

Abstract
Vasopressin signaling is critical for the regulation of urea transport in the inner medullary collecting duct (IMCD). Increased urea permeability is driven by a vasopressin-mediated elevation of cAMP that results in the direct phosphorylation of urea transporter (UT)-A1. The identification of cAMP-sensitive phosphorylation sites, Ser486 and Ser499, in the rat UT-A1 sequence was the first step in understanding the mechanism of vasopressin action on the phosphorylation-dependent modulation of urea transport. To investigate the significance of multisite phosphorylation of UT-A1 in response to elevated cAMP, we used highly specific and sensitive phosphosite antibodies to Ser486 and Ser499 to determine cAMP action at each phosphorylation site. We found that phosphorylation at both sites was rapid and sustained. Furthermore, the rate of phosphorylation of the two sites was similar in both mIMCD3 cells and rat inner medullary tissue. UT-A1 localized to the apical membrane in response to vasopressin was phosphorylated at Ser486 and Ser499. We confirmed that elevated cAMP resulted in increased phosphorylation of both sites by PKA but not through the vasopressin-sensitive exchange protein activated by cAMP pathway. These results elucidate the multisite phosphorylation of UT-A1 in response to cAMP, thus providing the beginning of understanding the intracellular factors underlying vasopressin stimulation of urea transport in the IMCD.
Keywords: vasopressin; urea transport; adenosine 3′,5′-cyclic monophosphate; urea transporter-A1
vasopressin regulation of urea transporter (UT)-A1 is a vital part of producing concentrated urine (12). We and others have shown that elevated intracellular cAMP, elicited by vasopressin, increased urea permeability and the abundance of phosphorylated (p)UT-A1 (20), suggesting that phosphorylation regulates urea transport. This hypothesis ultimately led to the identification of Ser486 in UT-A1 by phosphoproteomic analysis as a potential vasopressin-stimulated phosphorylation site (10). Our laboratory also identified Ser486 as a potential vasopressin-stimulated phosphorylation site and identified Ser499 as a second potential phosphorylation site (3). Using site-directed mutagenesis and transient transfection in heterologous expression systems, we showed that both Ser486 and Ser499 were phosphorylated in a cAMP-dependent manner (3).
The identification of multiple vasopressin-sensitive phosphorylation sites in UT-A1 is key to the regulation of transporter function. Multisite protein phosphorylation can provide a precise tool for dynamic regulation of downstream signaling. Furthermore, different phosphorylation profiles of a single protein might be linked to different cellular functions. A study (19) of another protein in the inner medullary collecting duct (IMCD), a water channel, aquaporin-2 (AQP2), also identified multiple vasopressin-sensitive phosphorylation sites in its protein sequence. Investigations have shown that each identified residue has a unique contribution to the localization of AQP2 and subsequent water permeability (19). Our initial investigations found that mutation of both Ser486 and Ser499 to alanines, but not either one alone, eliminated vasopressin stimulation of UT-A1 apical plasma membrane accumulation and urea transport, indicating that at least one of these serines must be phosphorylated to increase urea flux and UT-A1 accumulation in the apical plasma membrane (3). On account of these findings and the importance of posttranslational modifications in regulating another IMCD membrane protein, AQP2, there is a need for further investigation into the physiological significance of the two cAMP-sensitive phosphorylation sites in UT-A1 and the role that these sites may have in the localization and regulation of the transporter.
In addition to modifying activity and subcellular localization, multisite phosphorylation of UT-A1 may be a mechanism for introducing control by different signaling pathways. The cellular response to vasopressin, mediated by cAMP, is generally thought to occur through PKA; however, cAMP may have several downstream targets. Previous work has shown that only 50% of forskolin-stimulated urea flux is inhibited by H-89, a PKA inhibitor (8). This led to the investigation of PKA-independent pathways triggered by increased cAMP in the IMCD, such as those mediated by exchange protein activated by cAMP (Epac), a guanine nucleotide exchange factor for both Rap1 and Rap2 (7). We discovered that stimulation of Epac increases urea transport, accumulation of UT-A1 at the plasma membrane, and UT-A1 phosphorylation (22). Given its localization at the membrane of the renal epithelium (16), it is not surprising that Epac is involved in cAMP-mediated regulation of apical membrane processes, such as phosphorylation of UT-A1.
Thus far, a major difficulty in studying the cAMP-sensitive multisite phosphorylation of UT-A1 has been due to a lack of antibodies specific for the detection of phosphorylation at Ser486 and Ser499. Our group (11) has previously characterized an antibody that specifically recognizes phosphorylation of UT-A1 at Ser486, and, in the present study, we developed a highly specific and sensitive phosphosite antibody to Ser499. By combining these unique tools, we will elucidate the need for two vasopressin-sensitive phosphorylation sites for the regulation of UT-A1.
METHODS
Animals.
All animal protocols were approved by the Institutional Animal Care and Use Committee of Emory University. Male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA), weighing 100–150 g, received free access to water and standard rat chow (Test diet 5001, Purina) containing 23% protein.
Cells.
mIMCD3 cells (American Type Culture Collection) were grown in equal mixtures of DMEM and Ham's F-12 medium supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified atmosphere of 95% air-5% CO2. Cells were grown on six-well plates. At 60% confluence, cells were transfected with UT-A1 (wild type), UT-A1-S499A, or vehicle using Effectene (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Maximal protein expression occurred after 36 h.
Other experiments used a mIMCD3 cell line that stably expressed UT-A1 previously generated in our laboratory (11); mIMCD3 cells were grown under the same conditions as described above.
Tissue and cell sample preparation.
Previously generated stable mIMCD3-UT-A1 cells and/or cells transfected with UT-A1 (wild type) UT-A1-S486A, or UT-A1-S499A were grown to confluence in six-well plates. After the described treatments, cells were scraped from the surface and solubilized by boiling in Laemmli buffer.
