

Abstract
Meprin metalloproteases are abundantly expressed in the brush-border membranes of kidney proximal tubules. Meprins are implicated in ischemia-reperfusion (IR)-induced renal injury and diabetic nephropathy. The protein kinase A (PKA) signaling pathway modulates extracellular matrix metabolism in diabetic kidneys. The present study evaluated isoform-specific interactions between the catalytic subunit of PKA (PKA C) and meprins. To this end, cytosolic-enriched kidney proteins from meprin αβ double knockout mice, and purified forms of recombinant mouse PKA Cα, Cβ1, and Cβ2, were incubated with activated forms of either homomeric meprin A or meprin B. The cleaved protein products were subjected to SDS-PAGE and analyzed by Coomassie staining and Western blot analysis. While meprin A only cleaved PKA Cβ1, meprin B cleaved all three PKA C isoforms. Analysis of the proteolytic fragments by mass spectrometry revealed that meprin A and B cleave the PKA C isoforms at defined sites, resulting in unique cleavage products. Michaelis-Menten enzyme kinetics demonstrated that meprin B-mediated cleavage of PKA Cα occurs at a rate consistent with that of other physiologically relevant meprin substrates. Meprin cleavage decreased the kinase activity of PKA Cα, Cβ1, and Cβ2. PKA C levels were higher in diabetic kidneys, with evidence of in vivo fragmentation in wild-type diabetic kidneys. Confocal microscopy showed localization of meprin A in the glomeruli of diabetic kidneys. At 3 h post-IR, PKA C levels in proximal tubules decreased compared with distal tubules, which lack meprins. These data suggest that meprins may impact kidney injury, in part, via modulation of PKA signaling pathways.
Keywords: meprin metalloproteases, protein kinase A (PKA), enzyme processing, isoforms, diabetic nephropathy, ischemia-reperfusion
meprins are zinc metalloproteinases of the astacin family that are most abundantly expressed in the brush-border membranes (BBM) of kidney proximal tubules and small intestines. Expression of meprins was also documented in podocytes (33) and leukocytes (monocytes and macrophages) (48). Meprins are made of two subunits, α and β, which are encoded by two separate genes on human chromosome 6 and 18, respectively (6, 22). These result in two meprin protein isoforms, meprin A and meprin B. Meprin A is either a homo-oligomer of α-subunits or a hetero-oligomer of α- and β-subunits, while meprin B is a homo-oligomer of β-subunits (15). Both the level of expression and the localization of meprins are associated with different types of renal injury, including ischemia-reperfusion (IR)-induced acute kidney injury (7, 8, 34, 50, 52) and diabetic nephropathy (32, 36). In vitro studies showed that meprins are capable of proteolytically processing and/or degrading a variety of protein substrates, including extracellular matrix (ECM) proteins (e.g., collagen IV, nidogen-1, laminin, and fibronectin) (23, 27, 29, 52), bioactive proteins (e.g., gastrin, cholecystokinin, bradykinin, substance P, neurotensin, parathyroid hormone) (9, 27, 47, 57), pro-interleukin 18 (3), and the catalytic subunit of protein kinase A (PKA C) (9). However, data about the in vivo meprin targets and the impact that meprin proteolysis has on these substrates are still limited. Two cytoskeletal proteins, villin and actin, were recently identified as physiological meprin targets in the kidney (34). Identifying additional kidney meprin substrates is important in understanding the role of meprins in kidney disease.
PKA, a serine/threonine kinase, plays a central role in various cell signaling pathways, including growth factor-mediated regulation of cell cycle and survival. The holoenzyme of PKA is a heterotetramer consisting of two regulatory subunits (PKA R) and two catalytic subunits (PKA C). When activated by cAMP, the catalytic subunits are “unleashed” from the regulatory subunits, enabling them to phosphorylate various cellular substrates in the cytoplasm and nucleus. In the nucleus, PKA C phosphorylates the transcription factor, cAMP-responsive element binding protein (CREB), leading to activation of target genes. There are two PKA C isoforms, α and β, which have high sequence homology and are expressed in human, mouse, bovine, and porcine tissues. Both PKA Cα and PKA Cβ have splice variants which result in further PKA C diversity. The PKA Cα gene encodes two splice variants, Cα1 and Cα2 (11, 37, 42), while the PKA Cβ gene encodes 10 different splice variants (30). Among these, the PKA Cα1 and PKA Cβ1 isoforms are ubiquitously expressed while PKA Cβ2 is restricted to neuronal tissue in humans (20). Similarly, the mouse Cβ2 variant (which is homologous to the human Cβ4 variant) is highly expressed in lymphoid tissue. Recently, the PKA signaling pathway was shown to modulate ECM metabolism in kidney cells (44, 45). However, it is currently unclear how individual PKA C isoforms are regulated during this process.
There is evidence for a role for meprins in modulating the PKA signaling pathway. Alhanaty et al. (1, 2) first described proteolysis of the catalytic subunit of PKA Cα (Cα) by an enzyme present in preparations from the intestinal and renal BBMs, the two sites with the most abundant expression of meprin metalloproteinases (1, 2). These studies revealed that the proteolytic activity was specific for the free catalytic subunit and did not cleave either the undissociated, inactive form of the enzyme (R2C2) or the regulatory subunit (R). Degradation of the 40-kDa catalytic subunit was restricted and yielded a distinct 30-kDa fragment that was devoid of kinase activity. The specific recognition of the free catalytic PKA subunit suggested a distinct physiological role that could be regulatory in nature. However, a physiological interaction between meprins and PKA Cα could not be envisioned at that time because the active site of the kinase-splitting membranal proteinase (KSMP), which was later identified as meprin β, faced the exterior (lumen) of the intestinal brush border, while PKA C has an intracellular orientation. However, data from more recent studies showed that, under certain pathological conditions, such as ischemia-reperfusion, meprins are redistributed from the BBM to the cytosol and basolateral membranes of kidney proximal tubular cells (7, 34). This translocation would bring meprins into direct contact with PKA C isoforms, making these PKA C isoforms meprin targets in vivo under these conditions. Meprin interactions with the PKA pathway could thus play a role in the response to insults that cause renal injury. The objectives of the present study were 1) to evaluate the isoform-specific interactions between meprin metalloproteinases and PKA C isoforms, 2) to assess the impact of these interactions on PKA C activity, and 3) to examine the impact that meprins have on PKA C levels during ischemia-reperfusion-induced renal injury and diabetic nephropathy. This knowledge is important in understanding how meprins modulate acute and chronic renal injury.
MATERIALS AND METHODS
Purification and activation of recombinant meprins.
Recombinant forms of homomeric mouse meprin A (α-α) and rat meprin B (β-β) were purified from stably transfected human embryonic kidney (HEK)-293 cells and activated by incubating with trypsin in 20 mM Tris, 150 mM NaCl, pH 7.5, at 37°C for 30 min or 1 h for meprin A and meprin B, respectively, as previously described (34). Meprin activation was stopped by adding soybean trypsin inhibitor (STI) (42), and the trypsin and STI were removed by running the reaction mixtures through a Sephadex G25 column.
Extraction and fractionation of kidney proteins.
Kidney proteins were extracted from meprin αβ double-knockout (KO) mice which lack endogenous meprins. The mice were euthanized by CO2 asphyxiation followed by cervical dislocation. Kidneys were excised, decapsulated, wrapped in aluminum foil, snap-frozen in liquid nitrogen, and stored at −80°C until they were processed for protein extraction. Kidney proteins were extracted and fractionated into BBM- and cytosolic-enriched fractions using differential centrifugation, as previously described (24, 34). Briefly, kidneys were homogenized in nine volumes of ice-cold buffer (2 mM Tris·HCl, pH 7.0, with 10 mM mannitol) supplemented with EDTA-free protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN) and okadoic acid (0.1 mM final concentration). A 1 M stock of MgCl2 was added to a final concentration of 10 mM, and the homogenate was stirred at 4°C for 14 min. The homogenate was then centrifuged for 12 min at 1,500 g, and the sediment was discarded. The supernatant fraction was centrifuged for another 12 min at 15,000 g, and the resulting supernatant fluid was saved as the cytosolic-enriched fraction. The resulting sediment was suspended in five volumes of buffer and MgCl2 added to a final concentration of 10 mM. This was stirred at 4°C for 15 min and then centrifuged at 2,200 g for 12 min. The supernatant fluid from this centrifugation was transferred to a new tube, which was further centrifuged for 12 min at 15,000 g. The resulting pellet was suspended in an appropriate volume of RIPA buffer and saved as the BBM-enriched fraction. A cocktail of EDTA-free protease inhibitors (Roche Diagnostics) and okadoic acid (0.1 mM final concentration) was added to the RIPA buffer to prevent degradation of proteins after tissue disruption. The extracted kidney proteins were quantified by the Lowry protein assay method, using Bio-Rad's Protein reagent (Bio-Rad, Santa Cruz, CA) and stored at −80°C until used.
