

Abstract
An overview of recent advances in glycosylation with particular emphasis on mechanism is presented. The mounting evidence for both the existence of glycosyl oxocarbenium ions as fleeting intermediates in some reactions, and the crucial role of the associated in counter ion in others is discussed. The extremes of the SN1 and SN2 manifolds for the glycosylation reaction are bridged by a continuum of mechanisms in which it appears likely that most examples are located.
Keywords: oxocarbenium ions, counter ions, hydrogen bonding, kinetics
In 2010, a propos of glycosyl cations and the mechanism of chemical glycosylation, we surveyed critically the evidence for and against the existence of glycosyl oxocarbenium ions as intermediates in glycosylation reactions and discussed the influence of a number of factors, including protecting groups, solvents and additives on these reactions which are fundamental to glycoscience.1 We concluded that ‘glycosyl cations are likely to have a real existence in dichloromethane solution at low temperature and that their observation only awaits the development of a suitable “flash” technique for their generation in the probe of an NMR spectrometer’. In the update that follows we survey critically evidence presented in the intervening four years for the transient existence of glycosyl oxocarbenium ions in some glycosylation reactions, while stressing the important role of the counter ion in others. We begin by restating the general principle that glycosylation by nucleophilic substitution of a leaving group at the anomeric center of a glycosyl donor by a glycosyl acceptor in the presence of a promoter is best expressed in terms of a continuum of mechanisms2–4 spanning the entire range from pure SN1, with a discrete glycosyl oxocarbenium ion intermediate, through to pure concerted SN2 reactions. Concrete examples of both extremes exist, as set out previously,1 but most O-glycosylations are probably best viewed as SN1-like or SN2-like, that is, having most of the characteristics of one or the other of the two extremes without meeting the entire set of Ingoldian criteria that establish the limiting mechanisms.
Generation of Glycosyl Oxocarbenium Ions and Spectroscopic Evidence for Them
Continuing their work on the generation of carbocations in organic solution at low temperature by direct and indirect electrolytic methods Yoshida and coworkers reported the activation of 4-fluorophenyl tetra-O-benzyl-β-D-thioglucopyranoside by electrochemically generated tris(4-fluorophenyl)trisulfonium tetrakis(pentafluorophenyl)borate at a variety of temperatures below-48 °C in dichloromethane in a flow system (Scheme 2). After a mixing time of 0.17 s the reaction mixture was combined with methanol leading to the isolation of methyl tetra-O-benzylglucopyranoside in high yield, leading the authors to suggest that “a glycosyl cation somewhat stabilized by the co-ordination of ArSSAr” is formed by this process and that “the lifetime of the intermediate strongly depends on the temperature, and that the lifetime is on the order of a second even at −78 °C”.5
Scheme 2.

Generation of a Glycosyl Cation Equivalent by Flow Chemistry.
The electrochemical method for the activation of thioglycosides,6 and especially the in situ generation of glycosyl triflates (using tetrabutyl ammonium triflate as supporting electrolyte), has been developed into an iterative process enabling the one pot automated synthesis of oligosaccharides.7
In spite of these advances and the inference of a glycosyl cation stabilized by co-ordination to a disulfide with a lifetime of seconds at −78 °C, we are unaware of any other published spectroscopic detection of actual glycosyl oxocarbenium ions in organic solution at the present time. However, and although details are not yet available, it has been reported recently that a protected fructofuranosyl oxocarbenium ion and protected 2-deoxypyranosyl oxocarbenium ions have been generated in superacid media and characterized at low temperature by NMR spectroscopy.8–9 The details of this work are eagerly anticipated.
Evidence in support of the existence of conformationally equilibrating sialyl oxocarbenium ions on the activation of the corresponding thioglycosides with N-iodosuccinimide and triflic acid in the absence of an acceptor alcohol has been provided by the De Meo group; elimination to the glycal took place with loss of an axial or equatorial proton from C3 as determined with the help of deuterium labeled isotopomers.10
Computational Work on Glycosyl Oxocarbenium Ions
Satoh, Hüenberger, and their coworkers have conducted an extensive Quantum mechanical (QM) and molecular dynamics (MD) study of the tetra-O-methyl glucopyranosyl oxocarbenium ion in complex with the triflate anion making use of distance constraints to prevent collapse of the ion pair to the covalent triflates.11 They report that the conformation of the oxocarbenium ion is solvent dependent and that the predominant location of the counterion is conformation and thus solvent dependent. On this basis they formulate an alternative mechanism for glycosylation and a rationale for stereoselectivity titled ‘the conformer and counter ion distribution hypothesis’. According to this hypothesis the oxocarbenium ion adopts preferentially adopts a B2,5 conformation in more polar solvents with the counterion most closely associated with and sterically shielding the α-face; nucleophilic attack then takes place preferentially on the β-face (Scheme 3). In vacuum the 4H3 conformer is preferred, and because of the association of the counterion with the β-face of this conformer it affords α-selective reactions (Scheme 3).11 The solvent dependent conformational change of the oxocarbenium ion is attributed to increased stabilization of conformers with higher dipole moments by dielectric screening effects in the more polar solvents.
Scheme 3.

The Conformer and Counterion Distribution Hypothesis for Selectivity in the Tetra-O-glucopyranosyl Triflate Ion Pair.
Whitfield has discussed some of the difficulties associated with QM calculations of glycosylation reactions including apparent H-bonding of the incoming acceptor to oxygen atoms in the acceptor, 1,6-anhydro formation, and the formation of complexes between oxocarbenium ions and ether-type protecting groups.12 Preliminary attempts at the computational (QM) determination of transition states for the reaction of tetra-O-methyl α- and β-glucopyranosyl triflates with methanol have been reported.13 A lithium ion was included in the calculations at a mixed distance from the anomeric center so as to neutralize the charge on the departing triflate and prevent spontaneous collapse of any ion pairs.13 In spite of this artefact no ion pairs were identified as minima in these calculations which employed a very restricted number of explicit solvent molecules (CH2Cl2). All reactions studied were considered to be dissociative in so far as the C1-OTf bond was stretched to >2Å before any nucleophilic attack, nevertheless the nucleophile (MeOH) was found to be incorporated in all transition states located begging comparison with the mechanistic enzymologists’ exploded transition states discussed previously.1
Crich, Pratt and their coworkers computed (QM) transition states for the displacement of triflate from both anomers of 4,6-O-benzylidene-2,3-di-O-methyl gluco- and mannopyranosyl triflates by isopropanol.14 In both series displacement of the α-triflate was found to take place via a B2,5 conformation of the pyranosyl ring, while that of the β-triflates involved the a 4H3 conformation of the ring. In each case the C1-OTf and C1-O(H)iPr partial bonds at the transition states were 2.2Å or longer, again suggestive of exploded transition states (Fig. 1). Computed primary 13C kinetic isotope effects were in good agreement with experimentally determined values for the three of the four cases thereby providing strong support for loosely associative mechanisms in those cases. The single exception to the concordance between experimental and calculated KIE values was that of the displacement of the β-mannosyl triflate, which, with an experimental primary 13C KIE of 1.005 at 25 °C, is more consistent with a stepwise mechanism involving a discrete oxocarbenium ion intermediate.14 Bennet and coworkers studied the solvolysis of α-D-glucopyranosyl fluoride in hexafluoroisopropanol and on the basis of NMRdetermined KIE measurements suggest that the reaction either proceeds through a solvent-separated ion pair or via an SNi mechanism.15
Figure 1.

