

Abstract
Purpose
To mechanistically study and model the effect of lipids, either from food or self-emulsifying drug delivery systems (SEDDS), on drug transport in the intestinal lumen.
Methods
Simultaneous lipid digestion, dissolution/release, and drug partitioning were experimentally studied and modeled for two dosing scenarios: solid drug with a food-associated lipid (soybean oil) and drug solubilized in a model SEDDS (soybean oil and Tween 80 at 1:1 ratio). Rate constants for digestion, permeability of emulsion droplets, and partition coefficients in micellar and oil phases were measured, and used to numerically solve the developed model.
Results
Strong influence of lipid digestion on drug release from SEDDS and solid drug dissolution into food-associated lipid emulsion were observed and predicted by the developed model. 90 minutes after introduction of SEDDS, there was 9% and 70% drug release in the absence and presence of digestion, respectively. However, overall drug dissolution in the presence of food-associated lipids occurred over a longer period than without digestion.
Conclusion
A systems-based mechanistic model incorporating simultaneous dynamic processes occurring upon dosing of drug with lipids enabled prediction of aqueous drug concentration profile. This model, once incorporated with a pharmacokinetic model considering processes of drug absorption and drug lymphatic transport in the presence of lipids, could be highly useful for quantitative prediction of impact of lipids on bioavailability of drugs.
1. Introduction
Ingested lipids, either originating from food or used as delivery agents, can have significant effects on dissolution, solubility, transport, and bioavailability of orally delivered compounds. The influence of ingested lipids on compound absorption originates from several mechanisms. Colloidal structures formed during lipid digestion, and compound trafficking between these structures and aqueous medium, impact solubility and dissolution. The presence of lipids in the gastrointestinal (GI) tract increases transit time, and can also facilitate the transport of lipophilic compounds via the lymphatic pathway, enabling avoidance of first-pass metabolism (1). However, these effects are typically documented as empirical, compound-specific observations and not predictable (2, 3). Processes involved in enhancement of overall drug absorption by lipids: lipid digestion, drug dissolution/release, drug partitioning, and drug absorption, have frequently been studied in isolation and on disparate systems. Therefore, despite numerous studies about fat-rich food/drug interactions and lipid-based delivery systems, there is still an incomplete understanding – and thus a lack of general mechanistic modeling - of the influence of ingested lipids on in vivo oral bioavailability. The interconnected, dynamic processes that occur simultaneously during lipid digestion, and their dependence on dynamic system colloidal structure and composition, must be studied in a comprehensive, integrated fashion to enable quantitative prediction. Quantitative prediction of the impact of lipids on oral absorption would facilitate design of oral drug delivery systems and realization of the tremendous potential of lipids to impact bioavailability.
The goal of this study is to investigate quantitatively and model mathematically the influence of lipids originating either from i) food or ii) lipid based self-emulsifying drug delivery systems (SEDDS) on drug dissolution or drug release, respectively, in the presence of simultaneous digestion. Experimental studies were conducted in an in vitro simulated intestinal environment incorporating simulated intestinal fluids. The main differences between the cases of dosing food-associated lipids and SEDDS reflected in our experimental systems and models were: 1. the form of drug dosed (solid drug co-dosed with soybean oil as a model lipid, vs. drug dissolved in an oil/surfactant mixture as a model SEDDS formulation); 2. ratio of lipid to intestinal fluid volumes; 3. the simulated intestinal fluid composition (reflecting fed and fasted states, respectively). The extent of the lipid digestion was monitored experimentally by basic titration technique. Kinetics of drug dissolution/release and partitioning among colloidal phases (oil, micellar, aqueous) were analyzed by high performance liquid chromatography (HPLC) and electron paramagnetic resonance (EPR), respectively.
EPR (also called electron spin resonance, or ESR) is a non-invasive technique that makes it possible to detect and quantify paramagnetic molecules such as organic radicals. Because many systems of interest are not paramagnetic, a common approach is the spin labeling or spin probe technique, in which a stable radical such as a nitroxide is introduced into the system to act as a reporter on the local environment. This method has been widely employed in the study of condensed phases, membranes, and biopolymers (4–6), although its application in drug delivery studies has been hitherto rather limited (7). In the present application, spin probes are used to monitor the microenvironment in different phases, e.g., colloidal particles such as emulsion droplets and micelles, compared to aqueous media. Since EPR spectra are highly sensitive to changes in local polarity and viscosity, the spin probes in different phases can be resolved and quantified so that the partitioning and translocation of probes between different phases can be monitored and quantified in real-time. In this study, the spin probe TEMPOL benzoate (TB) was selected as a model for poorly water-soluble moderately lipophilic drug with an octanol/water partition coefficient (log P) of 2.46 (7).
Based on these quantitative analyses, we developed systems-based models that predict the rate at which drug enters the aqueous intestinal environment, after dosing either in solid form with food-associated lipids, or incorporated into a SEDDS formulation. These models include mathematical expressions that describe key parallel kinetic processes occurring in the GI tract upon co-dosing of a compound with lipids. The processes considered include compound dissolution (if solid drug is dosed), compound release (if dosed in SEDDS), lipid digestion, and compound partitioning into colloidal phases (Figure 1). In modeling both drug dissolution in the presence of food-associated lipids and drug release from SEDDS, the influence of colloids interacting with the drug on drug transport rates was explicitly taken into account. The ability of the models to predict the rate at which the drug enters the aqueous intestinal environment in the presence of the lipid digestion process was evaluated. Ultimately, the absorption process can be incorporated into these models to enable quantitative prediction of the impact of ingested lipids on oral compound absorption and bioavailability.
Figure 1.
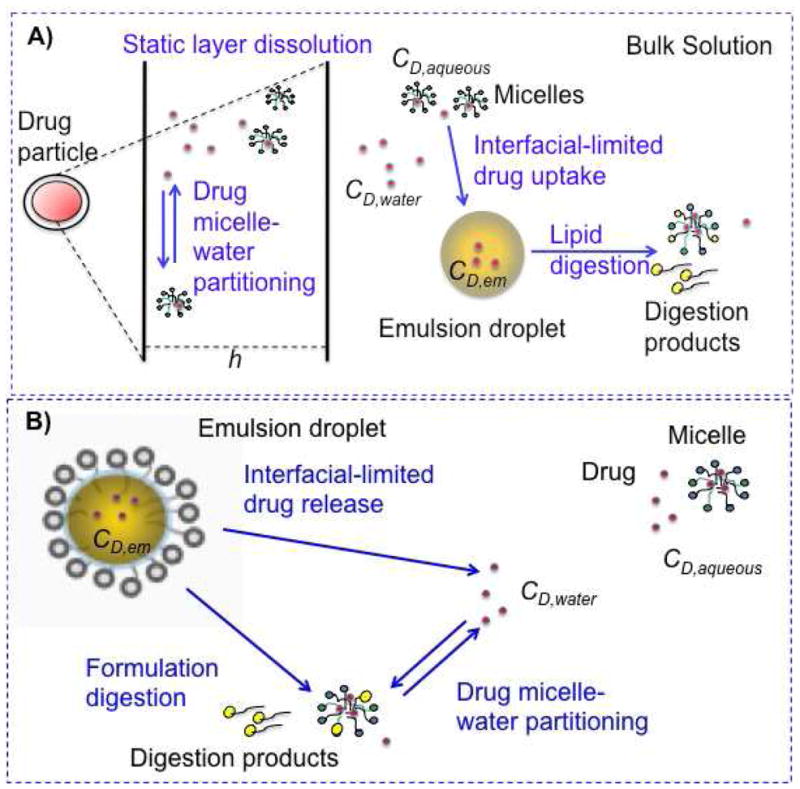
Schematic representation of two lipid systems studied: A) solid drug dosed with food-associated lipid, B) drug dosed in a lipid-based drug delivery system. Processes relevant to each system appear in blue text. CD,water: free drug concentration, CD,aqueous: free and micelle-associated drug concentration, CD,em: drug concentration in food-associated oil and formulation emulsions, h: static layer around the dissolving drug particles.
2. Materials and Methods
2.1 Materials
Trizma® maleate, sodium chloride (NaCl), calcium chloride didydrate CaCl2*2H2O, sodium azide (NaN3), sodium hydroxide (NaOH), sodium taurodeoxycholate (NaTDC or BS, cat. # T0875), L-alpha-phosphatidylcholine from egg yolk (PC), Type XVI-E (cat. # P3556), soybean oil, Tween 80, porcine pancreas lipases (cat. # L3126), 4-bromobenzeneboronic acid, and the model drug TB (cat # 371343) were purchased from Sigma-Aldrich Co., St. Louis, MO, USA. Polytetrafluoroethylene (PTFE) filters, nylon syringe filters as well as HPLC grade solvents utilized as mobile phases, methanol, water and trifluoroacetic acid (TFA), were purchased from Fisher Scientific, Pittsburgh, PA.