Evaluation of meprin cleavage of PKA C present in kidney proteins.
Western blot analysis was used to make initial determinations of the levels of PKA C in the different protein fractions and nonfractionated kidney proteins. The PKA C expression levels were shown to be highest in cytosolic-enriched kidney protein fractions. Subsequently, cytosolic-enriched kidney proteins were incubated with purified activated forms of homomeric mouse meprin A (α-α) and rat meprin B (β-β) in Tris buffer (20 mM Tris·HCl, 150 mM NaCl, pH 7.5) at 37°C for 0–3 h. Control reactions were incubated with Tris buffer without meprins and latent forms of the purified meprins. Cleavage products were subjected to electrophoretic separation on SDS polyacrylamide gels and fragmentation or degradation of PKA C evaluated by Western blot analysis with anti-PKA C-specific antibodies (BD Biosciences, San Jose, CA). Western blot analysis was also used to evaluate the levels of PKA C in cytosolic kidney protein extracts. To this end, cytosolic-enriched kidney protein fractions were extracted as previously described (34), and 60 μg of cytosolic kidney proteins from each sample were electrophoretically separated on 10% polyacrylamide gels. Proteins were transferred to nitrocellulose membranes and nonspecific binding sites blocked in 5% fat-free milk in Tris-buffered saline with 0.1% Tween-20 (TBS-T) for 1 h at room temperature. The membranes were then incubated with mouse monoclonal anti-PKA C antibodies (BD Biosciences) diluted 1:1,000 in 5% fat-free milk at room temperature for 1 h or overnight at 4°C. The membranes were washed three times for 10 min each in TBS-T. This was followed by incubation in secondary antibody (anti-rabbit IgG; Bio-Rad) diluted 1:15,000 for 1 h at room temperature or overnight at 4°C, then washed three times for 15 min each in TBS-T. Protein bands were detected by chemiluminescence and exposed to X-ray film, which was then developed. The intensities of protein bands on the X-ray film were quantified using Bio-Rad's GS800 calibrated densitometer and QuantityOne software (Bio-Rad, Hercules, CA).
Bacterial expression and purification of PKA Cα, Cβ1, and Cβ2.
The murine PKA Cα subunit was expressed and purified as described by Herbert et al. (18). N-terminal His6-tagged murine Cβ1 and Cβ2 subunits were purified as described previously (17, 46). Briefly, the N-terminal His6-tagged Cβ1 and Cβ2 subunits in pET28a vector were expressed in Escherichia coli [BL21(DE3)]. Cultures were grown at 37°C with shaking until the cells reached an optical density of 0.6–0.8. At this time, the temperature was lowered to 24°C, and protein expression was induced by adding 0.5 mM isopropyl β-d-1-thiogalactopyranoside. The cultures continued growing at 24°C for 6 h before being harvested and centrifuged at 5,150 g for 10 min. The resulting cell pellet was resuspended in lysis buffer (50 mM potassium phosphate, 20 mM Tris·HCl, 100 mM NaCl, 5 mM β-mercaptoethanol at pH 8.0). The cells were lysed using a Microfluidizer (Microfluidics) at 18,000 psi. The lysate was clarified by centrifugation at 31,000 g for 1 h in a Beckman JA20 rotor. The supernatant was then incubated with ProBond resin (Invitrogen, Grand Island, NY) and equilibrated in lysis buffer for 1 h at 4°C. The Ni2+ resin was then applied to a column and washed twice with lysis buffer, once with wash buffer (lysis buffer with 500 mM NaCl and 20 mM imidazole), and once more with lysis buffer. The protein was eluted using lysis buffer containing 250 mM imidazole. The eluted protein was combined and dialyzed overnight into buffer containing 20 mM potassium phosphate, 25 mM KCl, and 5 mM DTT at pH 7.25. This sample was loaded onto a prepacked Mono-S 10/100 (GE Healthcare) cation exchange column equilibrated in 25 mM potassium phosphate and 5 mM DTT at pH 7.25. The protein was eluted using a KCl gradient ranging from 0 to 1 M. The eluted protein was analyzed by SDS-PAGE gels stained with Coomassie to ensure purity, and all eluted protein was combined for the assays.
Determination of meprin cleavage of purified PKA C isoforms.
Activated forms of homomeric meprin A and meprin B were incubated with purified forms of three mouse PKA C isoforms (Cα, Cβ1, and Cβ2). The incubations were initially conducted in Tris buffer (20 mM Tris·HCl, 150 mM NaCl, pH 7.5) at 37°C for 0 to 6 h. Proteolysis of the PKA C proteins was stopped by adding SDS-PAGE sample buffer and boiling for 5 min. The products were electrophoretically separated and stained with Coomassie using Simply Blue reagent (Invitrogen, Carlsbad, CA). The levels of full-length PKA C and resulting cleaved PKA C products were quantified by optic densitometry using Bio-Rad's GS800 calibrated densitometer and QuantityOne software.
Determination of meprin B cleavage sites on PKA C isoforms.
To determine the meprin cleavage sites on the PKA C isoforms, purified forms of the mouse PKA C proteins to be tested were incubated with activated forms of homomeric meprin A and meprin B in Tris buffer (20 mM Tris·HCl, 150 mM NaCl, pH 7.5) at 37°C for 2 h. The cleavage products were subjected to electrophoresis and stained with Coomassie. Gel plugs containing the full-length PKA C proteins and cleaved PKA C protein products were cut out and trypsin digested. The gel plugs were first destained by incubating in 50 mM ammonium bicarbonate at 60°C for 30 min, and sequentially incubated in 50 mM ammonium bicarbonate with 50 and 75% acetonitrile, respectively. The gel slices were dried, then rehydrated in 10 mg/ml trypsin in 20 mM ammonium bicarbonate for 1 h at 4°C. Trypsin digestion was accomplished by incubation at 48°C for 4 h. Peptides in the gel plugs were extracted in 50% acetonitrile and 0.1% trifluoroacetic acid (TFA) at room temperature for 20 min. The extracted peptides were dried using a speed vacuum, resuspended in 0.5% TFA, processed through C18 Zip tips, and eluted in 0.1% TFA/50% acetonitrile. The digested peptides were separated by C18 nanoflow liquid chromatography-matrix-assisted laser desorption/ionization and identified by ProteinPilot software at the Pennsylvania State University College of Medicine Core Facility (Hershey, PA). Peptides identified in the full-length PKA C proteins but not present in the meprin-cleaved protein fragment represent the sequence that is cleaved off by meprins. Once putative cleavage sites were determined, the size of cleaved products was estimated from the amino acid sequence using ExPASy Proteomic Server tools (http://web.expasy.org/peptide_mass/) and compared with the size of protein bands observed on Coomassie-stained gels.
Evaluation of effects of meprin-mediated cleavage on PKA C activity.
To determine the impact that meprin cleavage has on the kinase activity of PKA C, purified PKA Cα, Cβ1, and Cβ2 were incubated with activated, purified recombinant meprin A or B for 2 h. The activity of each PKA C variant was measured using the Kinase-GLO luciferase assay (Promega) according to the manufacturer's instructions. Briefly, the purified PKA C proteins were diluted in kinase reaction buffer (40 mM Tris·HCl, pH 7.5, 20 mM MgCl2, 1 μM ATP, 0.1 mg/ml BSA) supplemented with 5 μM Kemptide substrate (Sigma-Aldrich), and the solution was incubated at room temperature for 5–30 min. Following incubation, an equal volume of Kinase-GLO reagent (containing GLO luciferase) was added to the solution and incubated for an additional 10 min at room temperature. Luminescence was measured using a M200 Pro multimode plate reader (Tecan). Because PKA C-mediated phosphorylation of Kemptide reduces the amount of ATP available to luciferase, an inverse relationship exists between kinase activity and luminescence. The kinase activity at each time point was normalized to the activity at time 0 (i.e., PKA C activity in the absence of meprin).
Determination of meprin B/PKA Cα enzyme kinetics.
To evaluate whether meprin B cleavage of PKA C is physiologically possible, we characterized the meprin B/PKA Cα enzyme kinetics. Varying concentrations of PKA Cα (0.7–8.0 μM) were incubated with 36 nM meprin B for 0–10 min. Substrate disappearance was determined using previously described methods (34). The Vmax and Km values were estimated by Michaelis-Menten kinetics using GraphPad Prism software and used to compute the kcat and kcat/Km ratio.
Experimental animals.