Computed Transition States for the Displacement of Triflate Anion by Isopropanol from 4,6-O-Benzylidene Protected α- and β-Mannopyranosyl Triflates
Pedersen, Bols and coworkers, following their discovery that a 4,6-O-(di-tert-butylsilylene) protected mannopyranosyl donor is β-selective on activation with N-iodosuccinimide and triflic acid at room temperature, computed (QM) the geometry of the corresponding oxocarbenium ion in the absence of any counterion.16 The minimum energy B2,5 conformation, which was favored in vacuum and in two implicit solvent models (CH2Cl2, MeCN) albeit by <1 kcal.mol−1, corresponds to the B2,5 conformer located earlier for 4,6-O-benzylidene protected mannosyl oxocarbenium ion by Whitfield17 (Fig. 2) and advocated by Crich and coworkers in explanation of their experimental results (vide supra).18–19
Figure 2.
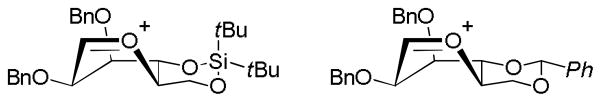
Low Energy B2,5 Conformers of the Silylene and Benzylidene Protected Mannosyl Oxocarbenium Ions
Contributing to the continued discussion on participation by remote ester groups in glycosylation reactions,20 Kalikanda and Li computed (QM) the relative energies of a series of 2-azido-2-deoxygalactopyranosyl oxocarbenium ions and their bridged counterparts derived by participation of a remote ester group. Not surprisingly, in view of the greater delocalization of the positive charge the bridged species uniformly were found to be the more stable.21 Stabilization by participating 3- and 4-O-acetyl groups were computed to be of comparable magnitude (≤ 1 kcal.mol−1 difference) while that afforded by a participating 6-O-acetate was found to be considerably smaller (Fig. 3).21
Figure 3.

Relative Stabilization of Galactosyl Oxocarbenium Ions by Participating Esters at the 3-, 4-, and 6-Positions.
In related experimental study, Yu and coworkers employed two peracetyl glucopyranosyl donors regioselectively trideuteriated in either the 2- or 4-O-acetyl groups to locate the origin of the bridge in the 3,6-di-O-acetyl-1,2,4-O-orthoacetyl-α-D-glucopyranose formed in low yield on activation. As the trideuteriomethyl group was retained in the product arising from use of the 4-O-(trideuterioacetyl) donor and not from its regioisomer at the 2-position, it was determined that the orthoester is formed through a mechanism involved initiation participation by the 4-O-ester followed by attack on the bridged cation by the O2 (Scheme 4).22 This experiment provides strong evidence for the possibility of remote participation from the 4-position at least for the case of the peracylated donors. 22
Scheme 4.
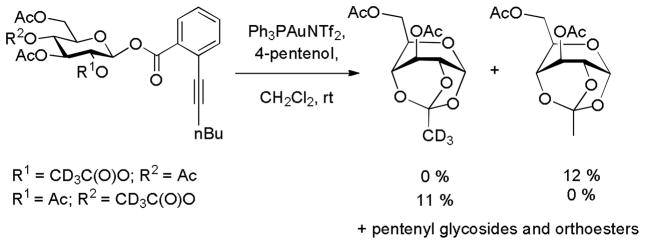
Formation of a Trideuterio Ortho Ester Supporting the Possibility of Remote Participation by Esters at the 4-Position in Peracylated Donors.
In addition to the earlier review by Whitfield,17 computational work on glycosylation reactions has been reviewed by Satoh and Kunada.23
Kinetic Studies
As part of a study on the regioselective activation of glycosyl acceptors by a diphenylborinic acid-derived catalyst, Taylor and coworkers investigated the kinetics of borinate-catalyzed glycosylation of methyl 6-O-(tert-butyldimethylsilyl)-α-D-glucopyranoside by acetobromoglucose promoted with silver oxide in acetonitrile. The formation of the observed β-glycoside was found to be first order in donor, acceptor and borinate, but zero order in silver oxide prompting them to advance direct SN2 displacement of the bromide by the acceptor-borinate complex at the phase boundary as the reaction mechanism (Scheme 5).24 Neighboring group participation was excluded from consideration on the grounds that the use of the alternative tetra-O-acetyl-β-D-glucopyranosyl chloride as donor resulted predominantly in 1,2-orthoester formation. 24
Scheme 5.

Kinetics and Mechanism of a Silver Oxide-Promoted Borinate-Catalyzed Glycosylation Reaction.
Tan and coworkers conducted a careful study of the methanol catalyzed spirocyclization of glycal epoxides leading to the preferential formation of the less stable of the two possible diastereomeric spiroketals. The results of a Hammett study strongly suggested that the reaction does not proceed with development of a high level of positive charge at the anomeric center in the transition state, consistent with an SN2-like mechanism. The reaction was found to be approximately second order in methanol and this, together with the information that the methanol reaction order was diminished for a comparable carbocyclic substrate lacking the ring oxygen, lead them to postulate a transition state involving hydrogen bonding of methanol to both the epoxide and the ring oxygens (Scheme 6).25
Scheme 6.

Hydrogen Bonding Catalysis in the Intramolecular Ring Opening of a Glycal Epoxide.
As discussed above, Crich, Pratt and their coworkers conducted an experimental and computational study of the 13C primary kinetic isotope effect on the displacement of α-triflates from 4,6-O-benzylidene protected gluco- and mannopyranosides by isopropanol (Fig. 1).14 In this manner the formation of benzylidene protected β-gluco- and mannopyranosides was found to take place via loose SN2 type transition states from the α-triflates. α-Glucopyranoside formation was found to occur in a similar manner from the less stable β-glucopyranosyl triflate thereby necessitating rapid interconversion of the α- and β-triflates in a Curtin-Hammett kinetic scheme. Formation of the α-mannoside was the exception to the rule in so far as the experimentally determined 13C KIE was not consistent with that computed for a bimolecular displacement but rather with a stepwise dissociative mechanism and the involvement of a discrete oxocarbenium ion intermediate. 14 Crich and coworkers subsequently developed the cyclization of a 2-O-(2-trimethylsilylmethyl)allyl ether onto the anomeric position of a mannosyl donor as a unimolecular clock reaction with which to investigate the molecularity of intermolecular glycosylation reactions.19 In this manner, and fully consistent with the above-mentioned KIE experiments, it was determined that the benzylidene-directed formation of β-mannosides is dependent on the concentration of the acceptor whilst that of the α-anomer is much less so indicating that the α- and β-anomers are formed by SN1 and SN2-like mechanisms, respectively (Scheme 7).
Scheme 7.

