi: 10.1016/0092-8674(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 30.Delbaldo C, Raymond E, Vera K, Hammershaimb L, Kaucic K, et al. Phase I and pharmacokinetic study of etaracizumab (Abegrin), a humanized monoclonal antibody against αvβ3 integrin receptor, in patients with advanced solid tumors. Investig New Drug. 2008;26:35–43. doi: 10.1007/s10637-007-9077-0. [DOI] [PubMed] [Google Scholar]
- 31.Scaringi C, Minniti G, Caporello P, Enrici RM. Integrin inhibitor cilengitide for the treatment of glioblastoma: a brief overview of current clinical results. Anticancer Res. 2012;32:4213–4223. [PubMed] [Google Scholar]
- 32.Reynolds LE, Wyder L, Lively JC, Taverna D, Robinson SD, et al. Enhanced pathological angiogenesis in mice lacking β3 integrin or β3 and β5 integrins. Nat Med. 2002;8:27–34. doi: 10.1038/nm0102-27. [DOI] [PubMed] [Google Scholar]
- 33.Taverna D, Moher H, Crowley D, Borsig L, Varki A, et al. Increased primary tumor growth in mice null for β3 or β3 and β5 integrins or selectins. Proc Natl Acad Sci USA. 2004;101:763–768. doi: 10.1073/pnas.0307289101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feng W, McCabe P, Mahabeleshwar GH, Samanath PR, Phillips DR, et al. The angiogenic response is dictated by β3 integrin on bone marrow-derived cells. J Cell Biol. 2008;182:1145–1157. doi: 10.1083/jcb.200802179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cretu A, Roth JM, Caunt M, Akalu A, Policarpio D, et al. Disruption of endothelial cell interactions with the novel HU177 cryptic collagen epitope inhibits angiogenesis. Clin Cancer Res. 2007;13:3068–3078. doi: 10.1158/1078-0432.CCR-06-2342. [DOI] [PubMed] [Google Scholar]
- 36.Akalu A, Roth JM, Caunt M, Policarpio D, Liebes L, et al. Inhibition of angiogenesis and tumor metastasis by targeting a matrix immobilized cryptic extracellular matrix epitope in laminin. Cancer Res. 2007;67:4353–4363. doi: 10.1158/0008-5472.CAN-06-0482. [DOI] [PubMed] [Google Scholar]
- 37.Contois LW, Nugent DP, Caron JM, Cretu A, Tweedie E, et al. Insulin-like growth factor binding protein-4 differentially inhibits growth factor-induced angiogenesis. J Biol Chem. 2012;287:179–189. doi: 10.1074/jbc.M111.267732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith LEH, Shen W, Perruzzi C, Soker S, Kinose F, et al. Regulation of vascular endothelial growth factor-dependent retinal neovascularization by insulin-like growth factor-1 receptor. Nat Med. 1999;12:1390–1395. doi: 10.1038/70963. [DOI] [PubMed] [Google Scholar]
- 39.Moreno MJ, Ball M, Andrade MF, Mcdermid A, Stanimirovic DB. Insulin-like growth factor binding protein-4 (IGFBP-4) is a novel anti-angiogenic and anti-tumorigenic mediator secreted by dibutyryl cyclic AMP (dB-cAMP)-differentiated glioblastoma cells. Glia. 2006;63:845–857. doi: 10.1002/glia.20345. [DOI] [PubMed] [Google Scholar]
- 40.Beattie J, McIntosh L, Walle V. Cross talk between the insulin-like growth factor (IGF) axis and membrane integrins to regulate cell physiology. J Cell Physiol. 2010;224:605–611. doi: 10.1002/jcp.22183. [DOI] [PubMed] [Google Scholar]
- 41.Rossello A, Orlandini E, Carelli P, Rapposelli S, Macchia M, et al. New N-arylsulfonyl-N-alkoxyaminoacetohydroxamic acids as selective inhibitors of gelatinase A (MMP2). Bioorg Med Chem. 2004;12:2441–2450. doi: 10.1016/j.bmc.2004.01.047. [DOI] [PubMed] [Google Scholar]
- 42.Freimark B, Clark D, Pernasetti F, Nickel J, Myszka D, et al. Targeting of humanized antibody D93 to sites of angiogenesis and tumor growth by binding to multiple epitopes on denatured collagens. Mol Immunol. 2007;44:3741–3750. doi: 10.1016/j.molimm.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 43.Mondy WL, Cameron D, Timmermans JP, De Clerck N, Sasov AC, et al. Micro-CT or corrosion casts for use in the computer-aided design of microvasculature. Tissue Eng Part C Methods. 2009;15:729–738. doi: 10.1089/ten.TEC.2008.0583. [DOI] [PubMed] [Google Scholar]
- 44.Felding-Habermann B, Mueller BM, Romerdahi CA, Cheresh DA. Involvement of integrin alpha v expression in human melanoma tumorigenicity. J Clin Investig. 1992;89:2018–2022. doi: 10.1172/JCI115811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Durai R, Davis M, Yang W, Yang SY, Seifalian A, et al. Biology of insulin-like growth factor binding protein-4 and its role in cancer. Int J Oncol. 2006;28:1317–1325. [PubMed] [Google Scholar]