Kidneys were removed, and the inner medulla was collected. For ex vivo treatments, inner medullary tissue was cut into small pieces (1 × 1 × 1 mm) and incubated in DMEM with vehicle or the designated treatment(s) for the indicated times at 37°C. For Western blot analysis, tissues were homogenized in ice-cold isolation buffer [10 mM triethanolamine, 250 mM sucrose (pH 7.6), 1 μg/ml leupeptin, and 2 mg/ml PMSF], and SDS was then added to a final concentration of 1%. Samples were then solubilized by boiling in Laemmli buffer.
Metabolic labeling with [32P]orthophosphate.
Metabolic labeling with [32P]orthophosphate was performed with mIMCD3-UT-A1 cells and inner medullary rat tissue as previously described (2, 3). Briefly, confluent cells or inner medulla pieces were incubated in phosphate-free DMEM containing 0.2 mCi/ml [32P]orthophosphate. After the 3-h labeling period, cells or tissues were incubated for a further 30 min with either vehicle or the designated treatment(s). Lysates were prepared, and UT-A1 was immunoprecipitated. Precipitated proteins were separated by SDS-PAGE, and radiolabeled UT-A1 was determined by autoradiography of the dried gel.
Primary antibodies.
Three primary antibodies were used in these experiments: 1) a polyclonal antibody to the COOH terminal of UT-A1 to assess total UT-A1 (14), 2) a phospho-specific antibody to UT-A1 phosphorylated at Ser486 (11), and 3) a new phospho-specific antibody to UT-A1 phosphorylated at Ser499. To acquire the 499 phospho-antibody, we commissioned PhosphoSolutions (Aurora, CO) to assist in the generation of the antibody. We designed an immunizing peptide prepared against our site of interest: SIRRRS(P)KVFGK[C]-NH2. Using this phosphopeptide, a rabbit was immunized, and serum was collected 8 wk later. To isolate the 499 phospho-specific antibody, the IgG fraction was collected from the serum and applied to a phospho-peptide affinity column. This column bound both the 499 phospho-specific antibody and a pan-specific antibody. These two antibodies were eluted from the phosphopeptide affinity column and then applied to a dephosphopeptide affinity column. This column bound the pan-specific antibody, allowing the 499 phospho-specific antibody to be collected in the flowthrough. The 499 phospho-specific antibody was first characterized at PhosphoSolutions by ELISA before further characterization in our laboratory, as described in results.
Western blot analysis.
Proteins (20 μg/lane) were size separated by SDS-PAGE on 10% gels and then electroblotted to polyvinylidene difluoride membranes (Immobilon, Millipore, Bedford, MA). After being blocked with 5% nonfat dry milk for 1 h, blots were incubated with primary antibody overnight at 4°C. Blots were washed three times in Tris-buffered saline with 0.5% Tween 20 and then incubated for 2 h with Alexa fluor 680-linked anti-rabbit IgG (Molecular Probes, Eugene, OR). Bound secondary antibody was visualized using infrared detection with the LI-COR Odyssey protein analysis system (Lincoln, NE), and densitometry of the desired band was collected. Blots were then stained with Coomassie, and densitometry of protein loading was determined by ImageJ. Densitometry values collected from the Coomassie staining were used to normalize Western blot data.
Immunohistochemistry.
Kidneys from rats that had been injected in the interperitoneal cavity with saline or vasopressin 45 min before they were perfusion fixed with 4% paraformaldehyde were embedded in paraffin and sectioned into 4-μm slices. Tissue slices were stained overnight with primary antibodies diluted in PBS (1:1,000 anti-COOH-terminal UT-A1, 1:100 Ser486-pUT-A1, and 1:200 Ser499-pUT-A1) and incubated for 2 h with Alexa fluor 488-linked goat anti-rabbit IgG (Invitrogen, Carlsbad, CA) for fluorescent confocal microscopy, as previously described (12). Slices for confocal micrographs were also treated with the nuclear stain 4′,6-diamidino-2-phenylindole. Confocal microscopy was performed with a Zeiss LSM 510 META ZEN confocal microscope and LSM ZEN imaging software.
Statistics.
Data are presented as means ± SE; comparisons were made with either an unpaired t-test with Welch's correction or one-way ANOVA and Fisher's least-significant-difference posttest. P values of <0.05 were considered significant.
RESULTS
Phospho-specific antibody to Ser499 in UT-A1.
We generated an antibody that specifically detects UT-A1 phosphorylation at Ser499. Forskolin treatment of mIMCD3 cells transiently transfected with UT-A1 revealed a significant increase in UT-A1 phosphorylation at Ser499 without a change in total protein abundance (Fig. 1A). To determine specificity to Ser499, mIMCD3 cells were transfected with a UT-A1 construct where Ser499 was changed to Ala499 by site-directed mutagenesis. Although replacing the serine at site 499 did not affect expression or protein structure, phosphorylation at site 499 was undetectable with the Ser499-pUT-A1 antibody in the mutated construct (Fig. 1B).
Fig. 1.