To evaluate in vivo interaction between meprins and PKA C, we obtained kidney tissue from mice with streptozotocin (STZ)-induced type 1 diabetes and IR-induced acute kidney injury. Two genotypes of mice were used; wild-type (WT) C57BL/6, which express high levels of both meprin A and B, and meprin αβ-double KO mice on a C57/BL/6 background, which have the genes for meprin α and β disrupted and are thus deficient in both meprin A and meprin B. The mice were housed at the Pennsylvania State University, College of Medicine, Hershey Medical Center, Animal Facility. The protocols used were approved by the Institutional Animal Care and Use Committee. The mice were maintained on a 12:12-h dark-light cycle and provided with rat chow and water ad libitum.
Induction of diabetes.
Type 1 diabetes was induced in 8-wk-old male mice by intraperitoneal injection of low-dose STZ (50 mg/kg body wt for 5 consecutive days). The blood glucose levels were measured at day 10 post-STZ injection. Mice with blood glucose levels of 250 mg/dl were considered diabetic. Body weights were monitored on a weekly basis. Blood was obtained by tail nicking at 4, 8, and 12 wk post-STZ injection for kidney phenotyping using blood urea nitrogen (BUN) as an indicator of kidney damage. We used BUN slides and a Vitros DT60 II analyzer (Ortho-Diagnostics, Rochester, NY). At 18 wk post-STZ injection, the mice were euthanized by isoflurane asphyxiation followed by cervical dislocation. Kidneys were cut out and decapsulated, and portions of the kidney tissue were fixed in Carnoy's fixative (60% ethanol/30% chloroform/10% acetic acid) overnight at 4°C, then transferred to 70% ethanol. The kidney tissue was paraffin embedded at the Histology Core Lab, at Penn State Hershey, and 5-μM cross sections were cut onto Superfrost slides for immunohistological analysis.
Induction of IR-associated renal injury.
Twelve-week-old mice were subjected to IR-induced renal injury using surgical procedures previously described (34). At 3 h post-IR, the mice were euthanized by exposure to isoflurane. The kidneys were excised, decapsulated, and kidney tissue was processed for paraffin embedding. BUN assays were done to confirm kidney injury.
Immunohistological staining and microscopic analysis.
The slide sections were deparaffinized through xylene, 100% ethanol, 95% ethanol, and water. Antigen unmasking was achieved by boiling in 10 mM sodium citrate buffer, pH 6.0, for 10 min. Nonspecific binding sites were blocked by incubation in 5% normal goat serum with 0.3% Triton-X-100 at room temperature for 1 h in a humidified chamber. Primary antibodies were diluted in PBS buffer with 1% BSA and 0.3% Triton-X-100 (PBS-T). The sections were incubated in the primary antibodies overnight at 4°C. Rabbit polyclonal anti-meprin A antibodies (HMC14), and rabbit polyclonal anti-meprin B antibodies (HMC77) were a gift from Dr. Judith Bond (Penn State Hershey Medical Center) and were diluted at 1:200 and 1:400, respectively. Mouse monoclonal anti-PKA C antibodies (BD Biosciences), were diluted 1:200. The slides were then rinsed three times in PBS for 10 min each and incubated in fluorophore-conjugated secondary antibodies (rabbit Alexa Fluor 488, for meprin A and meprin B; and mouse Alexa Fluor 555 for PKA C; Cell Signaling, Beverly MA) diluted 1:1,000 for 1 h at room temperature. 4,6-Diamidino-2-phenylindole was used for nuclei staining. Coverslips with Prolong antifade reagent (Life Technologies, Carlsbad CA) were mounted and allowed to dry at room temperature overnight. The tissue sections were evaluated for meprins and PKA C expression and localization using confocal microscopy (Carl Zeiss Microscopy, Thornwood, NY) and imaged using AxioVision Software.
Statistical analysis.
Data were analyzed using ANOVA on GraphPad Prism software. The kinase-GLO data were done in triplicate and repeated three times. P values ≤0.05 were considered significant.
RESULTS
Meprin B cleaves PKA C present in kidney lysates.
To determine whether homomeric meprin A (α-α) and meprin B (β-β) are capable of cleaving PKA C present in kidney lysates, purified forms of the meprins were initially incubated with cytosolic-enriched kidney proteins from meprin αβ double-KO mice (which lack endogenous meprins) and fragmentation/degradation was evaluated by Western blot analysis using anti-PKA-specific antibodies. Two PKA C protein bands were detected in the cytosolic kidney fractions, one migrating at ∼42 kDa (corresponding to the expected molecular masses of Cα and Cβ) and one migrating at ∼46 kDa (corresponding to a larger splice variant, Cβ4) (13, 51). The 42-kDa protein band was more intense, suggesting that the shorter PKA C isoforms predominate in kidney tissue. Following incubation with meprin B, a significant decrease in the intensities of the two protein bands was observed, suggesting that meprin B is capable of degrading each of the PKA C isoforms present in kidney lysates (Fig. 1). While levels of the 46-kDa PKA C species dropped below the limit of detection after 1 h, the intensity of the 42-kDa band decreased more gradually over a 3-h time period. In contrast, degradation was not observed when cytosolic kidney proteins were incubated with reaction buffer lacking meprins or with activated homomeric meprin A, indicating that the proteolytic processing of PKA C is isoform specific with respect to the meprin isoforms.
Fig. 1.

Representative immunoblot of the catalytic subunit of PKA (PKA C) present in cytosolic-enriched mouse kidney proteins (A) and associated normalized optic densities (OD) of the 2 PKA C bands detected (B). Activated purified forms of recombinant homomeric meprin A (α-α) and meprin B (β-β) were incubated with cytosolic-enriched kidney protein lysates from meprin αβ double-knockout mice (which lack endogenous meprins) for 0–3 h. The products were separated by electrophoresis and cleavage/degradation of PKA C evaluated by use of Western blot analysis with anti-PKA C-specific antibodies coupled with optic densitometry. Two PKA C protein bands (∼42 and ∼46 kDa) were detected. The intensities of the protein bands did not change in proteins incubated with buffer lacking meprins or with homomeric meprin A. In contrast, the protein band intensities decreased in a time-dependent manner in reactions containing activated purified meprin B.
Meprins cleave purified PKA C in an isoform-specific manner.
Based on the varying extent to which the PKA C isoforms were digested by meprin B, we next asked whether different PKA C isoforms are differentially cleaved by meprin A and meprin B. Because control experiments using purified PKA C isoforms showed that the anti-PKA C antibodies reacted with all three PKA C isoforms (Cα, Cβ1, Cβ2), we chose to use purified forms of PKA Cα, β1, and β2 for these studies. Therefore, to confirm isoform-specific interactions between meprins and the PKA C isoforms, activated forms of purified recombinant mouse homomeric meprin A (α-α) and rat meprin B (β-β) were incubated with purified recombinant forms of mouse PKA Cα, Cβ1, and Cβ2. Consistent with the data obtained from incubations with the kidney protein lysates, meprin B cleaved all three PKA C isoforms tested (Figs. 2 and 3). In contrast, meprin A only cleaved PKA Cβ1 (Figs. 2B and Fig. 5). Although both meprin A and meprin B cleaved PKA Cβ1, the size of the cleavage products were isoform specific, suggesting different cleavage sites. We initially compared the efficiency of cleavage/degradation of the PKA C isoforms by incubating equal molar concentrations of the purified PKA C proteins with equal molar concentrations of the activated meprins (Fig. 3). The data show that cleavage/degradation was least efficient for meprin B/PKA Cα, with 65% cleaved in 1 h. The efficiency of cleavage for meprin A/PKA C β1, meprin B/PKA Cβ1, and meprin B/PKA Cβ2 was comparable, at ∼85–90% each. To verify the cleavage specificity, a series of control experiments was conducted. In the first set of experiments, latent forms of meprin A and B were substituted for their activated counterparts. Under these conditions, cleavage/degradation of PKA C isoforms was not observed using either the kidney cell lysates or purified PKA C (Fig. 4). In the second set of experiments, activated meprins were pretreated with two meprin inhibitors, EDTA and actinonin, before incubation with either cytosolic kidney proteins or purified recombinant PKA C. As expected, neither the PKA C isoforms present in kidney lysates nor the purified PKA C isoforms were digested by the meprins in the presence of meprin inhibitors (Fig. 4). Together, these data suggest that the activated meprins are responsible for the observed cleavage/degradation of the PKA C isoforms.
Fig. 2.
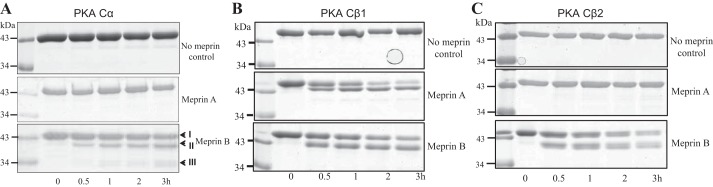
Representative Coomassie-stained gels of purified PKA C isoforms after incubation with activated meprins. The purified recombinant PKA C proteins were incubated with either meprin A or meprin B for 0–3 h. Control reactions were incubated with Tris buffer lacking meprins. Products were separated by electrophoresis and Coomassie stained. Lane 1 in each gel shows the protein ladder. A: PKA Cα cleavage was only observed in reactions with activated meprin B, and not homomeric meprin A, or control buffer lacking meprins. Band I represents full-length PKA Cα, and bands II and III represent the meprin B cleavage products. B: purified mouse PKA Cβ1 was cleaved by both meprin A and meprin B. However, the resulting products were meprin isoform-specific, suggesting different cleavage sites. C: only meprin B was capable of cleaving PKA Cβ2.