2.2 Preparation of the bio-relevant media
Simulated intestinal fluid compositions used in this study were similar to ones proposed earlier (8, 9), but modified slightly with respect to model bile component concentrations, salt concentrations, and pH considering physiological conditions for both fasted (10) and fed state systems (11) (Table I). The bio-relevant medium simulating the intestinal fasted state contained maleate buffer that consisted of 100 mM Trizma® maleate, 65 mM NaCl, 5 mM or 10 mM (fasted or fed) CaCl2*2H2O, and 3 mM NaN3 at pH 6.5. 5 mM NaTDC/1.25 mM PC and 12 mM NaTDC/4 mM PC were used in order to mimic fasted and fed state conditions, respectively. Model bile components were mixed with maleate buffer on a stirring plate at 37°C. In order to simulate SEDDS oral dosing in fasted state conditions, SEDDS formulation composed of soybean oil and Tween 80 at 1:1 weight ratio was added to fasted state simulated intestinal fluid at 1:100 volume ratio. To simulate the food-associated lipid intake, soybean oil was added to fed state simulated intestinal fluid at a final concentration of 50 mM in order to form a crude emulsion. Emulsification of soybean oil into fed state medium and formulation into fasted state medium was achieved by means of continuous magnetic stirring at 300 rpm and was verified by DLS (Brookhaven 90 Plus) measurements performed immediately upon sampling, before separation of phases might have occurred.
Table I.
Compositions of two separate systems studied: a) lipid drug delivery system dosed in fasted state, b) “food lipid” (soybean oil) in fed state bio-relevant media.
| a) Drug delivery system in fasted state | b) Food lipids in fed state | |||
|---|---|---|---|---|
| Maleate Buffer | pH 6.5 | pH 6.5 | ||
| Trizma® maleate | 100 mM | Trizma® maleate | 100 mM | |
| NaCl | 65 mM | NaCl | 65 mM | |
| CaCl2*2H2O | 5 mM | CaCl2*2H2O | 10 mM | |
| NaN3 | 3 mM | NaN3 | 3 mM | |
| NaOH | 40 mM | NaOH | 40 mM | |
|
| ||||
| Bile salt | NaTDC | 5 mM | NaTDC | 12 mM |
|
| ||||
| Phospholipid | Lecithin | 1.25 mM | Lecithin | 4 mM |
|
| ||||
| Ratio BS/PL | 4:1 | 3:1 | ||
|
| ||||
| Lipid Substrate | SEDDS formulation: Soybean oil:Tween 80 1:1 (w/w), 1:100 formulation dilution in fasted state solution | Soybean oil | 50 mM | |
2.3 Solubility measurements
Studies pertaining to in vitro drug dissolution, drug release and lipid digestion were performed with a model compound, the spin probe TB, in order to enable use of EPR to validate model simulations (in case of SEDDS) and to calculate partition coefficients (in case of food-associated lipid). The equilibrium solubility of the model drug TB was measured by adding excess amount of compound dissolved into water (used as a control), maleate buffer at pH 6.5, fasted and fed state bio-relevant media, SEDDS formulation, and soybean oil according to the shake-flask method. Samples were stirred at 37°C, 300 rpm for 72 hours on a hot stirring plate, and sampled every 24 hours (or until equilibrium solubility was attained). For determining aqueous solubility of TB in the presence of Tween 80 associated micelles (originating from SEDDS formulation), formulation was dispersed in fasted state bio-relevant medium at 1:100 dilution together with excess TB and stirred at 37°C, 300 rpm for 72 hours, and samples collected every 24 hours were filtered through 0.1 μm polytetrafluoroethylene (PTFE) syringe filters to remove emulsion droplets. The absence of emulsion droplets in the filtrate was validated by dynamic light scattering measurements. Samples of aqueous solutions were centrifuged at 1700 g and 37°C for 30 minutes, while the oil and formulation samples were centrifuged at 16100 g for 30 minutes. The supernatant was filtered through 0.45 μm nylon syringe filters, and properly diluted in isopropanol/methanol 2:1 (v/v) prior to measuring drug concentration by means of HPLC. Experiments were conducted in triplicate for each dissolving medium and standard error was calculated.
2.4 Drug dissolution
In dissolution experiments, relevant to the case of dosing drug as a solid together with food-associated lipids (Figure 1a), the solid model drug TB was added to a stirred beaker (0.003618 mmol/ml) containing the dissolving medium (maleate buffer at pH 6.5, or fed state bio-relevant medium with and without soybean oil) at 37°C and 250 rpm. Experiments in fed state bio-relevant medium with soybean oil were conducted in the presence of pancreatin extract, containing lipases, in order to assess the impact of the lipid digestion process on drug dissolution. Samples were withdrawn from the dissolution beaker at defined time intervals over a period of 3 hours. Dissolution only samples were filtered through 0.45 μm nylon syringe filters to remove undissolved drug; simultaneous dissolution and digestion samples were centrifuged at 1700 g and 37°C for 10 minutes instead of filtered to remove undissolved drug and not oil emulsions (12). All samples were analyzed via HPLC in order to determine the drug concentration in solution. Experiments were conducted in triplicate for each dissolving medium and standard error was calculated.
2.5 Drug release
Drug release from SEDDS formulations (Figure 1b) was tested in a stirred beaker at 37°C and 180 rpm in the absence and presence of pancreatin extract. In the absence of digestion, 400 μl of formulation including 50 mg/ml TB was added into 40 ml fasted state bio-relevant media. Over time, 2 ml samples were taken from the beaker and filtered through 0.1 μm polytetrafluoroethylene (PTFE) syringe filters to separate aqueous solution containing micelles from formulation emulsion droplets. Filtered solution was analyzed using HPLC for TB content. Standard error was calculated using two independent measurements. In separate experiments in the presence of digestion, 200 μl formulation including 7 mg/ml TB was added into 200 ml fasted state solution. 1 ml samples were taken from the beaker, digestion process was terminated by the addition of enzyme inhibitor, 4-BPB (4-bromophenacyl bromide), and samples were directly analyzed using EPR as described in Section 2.11.
2.6 Quantification of TB from solubility, dissolution, and release experiments using HPLC
The concentrations of the model drug TB in samples collected during solubility, dissolution, and release experiments were determined using HPLC with a photodiode detector (Shimadzu, Japan), and a wavelength of 232 nm. The analytical column used was Agilent Zorbax RX-C18 4.6 × 75 mm, 3.5 μm. The column temperature was maintained at 40°C, and the flow rate was 1 mL/min. The mobile phase contained distilled water with 0.15% TFA: methanol (70:10 to 10:70 over 16 min).
2.7 Digestion kinetics
For in vitro digestion of soybean oil, soybean oil (50 mM) was added into the fed state bio-relevant medium at 37°C, 250 rpm, and solution was equilibrated for 20 minutes prior to the addition of the model drug TB and the lipase enzyme. The lipid digestion and the dissolution processes were then commenced at the same time. In vitro lipolysis of SEDDS formulations (Figure 1b) was performed by addition of 180 μl SEDDS formulation into 18 ml fasted state simulated intestinal fluid (1:100 dilution ratio), prepared as described in Section 2.2, at 37°C and 250 rpm.
In all in vitro lipolysis experiments, the enzymatic hydrolysis of lipids was initiated by adding 2 ml pancreatin extract into the simulated intestinal fluids containing the lipid substrates (soybean oil or SEDDS formulation) (13). Pancreatin extract was prepared fresh for every in vitro lipolysis experiment following the procedure described by Sek et al. (13) Briefly, 2 g of porcine pancreatin powder were dissolved in 10 ml of maleate buffer (composition in Table I) and magnetically stirred for 15 minutes at room temperature. The solution was centrifuged at 1600g for 15 minutes at 5°C; the supernatant was collected and stored at 4°C until use. Measured enzyme activity of pancreatic lipase prepared in this manner from this supplier was similar to human pancreatic lipase activity in vivo (14). The change in pH was constantly monitored with a pH meter (Seven Multi pH meter, Mettler Toledo, Columbus, OH), and fatty acids produced due to the lipolysis reaction were titrated manually with 0.2 mM NaOH using a digital titration unit (VWR international, Plainfield, NJ). Fatty acid production was then calculated by measuring the total volume of NaOH added to maintain the pH at 6.5 during lipolysis. Digestion rate was related to the droplet surface area available to the enzyme, as explained in Section 2.8.1. Digestion rate constants were estimated via nonlinear regression fitting in MATLAB® using Equation 4.