Use of a Cation-Clock Reaction to Investigate the Relative Kinetics of Glycosylation.
The unexpected partial formation of the trans-fused tricyclic product from the cyclization reaction prompted the authors to suggest19 that any intermediate oxocarbenium ion must be able to access the B2,5 conformation consistent with an earlier suggestion from the same laboratory18 and a subsequent one from the Bols group16 (Scheme 3).
In an attempt to probe the influence of donor protecting groups on sialidation reactions Crich and coworkers developed and extended a concept reported earlier by Denekamp and Sandlers26–27 for simple glycosyl donors.28 In this method the energy required to fragment a series of glycosyl donors to the corresponding oxocarbenium ions in a mass spectrometer is related to the relative destabilization of the oxocarbenium ions by the different protecting group systems deployed. In this manner, by determining the minimum cone energies required for fragmentation in a TOF-ESI mass spectrometer (Fig. 4), it was determined that trans-fused oxazolidinones, and cyclic carbonates in the KDN series, are strongly electron-withdrawing. The consequent retardation of SN1-type mechanisms by the presence of such protecting groups favors associative processes leading to the high selectivities they typically exhibit.28
Figure 4.
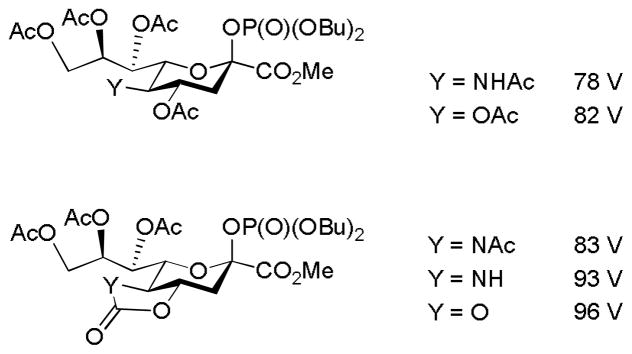
Minimum Mass Spectral Fragmentation Energies for a Series of Sialyl Phosphates Vary as a Function of Protecting Group.
With regard to the effect of concentration on the rate and stereoselectivity of glycosylation reactions, Kononov and coworkers have continued to investigate their concept of “supramers” (supramolecular isomers) and their influence on glycosylation reactions.29 According to this concept the degree and nature of association of reactants, solvents (and products) in a glycosylation reaction is concentration dependent and changes throughout the course of a reaction as the composition of the reaction mixture evolves; different supramers exhibit different reactivity and selectivity. This type of supramer effect is maximized with the type of sialyl donors used to illustrate it, because of their propensity toward hydrogen bonding. Importantly, it is hypothesized that the dependence of supramer composition on concentration exhibits critical points and that different supramers influence reaction rate and selectivity differently leading the authors to the conclusion that the manner of concentration dependence of stereoselectivity for a given glycosylation reaction may vary as a function of the concentration range studied. 29 At the very least the observations reported in this paper, together with the increased evidence for associative glycosylation reactions in general, underline the importance of the careful recording of reaction concentrations when reporting glycosylation reactions, caution against overgeneralized general reaction protocols, and stress the need for strict adherence to reported conditions when applying literature protocols. The influence of acceptor concentration in the stereoselectivigty of sialidation reactions has also been commented by Xing and coworkers.30
The DMTST-promoted rearrangement of S-(γ-hydroxypropyl) thioglycosides to the corresponding (γ-methyldisulfanylpropyl) glycosides has been demonstrated by crossover experiments to proceed substantially by an intramolecular pathway (Scheme 8). With non-participating protecting groups in the donor the reaction is considered to take place via an SNi mechanism in dichloromethane and via a glycosyl nitrilium ion when carried out in the presence of propionitrile.31 As noted above evidence for the intervention of a SNi mechanism in the solvolysis of α-D-glucopyranosyl fluoride by hexafluoroisopropanol has been provided from KIE measurements.15
Scheme 8.

Crossover Experiment Demonstrating the Intramolecular Rearrangement of S-(γ-hydroxypropyl) thioglycosides.
Echoing the earlier sentiments of Sinnott,32–33 Cumpstey has emphasized the limitations associated with the over-reliance on product-like stereoelectronic effects in the capture of glycosyl oxocarbenium ions for which early transition states are more appropriate.34
Protecting groups and influence of conformation on reactivity
It is becoming increasingly apparent that protecting groups can significantly affect the reactivity through their influence on the conformation of the donor and/or the incipient glycosyl oxocarbenium ion in addition to their more widely appreciated35 disarming effects arising from their electron-withdrawing ability. Extreme examples of this phenomenon are to be found in the super-armed donors reported by the Bols36 and Yamada37–38 groups employing multiple silyl ethers, whose high degree of reactivity is consistent with the enforcement of conformations containing multiple axial or pseudoaxial C-O known39–41 to be capable of stabilizing oxocarbenium ions while at the same time providing differential steric shielding of the α- and β-faces. Albeit not strictly a protecting group, the presence of the ester function in mannuronic acid donors has been shown in recently to support the inverted 1C4 conformation of covalent α-triflates. On the basis of NMR chemical shift arguments, it is hypothesized that this effect arises because of the ability of the axial ester group to stabilize the equatorial triflate through a non-classical hydrogen bond to the somewhat electron-deficient anomeric hydrogen atom (Fig. 5).42 Advances in the synthesis of the 1,2-cis-glycosides, including those of the mannuronate esters, have been reviewed recently.43–44
Figure 5.
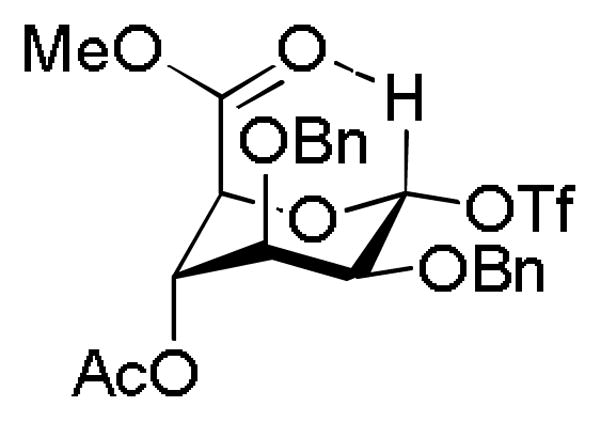
Proposed Non-Classical H-Bond Stabilizing the 1C4 Conformation of α-Glycosyl Triflates in the Mannuronic Acid Series.
The well-known disarming effect of a trans-fused benzylidene ring, attributed by Fraser-Reid to torsional effects,45–46 was rationalized by Bols as a combination of torsional effects and electronic disarming effects with the latter predominating and arising from the imposition of the tg conformation.47 Crich and coworkers, adapting the Bols method to the galactopyranose series, revealed the cis-fused alkylidene ring system also to be disarming pointing to the importance of a torsional component. cis-Fused model systems carrying methoxy substituents at the 6-position reaffirmed the strongly disarming character of the C6-O6 bond when locked in the tg conformation (Fig. 6).48 Pedersen and coworkers found a bicyclic 6-carbamannosyl donor to exhibit β-selectivity approaching that displayed by the now classical 4,6-O-benzylidene protected mannosyl donors, prompting them to suggest that the benzylidene effect is mainly torsional in origin and that the electronic component was previously overestimated.49 However, it was subsequently pointed out by Crich and coworkers that the replacement of an acetal oxygen in an alkylidene protected 4,6-diol by a methylene group, as in the Pedersen model, or even more so by a methoxymethylene group (Fig. 6) introduces numerous new torsional interactions. Consequently, such systems are more susceptible to changes in ring conformation than the acetals they model and therefore overestimate the torsional component of the benzylidene effect while underestimating the electronic component.48 A series of Wong-type competition glycosylations with arabinofuranosyl thioglycosides revealed the β-directing 3,5-O-di(tert-butyl) silylene acetal introduced earlier by the Boons50 and Crich51 laboratories to be strongly disarming consistent with the approximate tg conformation it imposes on the C4-C5 bond.52
Figure 6.