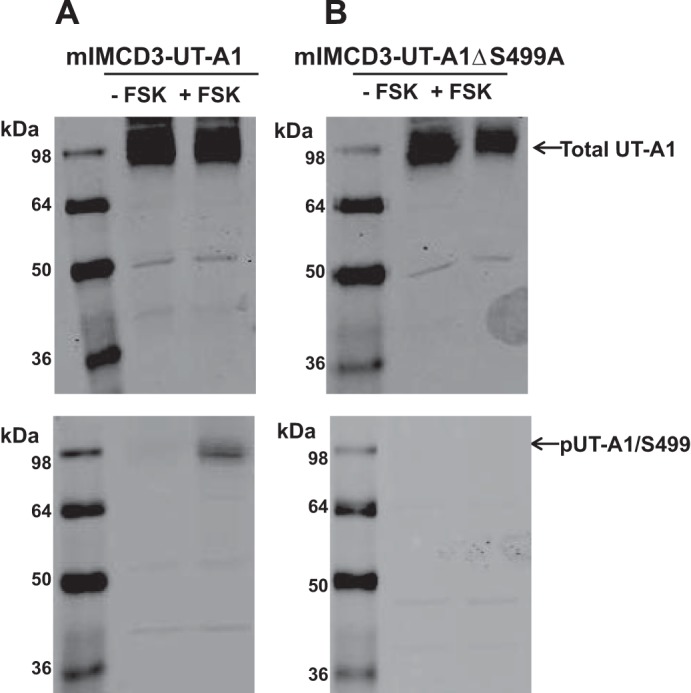
Phospho-antibody is specific for phosphorylation at urea transporter (UT)-A1 at Ser499 (S499). A: mIMCD3 cells transfected with UT-A1 (mIMCD3-UT-A1) were treated without (−) forskolin (FSK; vehicle) or with (+) FSK (10 μM) for 20 min at 37°C. After treatment, cells were lysed in RIPA buffer containing protease and phosphatase inhibitors. Shown is a representative Western blot that was probed with UT-A1 antibody (top) or the generated phospho-specific antibody for S499 [phosphorylated (p)UT-A1/S499; bottom]. B: mIMCD3 cells were transfected with UT-A1 containing a site-directed mutation at S499 to alter this residue to an alanine (mIMCD3-UT-A1ΔS499A). Cells were then treated with FSK in a similar manner to mIMCD3-UT-A1. Western blot analysis confirmed that UT-A1ΔS499A expression was identified by the UT-A1 antibody at the expected molecular weight (top) and that pUT-A1/S499 recognized phosphorylation at S499 only (bottom). n = 4.
Elevated cAMP increases UT-A1 phosphorylation at Ser486 and Ser499 at similar rates.
Using a mIMCD3 cell line that stably expresses UT-A1, the rate of cAMP-mediated phosphorylation at Ser486 and Ser499 was determined (Fig. 2). Using Western blot analysis on samples treated with forskolin for increasing periods of time, we observed that phosphorylation at both Ser486 and Ser499 was increased within 1 min of forskolin treatment (Fig. 2A). Extending the time of forskolin treatment revealed that maximum phosphorylation of both sites was reached at 2 min and remained stable for the remaining 20 min of observation (Fig. 2B). There was no variation in the rate of phosphorylation in response to increased cAMP between Ser486 and Ser499.
Fig. 2.
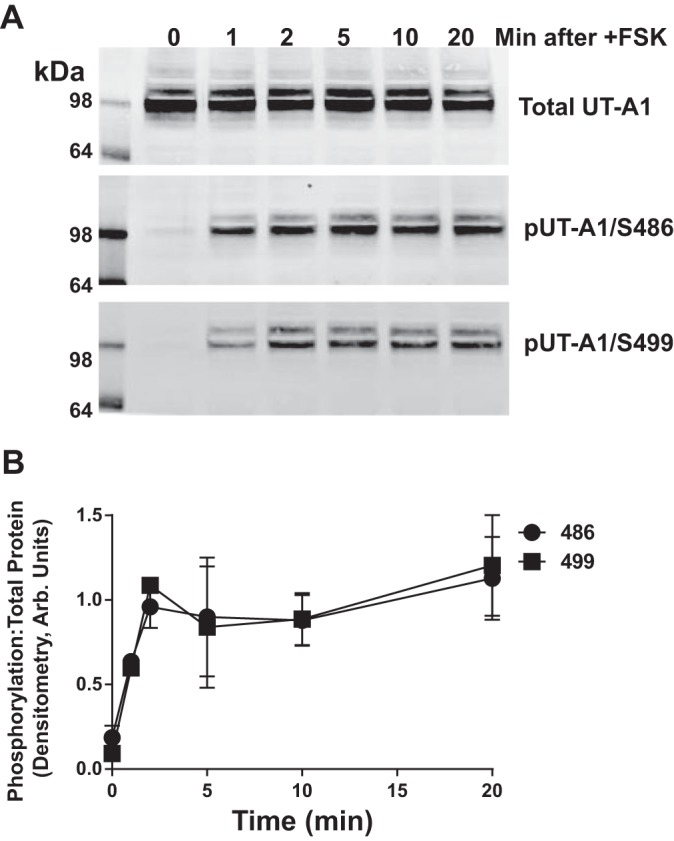
cAMP-mediated phosphorylation of UT-A1 at Ser486 (S486) and S499 occurs simultaneously. Stably transfected mIMCD3-UT-A1 cells were treated with FSK (10 μM) for increasing time increments. Lysate collected at each time point was subjected to Western blot analysis and probed for UT-A1 (top), phosphorylation at S486 (pUT-A1/S486, middle), and S499 (pUT-A1/S499; bottom). A: representative Western blots. B: time course of phosphorylation. Values are means ± SE; n = 4.
Generation of the Ser499-pUT-A1 antibody, in combination with the existing Ser486-pUT-A1 antibody, allowed us to investigate individual phosphorylation events in the whole renal medulla; a venture that has not been previously available. In rat medullary tissue, UT-A1 is glycosylated to different extents (4). Both glycosylation forms of the transporter (97 and 117 kDa) were phosphorylated (Fig. 3A). Specific phosphorylation at Ser486 and Ser499 were also detected in both glycosylated forms of UT-A1 (Fig. 3A). Total phosphorylation of UT-A1 in the rat inner medulla was increased with increasing periods of incubation with forskolin (Fig. 3B), similar to a previous report (23) in other models. UT-A1 phosphorylation at Ser486 and Ser499 were elevated in the inner medulla within 2 min and remained constant after 10 min of forskolin incubation (Fig. 3B).
Fig. 3.