2.8 Model development
Kinetic expressions describing the rate of drug entering the aqueous environment after dosing either a drug-loaded SEDDS or a solid drug co-administered with food lipids were developed considering major kinetic processes that occur simultaneously in vivo (Figure 1): formulation or lipid digestion, solid drug dissolution, drug release from formulation (i.e. SEDDS) emulsion droplets or drug uptake into food lipid emulsion droplets, and drug partitioning into micelles. Formulation or food-associated lipid was assumed to enter directly to small intestine upon oral administration.
2.8.1 Digestion
Digestion rate was expressed using a modification of equation recently proposed by Li et al. relating the rate of FA production to the droplet surface area available to the enzyme, assuming that the droplet size of oil-in-water emulsions decreases as digestion proceeds, and as fatty acids, products of digestion of oil and surfactant, leave oil droplets (15). In many cases, only a fraction of the total digestible FA present initially in the oil droplets is released because the lipolysis reaction is inhibited by FA formation in vitro (16). In order to take this into account, we modified the expression proposed by Li et al. to include inhibition proportional to the concentration of FA:
| (1) |
where CFA,aq is the concentration of FA produced during the lipid digestion and released in the aqueous phase, kdig is the digestion rate constant, Aem is the total oil droplet surface area per total solution volume, Vaq is the total solution volume (oil emulsions and aqueous phase), and kinh is the inhibition rate constant.
It is assumed that during digestion, oil droplets will shrink in size over time due to enzymatic reaction and this change in particle diameter, D(t) can be related to the fraction of fatty acids released from the droplets due to digestion. Hence, time dependent total surface area of the emulsion droplets, Aem(t), is
| (2) |
where, N is the number of oil droplets present, mFA,em is the moles of digestible fatty acids remaining in the emulsion droplet, mFA,0 is the initial moles of digestible fatty acids in the droplet, and D0 is the initial droplet diameter of emulsion droplets.
2.8.2 Solid drug dissolution
The dissolution kinetics was expressed according to a static layer model that takes into account the contribution of colloidal particles to the drug transport rate, derived by Higuchi (17). Assuming the presence of a single species of free colloid (micelles), no net changes in colloid number, and that solute/colloid interactions occur rapidly enough relative to other processes to be considered at equilibrium, the rate of drug dissolution in the aqueous phase (water and micelles) is:
| (3) |
where CD,aq is the drug concentration in the aqueous phase (mg/ml), Sp is the surface area of N dissolving drug particles (cm2), h is the stationary diffusion layer around a dissolving particle (cm), Vaq is the solution total volume (ml), Ds is the drug diffusion coefficient in water (cm2/s), CeqD,water is the drug solubility in water (mg/ml), CD,water is the free drug concentration in the total solution volume (mg/ml), Dm is the micelle diffusion coefficient in water (cm2/s), CeqD,micelle is the drug solubility in micelles (mg/ml), and CD,micelles is the drug concentration in micelles (mg/ml), where concentrations of drug in micelles are expressed as mass of drug in micelles per total volume of solution, at the concentration of micelles present in the solution. If the dissolving drug particles are assumed to be mono-dispersed and uniform spheres with an initial radius r0, and the total number of particles N does not vary with time, then the time-dependent expression for Sp can be written as:
| (4) |
where M0 is the initial mass of drug dosed (mg), Ms is the mass of undissolved drug (mg), and ρ is the drug density (mg/cm2) (18). Equilibrium solubility terms were determined experimentally by means of HPLC, as described in Section 2.3. The quantities CD,water and CD,micelles were calculated from the total measured drug in the aqueous phase (buffer and micelles) according to the partition coefficient Pm/w (see Section 2.8.4 for definition of partition coefficients). Diffusion coefficients for compounds and colloidal particles were calculated according to the Wilke-Chang and the Stokes-Einstein equations, respectively (Section 2.9).
Equation 3 was used to model solid drug dissolution data from experiments performed in bio-relevant media without lipids or lipid digestion. In experiments performed in the presence of a lipid substrate, oil droplets (soybean oil) are detected by DLS to be bigger than micelles (200 nm versus 5 nm), and consequently to have a calculated diffusion coefficient of 1.64*10−12 m2/s (according to the Stoke-Einstein equation, section 2.9) and an estimated diffusion time t through h (t =h2/D) of ~ 4 minutes, compared to ~ 6 seconds for micelles. Therefore, oil droplet contribution to the drug transport rate within the static diffusion layer h was neglected. However, some dissolved compound at each time t does transfer into the oil phase. Compound partitioning into oil droplets was considered using an interfacial barrier-limited model (19), rather than a diffusion-driven process as described by Equation 3. The rate (mg/cm2) of drug partitioning into the oil phase was expressed as:
| (5) |
where Aem is the surface area of N oil droplets (cm2), Prel is the permeability (cm/s), Vem is the total oil volume dispersed in the aqueous solution, CD,aq is the drug concentration (mg/ml) in the aqueous medium Vaq (buffer and micelles), and C′D,aq is the hypothetical aqueous drug concentration (mg/ml) in equilibrium with the drug concentration inside oil droplets CD,em (mg/ml). Calculation of C′D,aq as a function of the drug partition coefficient between the aqueous phase and the oil phase, Kaq/em, is explained in Section 2.8.4. Prel was experimentally determined by means of EPR during drug partitioning experiments in the absence of lipolysis (Section 2.11).
In experiments combining dissolution of the solid drug compound and in vitro lipolysis processes, specific parameters in the above expressions are expected to change, reflecting the dynamic nature of the digestion system. Aem is expected to decrease over time according to Equation 2, taking into account lipolysis kinetics (15) (Section 2.8.1). Furthermore, as the lipase enzyme digests the lipids, micelles are expected to evolve into new colloidal structures due to the interaction with lipid digestion products FA and monoglycerides (MG) liberated in solution. The dynamic evolution of colloidal structures affects their solubilization power CeqD,micelle in Equation 3 expressed as the quantity of dissolved drug associated with micelles per bulk volume. Therefore, the colloid solubilization power was affected by the extent of digestion and was no longer a constant during the dissolution process. The time-dependent colloid solubilization power, CeqD,micelle, was thus expressed as a function of the digestion kinetics as explained in detail in Section 2.8.4.
Combining the two kinetic expressions (Equations 3 and 5) describing the drug dissolution rate in the aqueous phase CD,aq, and the concurrent lipid digestion, the main equations utilized to describe FA and dissolved drug concentration profiles, respectively, are Equation 1 and:
| (6) |
2.8.3 Drug release from SEDDS formulation emulsions
In the case of dosing a lipid-based drug delivery system (SEDDS), drug is delivered already solubilized in emulsion droplets, and thus a process that occurs in parallel to formulation digestion is the drug release from inside of oil droplets to the surrounding aqueous media. Drug release was described using a kinetic expression developed by Higuchi et al. (19) considering interfacial resistance at the oil-water interface as a limiting barrier to compound transport, which is the same as Equation 5 used in Section 2.8.2.
However, along with the concentration gradient driven transport of compounds from inside the oil droplets into the aqueous media, there will be a compound transport from oil droplets into the aqueous media due to detachment of oil digestion products from the surface of the droplets. In order to account for compound transport due to digestion, drug release expression was modified to give:
| (7) |
where n is the moles of fatty acid produced per mole of formulation and Vmolar,em is averaged molar volume of the formulation content. C′D,aq is expressed as a function of CD,em and a partition coefficient, K, as described below in Section 2.8.4. CD,em is calculated using mass balance. Prel is calculated experimentally by means of drug release experiments in the absence of digestive enzyme. kdig is measured experimentally by in vitro lipolysis analysis, and time dependent change in emulsion droplet surface area, Aem, is calculated for each time point using Equations 2. Combining the two kinetic processes, digestion and release, the main equations used to describe aqueous fatty acid and drug concentration profiles, respectively, are Equations 2 and 7.
2.8.4 Definition of partition coefficient, K
C′D,aq, hypothetical aqueous concentration of the solute in equilibrium with compound concentration inside emulsion droplet, is defined by the formulation/oil-aqueous phase partition coefficient, Kem/aq, as:
| (8) |
where CeqD,em is the equilibrium solubility concentration of drug in oil emulsions based on emulsion volume, CeqD,aq is the equilibrium solubility concentration of drug in the aqueous phase based on the aqueous volume and Km/w is micelle-water partition coefficient. Using Equation 8, hypothetical drug concentration in aqueous phase ( ) can be calculated using values for Kem/aq and time dependent drug concentration inside emulsion droplets, CD,em. During digestion, we assume that digestion products that leave oil droplets incorporate into micelles, and as a result, the solubilization capacity of aqueous phase, CeqD,aq, increases and therefore Kem/aq changes over time.