The Stereochemical Component of the Disarming Influence of the C6-O6 Bond. [For the purposes of this scheme the conformational descriptors gt, gg, and tg employ O6 as the reference point and not C7]
To escape the torsional component of the 4,6-acetals and their carba-models, Kancharla and Crich studied the reactivity of a diastereomeric pair of monocyclic sialyl donors differing only in configuration at the 7-position.53 The natural 7S-isomer was found by NMR spectroscopy to adopt predominantly the gg-conformation of its C6-C7 bond and to be activated more rapidly than its 7R epimer which displays the gt conformation (Scheme 9). The gg conformation with the C7-O7 bond perpendicularly above the mean plane of the pyranose ring and optimally placed to stabilize any developing positive charge at the anomeric center was therefore found to be the most reactive conformation consistent with the observations in the conformationally locked systems (Fig. 6). The authors speculated that restriction of the conformation of the side chain in other monocyclic systems by adjacent protecting groups may play a role in controlling the reactivity and selectivity of glycosyl donors, and that, through the correct placement of suitable hydrogen bonding networks, that glycosidase and glycosyl transferase enzymes might also adopt such strategies.53
Scheme 9.

Dependence of Sialyl Donor Reactivity on the Configuration at C7. [For the purposes of this scheme the conformational descriptors gt, gg, and tg employ O6 as the reference point and not C7]
In highly α-selective conformationally-restricted sialyl donors such as the ones illustrated in Scheme 9, it has been demonstrated mass spectroscopically that the trans-fused oxazolidinone ring functions as a powerfully electron-withdrawing group that destabilizes the corresponding glycosyl oxocarbenium ion. Selectivity, therefore, is likely achieved via an associative SN2-like mechanism via displacement of either acetonitrile from a covalent nitrilium ion or a triflate anion from a glycosyl triflate.28 The use of the oxazolidinone-protected sialyl donors has recently enabled highly α-selective sialidation reactions to be conducted on polymer-supported acceptors.54
In an important advance Demchenko and coworkers demonstrated that remote picolinate esters and picolinyl ethers are able to direct glycosylation to the cis-face through hydrogen bonding to the incoming acceptor alcohol.55 It was found, for example, that a picolate ester or a picolinyl ether at the 3-position of a glucopyranosyl donor resulted in significantly higher β-selectivity than the corresponding 3-O-benzyl ether. Similarly, a 6-O-picolyl ester directs glucosylation to the β-face, while 4-O-picolyl ester results in α-glucopyranosylation. In the galactopyranose series the use of a a 4-O-picolyl ester results in very high selectivity for the formation of the β-anomeric product.55 The concept has been extended to the preparation of β-arabinofuranosides through the use of a 5-O-(2-quinolinecarboxylate) functionalized donor. 56 In the mannopyranosyl series optimal results for β-glycoside formation with primary acceptors were found with a monocyclic 3-O-picolyl donor, while secondary acceptors functioned best with a donor carrying both the 4,6-O-benzylidene group and a 3-O-picolyl ester (Scheme 10).57 The directing effect of the 3-O-picolyl ester in the benzylidene-protected donor is stark contrast to the corresponding 3-O-benzoate, which is strongly α-directing although for reasons that remain unclear.58–59 As these β-mannosylations can be conducted at room temperature, they present a significant advance over the classical benzylidene-directed methods.43,60 The manner in which remote esters direct glycosylation reactions has been reviewed by Nifantiev and co-workers.61
Scheme 10.

Remote Picolate Ester-Directed β-Mannosylation.
Counterion and Additive Effects
It is widely appreciated that counter ions introduced into glycosylation reactions as essential components of promoters or as additives influence reaction outcome. The influence of additives such as tetraalkylammonium bromides4,62 and tetramethylurea63 on glycosylation reactions has been studied for many years with covalent intermediates isolated and well-characterized in some cases. 64 Recent examples on the study of additives include the addition of sulfides and sulfoxides, which afford stable glycosyl sulfonium and oxysulfonium salts,30,65–66 and of dimethylformamide and other secondary amides, and even catalytic oxindoles,67 leading to the formation of glycosyl imidates.68–69 The additives area has been reviewed recently and as such we will not comment further on it here,70 but rather we will limit ourselves to counter ions.
The addition of anions into a glycosylation reaction mixture, either in the form of ionic salt-based promoters, in the form of triflic anhydride in the sulfoxide-type glycosylation reactions, or in the form of silyl triflate-type Lewis acidic promoters necessarily influences the reaction outcome. Such anions are intimately involved in glycosylation reaction mechanisms being a key component of the central ion pair/covalent equilibria (Scheme 1). Because of the widespread use of triflate-containing promoters and the spectroscopic identification of covalent glycosyl triflates,42,71–72 most discussions in recent years focus on the possibility of the involvement of glycosyl triflates. This is especially the case in the benzylidenedirected β-mannosylation reaction for which α-mannosyl triflates were identified as resting intermediates60,71–73 and as reactants in SN2-like displacements.14,19 It is reasonable to expect that for a given glycosyl oxocarbenium ion, the tightness of ion pairs with counter ions will vary inversely with the stability of the counter ion. Likewise, covalently bound intermediates will be stable to higher temperatures when the counter ion-generated on fragmentation is less stable. Indeed, glycosyl bromides and glycosyl iodides are frequently stable isolable substances at room temperature in the absence of moisture (pKa of HBr and HI: −9, −9.5, respectively).74 In this context it is pertinent to note that the pKa of triflic acid, one of the strongest acids known, is ~−1274 and that, as covalently bound glycosyl triflates have been amply demonstrated,34–35,37–38 any anion whose conjugate acid is less acidic than triflic acid will form observable covalent glycosyl adducts unless prevented from doing so by pervasive steric factors. Indeed, silver perchlorate is (HClO4 pKa ~−10)75 has been known since 1970 to form covalent glycosyl perchlorates on reaction with 3,4,6-tri-O-acetyl-2-chloro-2-deoxy gluco- and mannopyranosyl chloride. These crystalline glycosyl perchlorates, for which NMR spectra were recorded, were isolated by crystallization and found to decompose slowly at room temperature but to be stable in solution (SO2, ether and toluene) below 0 °C (Scheme 11).76 By way of comparison per-O-acetyl glucopyranosyl triflates were found by VT-NMR to decompose at ~ 0 °C,71 suggesting the glycosyl perchlorates to be at least as stable as comparable triflates. It is also pertinent that methyl triflate is a better methylating agent than methyl perchlorate, suggesting that the Me-OTf bond is weaker than the Me-OClO3 bond.77 It follows that the recently described β-mannosylation employing a 4,6-O-(di-tert-butylsilylene) protected mannosyl thioglycoside16 likely involves a covalent mannosyl perchlorate when the activation is achieved with HClO4 and NIS with reaction proceeding through a CIP rather than a naked glycsoyl oxocarbenium ion. Similarly, it is highly likely that a report on β-selective glucosylations employing a per-O-benzyl glucopyranosyl diphenylphosphate as donor in 1M solutions of lithium perchlorate in dichloromethane in the absence of other promoters involve the formation of a glucosyl perchlorate.78 Demchenko and coworkers have reported a relatively rare example of a carefully conducted comparison of the counterion effect, conducted with a 2-O-benzyl-3,4,6-tri-O-benzoyl-β-D-glucopyranosyl thiobenzoxazole as donor in dichloromethane at room temperature.79 The use of silver perchlorate or silver triflate as promoter gave highly α-selective reactions, whereas the employment of silver tetrafluoroborate or silver hexafluorophosphate resulted in far less selective reactions. Presumably, the α-selectivity observed with the perchlorate and triflate salts is the result of the reaction of transient β-glycosyl perchlorates and triflates that are in rapid equilibrium with the more stable α-anomers.79 The tetrafluoroborate and hexafluorophosphate anions on the other hand apparently do not participate in covalent adducts or tight ion pairs which, on the basis of previous work, would have been expected to yield glycosyl fluorides.80
Scheme 1.