Fig. 3.

Comparison of isoform-specific cleavage products following incubation of PKA Cα, Cβ1, and Cβ2 with activated meprins. Equal molar concentrations of the purified PKA C isoforms were incubated with equal molar concentrations of activated meprins for 2 h. The proteins were electrophoretically separated on 12% SDS-polyacrylamide gels and either Coomassie stained (I) or subjected to Western blot analysis using anti-PKA C antibodies (II). Cleavage products were isoform specific. The antibodies showed cross-reactivity with all 3 PKA C isoforms (α, β1, and β2). The intensity of the full-length PKA C protein band in each reaction was determined using optic densitometry and normalized to the levels in the control reaction lacking meprins. The bar graph represents the fraction of each PKA C isoform protein that is uncleaved after 1 h of incubation with meprins. The efficiency of cleavage was slowest in the meprin B/PKA Cα combination (∼65% cleaved) and comparable in the other 3 reactions (∼85–90% cleaved).
Fig. 5.

Putative meprin cleavage sites on mouse PKA C isoforms. The cleavage products were electrophoretically separated and Coomassie stained. The full-length proteins and meprin cleavage products were excised, trypsin digested, and subjected to mass spectrometry. PilotProtein software was used to compare the peptide maps in full-length PKA C isoforms and their corresponding meprin cleaved protein fragments to determine putative cleavage sites. A: meprin cleavage sites in PKA Cα. There are 2 meprin B cleavage sites in the PKA Cα sequence: the first is between Trp31 and Glu32 on the N terminus, and the second is between Phe328 and Asp329 on the C terminus. B: meprin cleavage sites in PKA Cβ1. Meprin A cleaves PKA Cβ1 at the N terminus between Arg29 and Lys30. Meprin B cleaves PKA Cβ1 at 2 sites: the first is between Lys22 and Ala23 near the N terminus, and the second is between Glu333 and Glu334 near the C terminus. C: meprin cleavage sites in PKA Cβ2. Meprin B cleaves PKA Cβ2 between Glu320 and Glu321. D: schematic diagram showing the domain architecture of each PKA C variant tested and the relative position of the meprin cleavage sites identified in A–C.
Fig. 4.
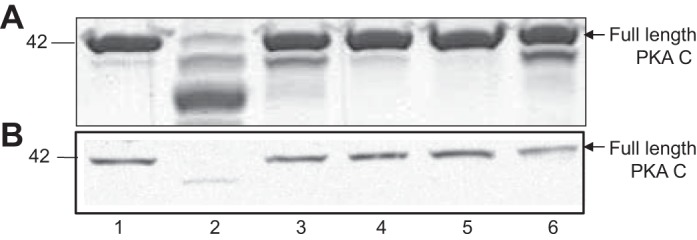
Meprin B cleavage of PKA C is specific and is blocked by preincubation with known meprin inhibitors. A: Coomassie-stained gel of purified PKA Cα. B: immunoblot of PKA C present in cytosolic-enriched kidney proteins from meprin αβ double-knockout mice. The following reactions were included: control buffer lacking meprins (lane 1), activated meprin B (lane 2), latent meprin B (lane 3), activated homomeric meprin A (lane 4), activated meprin B preincubated with EDTA (lane 5), and activated meprin B preincubated with actinonin (lane 6).
Identification of putative meprin cleavage sites on PKA C isoforms.
To identify putative cleavage sites on the PKA C proteins, the full-length PKA C proteins and the PKA C protein products resulting from meprin cleavage were excised from Coomassie-stained gels, trypsin digested, and subjected to mass spectrometry to identify cleavage sites. The peptide maps were analyzed using PilotProtein software. The data showed that meprin B cleaves PKA Cα at two sites. The first cleavage site is located near the N terminus, between Trp31 and Glu32 (site BI in Fig. 5A). A second site of cleavage is found near the C terminus, between Phe328 and Asp329 (site BII on Fig. 5A) and results in a 37.7-kDa product. Double cleavage of PKA Cα at sites BI and BII yields a 34.4-kDa product. Both meprin A and meprin B cleaved PKA Cβ1, but the cleavage sites were meprin isoform specific. Indeed, meprin A cleaves Cβ1 on the N-terminal end between Arg29 and Lys30, to yield a 37.5-kDa product (site A on Fig. 5B). In contrast, meprin B cleavage of PKA Cβ1 occurs at two sites: the first (site BI in Fig. 5B) is near the N terminus, between Lys22 and Ala23, and the second (site BII in Fig. 5B) occurs near the C terminus, between Glu333 and Glu334. Double cleavage produces a 36.3-kDa product. Finally, meprin B cleaves PKA Cβ2 at the C terminus, between Glu319 and Glu320, resulting in a 37.2-kDa fragment (site B in Fig. 5C). The meprin cleavage products observed on the Coomassie-stained gels are comparable to the sizes estimated using Expasy (http://web.expasy.org/cgi-bin/peptide_mass/peptide-mass.pl).
Meprin B cleavage of PKA cα occurs at a rate similar to other physiological meprin substrates.
Kinetic analysis of the cleavage reaction suggests that in vivo cleavage of PKA C by meprins is possible. Indeed, the Michaelis-Menten enzyme kinetics parameters for meprin B-mediated cleavage of PKA Cα were determined to be Vmax = 0.20 ± 0.03 μM/min; Km = 1.6 ± 0.8 μM; and kcat = 5.555 s−1, yielding a kcat/Km ratio = 3.47 × 106 M−1·s−1. These values are consistent with those previously determined for other physiologically relevant meprin substrates (3, 34) (Table 1).
Table 1.
Meprin B/PKA Cα enzyme kinetics
| Meprin B Substrate | Km, M × 10−6 | Vmax, M × 10−6/min | kcat, s−1 | kcat/Km, (M−1·s−1) × 106 | Source |
|---|---|---|---|---|---|
| PKA Cα | 1.6 ± 0.7 | 0.200 ± 0.03 | 5.555 | 3.47 | Present study |
| Actin | 0.73 ± 0.23 | 0.025 ± 0.002 | 0.625 | 0.86 | Ongeri et al., 2011 (10) |
| Pro-IL-18 | 1.31 ± 0.0001 | Not published | 6.790 | 5.1 | Barnejee et al., 2008 (19) |
| Villin | 1.17 ± 0.33 | 0.180 ± 0.02 | 150 | 128.20 | Ongeri et al. 2011 (10) |
Values are means ± SE. Varying concentrations of PKA Cα (0.7–8 μM) were incubated with 36 nM meprin B in Tris buffer (20 mM Tris·HCl, pH 7.5, 150 mM NaCl) at 37°C for 0–10 min. Equal volumes of product were removed at 1-min intervals, and the reaction was stopped by adding SDS sample buffer and boiling for 5 min. The products were electrophoretically separated, Coomassie stained, and band intensities were determined by optic densitometry using Bio-Rad's GS800 calibrated densitometer and QuantityOne software. The kinetic parameters were determined by plotting the rate of substrate disappearance (μM/min) using the Michaelis-Menten equation (GraphPad Prism software).
Meprin B cleavage of PKA C isoforms significantly reduces their kinase activity.
To determine whether meprin-mediated cleavage affects the activity of PKA C isoforms, recombinant mouse PKA Cα, β1, and β2 were preincubated with either meprin A or meprin B and their kinase activities were determined using a coupled luminescence assay (Fig. 6). Although meprin A cleaved PKA Cβ1 (see Fig. 2B), the proteolytic processing did not significantly alter its kinase activity. In contrast, meprin B-mediated cleavage resulted in substantial reductions in the activity of each of the PKA C isoforms, leading to an ∼11-fold decrease in PKA Cα activity (P = 1.0 × 10−4), an ∼22-fold decrease in PKA Cβ1 activity (P = 8.7 × 10−5), and an ∼4-fold reduction in PKA Cβ2 activity (P = 2.9 × 10−2) (Fig. 6). To ensure that the observed changes in activity were not due to global reductions in PKA C protein levels, total protein levels were measured in a subset of the reactions following meprin digestion. These experiments demonstrated that, although lower molecular weight species were clearly visible as the result of meprin-mediated cleavage, the total protein content did not vary substantially between reactions. Moreover, if the kinase activity was normalized to total protein content for a given reaction, the results were identical to those shown in Fig. 6 (data not shown). Together, these data indicate that meprin-mediated cleavage of PKA C has functional consequences on its kinase activity.