In order to calculate Kem/aq over time during digestion, a constant term, molar solubilization capacity of micelles, χ, was introduced and water partition coefficient, Km/w, was expressed as a function of amount of total surfactant that is incorporated into micelles (both endogenous and due to digestion), Csurf. Molar solubilization capacity of micelles, χ is defined as (20):
| (9) |
where cmc is the critical micelle concentration and Csurf is the time dependent molar concentration of total surfactant (bile salt, phospholipid, fatty acids) at a given point in time. We assume that as digestion products partition into bile salt/phospholipid (BS/PL) micelles, micelle composition changes, while the number of micelles remains constant. Total surfactant concentration, Csurf, is expressed as:
| (10) |
where CBS is bile salt concentration, CPL is phospholipid concentration, and CTween80 is amount of formulation component Tween 80 in aqueous phase (as opposed to formulation emulsions). Using Equation 11, micelle water partition coefficient, Km/w, is expressed as a function of the amount of surfactant that is associated with micelles and χ:
| (11) |
where CeqD,micelle is the equilibrium solubility of drug in micelles based on the total aqueous volume and CeqD,water is the equilibrium solubility of the drug in the water phase. Micelle molar solubilization capacity, χ, is a constant and can be experimentally determined, as well as the drug solubility at equilibrium, CeqD,water. Critical micelle concentration, cmc, is also a constant (21).
2.9 Estimation of the diffusion coefficients
Drug diffusion coefficient of TB, DD, was estimated using Wilke-Chang equation (22):
| (12) |
where DAB is the diffusivity of compound A in solvent B (cm2/s), ΨB is the constant that accounts for solvent/solvent interactions (2.6 for water), T is temperature (K), ηB is the viscosity of solvent B (cP), MWB is molecular weight of solvent B (g/mol), and VA is the molar volume of compound A. According to Equation 12, the diffusion coefficient of TB, DD, was 8.3743*10−6 cm2/s.
Diffusion coefficient of mixed micelles, Dm, was estimated using the Stokes-Einstein equation (23):
| (13) |
where DAB is the diffusivity of compound A in solvent B (cm2/s), k is the Boltzman constant (1.3806×10−23 m2 kg s−2 K−1), T is temperature (K), ηB is the viscosity of solvent B (Pa*s), and rA is the hydrodynamic radius of the particle (m). According to Equation 13, the diffusion coefficient of micelles, Dm, was 6.5578*10−11 cm2/s. In both diffusivity estimations, the viscosity of the solvent ηB was assumed to be equal to the water value at 37°C (0.00069244 Pa*s, or 0.69244 cP) and the temperature was fixed at the physiological value of 310°K.
2.10 Numerical solutions of the model differential equations
Differential equations pertaining to the developed mathematical model were solved using a built-in ordinary differential equation solver, ODE45, in MATLAB® by means of the Runge-Kutta numerical solution technique. Concentrations of the drug in the aqueous phase, of the drug inside formulation emulsions, and of the fatty acid produced via digestion in the aqueous phase were computed over time.
2.11 Electron Paramagnetic Resonance (EPR) measurements
In an effort to measure TB transport from aqueous solution into food lipids, EPR measurements of samples containing soybean oil in pre-lipolysis conditions (i.e. without lipase) were performed. Based on quantitative information obtained by EPR measurements for drug concentration in each phase (oil, micelle, aqueous), partition coefficients used in simulations of the impact of ingested food lipids (Section 2.8.4) were estimated. In addition, the applicability of the interfacial barrier-limited model to describe compound partitioning into oil droplets was validated. TB was dissolved in the bio-relevant medium before the addition of 50 mM soybean oil, after which samples were collected at different time points (up to 3 hours) and analyzed by EPR in order to follow drug partitioning between phases (aqueous and oil).
In order to measure drug release from SEDDS emulsion droplets into aqueous solution during digestion, EPR measurements were performed on samples collected at different time points (0, 5, 20, 50 minutes) during in vitro digestion experiments as explained in Section 2.7. Quantitative information obtained by EPR measurements for drug concentration in each phase during digestion (emulsion, micelle, and aqueous) was used as a means of model simulation validation for drug release from SEDDS during digestion.
Samples collected during experiments were analyzed immediately by EPR spectroscopy (9.1–9.9 GHz, X-Band; Bruker EMX) in an effort to perform real time quantitative non-invasive tracking of drug between emulsion droplets, aqueous and colloidal phases (micelles and vesicles). The parameters used during EPR measurements were as follows: microwave bridge frequency: 9.38 GHz, modulation frequency: 100kHz; microwave power: 1 mW; sweep width: 70 G, sweep time: 83.89 s; time constant: 327.68 ms; modulation amplitude: 1 G. Quantitative determination of the ratio of the spin probe in different environments over time was performed via multi-component fitting analysis of spectra using the Multicomponent EPR Labview module of Altenbach (24) to perform Simplex fitting of the spectrum.
3. Results
Lipid digestion kinetics and drug transport kinetics for both systems (Figure 1a and 1b) were studied experimentally. Simulations describing drug transport in presence of lipid digestion for both systems were run and validated using experimental data.
3.1 Ingested food lipids
3.1.1 Calculation of digestion kinetic constant
The digestion kinetic constants for lipolysis of 50 mM soybean oil in the fed state bio-relevant medium were determined according to Equation 2 as kdig = 3.6 ± 0.2×10−9 mmol/cm2s and kinh = 4.3 ± 0.3×10−4 1/s, based on experimental measurement of digestion kinetics (Figure 2). The excellent model fit (Equation 2) supports the theoretical description of digestion rate as proportional to emulsion droplet surface area and inhibited at a level proportional to the concentration of fatty acids produced.
Figure 2.
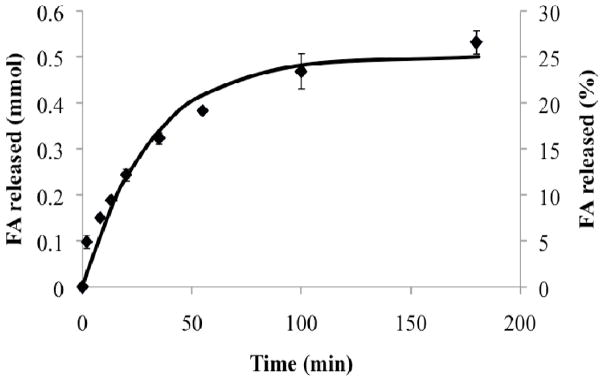
Experimental digestion profile (data points) of soybean oil in the fed state bio-relevant medium -measured during the dissolution experiment of TB – agreed well with the proposed digestion kinetics model (continuous line).
3.1.2 Drug dissolution without lipids
Dissolution kinetics and solubility values of the model drug TB were measured in maleate buffer, and in fed state bio-relevant medium by means of HPLC (Figure 3). Significant (8 fold) improvement in solubility was observed for TB in fed state bio-relevant medium relative to maleate buffer due to the presence of mixed micelle-forming species, NaTDC and lecithin (Table II). Improvement in dissolution kinetics was also evident from maleate buffer to fed state bio-relevant medium, as shown by the steeper slope of the initial part of the dissolution curve in medium containing micelles.
Figure 3.
Experimental dissolution profiles and simulations of the model drug TB in maleate buffer and in the fed state bio-relevant medium (n=3 for each dissolving medium).
Table II.
Experimental solubility values for the model drug TB in different media (n=3).
| Medium | Solubility (mg/ml) |
|---|---|
|
| |
| Maleate buffer-5 mM Ca2+ | 0.013 ± 0.003 |
| Maleate buffer-10 mM Ca2+ | 0.020 ± 0.001 |
| Fasted state bio-relevant medium | 0.096 ± 0.002 |
| Fed state bio-relevant medium | 0.145 ± 0.006 |
| Fasted state bio-relevant media with Tween 80 | 0.3958 ± 0.007 |
| Soybean oil | 217.8 ± 2.5 |
3.1.3 Drug dissolution in the presence of lipids
In dissolution experiments coupled with in vitro lipid digestion, dissolution kinetics of the model drug TB was determined as the digestion of 50 mM soybean oil - representing the lipid intake – proceeded (Figure 2). The fed state bio-relevant medium enriched by ingested lipids showed an approximately 4-fold further enhancement in solubilization power after 3 hours of dissolution-digestion, due to the high solubility of TB in soybean oil (Table II) and enhanced solubilization capacity of micelles. During lipolysis, TB concentration in solution (aqueous and oil phases) rose gradually over a time frame of approximately an hour, as opposed to the rapid increase to approximately 70% of the equilibrium solubility value in the case of dissolution experiments in fed state bio-relevant medium in the absence of lipid digestion (Figure 4). Simulations were run to test the ability of the proposed model to describe drug dissolution in the presence of simulated intestinal fluid and lipid digestion. Parameters used in simulations, determined as described above, are shown in Table III. Comparisons of simulation predictions with experimental dissolution profiles are (Figures 3 and 4) indicate favorable prediction of impact of bio-relevant media and lipid digestion on drug dissolution kinetics.