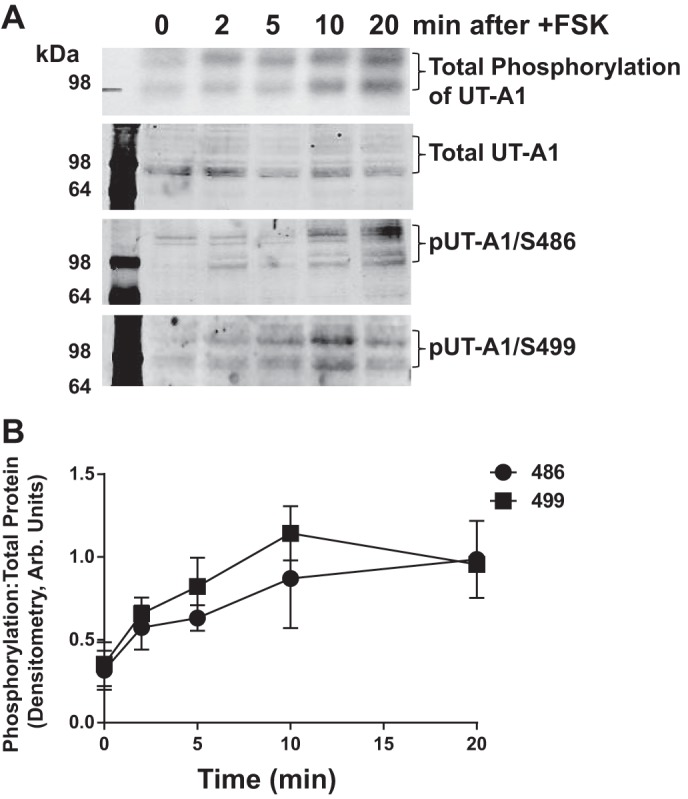
Phosphorylation of UT-A1 at S486 and S499 occur on the same time scale as total phosphorylation. Inner medullas were dissected from five Sprague Dawley rats, minced, and pooled together. Tissue was incubated in phosphate-free media containing [32P]orthophosphate (0.1 mCi/ml) for 2 h at 37°C. After tissues were loaded with phosphate, FSK (10 μM) was added to the pooled tissue, and equal volumes of tissue were collected and lysed at the indicated time points. Next, UT-A1 was immunoprecipitated. Autoradiography was used to determine total phosphorylation of UT-A1 (top). Western blot analysis was performed on the remaining samples, and immunoblot analysis was performed for total UT-A1, phosphorylation of UT-A1 at S486 (pUT-A1/S486), and phosphorylation of UT-A1 at S499 (pUT-A1/S499). A: representative autoradiographic images and Western blots, where brackets indicate the multiple glycosylation forms of UT-A1 observed in the tissue. B: time course of phosphorylation. Values are means ± SE; n = 5.
UT-A1 phosphorylation at Ser486 and Ser499 sites occur independently of one another.
Multisite phosphorylation of a protein may depend on a hierarchical organization where phosphorylation of one site is subordinate to the phosphorylation of a separate site. Conversely, phosphorylation of multiple sites may occur in an independent manner. Using site-directed mutagenesis, we found that forskolin increased phosphorylation of the UT-A1 Ser499 site in the absence of an active Ser486 site (Fig. 4). Furthermore, forskolin stimulated phosphorylation at Ser486 despite rendering the Ser499 site inactive (Fig. 4).
Fig. 4.
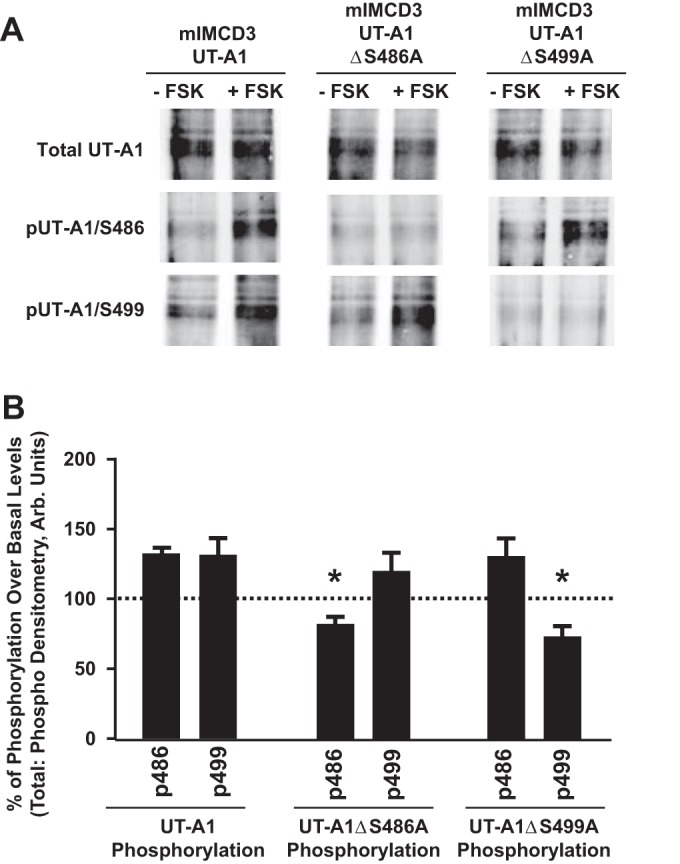
Elevated cAMP stimulates independent phosphorylation of UT-A1 residues S486 and S499. A: mIMCD3 cells were transfected with 1) unaltered UT-A1 (mIMCD3-UT-A1), 2) UT-A1 containing a site-directed mutation at S486 (mIMCD3-UT-A1ΔS486A), or 3) UT-A1 containing a site-directed mutation at S499 (mIMCD3-UT-A1ΔS499A). Thirty-six hours after transfection, cells were incubated with either vehicle (−FSK) or FSK (+FSK; 10 μM) for 20 min at 37°C. After treatment, cells were lysed in RIPA buffer containing protease and phosphatase inhibitors. UT-A1 was enriched in all samples by performing immunoprecipitation with UT-A1 antibody. Shown are representative Western blots probed with UT-A1 antibody (top), the phospho-specific antibody for site S486 (pUT-A1/S486; middle), or the phospho-specific antibody for site S499 (pUT-A1/S499; bottom). B: amount of detected phosphorylation relative to untreated samples, which were normalized to 100%. Values are means ± SE; n = 3. *P < 0.05 (significant difference).