Fig. 6.
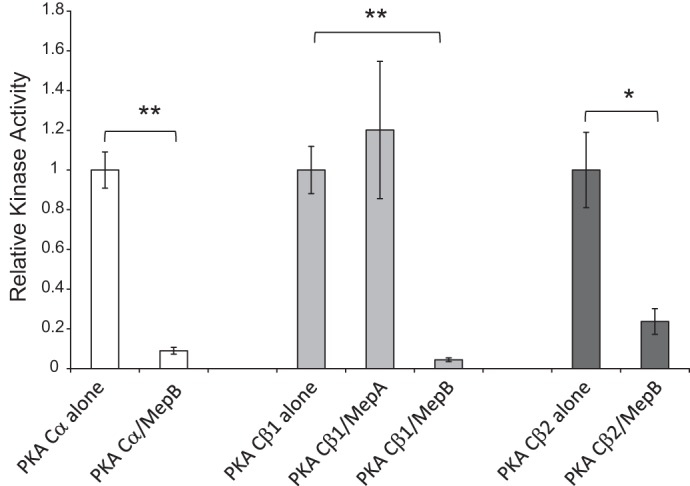
Impact of meprin cleavage on the kinase activity of PKA C isoforms. The activity of those PKA C isoforms that were cleaved by meprins was measured following incubation with either meprin A or meprin B for 2 h. The kinase activity was normalized to the activity at time 0 (i.e., activity of the PKA C isoform in the absence of meprins). Meprin B cleavage of all 3 PKC isoforms resulted in PKA C fragments with a significantly decreased kinase activity. White bars, PKA Cα; light gray bars, PKA Cβ1; dark gray bars, PKA Cβ2. Error bars represent SE of at least 3 independent experiments. *P < 0.05. **P < 0.01 (see the text for specific P values).
Meprin A and PKA C are localized in glomeruli of diabetic mouse kidneys.
To determine whether meprin-mediated cleavage of PKA C could occur under physiological conditions, we examined the localization of each meprin isoform and PKA C in mouse kidney tissues using immunofluorescence. To this end, WT and meprin αβ double-KO mice were either treated with low-dose STZ to induce type 1 diabetes or subjected to IR-induced kidney injury. Kidney tissue sections were then stained with anti-meprin and anti-PKA C antibodies and imaged using confocal microscopy. These data showed that meprin A and PKA C both localized in the glomeruli of diabetic mouse kidneys (Fig. 7A). Expression of meprin A was not observed in the glomeruli of kidney sections from nondiabetic control mice, suggesting an association between glomerular meprin A expression and diabetic nephropathy. Surprisingly, meprin B localization was not observed in the glomeruli of control or diabetic kidneys (Fig. 7B).
Fig. 7.

Immunolocalization of meprins (green) and PKA C (red) in the glomeruli (GM) of kidney tissue from wild-type mice with streptozotocin (STZ)-induced type 1 diabetes. Male mice were injected with low-dose STZ at age 8 wk to induce type 1 diabetes. The mice were euthanized at 18 wk post-STZ injection, and the kidney tissue was fixed in Carnoy's fixative and paraffin embedded. Five-micrometer cross sections of kidney tissue were cut onto Superfrost slides. Fluorescence immunohistochemical staining with either anti-meprin B (HMC77) or anti-meprin A (HMC14) and anti-PKA C antibodies was used to evaluate expression of meprins and PKA C. 4,6-Diamidino-2-phenylindole (DAPI) was used to stain the nuclei (blue). In control mice, PKA C was ubiquitously expressed in the kidney tissue, while both meprin A and meprin B expression was restricted to the brush-border membrane of proximal tubules. In kidneys from mice with STZ-induced type 1 diabetes, localization of meprin A (A), but not meprin B (B), was observed in the glomeruli. Tubules adjacent to the renal corpuscle are labeled T.
Cytosolic/soluble kidney PKA C levels increased in diabetic kidneys.
To further explore the impact of meprins on PKA C levels during STZ-induced diabetes, cytosolic-enriched kidney protein extracts were obtained from WT and meprin αβ KO mice and evaluated by Western blot analysis. Protein samples from diabetic kidneys showed higher levels of the 42-kDa PKA C species relative to nondiabetic controls (Fig. 8, A and B), with diabetic mice exhibiting an approximately twofold increase in PKA C levels (Fig. 8AII). This difference was even more pronounced in kidney proteins obtained from meprin αβ KO mice, with diabetic mice exhibiting approximately fivefold higher levels of the 42-kDa PKA C species than nondiabetic control counterparts (≤0.05). Although a similar increase in protein levels was not observed for the 46-kDa PKA C species (Fig. 8AIII), this isoform did appear to be fragmented following STZ treatment in WT, but not in meprin αβ KO mice (Fig. 8AI). In vivo fragmentation of the 42-kDa PKA C species was also observed in protein extracts from diabetic kidneys. Indeed, the levels of a 37-kDa PKA C fragment were significantly higher (P ≤ 0.01) in diabetic WT kidneys compared with nondiabetic WT controls (Fig. 8BII). While a small amount of fragmentation was also observed in protein extracts from meprin αβ KO kidneys, there was no significant difference between diabetic and nondiabetic mice for this genotype. Importantly, the 37-kDa fragment was of comparable size to the PKA C protein fragment obtained when purified PKA Cα and PKA C β2 were incubated with activated meprin B (Figs. 2 and 3).
Fig. 8.

Representative immunoblot for PKA C in cytosolic-enriched kidney protein fractions obtained from wild-type (WT) and meprin αβ knockout (KO) mice on a C57BL/6 background at 18 wk post-STZ injection. A: Western blot analysis coupled with optic densitometry was used to compare the PKA C levels. Two PKA C isoforms were detected: a dominant 42-kDa band and a less abundant 46-kDa band (I). While the levels of the 42-kDa PKA C band were significantly higher in diabetic kidneys compared with nondiabetic control counterparts for both genotypes (P ≤ 0.05), the fold-increase was much higher in the meprin αβ KO mice (II). Although there was no significant change in the levels of the 46-kDa band, a cleavage product was observed in STZ-treated WT mice, but not meprin αβ KO mice. B: in vivo fragmentation of the 42-kDa PKA C species produced a 37-kDa product. The fragmentation was more pronounced in diabetic kidneys from WT mice than in meprin αβ KO mice.
PKA C protein levels decreased in meprin-expressing tubules after IR-induced renal injury.
Immunofluorescence staining of kidney tissues with anti-meprin and anti-PKA C antibodies showed a negative correlation between expression of meprins and tubular PKA C levels in kidneys subjected to IR-induced renal injury. At 3 h post-IR, PKA C levels were lower in the proximal tubules which express meprins compared with the distal tubules which lack meprins (Fig. 9, A and B). In contrast, the PKA C levels and expression patterns were comparable in proximal and distal tubules in kidneys from sham-operated control mice. As previously observed (7, 34), both meprin A and meprin B redistributed to the cytosol in IR. Together, these data suggest that the redistribution of meprins into the cytosol in IR may contribute to the lower PKA C levels observed in the meprin-expressing tubular cells. In kidneys subjected to IR-induced renal injury, meprins were not present in the glomeruli. Additionally, meprins were not detected in either the αβ KO kidney tissue or the negative controls without primary antibody. However, Western blot analysis of cytosolic-enriched kidney proteins did not show significant differences in the levels of either the 42- or 46-kDa PKA C isoforms (Fig. 10).
Fig. 9.

Immunolocalization of meprins (green) and PKA C (red) in the tubules of kidney tissue from WT mice with ischemia-reperfusion (IR)-induced renal injury. IR renal injury was induced in 12-wk-old male mice by surgical clamping of renal arteries for 26 min. The mice were euthanized at 3 h post-IR, and the tissue was paraffin embedded. Immunofluorescence staining with anti-meprin antibodies (green) and anti-PKA C antibodies (red) was used to localize the proteins, and imaged by confocal microscopy. DAPI was used to stain the nuclei (blue). Expression of meprins was restricted to the brush-border membrane of proximal tubules (PT) in sham-operated control mice, and the PKA C expression pattern was comparable in proximal and distal tubules (DT) in kidney tissue. However, at 3 h post-IR, meprins were redistributed to the cytosol of proximal tubule cells, and the levels of PKA C were lower in the meprin-expressing proximal tubules compared with the distal tubules (lacking meprins).
Fig. 10.

Representative immunoblot for PKA C in cytosolic-enriched kidney proteins from WT mice kidneys at 3 h post IR-induced renal injury (I). Densitometric analysis of the 42-kDa PKA C isoform (II) and that for the 46-kDa PKA C isoform (III) are shown. There was no significant change in the total kidney PKA C levels after IR demonstrated by Western blotting.