A General Glycosylation Mechanism.
Scheme 11.

Covalent Glycosyl Perchlorates and their Stabilities and Reactivities
The question of the possibility of the involvement of glycosyl triflates in reactions in which thioglycosides are activated in the presence of acceptor alcohols by systems using only catalytic amounts of triflic acid is an interesting one that turns on the relative nucleophilicity of the triflate anion and alcohols toward oxocarbenium ions.16 Notwithstanding recent criticisms of the hard and soft acid and base principle,81 it is reasonable to suggest that the triflate anion might outcompete the softer alcohol for reaction with the hard oxocarbenium leading to the formation of the glycosyl triflate. Support for this argument can be drawn from the recent QM computational determination that triflate anion has a higher intrinsic reactivity index than methanol.82
The importance of the correct choice of counter-ion in glycosylation reactions is brought home in a striking manner by the recent solution of the 2-deoxy-β-glycopyranoside problem83 by the Bennett group.84–85 Thus, reaction of otherwise perbenzylated 2-deoxyglycopyranoses with potassium hexamethyldisilazide with toluenesulfonic anhydride in THF at −78 °C was shown spectroscopically to afford the α-glycosyl toluenesulfonates, related to the ones previously prepared by Schuerch and coworkers albeit by a more cumbersome method.86 Subsequent treatment of the covalent toluenesulfonates with the potassium alkoxides of the desired acceptors resulted in the formation of the β-glycosides in excellent yield and selectivity. The sequence functions for both 2-deoxy and 2,6-dideoxy donors and is compatible with the use of phenols, primary and secondary alcohols as acceptors (Scheme 12).85 It is remarkable that each of the long-touted three major problems in glycosylation – β-mannopyranosylation, 2-deoxy-β-glycopyranosylation, and α-sialidation – have now each been solved by methods that likely involve the SN2-like displacement of covalent glycosyl intermediates.
Scheme 12.

Stereoselective Synthesis of a2-Deoxy-β-glycopyranoside via a α-Glycosyl Toluenesulfonate.
Schmidt has developed a method for the synthesis of β-gluco- and β-galactopyranosides in the absence of participating groups from trichloroacetimidates that depends on the addition of either phenylboron difluoride or diphenylboron fluoride that is predicated on the formation of a borate complex with the acceptor. This complex then serves to activate the imidate and deliver the acceptor concomitantly through an eight-membered cyclic transition state (Scheme 13).87 Substituting thiols for alcohols in this process gives rise to thioglycosides with inversion of configuration.88 The use of binol-based chiral phosphoric acids as promoters for glycosylations with trichloroacetimidate-type donors enables enantioselectivity in couplings to racemic acceptors. A twelve-membered cyclic transition state is invoked in which the phosphoric acid hydrogen simultaneously bonds to the donor and acceptor and preorganizes the latter for an SN2-type displacement.89
Scheme 13.

β-Glycopyranoside Synthesis from Trichloroacetimidates with the Aid of a Lewis-Acidic Boron Complex.-
A more complex variant on this theme relies on the co-operation of two organocatalysts – a thiourea and diaryl phosphoric acid, which are considered to assemble with the donor and acceptor into a four-component scaffold that affords β-selective glycosylations from armed trichloroacetimidates (Scheme 14).90 The same thiourea has also been employed as a catalyst for the selective addition of alcohols to glycals resulting in the formation of 2-deoxy-α-glycosides, although in this instance it is considered to function merely as a mild protic acid.91–92 In related work chiral binol-derived phosphoric acid catalysts have been employed to promote cyclization of 1-(γ-hydroxypropyl)glycals and their homologs to spiroketals. It was demonstrated that the anomeric selectivity of the reaction is dependent on the chirality of the catalyst, and cyclic hydrogen bonded transition state was invoked to explain the phenomenon.93
Scheme 14.

Co-operative Catalysis in the Synthesis of β-Glycopyranosides.
Several groups have published on the development of photocatalytic methods for glycosylation. Thus, both the Bowers and Ragains laboratories have employed ruthenium and iridium-based photocatalysts to activate electron-rich thioglycosides and selenoglycosides for coupling to alcohols under irradiation by a CFL. Both laboratories employed either carbon tetrabromide or bromotrichloromethane as an auxiliary oxidant and the exact mechanisms these reactions remain unclear.94–95 In a related process it has also been demonstrated that anomeric hemiacetals may be converted to glycosyl bromides by white light irradiation in the presence of a ruthenium-based photocatalyst and carbon tetrabromide.96 The Ragains group further demonstrated that diphenyl diselenide can serve as a simple organic photocatalyst for the activation of selenoglycosides in the presence of carbon tetrabromide. In the this case the mechanism is considered to involve homolytic abstraction of a bromine atom from the tetrabromide by a phenyl selenyl radical to give phenylselenyl bromide, which then serves as the promotor in a more traditional mechanism.95 Although not catalytic, the Crich and Toshima laboratories also reported methods for the white light photochemical activation of selenoglycosides and thioglycosides employing, respectively, N-methylquinolinium hexfluorophosphate and DDQ as the photoactivated oxidants.97–98
Non-Traditional Methods
A second elegant solution to the 2-deoxy-β-glycopyranoside problem has been advanced based on the alkylation of anomeric hemiacetals with secondary electrophiles. Related to earlier work on anomeric alkylation by the Schuerch,99 Schmidt,100 and Kovac laboratories101 the method relies on the displacement of triflate esters with anomeric sodium alkoxides in dioxane in the presence of 18-crown-6 (Scheme 15).102
Scheme 15.