Figure 4.
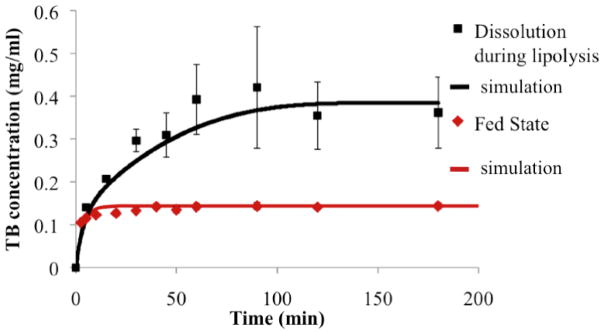
Dissolution profiles of the model drug TB in the fed state bio-relevant medium and during the digestion of 50 mM of soybean oil (n=3 for each dissolving medium).
Table III.
Input parameters to the developed models
| Input parameters | a) Release from SEDDS | b) Drug dissolution from solid dosage form |
|---|---|---|
| Physiological parameters | Value | Value |
| Bile salt concentration | 5×10−3 mmol/ml | 12×10−3 mmol/ml |
| Phospholipid concentration | 1.25×10−3 mmol/ml | 4×10−3 mmol/ml |
| Critical micelle concentration (21) | 1.6×10−3 mmol/ml | 1.6×10−3 mmol/ml |
| Formulation and drug parameters | ||
| Mean droplet diameter of oil emulsions (10) | 403 nm | 386 nm |
| Molecular weight of TB | 276.35 mg/mmol | 276.35 mg/mmol |
| Number of digestible FA per mole oil emulsion (15) | 2 | 2 |
| Initial concentration of TB in a) formulation volume, b) solution volume | 0.02533 mmol/ml | 0.003618 mmol/ml |
| Initial volume ratio of a) formulation, b) oil in intestinal lumen | 1:100 | 4.8:100 |
| Concentration of Tween 80 associated in BS/PL micelles | 8.14 mmol/ml | - |
| (Molar) solubilization of TB in BS/PL micelles | 0.2115 mmol/ml | 0.2115 mmol/ml |
| Solubility of TB in buffer (5mM Ca2+/10mM Ca2+) | 4.7×10−5 mmol/ml | 7.2×10−5 mmol/ml |
| Solubility of TB in a) fasted, b) fed bio-relevant media | 3.4×10−4mmol/ml | 4.5×10−4 mmol/ml |
| Solubility of TB in a) formulation, b) soybean oil | 0.579 mmol/ml | 0.788 mmol/ml |
| Stationary diffusion layer, h1 | - | 20 μm |
| Particle size of TB2 | - | 20 μm |
| Kinetic parameters | ||
| Diffusion coefficient of TB, DD | - | 8.3743*10−10 m2/s |
| Diffusion coefficient of micelles, Dm | - | 6.5578*10−11 m2/s. |
| Formulation or oil digestion kinetic constant, kdig | 4.7×10−9 mmol/cm2s | 3.6×10−9mmol/cm2 s |
| Formulation or oil digestion inhibition kinetic constant, kinh | 2.8×10−4 1/s | 4.3×10−4 1/s |
| a) TB release constant from formulation, b) TB uptake constant into oil, Prel | 5.55×10−9 cm/s | 3.52×10−8 cm/s |
Notes -
assumed to be equal to initial particles’ radius (25);
estimated based on particles’ separation by controlled sieve mesh; all the other parameters were calculated as explained in the text.
3.2. SEDDS
3.2.1 Calculation of digestion kinetics constant
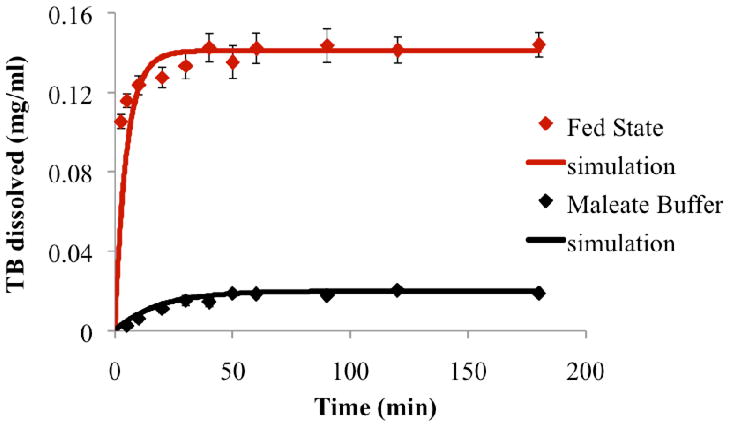
A digestion rate constant (kdig) of 4.7 ± 0.2 ×10−9 mmol/cm2s and a digestion inhibition constant (kinh) of 2.8 ± 0.1×10−4 1/s were calculated from experimental in vitro SEDDS digestion profiles (Figure 5) using Equation 2. Favorable model agreement again supports the theoretical description of emulsion digestion as proportional to total droplet surface area.
Figure 5.
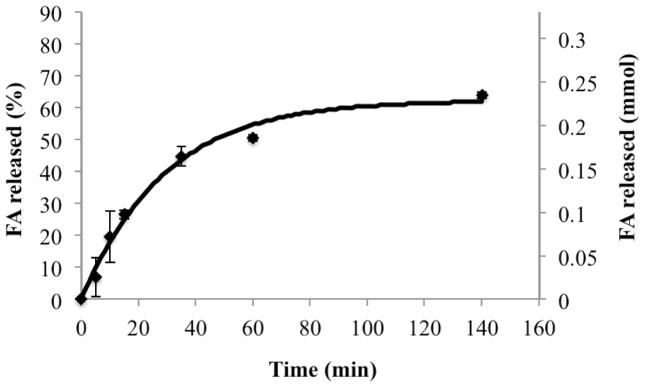
Comparison of model simulation results (continuous line) with experimental results (data points) showing rate of digestion in terms of rate of free fatty acid production.
3.2.2 Experimental drug release profile and calculation of drug release constant from SEDDS
Drug release kinetics studied in the absence of digestive enzyme revealed an initial “burst” release of the drug in the aqueous media upon dispersion of the oil/surfactant/drug mixture. Over 18 hours, 14% of drug was released (Figure 6). First order oil-water interface permeability constant, Prel, was calculated from experimental release data using Equation 9 as 5.55 ± 1.4 × 10−9 cm/s.
Figure 6.
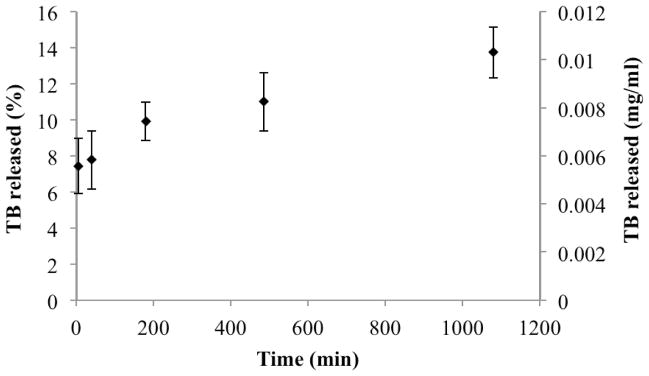
In vitro drug release from SEDDS in the absence of digestion.
TB aqueous solubility (TB solubility in fasted state simulated intestinal fluid) changes once the formulation is dispersed in FaSSIF and Tween 80 molecules get associated with FaSSIF micelles. Therefore, aqueous solubility in the presence of Tween 80 associated micelles was also measured (Table II). Other constant parameters, CeqD,water and CeqD,em, which refer to TB solubilities in maleate buffer and in formulation, respectively, were also measured experimentally. Total surfactant concentration, Csurf, together with CeqD,water and CeqD,em were used to calculate the molar solubilization constant, χ, using Equation 11 as 0.2115 ± 0.008 mmol/ml. Table II summarizes equilibrium solubility values that were measured in each phase.
3.2.3 Simulation of drug release from SEDDS during digestion
Kinetic constants calculated separately for each process, formulation digestion and drug release from formulation in the absence of digestive enzyme, were used to solve the developed mathematical model considering both processes occurring simultaneously in order to predict their synergistic effect on released drug concentration from SEDDS over time during digestion. Input parameters to the developed model are given in Table III.
3.2.4 Comparison of model predictions with in vitro experimental results
Multi-component fitting analysis of EPR spectra collected over the course of digestion allowed quantitative determination of the ratio of the spin probe in each phase over time. Model predictions for the amount of drug in the aqueous phase (buffer and micelles), CD,aq, over time in the presence of digestion of a formulation with 7 mg/ml drug load compared favorably with combined release and digestion experiments analyzed by EPR.