DISCUSSION
Meprins, metalloproteinases that are highly expressed in the BBM of kidney proximal tubules and small intestines, were shown to play a role in the pathology of acute and chronic kidney injury. The current study demonstrates isoform-specific interactions between meprins and the catalytic subunit of protein kinase A (PKA C). Using purified recombinant forms of three PKA C isoforms, PKA Cα, Cβ1, and Cβ2, and two meprin isoforms, meprin A and B, the present study demonstrates that activated forms of meprins cleave PKA C in an isoform-specific manner. While homomeric meprin A (α-α) only cleaved PKA Cβ1, meprin B (β-β) cleaved PKA Cα, Cβ1, and Cβ2. The cleavage was meprin specific and was blocked by preincubation with meprin inhibitors. Proteolysis of PKA C by proteases present in the intestinal brush-border membrane (then referred to as KSMP and later determined to be meprin B) was first reported by De Jonge et al. (10). Although at the time this interaction was not thought to be physiologically significant because of the nonoverlapping localization of meprins and PKA C in proximal tubules, recent data have demonstrated that meprins are redistributed to the cytosol under certain pathological conditions (7, 34), raising the possibility that meprins can cleave PKA C in vivo. Meprin interactions with the PKA pathway could thus play a role in the response to insults that cause renal injury. In addition to modulating injury in tubular cells where meprins are most abundant, expression of meprins in leukocytes (48) and podocytes (33) suggests that meprins could modulate ECM metabolism in renal glomeruli and may thus play a role in the ECM buildup and fibrosis associated with diabetic nephropathy. Indeed, while PKA-mediated signaling occurs in many cell types, the downstream modulators of the PKA-transforming growth factor (TGF)-β1-induced response are cell-type specific and depend on the cellular environment. A role for meprin B in the cAMP-PKA signaling pathway could thus be significant in kidney disease conditions associated with fibrosis. Data from the current study show localization of meprin A and PKA C in the glomeruli of diabetic mouse kidneys. Interestingly, glomerular localization of meprin B was not observed in diabetic kidneys. This suggests that in mice the meprin A isoform could play a role in the glomerular pathology associated with diabetes. Future studies should evaluate the impact of heteromeric meprin A (α-β), as this is the meprin A isoform that is membrane bound. To our knowledge, this is the first study to document glomerular expression of meprin A in the mouse and supports the findings in rat kidneys (33). A second important observation from the fluorescence analysis of native tissue was the negative correlation between meprin expression and tubular PKA C levels in kidneys subjected to IR-induced renal injury. While Western blot analysis did not show an overall decrease in total PKA C levels after IR, it is possible that the decrease in proximal tubule PKA C levels is balanced by an increase in distal tubule levels. Data from this study suggest that the tubular injury associated with meprins may be due in part to decreased PKA C levels, which ultimately would impact the PKA signaling pathway response to hypoxia in meprin-expressing cells. Previous studies have shown that the meprin-expressing S3 segment of proximal tubules have more injury in IR compared with the distal tubular cells lacking meprins (55). Additionally, pretreatment with meprin inhibitors and disruption of the meprin genes protect mice from IR-induced renal injury (7). Our data also provide evidence for in vivo fragmentation of PKA C in DN, yielding cleavage products comparable in size to the products obtained when purified PKA C is cleaved by purified activated meprin B. Indeed, the intensity of a 37-kDa PKA C fragmentation product increased nearly twofold in kidney proteins obtained from diabetic WT mice. Although this fragment was also present at low levels in kidney proteins extracted from meprin αβ KO mice, its intensity did not change following induction of diabetes in these mice. Together, these data suggest that another protease that is not responsive to diabetic nephropathy may cleave a small portion of PKA C under basal conditions, but meprins activated during diabetic nephropathy further cleave PKA C, significantly increasing the intensity of the 37-kDa fragment. Taken together, data from the present study provide evidence that in vivo interaction between meprins and PKA C is possible under pathological conditions such as diabetic nephropathy and IR. Further studies are needed to establish the source of glomerular meprin A. It is probable that progression of diabetic nephropathy induces meprin expression in podocytes. Alternatively, since leukocytes express meprins, glomerular leukocyte infiltration could contribute to the meprin proteins.
The mass spectrometry analysis of the peptide sequence maps of full-length PKA C and its meprin-mediated cleavage products suggests that cleavage by meprins removes important functional elements from the three PKA C isoforms. Meprin B cleaves PKA Cα and Cβ1 at both the N and C termini. Similarly, meprin B cleaves the C-terminal tail of PKA Cβ2. The C-terminal tail segments of C include the “FDDY” motif, which forms part of the binding pocket for ATP. Previous mutational analysis of the C tail showed that loss of these residues renders the enzyme inactive (4), and this would affect all downstream signaling. Also, loss of the C tail would likely prevent possible protein binding interactions with both regulatory proteins, such as PDK1 (38), and protein substrates. Indeed, the cleaved C-tail region contains the binding site for PDK1 as well as the “PxxP” motif, which is thought to be a protein binding site (16). Although the “PxxP” motif is not removed by meprin B-mediated cleavage, removal of portions of the C tail may prevent normal protein binding interactions with this motif. Additionally, the C-terminal tail contains a site that is constitutively phosphorylated and resistant to phosphatases (Ser339), and this phosphate may be important for proper activity of the enzyme (20, 25). On the other hand, loss of the N-terminal residues, as occurs in Cα following meprin B-mediated cleavage and Cβ1 following both meprin A- and meprin B-mediated cleavage, could prevent binding to the protein A-kinase interacting protein (AKIP). AKIP binds at the N terminus of the C-subunit between residues 15 and 30, and the binding is believed to be important in localizing the C-subunit to the nucleus (14, 26, 43). The N terminus also contains several modifications that would be lost after this cleavage, including myristoylation of Gly1, deamination of Asn2, and phosphorylation of Ser10 (16, 35, 49).
While the impact of meprin-mediated cleavage of PKA C on kidney cell function has not been studied, loss of kinase activity following cleavage by meprin B present in bovine membrane intestinal preparations was reported (9). The present study also shows a significant decrease of PKA Cα, Cβ1, and Cβ2 activity following incubation with activated forms of meprin B, confirming the predictions made from the mass spectrometry peptide map analysis data. Loss of kinase activity could impact cellular mechanisms mediated by the PKA pathway in kidneys, including TGF-β1-mediated regulation of the metabolism and catabolism of ECM proteins (19, 31, 44, 45, 54, 56), as well as the production of inflammatory response mediators in diabetic nephropathy (28). The impact on ECM metabolism is significant because thickening of the glomerular basement membrane (GBM) and tubulointerstitial fibrosis, two key pathological changes in diabetic kidneys, are a direct consequence of ECM protein accumulation (12). Previous studies showed that meprin deficiency is associated with renal ECM protein build-up and tubulointerstitial fibrosis (40). Meprin β is also downregulated in collagen IVA3 KO mice that develop Alport's syndrome (41). A common mouse model for studying type 2 diabetes, db/db mice, manifested decreased meprin α and meprin β gene and protein expression before the development of clinical diabetic nephropathy (32). The Michaelis-Menten kinetics data further suggest that PKA Cα is a potentially relevant physiological substrate for meprin B. The Km and kcat values obtained for meprin B were comparable to those for previously identified meprin B substrates (3, 34).
An interesting observation in the present study was meprin B-mediated processing of murine PKA Cβ2 (also known as Cβ4, which is homologous to the human Cβ4 variant). The mouse PKA Cβ2 variant is highly expressed in lymphoid tissue (13), suggesting a role in the immune response. This interaction is relevant because expression of meprins has been documented in monocytes and macrophages (48), making a case for meprin modulation of the immune response. Meprins were also shown to impact infiltration of leukocytes and cleave proinflammatory cytokines such as pro-IL-18. Furthermore, meprins have been shown to cleave proteins present in neural tissue, where PKA Cβ2 is abundantly expressed. For instance, meprin B cleavage of amyloid β peptides was demonstrated in vitro and in vivo (5, 21), suggesting a role in Alzheimer's disease (AD). Interestingly, a signaling axis involving the β-adrenergic receptor, PKA, and JNK has also recently been implicated in the etiology of AD (53). Further in vivo work with meprin KO mice will advance knowledge of meprin-PKA C interactions and elucidate the underlying mechanisms in physiological and pathological states.