2-Deoxy-β-glycoside Synthesis by Anomeric Alkylation.
The reaction of alcohols with cyclic π-allyl palladium complexes derived from 6-tert-butoxycarboxy-2H-pyran-3(6H)-ones to give unsaturated glycoside with very high diastereoselectivity developed in the O’Doherty laboratory has evolved to the stage at which it must be considered competitive with classical glycosylation methods, certainly for the unnatural enantiomers and even in some cases for the natural ones. The essential 6-tert-butoxycarbonyl-dihydropyran-2-one blocks building are rapidly assembled in either enantiomeric form by Noyori reduction of 2-acylfurans followed by Achmatowicz rearrangement and installation of the Boc group.103 The method is illustrated (Scheme 16) by the synthesis of the lipophilic trisaccharide Cleistrioside 5 from one such common building block.104 In this sequence the α-selective quenching the of the π-allyl palladium complex is followed by stereoselective Luche reduction of the enone and subsequent osmium tetroxide-mediated cis-dihydroxylation to give a α-L-rhamnopyranoside in high yield and selectivity. Iterations of the same basic theme with modifications in the protecting groups steps eventually afford the trisaccharide.
Scheme 16.

De Novo Synthesis of a Complex Trisaccharide.
Conclusion
In the brief few years since we last opined à propos of glycosyl oxocarbenium ions, considerable evidence has been presented in support of their existence as transient intermediates in some glycosylation reactions. Preliminary spectroscopic evidence for the existence of protected fructofuranosyl and 2-deoxyglycopyranosyl oxocarbenium ions at low temperature in superacid-media has now been presented. It remains true, however, that unequivocal spectroscopic evidence for such a glycosyl oxocarbenium ion in organic solution has yet to be presented. It also can be said that a substantial and growing body of work supports the importance of counter ions and the role of contact ion pairs or loosely concerted associative transition states in many glycosylation reactions. Most notably in this respect, solutions to the long-standing problems of β-mannosylation, α-sialidation, and 2-deoxy-β-glycosylation have been located in which the counter ion, either alone or in combination with strongly electron-withdrawing and oxocarbenium ion destabilizing protecting groups, plays a major role. The concept of donor-acceptor hydrogen bonding as a stereodirecting force in glycosylation has matured into an experimental fact and notably provides a second solution to the β-mannosylation problem. The general glycosylation mechanism of Scheme 1 remains valid and, while evidence has been presented for examples of simple glycosylation reactions that approach the two extremes of the SN1 and SN2 mechanisms, it is likely that most such reactions take place somewhere along the continuum that separates the two.
Highlights.
Mechanistic and spectroscopic evidence for glycosyl oxocarbenium ions
Computational work on glycosyl oxocarbenium ions
Kinetic studies on glycosylation reactions
Counterion and additive effects
Non-traditional methods
Acknowledgments
DC thanks the NIH (GM62160) for continued and generous support of his programs in glycochemistry.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bohé L, Crich D. CR Chimie. 2011;14:3–16. [Google Scholar]
- 2.Rhind-Tutt AJ, Vernon CA. J Chem Soc. 1960:4637–4644. [Google Scholar]
- 3.Lucas TJ, Schuerch C. Carbohydr Res. 1975;39:39–45. doi: 10.1016/s0008-6215(00)82649-x. [DOI] [PubMed] [Google Scholar]
- 4.Lemieux RU, Hendriks KB, Stick RV, James K. J Am Chem Soc. 1975;97:4056–4062. [Google Scholar]
- 5.Saito K, Ueoka K, Matsumoto K, Suga S, Nokami T, Yoshida J-i. Angew Chem Int Ed. 2011;50:5153–5156. doi: 10.1002/anie.201100854. [DOI] [PubMed] [Google Scholar]
- 6.Nokami T, Saito K, Yoshida J-i. Carbohydr Res. 2012;363:1–6. doi: 10.1016/j.carres.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 7.Nokami T, Hayashi R, Saigusa Y, Shimizu A, Liu C-Y, Mong K-KT, Yoshida J-i. Org Lett. 2013;15:4520–4523. doi: 10.1021/ol402034g. [DOI] [PubMed] [Google Scholar]
- 8.Akien GR, Subramaniam B. 245th ACS National Meeting & Exposition,; New Orleans. 2013; p. CARB-105. [Google Scholar]
- 9.Akien GR, Subramaniam B. 247th ACS National Meeting & Exhibition; Dallas. 2014; p. CARB-96. [Google Scholar]
- 10.De Meo C, Wallace CE, Geringer SA. Org Lett. 2014;14:2676–2679. doi: 10.1021/ol500917k. [DOI] [PubMed] [Google Scholar]
- 11.Satoh H, Hansen HS, Manabe S, van Gunsteren WF, Hunenberger PH. J Chem Theor Comput. 2010;6:1783–1797. doi: 10.1021/ct1001347. [DOI] [PubMed] [Google Scholar]
- 12.Whitfield DM. Carbohydr Res. 2012;356:191–195. doi: 10.1016/j.carres.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Whitfield DM. Carbohydr Res. 2012;356:180–190. doi: 10.1016/j.carres.2012.03.040. [DOI] [PubMed] [Google Scholar]
- 14.Huang M, Garrett GE, Birlirakis N, Bohé L, Pratt DA, Crich D. Nat Chem. 2012;4:663–667. doi: 10.1038/nchem.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan J, Tang A, Bennet AJ. J Am Chem Soc. 2012;134:1212–1220. doi: 10.1021/ja209339j. [DOI] [PubMed] [Google Scholar]
- 16.Heuckendorff M, Bendix J, Pedersen CM, Bols M. Org Lett. 2014;16:1116–1119. doi: 10.1021/ol403722f. [DOI] [PubMed] [Google Scholar]
- 17.Whitfield DM. Adv Carbohydr Chem Biochem. 2009;62:83–159. doi: 10.1016/S0065-2318(09)00004-3. [DOI] [PubMed] [Google Scholar]