UT-A1 phosphorylated at Ser486 and Ser499 in response to vasopressin is localized to the apical membrane of the IMCD.
Because a majority of cAMP in the IMCD is synthesized in response to vasopressin (15), phosphorylation of UT-A1 at Ser486 and Ser499 is likely a downstream effect of this hormone. Inner medullary tissue from vasopressin-treated rats had increased phosphorylation of both glycoprotein forms of UT-A1 at these two cAMP-sensitive sites (Fig. 5).
Fig. 5.
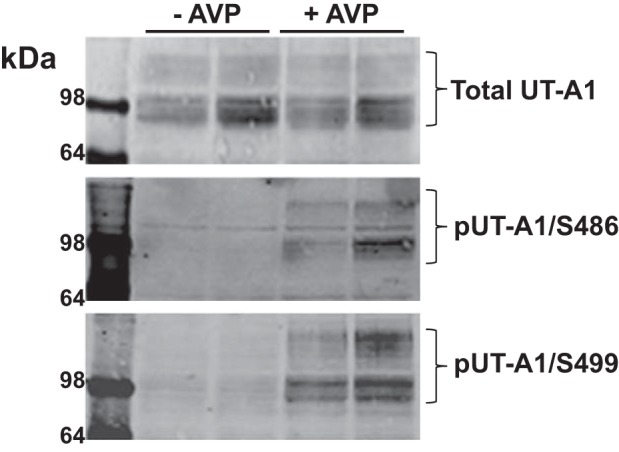
Elevated vasopressin increases phosphorylation of UT-A1 at S486 and S499 in the rat inner medulla. Sprague Dawley rats were injected without (−) arginine vasopressin (AVP; saline) or with (+) 5 nmol AVP 45 min before kidneys were removed and the inner medulla was collected and subsequently homogenized. The resulting lysates were subjected to Western blot analysis for the determination of vasopressin effects on total UT-A1, phosphorylation of UT-A1 at S486 (pUT-A1/S486), and phosphorylation of UT-A1 at S499 (pUT-A1/S499). Representative blots are shown with the glycosylated forms of UT-A1 highlighted by brackets. Each lane represents the inner medullas from one animal. n = 4.
To assess the cellular location of UT-A1 after cAMP-mediated phosphorylation in vivo, kidneys from vasopressin-treated rats were examined histologically. Vasopressin increased total UT-A1 accumulation at the apical plasma membrane of the IMCD (Fig. 6), supporting findings of a previous report (13). These images show that UT-A1 phosphorylated at Ser499 was predominately expressed at the apical plasma membrane (Fig. 6). UT-A1 phosphorylated at Ser486 was also found predominately at the apical plasma membrane (Fig. 6), consistent with our previous findings (11).
Fig. 6.

Phosphorylated UT-A1 is located at the apical surface of the inner medullary collecting duct. Sprague-Dawley rats were injected with either saline (sham treatment) or vasopressin (5 nmol) 45 min before kidneys were perfusion fixed and embedded in paraffin. Immunofluorescence was performed on 4-μm sections, and the location of total UT-A1, UT-A1 phosphorylated at S486, and UT-A1 phosphorylated at S499 was determined by confocal microscopy. n = 5.
Phosphorylation of UT-A1 at Ser486 and Ser499 is mediated by PKA.
The common kinase that amplifies cAMP signaling is PKA. Using mIMCD3-UT-A1 cells, we observed that inhibition of PKA by H-89 reduced total phosphorylation of UT-A1 (Fig. 7). Forskolin-treated cells showed that the addition of H-89 not only inhibited phosphorylation of UT-A1 at both Ser486 and Ser499 but also damped basal phosphorylation at these sites as well (Fig. 7).
Fig. 7.
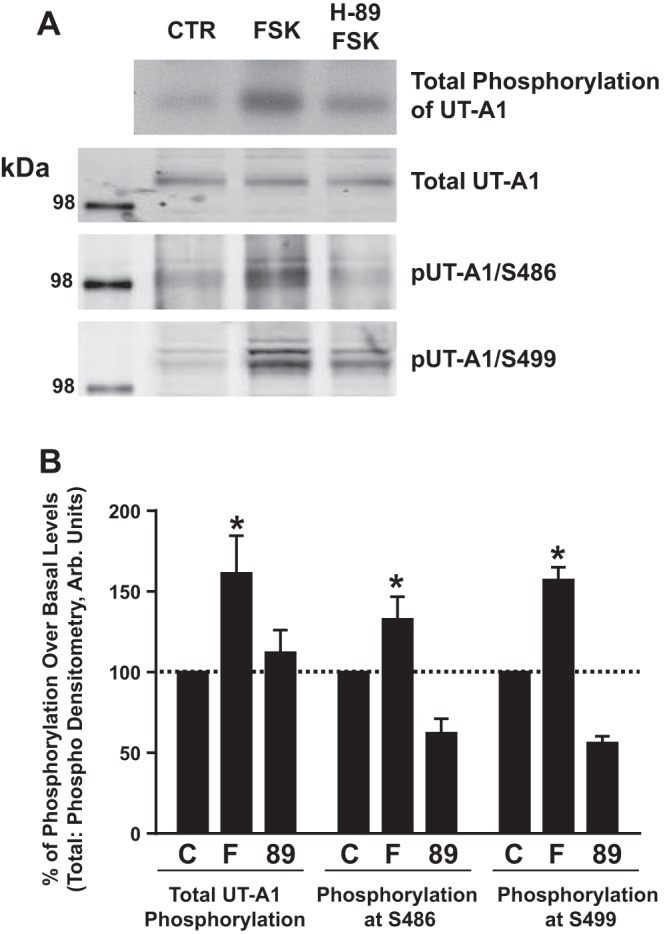
PKA phosphorylates UT-A1 at S486 and S499. Stably transfected mIMCD3-UT-A1 cells were first treated with phosphate-free media containing [32P]orthophosphate (0.1 mCi/ml) for 2 h at 37°C and then treated with either 1) vehicle [control (CTR; C)], 2) FSK (10 μM; F), or 3) H-89 (5 μM; 89) and FSK for 20 min at 37°C. Cells were lysed and subjected to Western blot analysis and autoradiography. A: blots and film from a representative experiment probed with UT-A1, pUT-A1/S486, and pUT-A1/S499 antibodies. B: amount of detected phosphorylation relative to untreated samples, which were normalized to 100%. Values are means ± SE; n = 5. *P < 0.05 (significant difference).