GRANTS
This work was supported by the National Institute of General Medical Sciences (NIGMS) under Award SC3GM102049-01 to E. M. Ongeri and Title III Start-up funds from the College of Arts and Sciences at North Carolina A&T State University to R. H. Newman. Also acknowledged is support from NIGMS Grants GM19301 (S. S. Taylor) and F31GM099415 (A. C. Bastidas). Additionally, A. Bastidas was funded by a Ford Foundation Diversity Fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J.-M.V.N., R.H.N., S.S.T., and E.M.O. provided conception and design of research; J.-M.V.N., A.C.B., R.H.N., and E.M.O. performed experiments; J.-M.V.N., R.H.N., and E.M.O. analyzed data; J.-M.V.N., A.C.B., R.H.N., S.S.T., and E.M.O. interpreted results of experiments; J.-M.V.N., R.H.N., and E.M.O. prepared figures; J.-M.V.N. and E.M.O. drafted manuscript; J.-M.V.N., A.C.B., R.H.N., S.S.T., and E.M.O. edited and revised manuscript; J.-M.V.N., A.C.B., R.H.N., S.S.T., and E.M.O. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Judith S. Bond, Department of Biochemistry and Molecular Biology, Pennsylvania State University College of Medicine (Hershey, PA), for the meprin knockout mice and critiques of the manuscript. Many thanks to Dr. Timothy Keifer (Biochemistry and Molecular Biology, Penn State Hershey) for help with purification of recombinant meprin proteins, and Dr. Bruce Stanley and Anne Stanley (Proteins and Mass Spectrometry Core Lab, Penn State Hershey) for help with the mass spectrometry analysis. Many thanks also to Dr. Dennis R. LaJeunesse for giving us access to the confocal microscope laboratory at the Joint School of Nanosciences and Nanoengineering.
REFERENCES
- 1.Alhanaty E, Patinkin J, Tauber-Finkelstein M, Shaltiel S. Degradative inactivation of cyclic AMP-dependent protein kinase by a membranal proteinase is restricted to the free catalytic subunit in its native conformation. Proc Natl Acad Sci USA 78: 3492–3495, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alhanaty E, Shaltiel S. Limited proteolysis of the catalytic subunit of cAMP-dependent protein kinase–a membranal regulatory device? Biochem Biophys Res Commun 89: 323–332, 1979. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee S, Bond JS. Prointerleukin-18 is activated by meprin beta in vitro and in vivo in intestinal inflammation. J Biol Chem 283: 31371–31377, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batkin M, Schvartz I, Shaltiel S. Snapping of the carboxyl terminal tail of the catalytic subunit of PKA onto its core: characterization of the sites by mutagenesis. Biochemistry 39: 5366–5373, 2000. [DOI] [PubMed] [Google Scholar]
- 5.Bien J, Jefferson T, Causevic M, Jumpertz T, Munter L, Multhaup G, Weggen S, Becker-Pauly C, Pietrzik CU. The metalloprotease meprin beta generates amino terminal-truncated amyloid beta peptide species. J Biol Chem 287: 33304–33313, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bond JS, Rojas K, Overhauser J, Zoghbi HY, Jiang W. The structural genes, MEP1A and MEP1B, for the alpha and beta subunits of the metalloendopeptidase meprin map to human chromosomes 6p and 18q, respectively. Genomics 25: 300–303, 1995. [DOI] [PubMed] [Google Scholar]
- 7.Bylander J, Li Q, Ramesh G, Zhang B, Reeves WB, Bond JS. Targeted disruption of the meprin metalloproteinase β gene protects against renal ischemia-reperfusion injury in mice. Am J Physiol Renal Physiol 294: F480–F490, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Carmago S, Shah SV, Walker PD. Meprin, a brush-border enzyme, plays an important role in hypoxic/ischemic acute renal tubular injury in rats. Kidney Int 61: 959–966, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Chestukhin A, Litovchick L, Muradov K, Batkin M, Shaltiel S. Unveiling the substrate specificity of meprin beta on the basis of the site in protein kinase A cleaved by the kinase splitting membranal proteinase. J Biol Chem 272: 3153–3160, 1997. [DOI] [PubMed] [Google Scholar]
- 10.De Jonge H, Schmeeda H, Shaltiel S. Orientation of the brush-border membranal proteinase which specifically splits the catalytic subunit of cAMP-dependent protein kinase. Eur J Biochem 169: 503–509, 1987. [DOI] [PubMed] [Google Scholar]
- 11.Desseyn JL, Burton KA, McKnight GS. Expression of a nonmyristylated variant of the catalytic subunit of protein kinase A during male germ-cell development. Proc Natl Acad Sci USA 97: 6433–6438, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falk RJ, Scheinman JI, Mauer SM, Michael AF. Polyantigenic expansion of basement membrane constituents in diabetic nephropathy. Diabetes 32, Suppl 2: 34–39, 1983. [DOI] [PubMed] [Google Scholar]
- 13.Funderud A, Henanger HH, Hafte TT, Amieux PS, Orstavik S, Skalhegg BS. Identification, cloning and characterization of a novel 47 kDa murine PKA C subunit homologous to human and bovine Cbeta2. BMC Biochem 7: 20, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao N, Hibi Y, Cueno M, Asamitsu K, Okamoto T. A-kinase-interacting protein 1 (AKIP1) acts as a molecular determinant of PKA in NF-kappaB signaling. J Biol Chem 285: 28097–28104, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorbea CM, Marchand P, Jiang W, Copeland NG, Gilbert DJ, Jenkins NA, Bond JS. Cloning, expression, and chromosomal localization of the mouse meprin beta subunit. J Biol Chem 268: 21035–21043, 1993. [PubMed] [Google Scholar]
- 16.Gould CM, Kannan N, Taylor SS, Newton AC. The chaperones Hsp90 and Cdc37 mediate the maturation and stabilization of protein kinase C through a conserved PXXP motif in the C-terminal tail. J Biol Chem 284: 4921–4935, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guthrie CR, Skalhegg BS, McKnight GS. Two novel brain-specific splice variants of the murine Cbeta gene of cAMP-dependent protein kinase. J Biol Chem 272: 29560–29565, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Herberg FW, Bell SM, Taylor SS. Expression of the catalytic subunit of cAMP-dependent protein kinase in Escherichia coli: multiple isozymes reflect different phosphorylation states. Protein Eng 6: 771–777, 1993. [DOI] [PubMed] [Google Scholar]
- 19.Hui L, Hong Y, Jingjing Z, Yuan H, Qi C, Nong Z. HGF suppresses high glucose-mediated oxidative stress in mesangial cells by activation of PKG and inhibition of PKA. Free Radic Biol Med 49: 467–473, 2010. [DOI] [PubMed] [Google Scholar]
- 20.Humphries KM, Deal MS, Taylor SS. Enhanced dephosphorylation of cAMP-dependent protein kinase by oxidation and thiol modification. J Biol Chem 280: 2750–2758, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Jefferson T, Causevic M, auf dem Keller U, Schilling O, Isbert S, Geyer R, Maier W, Tschickardt S, Jumpertz T, Weggen S, Bond JS, Overall CM, Pietrzik CU, Becker-Pauly C. Metalloprotease meprin beta generates nontoxic N-terminal amyloid precursor protein fragments in vivo. J Biol Chem 286: 27741–27750, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang W, Dewald G, Brundage E, Mucher G, Schildhaus HU, Zerres K, Bond JS. Fine mapping of MEP1A, the gene encoding the alpha subunit of the metalloendopeptidase meprin, to human chromosome 6P21. Biochem Biophys Res Commun 216: 630–635, 1995. [DOI] [PubMed] [Google Scholar]
- 23.Kaushal GP, Walker PD, Shah SV. An old enzyme with a new function: purification and characterization of a distinct matrix-degrading metalloproteinase in rat kidney cortex and its identification as meprin. J Cell Biol 126: 1319–1327, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kenny AJ, Ingram J. Proteins of the kidney microvillar membrane. Purification and properties of the phosphoramidon-insensitive endopeptidase ('endopeptidase-2') from rat kidney. Biochem J 245: 515–524, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keshwani MM, Klammt C, von Daake S, Ma Y, Kornev AP, Choe S, Insel PA, Taylor SS. Cotranslational cis-phosphorylation of the COOH-terminal tail is a key priming step in the maturation of cAMP-dependent protein kinase. Proc Natl Acad Sci USA 109: E1221–E1229, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.King CC, Sastri M, Chang P, Pennypacker J, Taylor SS. The rate of NF-kappaB nuclear translocation is regulated by PKA and A kinase interacting protein 1. PloS one 6: e18713, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohler D, Kruse M, Stocker W, Sterchi EE. Heterologously overexpressed, affinity-purified human meprin alpha is functionally active and cleaves components of the basement membrane in vitro. FEBS Lett 465: 2–7, 2000. [DOI] [PubMed] [Google Scholar]