4.0 Discussion
In the present study, we investigated the effects of ingested lipids and the lipid digestion process on drug transport in a simulated intestinal lumen considering two different sources of lipids, food-associated lipids and lipid-based drug delivery systems (SEDDS).
The presence of lipids and their digestion products alter greatly the intestinal lumen composition, which consequently influences compounds’ solubility, dissolution kinetics and release kinetics in the GI fluids. The effects of dynamic lipid digestion on drug transport were investigated. For both food lipids and lipid-based drug delivery systems, the digestion rate was described using an equation that relates it to the total droplet surface area available to the enzyme and takes into account the lipase inhibition due to the accumulation of FA at the oil/water interface. FA accumulation at the oil/water interface is related to the degree of their solubility in the aqueous phase. FA having short and medium chains were reported to not inhibit or to inhibit less the lipase than long chain FA (15). Similarly, presence of higher concentration of bile salt/phospholipids in the aqueous phase may decrease the level of FA accumulation at the interface by increasing the solubilization capacity of the medium. In addition, FA inhibition action described here mainly characterizes the in vitro digestion systems, while it is expected to be less relevant in vivo where FA are continuously removed by absorption. Based on this equation, digestion rate constants kdig were calculated as 3.6*10−9 mmol/sec*cm2 and 4.7*10−9 mmol/sec*cm2 for food lipids and lipid-based drug delivery systems, respectively. The presence of surfactant Tween 80 at the oil-aqueous interface in lipid based drug delivery systems compared to BS/PL surfactants at the food lipid emulsions surface may contribute to the differences in digestion rates of the two systems, since the interface is considered to be the site of the enzyme-lipid substrate interaction (26). Another difference between the two systems studied is the concentration of BS/PL forming micelles in the fasted and fed bio-relevant media. On the other hand, a higher digestion rate of food-associated lipid suspension compared to self-emulsifying systems including the same oil component (Sesame oil: Maisine 35-1, 1:1) was reported previously (12). It should be noted that measured digestion profiles were based on titration of FA, and it has recently been reported that titration of FA can lead to underestimation of extent of digestion due to only a portion of the FA being ionized (27). Thus, a more rigorous analysis of digestion rate may affect calculated digestion rate constants.
Our investigations regarding the dissolution rates of TB showed that the dissolution enhancement did not follow the same trend as solubility. The dissolution kinetics was faster in the fed state bio-relevant medium compared to maleate buffer, following the same trend as in solubility. During food lipid digestion, there was an approximately 4-fold enhancement in solubility, comparing equilibrium solubility in fed state medium to drug dissolved at the conclusion of combined dissolution and digestion experiments. However, during food lipid digestion, the drug dissolution rate from solid dosage form was not enhanced to the same extent as solubility. The initial drug dissolution rates (first 5 minutes of dissolution experiment) were comparable in the presence and the absence of the digestion process (0.14 mmol/min*ml vs. 0.12 mmol/min*ml respectively). However, the overall drug dissolution in the presence of digestion was more gradual over a longer period of time than without digestion, despite the observed increased solubility (approximately 4-fold) due to the presence of oil droplets and lipid digestion products. This was in agreement with previous studies showing that the solubility of lipophilic drugs in simulated intestinal fluids increased proportionally to an increased content of surfactants, while the dissolution rates did not increase proportionally (18, 28). The lack of a direct proportionality between the enhancement in solubility and the enhancement in dissolution kinetics after food intake was also confirmed by a study (29) that employed real human intestinal fluids obtained under fed conditions. Thus, the present observations support the in vivo relevance of including in vitro lipolysis models in drug dissolution tests. The lack of significant improvement in dissolution kinetics of TB observed during the lipid digestion process might be related to the relative kinetics of partitioning between the dissolved drug and soybean oil droplets, and associated impact on concentration of drug in the aqueous solution, and thus the driving force for dissolution. Further investigations carried out by means of EPR revealed that indeed the transport of dissolved TB from the fed state bio-relevant medium into soybean oil droplets occurred over a time scale similar to that of drug dissolution (3 hours) (data not shown). These results support the necessity of modifying the classical Noyes-Whitney equation in order to include explicitly: i) the role of colloidal particles in mass transport, and ii) the partitioning process between dissolved drug and oil droplets.
In the absence of digestion, studies of in vitro drug release from SEDDS (Figure 1b) indicated that there is a certain amount of drug in the aqueous phase at time 0. This may be due to a certain amount of the surfactant, Tween 80, associating with micelles upon emulsification of the SEDDS formulation in the fasted state simulated medium. Due to this association, a certain amount of drug that is initially dissolved in the surfactant component of the formulation may also associate with micelles. This association is, in fact, in agreement with preliminary Small-angle neutron scattering (SANS) analysis of the system, where it was shown that there was an alteration in BS/PL micelle composition and possibly size and structure with the addition of formulation into the aqueous system (data not shown). In the absence of digestion, percentage of released drug at 90 minutes upon introduction of the formulation was 9%, whereas in the presence of formulation digestion this value was 70%. The significantly higher amount of drug released due to digestion can be explained by an active digestion facilitated release mechanism where the drug associated with the digestion products directly partition into the endogenous BS/PL micelles formed in the presence of digestion products. Thus, this active transport mechanism was taken into account while expressing drug release from formulations in the presence of digestion (Equation 7). Drug solubilization/release into aqueous phase during in vitro digestion was previously studied using static (27, 30, 31) and dynamic lipolysis models (12, 32). Fatouros et al. (12) studied drug release profiles of the model drug Probucol (logP 8.92), during dynamic in vitro lipolysis, formulated with a SNEDDS (Sesame oil: Maisine 35-1: Cremophor RH 40: Ethanol, 30: 30: 30: 10) and SMEDDS (Sesame oil: Maisine 35-1: Cremophor RH 40: Ethanol, 26.7: 26.7: 26.7: 20) formulations with droplet particle size of 45.0 ± 3.4 nm and 4.58 ± 0.84 μm, respectively. Drug release from these formulations ranged between 70–95% at 60 minutes into digestion, which is similar to our findings. The main difference in release profiles between our findings and what has been previously reported is the earlier leveling off of drug release, which is about 0–15 minutes for Fatouros et al., as opposed to 50 minutes in our studies. This discrepancy, while likely due in part to differences in formulation composition, might originate in part from differences in analysis of drug concentration techniques. The most conventional way of measuring drug partitioning during digestion so far has involved sampling during in vitro digestion, centrifugation of collected samples in order to separate aqueous phase from the formulation phase, and quantification of drug concentration in the aqueous phase (12). While this method has proven to give, in many instances, reliable information about the degree of drug release and partitioning and possible performance of the specific drug delivery system, it may result in misinterpretation of time profiles of this dynamic process since the time spent during centrifugation has been as much as 135 minutes. In our study, EPR was used as a non-invasive, online method to monitor model drug distribution in different phases. EPR spectroscopy offers advantages of a non-invasive, real time method for analysis of the amount of compound in each phase (water, micelles, and formulation) during in vitro digestion, and is thus a powerful tool to monitor drug delivery processes (7, 33). Our results on both of the systems studied, food lipids and lipid based drug delivery systems, indicate strong influences of the presence of lipids and lipid digestion on drug transport. During food lipid digestion, the drug dissolution rate from solid dosage form did not increase at the same extent as solubility (approximately 4-fold enhancement), as noted above. Whereas in the case of lipid drug delivery systems, the presence of digestion led to an increase in both the rate and extent of drug release from lipid delivery system emulsion.
Recently, Sugano (34) summarized the effects of fed state intestinal conditions on oral drug absorption via interactions with bile micelles. In particular, the interactions between compounds and bile micelles present in the GI fluids were proposed as the theoretical basis for food effects on co-administered compounds, supporting the central role of micelles in overall oral absorption. Our modeling approach included lipids and dynamic lipid digestion products interacting with endogenous colloidal particles and drug compounds in addition to bile micelles. Furthermore, the model aimed to unify the mechanism behind the observed effects of ingested lipids on two processes - drug dissolution and drug release – based upon fundamental principles of mass transport across oil-water-micelle interfaces as lipolysis proceeds. The significance of predicting drug transport between different colloidal phases present during lipid digestion lies in the assumption that simultaneous absorption occurs in vivo, and that this absorption process is driven by aqueous drug concentration. Although overall drug absorptive flux has been related to total drug concentration (35), multiple literature reports indicate that drug absorption is driven by the concentration of drug in the aqueous phase (33, 35, 36).