Activation of the Epac pathway does not affect phosphorylation of UT-A1 at Ser486 and Ser499.
Vasopressin also activates an Epac signaling pathway that has been proven to stimulate urea transport, UT-A1 phosphorylation, and UT-A1 accumulation in the plasma membrane of rat IMCDs (22). Consistent with our previous study, total UT-A1 phosphorylation was significantly increased above basal levels when Epac was specifically stimulated pharmacologically with Sp-8-pCPT-2′-O-methyl-cAMPS in mIMCD3-UT-A1 cells (Fig. 8). There was no change in phosphorylation at either Ser486 and Ser499 after Epac stimulation in this cell line (Fig. 8).
Fig. 8.
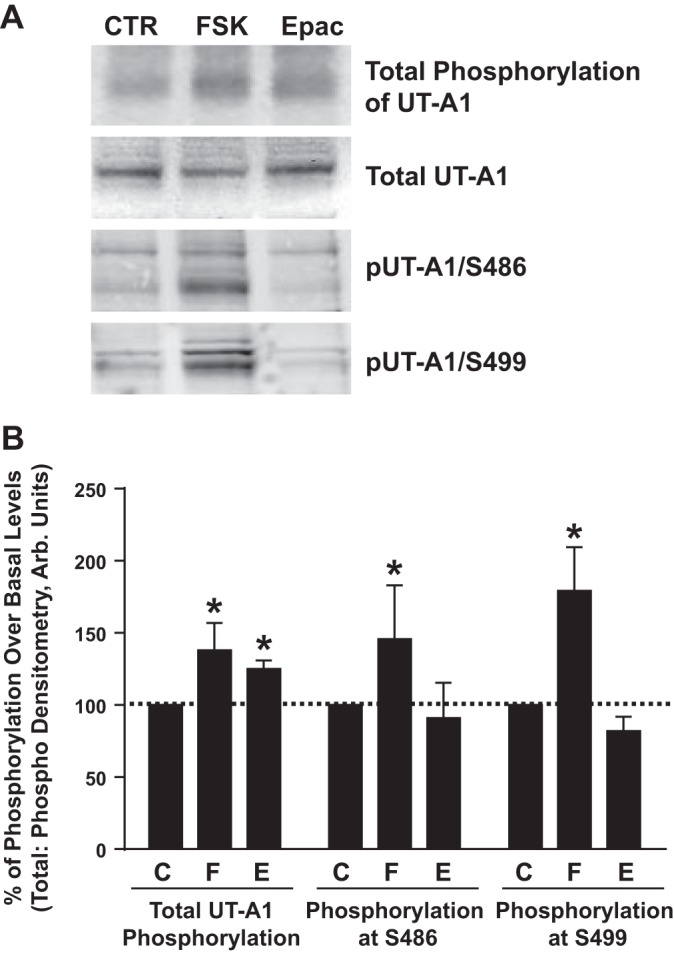
Phosphorylation of UT-A1 at S486 and S499 is independent of exchange protein activated by cAMP (Epac). After treatment with phosphate-free media containing [32P]orthophosphate (0.1 mCi/ml) for 2 h at 37°C, stably transfected mIMCD3-UT-A1 cells were treated with either 1) vehicle (CTR; C), 2) FSK (10 μM; F), or 3) an activator of the Epac pathway (Sp-8-pCPT-2′-O-methyl-cAMPS; 75 μM; E) for 20 min at 37°C. Cells were lysed and subjected to Western blot analysis and autoradiography. A: blots from a representative experiment probed with UT-A1, pUT-A1/S486, and pUT-A1/S499 antibodies. B: amount of detected phosphorylation relative to untreated samples, which were normalized to 100%. Values are means ± SE; n = 5. *P < 0.05 (significant difference).
DISCUSSION
A current challenge in understanding how urine is concentrated is the absence of a detailed knowledge of how vasopressin signaling networks integrate and transmit signals to specific transport proteins in the inner medulla. Although phosphorylation is not the only regulatory system in the IMCD, it is intimately tied to many other intracellular systems that ultimately control water and urea movement across the cell membrane. Therefore, an important step in exploring responsiveness to vasopressin signaling is the identification and characterization of phosphorylation sites on the proteins that regulate water and urea reabsorption, AQP2 and UT-A1. Multisite phosphorylation can modulate the biological activity and subcellular location of target proteins like AQP2 and UT-A1 as well as determine the extent and duration of cellular response; thus, further clarification of the role of each phosphorylation site on these inner medullary transporters is needed.