- 18.Moumé-Pymbock M, Crich D. J Org Chem. 2012;77:8905–8912. doi: 10.1021/jo3011655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang M, Retailleau P, Bohé L, Crich D. J Am Chem Soc. 2012;134:14746–14749. doi: 10.1021/ja307266n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Komarova BS, Orekhova MV, Tsvetkov YE, Nifantiev NE. Carbohydr Res. 2014;384:70–86. doi: 10.1016/j.carres.2013.11.016. [DOI] [PubMed] [Google Scholar]
- 21.Kalikanda J, Li Z. J Org Chem. 2011;76:5207–5218. doi: 10.1021/jo1025157. [DOI] [PubMed] [Google Scholar]
- 22.Ma Y, Lian G, Li Y, Yu B. Chem Commun. 2011;47:7515–7517. doi: 10.1039/c1cc11680k. [DOI] [PubMed] [Google Scholar]
- 23.Satoh H, Nukada T. Trends in Glycosci Glycobiol. 2014;26:11–27. [Google Scholar]
- 24.Gouliaras C, Lee D, Chan L, Taylor MS. J Am Chem Soc. 2011;133:13926–13929. doi: 10.1021/ja2062715. [DOI] [PubMed] [Google Scholar]
- 25.Wurst JM, Liu G, Tan DS. J Am Chem Soc. 2011;133:7916–7925. doi: 10.1021/ja201249c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Denekamp C, Sandlers Y. J Mass Spectrom. 2005;40:765–771. doi: 10.1002/jms.848. [DOI] [PubMed] [Google Scholar]
- 27.Denekamp C, Sandlers Y. J Mass Spectrom. 2005;40:1055–1063. doi: 10.1002/jms.880. [DOI] [PubMed] [Google Scholar]
- 28.Kancharla PK, Navuluri C, Crich D. Angew Chem Int Ed. 2012;51:11105–11109. doi: 10.1002/anie.201204400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kononov LO, Malysheva NN, Orlova AV, Zinin AI, Laptinskaya TV, Kononova EG, Kolotyrkina NG. Eur J Org Chem. 2012:1926–1934. [Google Scholar]
- 30.Zhang X-t, Gu Z-y, Xing G-w. Carbohydr Res. 2014;388:1–7. doi: 10.1016/j.carres.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 31.Hoang KLM, Bai Y, Ge X, Liu XW. J Org Chem. 2013;78:5196–5204. doi: 10.1021/jo400020q. [DOI] [PubMed] [Google Scholar]
- 32.Sinnott ML. In: The Anomeric Effect and Associated Stereoelectronic Effects. Thatcher GRJ, editor. Vol. 539. ACS; Washington: 1993. pp. 97–113. [Google Scholar]
- 33.Sinnott ML. Adv Phys Org Chem. 1988;24:113–204. [Google Scholar]
- 34.Cumpstey I. Org Biomol Chem. 2012;10:2503–2508. doi: 10.1039/c2ob06696c. [DOI] [PubMed] [Google Scholar]
- 35.Fraser-Reid B, Lopez C. Top Curr Chem. 2011;301:1–29. doi: 10.1007/128_2010_105. [DOI] [PubMed] [Google Scholar]
- 36.Jensen HH, Pedersen CM, Bols M. Chem Eur, J. 2007;13:7576–7582. doi: 10.1002/chem.200700947. [DOI] [PubMed] [Google Scholar]
- 37.Okada Y, Nagata O, Taira M, Yamada H. Org Lett. 2007;9:2755–2758. doi: 10.1021/ol070720b. [DOI] [PubMed] [Google Scholar]
- 38.Okada Y, Mukae T, Okajimia K, Taira M, Fujita M, Yamada H. Org Lett. 2007;9:1573–1576. doi: 10.1021/ol070427b. [DOI] [PubMed] [Google Scholar]
- 39.Smith DM, Woerpel KA. Org Biomol Chem. 2006;4:1195–1201. doi: 10.1039/b600056h. [DOI] [PubMed] [Google Scholar]
- 40.Walvoort MTC, Dinkelar J, van den Bos LJ, Lodder G, Overklee] HS, Codée JDC, van der Marel GA. Carbohydr Res. 2010;345:1252–1263. doi: 10.1016/j.carres.2010.02.027. [DOI] [PubMed] [Google Scholar]
- 41.Jensen HH, Bols M. Acc Chem Res. 2006;39:259–265. doi: 10.1021/ar050189p. [DOI] [PubMed] [Google Scholar]
- 42.Rönols J, Walvoort MTC, van der Marel GA, Codée JDC, Widmalm G. Org Biomol Chem. 2013;11:8127–8134. doi: 10.1039/c3ob41747f. [DOI] [PubMed] [Google Scholar]
- 43.Christina AE, van der Marel GA, Codée JDC. In: Modern Synthetic Methods in Catbohydrate Chemistry; From Monosaccharides to Complex Glycoconjugates. Werz DB, Vidal S, editors. Wiley; Weinheim: 2014. pp. 97–124. [Google Scholar]
- 44.Crotti S, Adamo R. Curr Org Synth. 2013:501–524. [Google Scholar]
- 45.Fraser-Reid B, Wu ZC, Andrews W, Skowronski E. J Am Chem Soc. 1991;113:1434–1435. [Google Scholar]
- 46.Andrews CW, Rodebaugh R, Fraser-Reid B. J Org Chem. 1996;61:5280–5289. doi: 10.1021/jo961115h. [DOI] [PubMed] [Google Scholar]
- 47.Jensen HH, Nordstrom M, Bols M. J Am Chem Soc. 2004;126:9205–9213. doi: 10.1021/ja047578j. [DOI] [PubMed] [Google Scholar]
- 48.Moumé-Pymbock M, Furukawa T, Mondal S, Crich D. J Am Chem Soc. 2013;135:14249–14255. doi: 10.1021/ja405588x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gylling Frihed T, Walvoort MTC, Codée JDC, van der Marel GA, Bols M, Pedersen CM. J Org Chem. 2013;78:2191–2205. doi: 10.1021/jo302455d. [DOI] [PubMed] [Google Scholar]
- 50.Zhu X, Kawatkar SP, Rao Y, Boons GJ. J Am Chem Soc. 2006;128:11948–11957. doi: 10.1021/ja0629817. [DOI] [PubMed] [Google Scholar]
- 51.Crich D, Pedersen CM, Bowers AA, Wink DJ. J Org Chem. 2007;72:1553–1565. doi: 10.1021/jo061440x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liang XY, Bin HC, Yang JS. Org Lett. 2013;15:2834–2837. doi: 10.1021/ol401166x. [DOI] [PubMed] [Google Scholar]
- 53.Kancharla PK, Crich D. J Am Chem Soc. 2013;135:18999–19007. doi: 10.1021/ja410683y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Corcilius L, Payne RJ. Org Lett. 2013;15:5794–5797. doi: 10.1021/ol402845e. [DOI] [PubMed] [Google Scholar]
- 55.Yasomanee JP, Demchenko AV. J Am Chem Soc. 2012;134:20097–20102. doi: 10.1021/ja307355n. [DOI] [PubMed] [Google Scholar]
- 56.Liu QW, Bin HC, Yang JS. Org Lett. 2013;15:3974–3977. doi: 10.1021/ol401755e. [DOI] [PubMed] [Google Scholar]
- 57.Pistorio SG, Yasomanee JP, Demchenko AV. Org Lett. 2014;16:716–719. doi: 10.1021/ol403396j. [DOI] [PubMed] [Google Scholar]
- 58.Crich D, Cai W, Dai Z. J Org Chem. 2000;65:1291–1297. doi: 10.1021/jo9910482. [DOI] [PubMed] [Google Scholar]