- 46.Yu JZ, Warycha MA, Christos PJ, Darvishian F, Yee H, et al. Assessing the clinical utility of measuring insulin-like growth factor binding proteins in tissues and sera from melanoma patients. J Transl Med. 2008;6:70. doi: 10.1186/1479-5876-6-70. doi:10.1186/1479-5876-6-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Estrada Y, Dong J, Ossowski L. Positive crosstalk between Erk and p38 in melanoma stimulates migration and in vivo proliferation. Pigment Cell Melanoma Res. 2009;22:66–76. doi: 10.1111/j.1755-148X.2008.00520.x. [DOI] [PubMed] [Google Scholar]
- 48.Hennig T, Mogensen C, Kirsch J, Pohl U, Gloe T, et al. Shear stress induces the release of an endothelial elastase: role in integrin αvβ3-mediated FGF-2 release. Vasc Res. 2011;48:453–464. doi: 10.1159/000327009. [DOI] [PubMed] [Google Scholar]
- 49.Nakamura M, Miyamoto S, Maeda H, Ishii G, Hasebe T, et al. Matrix metalloproteinase-7 degrades all insulin-like growth factor binding proteins and facilitates insulin-like growth factor bioavailability. Biochem Biophys Res Commun. 2005;333:1011–11016. doi: 10.1016/j.bbrc.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 50.Prudova A, auf dem Keller U, Butler GS, Overall CM. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol Cell Proteomics. 2010 doi: 10.1074/mcp.M000050-MCP201. doi:10.1074/mcp.M000050-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Denkert C, Siegert A, Leclere A, Turznski A, Hauptmann S. An inhibitor of stress-activated MAP-kinase reduces invasion and MMP-2 expression of malignant melanoma cells. Clin Exp Metastasis. 2002;19:79–85. doi: 10.1023/a:1013857325012. [DOI] [PubMed] [Google Scholar]
- 52.Gomes LR, Terra LF, Wailemann RA, Labriola L, Sogayar MC. TGF-β1 modulates the homeostasis between MMPs and MMP inhibitors through p38 MAPK and ERK1/2 in highly invasive breast cancer cells. BMC Cancer. 2012 doi: 10.1186/1471-2407-12-26. doi:10.1186/1471-2407-12-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reynolds AR, Reynolds LE, Nagel TE, Robinson SD, Hicklin DJ, et al. Elevated FLK1 (vascular endothelial growth factor receptor 2) signaling mediates enhanced angiogenesis in β3-integrin-deficient mice. Cancer Res. 2004;64:8643–8650. doi: 10.1158/0008-5472.CAN-04-2760. [DOI] [PubMed] [Google Scholar]
- 54.Waston RA, Pitchford SC, Reynolds LE, Direkze N, Alison MR, et al. Deficiency of bone marrow β3-integrin enhances non-functional neovascularization. Am J Pathol. 2010;220:435–445. doi: 10.1002/path.2660. [DOI] [PubMed] [Google Scholar]
- 55.Steri V, Ellison TS, Gontarczyk AM, Weilbaecher K, Scheider JG, et al. Acute depletion of endothelial β3-integrin transiently inhibits tumor growth and angiogenesis in mice. Circ Res. 2014;114:79–91. doi: 10.1161/CIRCRESAHA.114.301591. [DOI] [PubMed] [Google Scholar]
- 56.Robinson SD, Hodivala-Dilke KM. The role of β3-integrins in tumor angiogenesis: context is everything. Curr Opin Cell Biol. 2011;23:630–670. doi: 10.1016/j.ceb.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 57.Friedlander M, Brooks PC, Shaffer RW, Kincaid CM, Varner JA, et al. Definition of two angiogenic pathways by distinct αv integrins. Science. 1996;270:1500–1502. doi: 10.1126/science.270.5241.1500. [DOI] [PubMed] [Google Scholar]
- 58.Lorger M, Krueger JS, O’Neal M, Staflin K, Felding-Habermann B. Activation of tumor cell integrin αvβ3 controls angiogenic and metastatic growth in the brain. Proc Natl Acad Sci USA. 2009;106:10666–10671. doi: 10.1073/pnas.0903035106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gloe T, Sohn HY, Meininger GA, Pohl U. Shear stress-induced release of basic fibroblast growth factor from endothelial cells is mediated by matrix interaction via integrin αvβ3. J Biol Chem. 2002;277:23453–23458. doi: 10.1074/jbc.M203889200. [DOI] [PubMed] [Google Scholar]
- 60.Stromblad S, Becker JC, Yebra M, Brooks PC, Cheresh DA. Suppression of p53 activity and p21WAF1/CIP1 expression by vascular cell integrin αvβ3 during angiogenesis. J Clin Investig. 1996;98:426–433. doi: 10.1172/JCI118808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bao W, Stomblad S. Integrin αv-mediated inactivation of p53 controls a MEK1-dependent melanoma cell survival pathway in three-dimensional collagen. J Cell Biol. 2004;167:745–756. doi: 10.1083/jcb.200404018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Teodora JG, Parker AE, Zhu X, Green MR. P53-mediated inhibition of angiogenesis through up-regulation of a collagen prolyl hydroxylase. Science. 2006;313:968–997. doi: 10.1126/science.1126391. [DOI] [PubMed] [Google Scholar]