A considerable amount of work has already justifiably focused on the vasopressin-sensitive phosphorylation sites of AQP2. This water channel has four vasopressin-sensitive phosphorylation sites: Ser256, Ser261, Ser264, and Ser269 (1, 10). Following identification of these sites, further work revealed that Ser256 and Ser261 are involved in AQP2 trafficking to the apical plasma membrane (9) and that elevated cAMP levels result in increased initial phosphorylation of AQP2 at Ser256 followed by increased Ser264 and Ser269 phosphorylation and reduced Ser261 phosphorylation (9, 18, 21). These posttranslational modifications are key for the steady-state redistribution of AQP2 between vesicles and the apical plasma membrane by regulated exocytosis and endocytosis (19). Due to these and many additional nuances of the multiple vasopressin-sensitive phosphorylation sites of AQP2, it is safe to conclude that each residue has a unique contribution to the localization and ultimately function of AQP2. This advanced body of work on AQP2 led us to investigate whether there were similar differences between the two vasopressin-sensitive phosphorylation sites, Ser486 and Ser499, in UT-A1.
Surprisingly, we did not find any difference between Ser486 and Ser499 in terms of the time course of phosphorylation. Both Ser486 and Ser499 were rapidly phosphorylated, and the phosphorylation was sustained for at least 20 min. Further investigation indicated that phosphorylation at Ser486 and Ser499 comprised independent posttranslational events that were not hierarchical in the presence of elevated cAMP. Different phosphorylations could be linked to distinct protein functions or graded effects of a single function (6). Our earlier work showed that phosphorylation of either Ser486 and Ser499 is sufficient for activation, whereas maximal urea permeability is achieved when both sites are phosphorylated (3). In addition to demonstrating that Ser486 and Ser499 are distinct targets for phosphorylation, our data suggest that the two phosphorylation sites provide a possible feedback amplification mechanism to fully activate urea transport. Such a mechanism could result in a graded response and thus allow even low levels of vasopressin to transport measured urea across the IMCD. This is certainly plausible from a physiological standpoint and warrants further future investigation.
The present results also showed that UT-A1 phosphorylated at either Ser486 or Ser499 was localized to the apical membrane in response to elevated vasopressin. Multisite phosphorylation can regulate membrane trafficking and spatiotemporal organization of membrane-associated channels like AQP2. Previously, we found that ablation of both Ser486 and Ser499 in UT-A1 prevented membrane accumulation (3); however, we were unable to determine specific cellular localization of these phosphorylated forms of the transporter until now. Our group has previously found that the large, intracellular loop region of UT-A1 (between transmembrane helixes 6 and 7) containing the phosphorylation sites Ser486 and Ser499 also binds snapin, a component of the soluble N-ethylmaleimide-sensitive factor-activating protein receptor (SNARE) fusion machinery that recruits UT-A1 from intracellular vesicles into the plasma membrane (17). Although the precise location of the snapin-binding site within the 72 amino acids that comprise the examined UT-A1 loop region is unknown, it is likely that the binding site is close to Ser486 and Ser499, suggesting a possible relationship between UT-A1 phosphorylation and snapin binding. Other work has demonstrated the importance of phosphorylation events serving as a biochemical switch for interactions between N-type Ca2+ channels and SNARE protein complexes (5), thus supporting the idea that phosphorylation at Ser486 and Ser499 may regulate UT-A1 localization by controlling its interaction with the necessary exocytotic machinery for trafficking to the apical membrane.
In addition to PKA, our group has reported that another cAMP-stimulated pathway mediated by Epac activity can also increase total UT-A1 phosphorylation (22). The potential for multiple pathways to target the same substrate sites on UT-A1 could result in a complex regulation of the functional status of the transporter compared with the action of a single kinase. Similarly, the presence of two independent phosphorylation sites like Ser486 and Ser499 could indicate that each site is the target of two completely different pathways. Our present results confirm that PKA phosphorylates both Ser486 and Ser499, ruling out the later assumption. The present study also revealed that Epac does not result in the increased the phosphorylation of either Ser486 or Ser499. We can therefore conclude that Epac-mediated phosphorylation of UT-A1, presumably via the MEK-ERK pathway (22), must occur at another site in the transporter, further adding to the complexity of this multitargeted phosphoprotein.
In summary, our recent development of phosphosite-specific antibodies to Ser486 and Ser499 in UT-A1 allowed us to analyze the regulation of the transporter by cAMP-sensitive phosphorylation signaling events in the IMCD. Without the development of these tools, experimental design only allowed for the observation of global phosphorylation events, which was not useful in understanding the physiological significance of multisite phosphorylation of UT-A1. We found that 1) Ser486 and Ser499 phosphorylations are independent from one another and occur at the same rate; 2) modification of UT-A1 by phosphorylation at Ser486 and Ser499 in response to vasopressin results in apical membrane accumulation of the transporter; 3) both Ser486 and Ser499 are targeted by the same kinase, PKA; and 4) despite increased total phosphorylation of UT-A1, the cAMP-activated Epac pathway does not result in the phosphorylation of UT-A1 at Ser486 and Ser499. These findings are the first to elucidate the multisite phosphorylation of UT-A1 in response to vasopressin. Given that multiple phosphorylation events can potentially provide a fastidious tool for dynamic regulation of urea transport, our results provide the framework for understanding the physiological significance of urea handling by the IMCD.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grants K01-DK-82733 and R03-DK-91501 and a Satellite Healthcare Coplan Grant (to M. A. Blount) and by NIDDK Grants R01-DK-89828, R01-DK-41707, and R21-DK-91147 (to J. M. Sands).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: C.A.H. and M.A.B. conception and design of research; C.A.H., L.N.B., R.J.O., D.A.G., F.E.P., J.H.S., and M.A.B. performed experiments; C.A.H., L.N.B., R.J.O., D.A.G., F.E.P., and M.A.B. analyzed data; C.A.H., J.M.S., and M.A.B. interpreted results of experiments; C.A.H., L.N.B., R.J.O., D.A.G., F.E.P., J.H.S., and M.A.B. prepared figures; C.A.H., J.M.S., and M.A.B. drafted manuscript; C.A.H., L.N.B., R.J.O., D.A.G., J.H.S., J.M.S., and M.A.B. edited and revised manuscript; C.A.H., L.N.B., R.J.O., D.A.G., F.E.P., J.H.S., J.M.S., and M.A.B. approved final version of manuscript.
