

Abstract
Mutation of threonine for isoleucine at codon 73 (I73T) in the human surfactant protein C (hSP-C) gene (SFTPC) accounts for a significant portion of SFTPC mutations associated with interstitial lung disease (ILD). Cell lines stably expressing tagged primary translation product of SP-C isoforms were generated to test the hypothesis that deposition of hSP-CI73T within the endosomal system promotes disruption of a key cellular quality control pathway, macroautophagy. By fluorescence microscopy, wild-type hSP-C (hSP-CWT) colocalized with exogenously expressed human ATP binding cassette class A3 (hABCA3), an indicator of normal trafficking to lysosomal-related organelles. In contrast, hSP-CI73T was dissociated from hABCA3 but colocalized to the plasma membrane as well as the endosomal network. Cells expressing hSP-CI73T exhibited increases in size and number of cytosolic green fluorescent protein/microtubule-associated protein 1 light-chain 3 (LC3) vesicles, some of which colabeled with red fluorescent protein from the gene dsRed/hSP-CI73T. By transmission electron microscopy, hSP-CI73T cells contained abnormally large autophagic vacuoles containing organellar and proteinaceous debris, which phenocopied ultrastructural changes in alveolar type 2 cells in a lung biopsy from a SFTPC I73T patient. Biochemically, hSP-CI73T cells exhibited increased expression of Atg8/LC3, SQSTM1/p62, and Rab7, consistent with a distal block in autophagic vacuole maturation, confirmed by flux studies using bafilomycin A1 and rapamycin. Functionally, hSP-CI73T cells showed an impaired degradative capacity for an aggregation-prone huntingtin-1 reporter substrate. The disruption of autophagy-dependent proteostasis was accompanied by increases in mitochondria biomass and parkin expression coupled with a decrease in mitochondrial membrane potential. We conclude that hSP-CI73T induces an acquired block in macroautophagy-dependent proteostasis and mitophagy, which could contribute to the increased vulnerability of the lung epithelia to second-hit injury as seen in ILD.
Keywords: surfactant protein, autophagy, pulmonary fibrosis, alveolar epithelium, amphisome
human surfactant protein c (hSP-C) is a hydrophobic protein produced exclusively by alveolar type 2 (AT2) epithelial cells and plays an important role in the modulation of lung mechanics by its direct effects on alveolar surface tension (73). The hSP-C gene (SFTPC) encodes a primary translation product (proSP-C) containing four distinct structural and functional domains, is posttranslationally processed and trafficked as a type II integral membrane protein to specialized AT2 lysosomal-related organelles (LROs) termed lamellar bodies (LBs), and is released via regulated exocytosis together with other surfactant proteins and phospholipids (8). The NH2 flanking propeptide (residues 1–23), which resides on the cytosolic side, contains a PPDY motif and a lysine residue (K6) required for interaction with the E3 ubiquitin ligase Nedd4-2 and monoubiquitination, respectively, and functions as the critical targeting motif for post-Golgi trafficking to LBs (33). The mature SP-C domain (residues 24–59) is a membrane spanning α-helix that acts as a noncleavable signal anchor peptide and is ultimately secreted as a 3.7-kDa form in tight association with surfactant phospholipids (8, 73). The COOH propeptide (residues 59–197), which resides in the lumen, contains at least two distinct domains. The human BRICHOS domain of SP-C (residues 94–197) is involved in the proper folding of the proprotein within the endoplasmic reticulum (ER) lumen and has high homology to a number of proteins whose family members are most prominently linked to neurodegenerative diseases (56). Studies suggest that the BRICHOS domain binds to the transmembrane segment of SP-C to prevent β-sheet formation (74, 75). The more proximal portion of the COOH region (residues 59–93), joining the mature SP-C peptide to the BRICHOS domain, has been termed the “linker domain” (74–75).