Several studies (17, 23, 37) have shown that the classic Noyes-Whitney equation might have to be modified when the dissolution of solid compound takes place in solutions containing solubilizing agents. Previously, we have demonstrated that a model considering micelle-drug partitioning as a pseudo-equilibrium process and an unstirred boundary layer surrounding dissolving particles across which drugs and micelles diffuse could effectively describe drug dissolution in simulated intestinal fluids (18). However, this experimental study and modeling of dissolution of solid compounds did not include the presence of lipid emulsions and the lipid digestion process. In this study, therefore, ingested lipids and the lipolysis process were included in order to more closely mimic the in vivo dynamic conditions of the GI fluids after lipid intake. The proposed model presented herein took into account the mass transport of the model drug TB between the aqueous phase – containing micelles – and the oil droplets during oil digestion (19, 38).
Close comparison between simulations of drug release/dissolution during digestion and experimental results (Figures 4 and 7) supports the validity of expressions for kinetic processes for both systems. In addition, several assumptions made solving the model such as increased aqueous solubilization directly related to free fatty acid concentration, and shrinkage of emulsion droplet size proportional to the amount of fatty acid leaving droplets, were supported by the reasonable accuracy of simulation predictions. However, coalescence during digestion of lipid-based systems was reported previously (15). In cases where oil droplet flocculation and coalescence is prominent during digestion, the model assumption that droplets decrease in size proportional to fatty acid leaving the droplets may not be valid. Lipid digestion was also associated with the formation of several liquid crystalline phases at the water-lipid interface at different stages of the lipolysis (39). Liquid crystalline formation was also correlated with the type of the oil digested (long chain vs medium or short chain triglyceride) (40). Our model was tested using a single long chain triglyceride, soybean oil. It should be noted that based on the lipid type, modification on the droplets surface structure (liquid crystal phases) might occur during digestion, in which case model validity might potentially change. Furthermore, we assumed that the enhanced drug solubilization observed during the lipid digestion was directly proportional to the surfactants’ concentration. Simple micelle inclusion of FA may be an over-simplification of the more complex colloidal system formation during intestinal lipolysis. However, lacking insight into how colloidal species evolve over time, a first approximation was used to model mathematically the enhancement in solubilization capacity of such colloidal systems due to digestion. Increased solubilization capacity of aqueous phase during digestion in some cases was linked to a supersaturation phenomenon, (41) where an initial supersaturation status is followed by drug precipitation. At the drug load employed in this study, we did not observe a supersaturation as suggested by the lack of precipitate in samples upon centrifugation. Therefore, we did not consider supersaturation and precipitation processes in the presented model. However, higher initial drug loading in SEDDS formulation might potentially induce a supersaturation stage in the aqueous phase upon drug release during digestion. These results substantiate the concept that mechanistic studies based on physiologically relevant in vitro experiments can provide a better prediction of drug dissolution and release in the presence of ingested lipids; they can ultimately be combined with permeability studies and pharmacokinetic models, enabling prediction of the overall impact on drug absorption and bioavailability.
Figure 7.
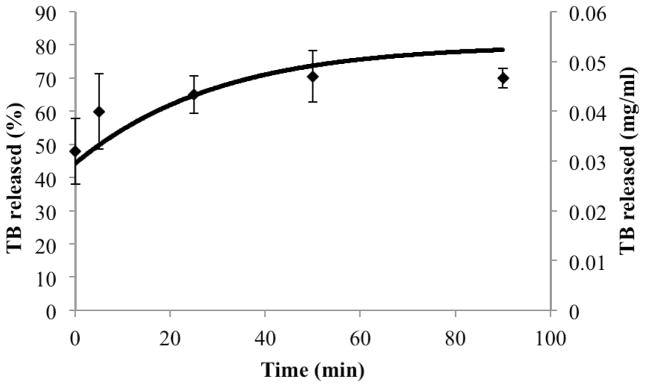
Comparison of model simulation results (continuous line) with experimental results (data points) pertaining to drug release from inside of emulsion droplets to the outside aqueous media during formulation digestion.
5.0 Conclusions
A physical model considering the simultaneous diffusion of free solute and micelle-solubilized solute across the aqueous boundary layer surrounding a dissolving drug particle, the partitioning and permeation of drug across the oil-water interface of emulsion droplet surfaces, and the lipid digestion process, adequately describes the influence of ingested lipids on both: 1. solid drug dissolution when dosed with food-associated lipids, and 2. drug release from SEDDS in simulated intestinal fluids. It is noted that the proposed model does not address other relevant factors affecting drug absorption – such as lymphatic drug transport, drug permeability across the intestinal wall, and drug metabolism. Nevertheless, this modeling approach can be utilized in a system-based model that incorporates the aforementioned factors and pharmacokinetics in order to enable quantitative prediction of impact of the presence of fat-rich food or a lipid based drug delivery system in the GI tract on bioavailability of drugs.
Supplementary Material
Acknowledgments
The first and third authors were supported by a National Science Foundation Career Award (grant # CBET-0748048).
Abbreviations
- BS
bile salts
- EPR
electron paramagnetic resonance
- FA
fatty acids
- GI
gastrointestinal
- HPLC
high performance liquid chromatography
- MG
monoglycerides
- NaTDC
sodium taurodeoxycholate
- PC
L-alpha-phosphatidylcholine
- PL
phospholipids
- SEDDS
self-emulsifying drug delivery systems
- TB
TEMPOL benzoate
References
- 1.Porter CJH, Charman WN. In vitro assessment of oral lipid based formulations. Adv Drug Deliv Rev. 2001;50(Supplement 1):S127–S47. doi: 10.1016/s0169-409x(01)00182-x. [DOI] [PubMed] [Google Scholar]
- 2.DiSanto AR, Golden G. Effect of Food on the Pharmacokinetics of Clozapine Orally Disintegrating Tablet 12.5mg: A Randomized, Open-Label, Crossover Study in Healthy Male Subjects. Clinical Drug Investigation. 2009;29(8):539–49. doi: 10.2165/00044011-200929080-00004. [DOI] [PubMed] [Google Scholar]
- 3.Chow H-HS, Hakim IA, Vining DR, Crowell JA, Ranger-Moore J, Chew WM, et al. Effects of dosing condition on the oral bioavailability of green tea catechins after single-dose administration of polyphenon E in healthy individuals. Anglais. 2005;11(12):4627–33. doi: 10.1158/1078-0432.CCR-04-2549. [DOI] [PubMed] [Google Scholar]
- 4.Berliner L, Spin J, Labeling I. Theory and Application. New York: Academic Press Inc; 1976. [Google Scholar]
- 5.Hubbell W, Altenbach C. Site-Directed Spin Labeling of Membrane Proteins. In: White S, editor. Membrane Protein Structure. Springer; New York: 1994. pp. 224–48. [Google Scholar]
- 6.Melanson M, Sood A, Torok F, Torok M. Introduction to spin label electron paramagnetic resonance spectroscopy of proteins. Biochem Mol Biol Educ. 2013;41(3) doi: 10.1002/bmb.20677. [DOI] [PubMed] [Google Scholar]
- 7.Rübe A, Klein S, Mäder K. Monitoring of in vitro fat digestion by electron paramagnetic resonance spectroscopy. Pharm Res. 2006;23(9):2024–9. doi: 10.1007/s11095-006-9069-3. [DOI] [PubMed] [Google Scholar]
- 8.Jantratid E, Janssen N, Reppas C, Dressman J. Dissolution media simulating conditions in the proximal human gastrointestinal tract: An update. Pharm Res. 2008;25(7):1663–76. doi: 10.1007/s11095-008-9569-4. [DOI] [PubMed] [Google Scholar]
- 9.Sek L, Porter CJH, Kaukonen AM, Charman WN. Evaluation of the in-vitro digestion profiles of long and medium chain glycerides and the phase behaviour of their lipolytic products. Journal of Pharmacy and Pharmacology. 2002;54(1):29–41. doi: 10.1211/0022357021771896. [DOI] [PubMed] [Google Scholar]
- 10.Buyukozturk F, Benneyan JC, Carrier RL. Digestion of self-emulsifying drug delivery systems: kinetics and potential impact on lymphatic transport. 2013 Manuscript submitted for publication. [Google Scholar]