REFERENCES
- 1.Bansal AD, Hoffert JD, Pisitkun T, Hwang S, Chou CL, Boja ES, Wang G, Knepper MA. Phosphoproteomic profiling reveals vasopressin-regulated phosphorylation sites in collecting duct. J Am Soc Nephrol 21: 303–315, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bessay EP, Zoraghi R, Blount MA, Grimes KA, Beasley A, Francis SH, Corbin JD. Phosphorylation of phosphodiesterase-5 is promoted by a conformational change induced by sildenafil, vardenafil, or tadalafil. Front Biosci 12: 1899–1910, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Blount MA, Mistry AC, Fröhlich O, Price SR, Chen G, Sands JM, Klein JD. Phosphorylation of UT-A1 urea transporter at serines 486 and 499 is important for vasopressin-regulated activity and membrane accumulation. Am J Physiol Renal Physiol 295: F295–F299, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradford AD, Terris JM, Ecelbarger CA, Klein JD, Sands JM, Chou CL, Knepper MA. 97- and 117-kDa forms of collecting duct urea transporter UT-A1 are due to different states of glycosylation. Am J Physiol Renal Physiol 281: F133–F143, 2001. [DOI] [PubMed] [Google Scholar]
- 5.Catterall WA. Interactions of presynaptic Ca2+ channels and snare proteins in neurotransmitter release. Ann NY Acad Sci 868: 144–159, 1999. [DOI] [PubMed] [Google Scholar]
- 6.Cohen P. The regulation of protein function by multisite phosphorylation–a 25 year update. Trends Biochem Sci 25: 596–601, 2000. [DOI] [PubMed] [Google Scholar]
- 7.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396: 474–477, 1998. [DOI] [PubMed] [Google Scholar]
- 8.Fröhlich O, Klein JD, Smith PM, Sands JM, Gunn RB. Regulation of UT-A1-mediated transepithelial urea flux in MDCK cells. Am J Physiol Cell Physiol 291: C600–C606, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Hoffert JD, Nielsen J, Yu MJ, Pisitkun T, Schleicher SM, Nielsen S, Knepper MA. Dynamics of aquaporin-2 serine-261 phosphorylation in response to short-term vasopressin treatment in collecting duct. Am J Physiol Renal Physiol 292: F691–F700, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Hoffert JD, Pisitkun T, Wang G, Shen RF, Knepper MA. Quantitative phosphoproteomics of vasopressin-sensitive renal cells: regulation of aquaporin-2 phosphorylation at two sites. Proc Natl Acad Sci USA 103: 7159–7164, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klein JD, Blount MA, Fröhlich O, Denson CE, Tan X, Sim JH, Martin CF, Sands JM. Phosphorylation of UT-A1 on serine 486 correlates with membrane accumulation and urea transport activity in both rat IMCDs and cultured cells. Am J Physiol Renal Physiol 298: F935–F940, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein JD, Blount MA, Sands JM. Molecular mechanisms of urea transport in health and disease. Pflügers Arch 464: 561–572, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klein JD, Frohlich O, Blount MA, Martin CF, Smith TD, Sands JM. Vasopressin increases plasma membrane accumulation of urea transporter UT-A1 in rat inner medullary collecting ducts. J Am Soc Nephrol 17: 2680–2686, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Klein JD, Price SR, Bailey JL, Jacobs JD, Sands JM. Glucocorticoids mediate a decrease in AVP-regulated urea transporter in diabetic rat inner medulla. Am J Physiol Renal Physiol 273: F949–F953, 1997. [DOI] [PubMed] [Google Scholar]
- 15.Knepper MA, Nielsen S, Chou CL, DiGiovanni SR. Mechanism of vasopressin action in the renal collecting duct. Semin Nephrol 14: 302–321, 1994. [PubMed] [Google Scholar]
- 16.Li Y, Konings IB, Zhao J, Price LS, de Heer E, Deen PM. Renal expression of exchange protein directly activated by cAMP (Epac) 1 and 2. Am J Physiol Renal Physiol 295: F525–F533, 2008. [DOI] [PubMed] [Google Scholar]
- 17.Mistry AC, Mallick R, Klein JD, Sands JM, Fröhlich O. Functional characterization of the central hydrophilic linker region of the urea transporter UT-A1: cAMP activation and snapin binding. Am J Physiol Cell Physiol 298: C1431–C1437, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moeller HB, MacAulay N, Knepper MA, Fenton RA. Role of multiple phosphorylation sites in the COOH-terminal tail of aquaporin-2 for water transport: evidence against channel gating. Am J Physiol Renal Physiol 296: F649–F657, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moeller HB, Olesen ET, Fenton RA. Regulation of the water channel aquaporin-2 by posttranslational modification. Am J Physiol Renal Physiol 300: F1062–F1073, 2011. [DOI] [PubMed] [Google Scholar]
- 20.Sands JM, Blount MA, Klein JD. Regulation of renal urea transport by vasopressin. Trans Am Clin Climatol Assoc 122: 82–92, 2011. [PMC free article] [PubMed] [Google Scholar]
- 21.Tamma G, Robben JH, Trimpert C, Boone M, Deen PM. Regulation of AQP2 localization by S256 and S261 phosphorylation and ubiquitination. Am J Physiol Cell Physiol 300: C636–C646, 2011. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Klein JD, Blount MA, Martin CF, Kent KJ, Pech V, Wall SM, Sands JM. Epac regulates UT-A1 to increase urea transport in inner medullary collecting ducts. J Am Soc Nephrol 20: 2018–2024, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang C, Sands JM, Klein JD. Vasopressin rapidly increases phosphorylation of UT-A1 urea transporter in rat IMCDs through PKA. Am J Physiol Renal Physiol 282: F85–F90, 2002. [DOI] [PubMed] [Google Scholar]