- 28.Kreisberg JI, Kreisberg SH. High glucose activates protein kinase C and stimulates fibronectin gene expression by enhancing a cAMP response element. Kidney Int Suppl 51: S3–S11, 1995. [PubMed] [Google Scholar]
- 29.Kruse MN, Becker C, Lottaz D, Kohler D, Yiallouros I, Krell HW, Sterchi EE, Stocker W. Human meprin alpha and beta homo-oligomers: cleavage of basement membrane proteins and sensitivity to metalloprotease inhibitors. Biochem J 378: 383–389, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kvissel AK, Orstavik S, Oistad P, Rootwelt T, Jahnsen T, Skalhegg BS. Induction of Cbeta splice variants and formation of novel forms of protein kinase A type II holoenzymes during retinoic acid-induced differentiation of human NT2 cells. Cell Signal 16: 577–587, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Li XC, Carretero OA, Shao Y, Zhuo JL. Glucagon receptor-mediated extracellular signal-regulated kinase 1/2 phosphorylation in rat mesangial cells: role of protein kinase A and phospholipase C. Hypertension 47: 580–585, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mathew R, Futterweit S, Valderrama E, Tarectecan AA, Bylander JE, Bond JS, Trachtman H. Meprin-α in chronic diabetic nephropathy: interaction with the renin-angiotensin axis. Am J Physiol Renal Physiol 289: F911–F921, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Oneda B, Lods N, Lottaz D, Becker-Pauly C, Stocker W, Pippin J, Huguenin M, Ambort D, Marti HP, Sterchi EE. Metalloprotease meprin beta in rat kidney: glomerular localization and differential expression in glomerulonephritis. PloS one 3: e2278, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ongeri EM, Anyanwu O, Reeves WB, Bond JS. Villin and actin in the mouse kidney brush-border membrane bind to and are degraded by meprins, an interaction that contributes to injury in ischemia-reperfusion. Am J Physiol Renal Physiol 301: F871–F872, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pepperkok R, Hotz-Wagenblatt A, Konig N, Girod A, Bossemeyer D, Kinzel V. Intracellular distribution of mammalian protein kinase A catalytic subunit altered by conserved Asn2 deamidation. J Cell Biol 148: 715–726, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Red Eagle AR, Hanson RL, Jiang W, Han X, Matters GL, Imperatore G, Knowler WC, Bond JS. Meprin beta metalloprotease gene polymorphisms associated with diabetic nephropathy in the Pima Indians. Human Genet 118: 12–22, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Reinton N, Orstavik S, Haugen TB, Jahnsen T, Tasken K, Skalhegg BS. A novel isoform of human cyclic 3′,5′-adenosine monophosphate-dependent protein kinase, c alpha-s, localizes to sperm midpiece. Biol Reprod 63: 607–611, 2000. [DOI] [PubMed] [Google Scholar]
- 38.Romano RA, Kannan N, Kornev AP, Allison CJ, Taylor SS. A chimeric mechanism for polyvalent trans-phosphorylation of PKA by PDK1. Protein Sci 18: 1486–1497, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sacks FM, Hermans MP, Fioretto P, Valensi P, Davis T, Horton E, Wanner C, Al-Rubeaan K, Aronson R, Barzon I, Bishop L, Bonora E, Bunnag P, Chuang LM, Deerochanawong C, Goldenberg R, Harshfield B, Hernandez C, Herzlinger-Botein S, Itoh H, Jia W, Jiang YD, Kadowaki T, Laranjo N, Leiter L, Miwa T, Odawara M, Ohashi K, Ohno A, Pan C, Pan J, Pedro-Botet J, Reiner Z, Rotella CM, Simo R, Tanaka M, Tedeschi-Reiner E, Twum-Barima D, Zoppini G, Carey VJ. Association between plasma triglycerides and high-density lipoprotein cholesterol and microvascular kidney disease and retinopathy in type 2 diabetes mellitus: a global case-control study in 13 countries. Circulation 129: 999–1008, 2014. [DOI] [PubMed] [Google Scholar]
- 40.Sadlier DM, Connolly SB, Kieran NE, Roxburgh S, Brazil DP, Kairaitis L, Wang Y, Harris DC, Doran P, Brady HR. Sequential extracellular matrix-focused and baited-global cluster analysis of serial transcriptomic profiles identifies candidate modulators of renal tubulointerstitial fibrosis in murine adriamycin-induced nephropathy. J Biol Chem 279: 29670–29680, 2004. [DOI] [PubMed] [Google Scholar]
- 41.Sampson NS, Ryan ST, Enke DA, Cosgrove D, Koteliansky V, Gotwals P. Global gene expression analysis reveals a role for the alpha 1 integrin in renal pathogenesis. J Biol Chem 276: 34182–34188, 2001. [DOI] [PubMed] [Google Scholar]
- 42.San Agustin JT, Lesly JD, Nuwaysir LM, Witman GB. The catalytic subunit of the cAMP-dependent protein kinase of ovine sperm flagella has a unique amino-terminal sequence. J Biol Chem 273: 24874–24883, 1998. [DOI] [PubMed] [Google Scholar]
- 43.Sastri M, Barraclough DM, Carmichael PT, Taylor SS. A-kinase-interacting protein localizes protein kinase A in the nucleus. Proc Natl Acad Sci USA 102: 349–354, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh LP, Andy J, Anyamale V, Greene K, Alexander M, Crook ED. Hexosamine-induced fibronectin protein synthesis in mesangial cells is associated with increases in cAMP responsive element binding (CREB) phosphorylation and nuclear CREB: the involvement of protein kinases A and C. Diabetes 50: 2355–2362, 2001. [DOI] [PubMed] [Google Scholar]
- 45.Singh LP, Crook ED. Hexosamine regulation of glucose-mediated laminin synthesis in mesangial cells involves protein kinases A and C. Am J Physiol Renal Physiol 279: F646–F654, 2000. [DOI] [PubMed] [Google Scholar]
- 46.Steichen JM, Kuchinskas M, Keshwani MM, Yang J, Adams JA, Taylor SS. Structural basis for the regulation of protein kinase A by activation loop phosphorylation. J Biol Chem 287: 14672–14680, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sterchi EE, Naim HY, Lentze MJ, Hauri HP, Fransen JA. N-benzoyl-l-tyrosyl-p-aminobenzoic acid hydrolase: a metalloendopeptidase of the human intestinal microvillus membrane which degrades biologically active peptides. Arch Biochem Biophys 265: 105–118, 1988. [DOI] [PubMed] [Google Scholar]
- 48.Sun Q, Jin HJ, Bond JS. Disruption of the meprin alpha and beta genes in mice alters homeostasis of monocytes and natural killer cells. Exp Hematol 37: 346–356, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tholey A, Pipkorn R, Bossemeyer D, Kinzel V, Reed J. Influence of myristoylation, phosphorylation, and deamidation on the structural behavior of the N-terminus of the catalytic subunit of cAMP-dependent protein kinase. Biochemistry 40: 225–231, 2001. [DOI] [PubMed] [Google Scholar]
- 50.Trachtman H, Valderrama E, Dietrich JM, Bond JS. The role of meprin A in the pathogenesis of acute renal failure. Biochem Biophys Res Commun 208: 498–505, 1995. [DOI] [PubMed] [Google Scholar]
- 51.Uhler MD, Chrivia JC, McKnight GS. Evidence for a second isoform of the catalytic subunit of cAMP-dependent protein kinase. J Biol Chem 261: 15360–15363, 1986. [PubMed] [Google Scholar]
- 52.Walker PD, Kaushal GP, Shah SV. Meprin A, the major matrix degrading enzyme in renal tubules, produces a novel nidogen fragment in vitro and in vivo. Kidney Int 53: 1673–1680, 1998. [DOI] [PubMed] [Google Scholar]
- 53.Wang D, Fu Q, Zhou Y, Xu B, Shi Q, Igwe B, Matt L, Hell JW, Wisely EV, Oddo S, Xiang YK. Beta2 adrenergic receptor, protein kinase A (PKA) and c-Jun N-terminal kinase (JNK) signaling pathways mediate tau pathology in Alzheimer disease models. J Biol Chem 288: 10298–10307, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang L, Zhu Y, Sharma K. Transforming growth factor-beta1 stimulates protein kinase A in mesangial cells. J Biol Chem 273: 8522–8527, 1998. [DOI] [PubMed] [Google Scholar]
- 55.Weinberg JM, Buchanan DN, Davis JA, Abarzua M. Metabolic aspects of protection by glycine against hypoxic injury to isolated proximal tubules. J Am Soc Nephrol 1: 949–958, 1991. [DOI] [PubMed] [Google Scholar]
- 56.Xu Y, Osborne BW, Stanton RC. Diabetes causes inhibition of glucose-6-phosphate dehydrogenase via activation of PKA, which contributes to oxidative stress in rat kidney cortex. Am J Physiol Renal Physiol 289: F1040–F1047, 2005. [DOI] [PubMed] [Google Scholar]
- 57.Yamaguchi T, Fukase M, Sugimoto T, Kido H, Chihara K. Purification of meprin from human kidney and its role in parathyroid hormone degradation. Biol Chem Hoppe Seyler 375: 821–824, 1994. [PubMed] [Google Scholar]