- 11.Di Maio S, Carrier RL. Gastrointestinal contents in fasted state and post-lipid ingestion: In vivo measurements and in vitro models for studying oral drug delivery. Journal of Controlled Release. 2011;151(2):110–22. doi: 10.1016/j.jconrel.2010.11.034. [DOI] [PubMed] [Google Scholar]
- 12.Fatouros DG, Nielsen FS, Douroumis D, Hadjileontiadis LJ, Mullertz A. In vitro–in vivo correlations of self-emulsifying drug delivery systems combining the dynamic lipolysis model and neuro-fuzzy networks. European Journal of Pharmaceutics and Biopharmaceutics. 2008;69(3):887–98. doi: 10.1016/j.ejpb.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 13.Sek L, Porter CJH, Charman WN. Characterization and quantification of medium chain and long chain triglycerides and their in vitro digestion products, by HPTLC coupled with in situ densitometric analysis. Journal of Pharmaceutical and Biomedical Analysis. 2001;25(3–4):651–61. doi: 10.1016/s0731-7085(00)00528-8. [DOI] [PubMed] [Google Scholar]
- 14.Carrière F, Renou C, Lopez V, De Caro J, Ferrato F, Lengsfeld H, et al. The specific activities of human digestive lipases measured from the in vivo and in vitro lipolysis of test meals. Gastroenterology. 2000;119(4):949–60. doi: 10.1053/gast.2000.18140. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, McClements DJ. New Mathematical Model for Interpreting pH-Stat Digestion Profiles: Impact of Lipid Droplet Characteristics on in Vitro Digestibility. Journal of Agricultural and Food Chemistry. 2010;58(13):8085–92. doi: 10.1021/jf101325m. [DOI] [PubMed] [Google Scholar]
- 16.Reis P, Holmberg K, Watzke H, Leser M, Miller R. Lipases at interfaces: a review. Adv Colloid Interface Sci. 2009;147:237–50. doi: 10.1016/j.cis.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 17.Higuchi WI. Effects of interacting colloids on transport rates. Journal of Pharmaceutical Sciences. 1964;53(5):532–5. doi: 10.1002/jps.2600530516. [DOI] [PubMed] [Google Scholar]
- 18.Gamsiz E, Ashtikar M, Crison J, Woltosz W, Bolger M, Carrier R. Predicting the Effect of Fed-State Intestinal Contents on Drug Dissolution. Pharm Res. 2010;27(12):2646–56. doi: 10.1007/s11095-010-0264-x. [DOI] [PubMed] [Google Scholar]
- 19.Bikhazi AB, Higuchi WI. Interfacial barrier limited interphase transport of cholesterol in the aqueous polysorbate 80—hexadecane system. Journal of Pharmaceutical Sciences. 1970;59(6):744–8. doi: 10.1002/jps.2600590603. [DOI] [PubMed] [Google Scholar]
- 20.Rangel-Yagui C, Pessoa AJ, LCT Micellar solubilization of drugs. J Pharm Pharm Sci. 2005;8(2):147–65. [PubMed] [Google Scholar]
- 21.Spivak W, Morrison C, Devinuto D, Yuey W. Spectrophotometric determination of the critical micellar concentration of bile salts using bilirubin monoglucuronide as a micellar probe. Utility of derivative spectroscopy. doi: 10.1042/bj2520275. (0264-6021 (Print)) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J, Carr PW. Accuracy of Empirical Correlations for Estimating Diffusion Coefficients in Aqueous Organic Mixtures. Analytical Chemistry. 1997;69(13):2530–6. doi: 10.1021/ac961005a. [DOI] [PubMed] [Google Scholar]
- 23.Crison JR, Shah VP, Skelly JP, Amidon GL. Drug dissolution into micellar solutions: Development of a convective diffusion model and comparison to the film equilibrium model with application to surfactant-facilitated dissolution of carbamazepine. Journal of Pharmaceutical Sciences. 1996;85(9):1005–11. doi: 10.1021/js930336p. [DOI] [PubMed] [Google Scholar]
- 24.Altenbach C. 2013 Jan 25; Available from: https://sites.google.com/site/altenbach/labview-programs/epr-programs.
- 25.Johnson KC. Intellipharm L, editor. Intellipharm. http://www.intellipharm.com.
- 26.Ransac S, Ivanova M, Panaiotov I, Verger R. Monolayer Techniques for Studying Lipase Kinetics. 1998:279–302. doi: 10.1385/1-59259-581-2:279. [DOI] [PubMed] [Google Scholar]
- 27.Williams HD, Sassene P, Kleberg K, Bakala-N’Goma J-C, Calderone M, Jannin V, et al. Toward the establishment of standardized in vitro tests for lipid-based formulations, part 1: Method parameterization and comparison of in vitro digestion profiles across a range of representative formulations. Journal of Pharmaceutical Sciences. 2012;101(9):3360–80. doi: 10.1002/jps.23205. [DOI] [PubMed] [Google Scholar]
- 28.Horter D, Dressman JB. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 1997;25(1):3–14. doi: 10.1016/s0169-409x(00)00130-7. [DOI] [PubMed] [Google Scholar]
- 29.Persson E, Gustafsson A-S, Carlsson A, Nilsson R, Knutson L, Forsell P, et al. The Effects of Food on the Dissolution of Poorly Soluble Drugs in Human and in Model Small Intestinal Fluids. Pharm Res. 2005;22(12):2141–51. doi: 10.1007/s11095-005-8192-x. [DOI] [PubMed] [Google Scholar]
- 30.Kaukonen AM, Boyd BJ, Charman WN, Porter CJH. Drug Solubilization Behavior During in Vitro Digestion of Suspension Formulations of Poorly Water-Soluble Drugs in Triglyceride Lipids. Pharm Res. 2004;21(2):254–60. doi: 10.1023/b:pham.0000016283.87709.a9. [DOI] [PubMed] [Google Scholar]
- 31.Fernandez S, Chevrier S, Ritter N, Mahler B, Demarne F, Carrière F, et al. In Vitro Gastrointestinal Lipolysis of Four Formulations of Piroxicam and Cinnarizine with the Self Emulsifying Excipients Labrasol® and Gelucire® 44/14. Pharm Res. 2009;26(8):1901–10. doi: 10.1007/s11095-009-9906-2. [DOI] [PubMed] [Google Scholar]
- 32.Dahan A, Hoffman A. Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs. Journal of Controlled Release. 2008;129:1–10. doi: 10.1016/j.jconrel.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 33.Lurie DJ, Mäder K. Monitoring drug delivery processes by EPR and related techniques—principles and applications. Advanced Drug Delivery Reviews. 2005;57(8):1171–90. doi: 10.1016/j.addr.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 34.Sugano K, Kataoka M, da Costa Mathews C, Yamashita S. Prediction of food effect by bile micelles on oral drug absorption considering free fraction in intestinal fluid. Eur J Pharm Sci. 2010;40(2):118–24. doi: 10.1016/j.ejps.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 35.Vertzoni M, Markopoulos C, Symillides M, Goumas C, Imanidis G, Reppas C. Luminal Lipid Phases after Administration of a Triglyceride Solution of Danazol in the Fed State and Their Contribution to the Flux of Danazol Across Caco-2 Cell Monolayers. Molecular Pharmaceutics. 2012;9(5):1189–98. doi: 10.1021/mp200479f. [DOI] [PubMed] [Google Scholar]
- 36.MacGregor KJ, Embleton JK, Lacy JE, Perry EA, Solomon LJ, Seager H, et al. Influence of lipolysis on drug absorption from the gastro-intestinal tract. Advanced Drug Delivery Reviews. 1997;25(1):33–46. [Google Scholar]
- 37.Singh P, Desai SJ, Flanagan DR, Simonelli AP, Higuchi WI. Mechanistic study of the influence of micelle solubilization and hydrodynamic factors on the dissolution rate of solid drugs. Journal of Pharmaceutical Sciences. 1968;57(6):959–65. doi: 10.1002/jps.2600570608. [DOI] [PubMed] [Google Scholar]
- 38.Surpuriya V, Higuchi WI. Interfacially controlled transport of micelle-solubilized sterols across an oil/water interface in two ionic surfactant systems. Journal of Pharmaceutical Sciences. 1972;61(3):375–9. doi: 10.1002/jps.2600610311. [DOI] [PubMed] [Google Scholar]
- 39.Fatouros D, Deen GR, Arleth L, Bergenstahl B, Nielsen F, Pedersen J, et al. Structural Development of Self Nano Emulsifying Drug Delivery Systems (SNEDDS) During In Vitro Lipid Digestion Monitored by Small-angle X-ray Scattering. Pharm Res. 2007;24(10):1844–53. doi: 10.1007/s11095-007-9304-6. [DOI] [PubMed] [Google Scholar]
- 40.Kossena GA, Charman WN, Boyd BJ, Porter CJH. Influence of the intermediate digestion phases of common formulation lipids on the absorption of a poorly water-soluble drug. Journal of Pharmaceutical Sciences. 2005;94(3):481–92. doi: 10.1002/jps.20260. [DOI] [PubMed] [Google Scholar]
- 41.Anby MU, Williams HD, McIntosh M, Benameur H, Edwards GA, Pouton CW, et al. Lipid Digestion as a Trigger for Supersaturation: Evaluation of the Impact of Supersaturation Stabilization on the in Vitro and in Vivo Performance of Self-Emulsifying Drug Delivery Systems. Molecular Pharmaceutics. 2012;9(7):2063–79. doi: 10.1021/mp300164u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.