- 59.Crich D, Hu T, Cai F. J Org Chem. 2008;73:8942–8953. doi: 10.1021/jo801630m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crich D. Acc Chem Res. 2010;43:1144–1153. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- 61.Komarova BS, Ustyuzhania NE, Tsvetkov YE, Nifantiev NE. In: Modern Synthetic Methods in Catbohydrate Chemistry; From Monosaccharides to Complex Glycoconjugates. Werz DB, Vidal S, editors. Wiley; Weinheim: 2014. pp. 125–160. [Google Scholar]
- 62.Kaeothip S, Yasomanee JP, Demchenko AV. J Org Chem. 2012;77:291–299. doi: 10.1021/jo2019174. [DOI] [PubMed] [Google Scholar]
- 63.Haines AH. Adv Carbohydr Chem Biochem. 1976;33:11–109. [Google Scholar]
- 64.Bock K, Guzman JFB, Refn S. Carbohydr Res. 1992;232:353–357. [Google Scholar]
- 65.Nokami T, Nozaki Y, Saigusa Y, Shibuya A, Manabe S, Ito Y, Yoshida J-i. Org Lett. 2011;13:1544–1547. doi: 10.1021/ol200242u. [DOI] [PubMed] [Google Scholar]
- 66.Nokami T. Trends in Glycosci Glycobiol. 2012;24:203–214. [Google Scholar]
- 67.Nigudkar SS, Stine KJ, Demchenko AV. J Am Chem Soc. 2014;136:921–923. doi: 10.1021/ja411746a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lu SR, Lai YH, Chen JH, Liu CY, Mong KKT. Angew Chem Int Ed. 2011;50:7315–7320. doi: 10.1002/anie.201100076. [DOI] [PubMed] [Google Scholar]
- 69.Ingle AB, Chao C-S, Hung W-c, Mong K-KT. Org Lett. 2013;15:5290–5293. doi: 10.1021/ol402519c. [DOI] [PubMed] [Google Scholar]
- 70.Mulani SK, Hung WC, Ingle AB, Shiau KS, Mong KKT. Org Biomol Chem. 2014;12:1184–1197. doi: 10.1039/c3ob42129e. [DOI] [PubMed] [Google Scholar]
- 71.Crich D, Sun S. J Am Chem Soc. 1997;119:11217–11223. [Google Scholar]
- 72.Walvoort MTC, van der Marel GA, Overkleeft HS, Codée JDC. Chem Sci. 2013;4:897–906. [Google Scholar]
- 73.Aubry S, Sasaki K, Sharma I, Crich D. Top Curr Chem. 2011;301:141–188. doi: 10.1007/128_2010_102. [DOI] [PubMed] [Google Scholar]
- 74.Raamat E, Kaupmees K, Ovsjannikov G, Trummal A, Kütt A, Saame J, Koppel I, Kaljurand I, Lipping L, Rodima T, Pihl V, Koppel IA, Leito I. J Phys Org Chem. 2013;26:162–170. [Google Scholar]
- 75.Brownstein S, Stillman AE. J Phys Chem. 1959;63:2061–2062. [Google Scholar]
- 76.Igarashi K, Honma T, Irisawa J. Carbohydr Res. 1970;15:329–337. [Google Scholar]
- 77.Kevill DN, Lin GML. Tetrahedron Lett. 1978;19:949–952. [Google Scholar]
- 78.Waldmann H, Böhm G, Schmid U, Röttele H. Angew Chem Int Ed. 1994;33:1944–1946. [Google Scholar]
- 79.Hasty SJ, Ranade SC, Demchenko AV. Reports Org Chem. 2014;4:1–10. [Google Scholar]
- 80.Matsumoto K, Ueoka K, Suzuki S, Suga S, Yoshida J-i. Tetrahedron. 2009;65:10901–10907. [Google Scholar]
- 81.Mayr H, Breugst M, Ofial AR. Angew Chem Int Ed. 2011;50:6470–6505. doi: 10.1002/anie.201007100. [DOI] [PubMed] [Google Scholar]
- 82.Kiyooka S-i, Kaneno D, Fujiyama R. Tetrahedron. 2013;69:4247–4258. [Google Scholar]
- 83.Borovika A, Nagorny P. J Carbohydr Chem. 2012;31:255–283. [Google Scholar]
- 84.Chu AHA, Nguyen SH, Sisel JA, Minciunescu A, Bennett CS. Org Lett. 2013;15:2566–2569. doi: 10.1021/ol401095k. [DOI] [PubMed] [Google Scholar]
- 85.Issa JP, Bennett CS. J Am Chem Soc. 2014;136:5740–5744. doi: 10.1021/ja500410c. [DOI] [PubMed] [Google Scholar]
- 86.Eby R, Schuerch C. Carbohydr Res. 1974;34:79–90. [Google Scholar]
- 87.Kumar A, Kumar V, Dere RT, Schmidt RR. Org Lett. 2011;13:3612–3615. doi: 10.1021/ol201231v. [DOI] [PubMed] [Google Scholar]
- 88.Kumar A, Schmidt RR. Eur J Org Chem. 2012:2715–2719. [Google Scholar]
- 89.Kimura T, Sekine M, Takahashi D, Toshima K. Angew Chem Int Ed. 2013;52:12131–12134. doi: 10.1002/anie.201304830. [DOI] [PubMed] [Google Scholar]
- 90.Geng Y, Kumar A, Faidallah HM, Albar HA, Mhkalid IA, Schmidt RR. Angew Chem Int Ed. 2013;52:10089–10092. doi: 10.1002/anie.201302158. [DOI] [PubMed] [Google Scholar]
- 91.Balmond EI, Coe DM, Galan MC, McGarrigle EM. Angew Chem Int Ed. 2012;51:9152–9155. doi: 10.1002/anie.201204505. [DOI] [PubMed] [Google Scholar]
- 92.Balmond EI, Galan MC, McGarrigle EM. Synlett. 2013;24:2335–2339. [Google Scholar]
- 93.Sun Z, Winschel GA, Borovika A, Nagorny P. J Am Chem Soc. 2012;134:8074–8077. doi: 10.1021/ja302704m. [DOI] [PubMed] [Google Scholar]
- 94.Wever WJ, Cinelli MA, Bowers AA. Org Lett. 2013;15:30–33. doi: 10.1021/ol302941q. [DOI] [PubMed] [Google Scholar]
- 95.Spell M, Wang X, Wahba AE, Conner E, Ragains J. Carbohydr Res. 2013;369:42–47. doi: 10.1016/j.carres.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 96.Yuan X, Cheng S, Shi Y, Xue W. Synthesis. 2014;46:331–335. [Google Scholar]
- 97.Cumpstey I, Crich D. J Carbohyd Chem. 2011;30:469–485. [Google Scholar]
- 98.Nakanishi M, Takahashi D, Toshima K. Org Biomol Chem. 2013;11:5079–5082. doi: 10.1039/c3ob41143e. [DOI] [PubMed] [Google Scholar]
- 99.Srivastava VK, Schuerch C. Tetrahedron Lett. 1979;20:3269–3272. [Google Scholar]
- 100.Schmidt RR. Angew Chem Int Ed. 1986;25:212–235. [Google Scholar]
- 101.Hodosi G, Kováč P. J Am Chem Soc. 1997;119:2335–2336. [Google Scholar]
- 102.Zhu D, Baryal KN, Adhikara S, Zhu J. J Am Chem Soc. 2014;136:3172–3175. doi: 10.1021/ja4116956. [DOI] [PubMed] [Google Scholar]
- 103.Cuccarese MF, Li JJ, O’Doherty GA. In: Modern Synthetic Methods in Catbohydrate Chemistry; From Monosaccharides to Complex Glycoconjugates. Werz DB, Vidal S, editors. Wiley; Weinheim: 2014. pp. 1–28. [Google Scholar]
- 104.Shi P, Silva MC, Wang HYL, Wu B, Akhmedov NG, Li M, Beuning PJ, O’Doherty GA. ACS Med Chem Lett. 2012;3:1086–1090. doi: 10.1021/ml300303g. [DOI] [PMC free article] [PubMed] [Google Scholar]