The threonine for isoleucine missense substitution at residue 73 (hSP-CI73T) located within the proSP-C linker domain is the most frequently found mutation in humans, accounting for over 30% of all SFTPC mutations associated with diffuse parenchymal lung disease, in both de novo (sporadic) as well as inherited (autosomal dominant) cases (12, 14, 16, 52, 67). Histological and biochemical analyses of lungs from these patients are typically marked by a pattern of interstitial pneumonitis, which is often accompanied by evidence of alveolar accumulation of phospholipids, protein, and macrophages. Moreover, an accumulation of COOH-terminal proSP-C cleavage products in the bronchoalveolar lavage has also been observed in some patients with hSP-CI73T (23) although the consequences of their presence for surfactant function or contribution to disease progression are unknown.
The hSP-CI73T missense substitution is one on a growing list of monogenetic mutations of genes expressed in lung epithelia [SFTPC, SFTPA, and ATP binding cassette class A3 (ABCA3)] that are recognized as important causes of inherited pulmonary disorders in both children and adults (24, 38). The majority of SFTPC mutations that were first described were localized within the distal COOH-terminal flanking region of the BRICHOS domain. BRICHOS mutations have been shown in vitro to cause the protein to misfold and aggregate, inducing the classical unfolded protein response (UPR), ER-associated degradation (ERAD), ER stress, and apoptosis (8, 39–41, 46, 47). In contrast, mutations in the more proximal proSP-C COOH linker domain (termed non-BRICHOS) such as hSP-CI73T appear to adversely affect epithelial cell function through other mechanisms that do not involve misfolding, aggregation, or induction of ER stress. Transient expression of these non-BRICHOS mutants in a variety of in vitro models is associated with their abnormal routing directly to the plasma membrane (4, 8, 12) with subsequent reinternalization and deposition of aberrantly processed proSP-C intermediates within early endosomal antigen 1 (EEA-1)-positive compartments that are associated with impaired uptake and degradation of exogenous surfactant lipid (4).
Macroautophagy (hereafter used interchangeably with autophagy) is a highly conserved cellular catabolic process originally described as an inducible response to nutrient deprivation for the mobilization of endogenous energy components (amino acids and lipids), which more recently has emerged as a major cellular quality control pathway for the targeted removal of both long-lived, aggregation-prone proteins (e.g., huntingtin, α-synuclein, and neuroserpins) and dysfunctional organelles (e.g., mitochondria) (32). Autophagy is a dynamic process of intracellular membrane assembly and vacuolarization consisting of multiple highly orchestrated discrete stages controlled by at least 30 known autophagy (Atg) genes (32, 34). Sequestration of autophagy substrates from the cytosol first involves Atg-dependent formation of an isolation membrane, which, through the actions of additional Atg gene products, such as Atg5/12 and Atg8 (microtubule-associated protein 1 light-chain 3, LC3), eventually self-encloses to form the characteristic double-membrane autophagosome (32). Further maturation of this autophagic vacuole has been shown to involve an additional intermediate step characterized by fusion of the autophagosome with a variety of early (EEA-1; Rab5 positive) and late endosomal (LE) (Rab7 positive) compartments to yield a hybrid intermediate organelle termed the amphisome (10, 34). The autophagic process is completed by a final fusion of autophagic vacuoles (autophagosomes/amphisomes), with lysosomes resulting in degradation of captured substrates mediated by lysosomal hydrolases and an acidic intraluminal pH (44). The critical role played by autophagy in cellular homeostasis is underscored by recent data from lysosomal storage diseases, which exhibit alterations in both macromolecular autophagy and mitophagy, resulting in defective macromolecular substrate removal (degradation) and accumulation of dysfunctional mitochondria, together contributing to cellular dysfunction and disease pathogenesis (37, 59, 60).
Given the prior work showing the terminal deposition of the hSP-CI73T mutant within the endosomal network, we hypothesized that the expression of this mistrafficked non-BRICHOS SFTPC mutant would promote a more global epithelial cell dysfunction via impairment of macroautophagy. In this study, we show that stable expression of hSP-CI73T produces a distal block in cellular autophagy at the level of LE/autophagosome maturation, leading to impaired clearance of aggregation-prone autophagy substrates and accompanied by a functional impairment of mitophagy with resultant accumulation of damaged mitochondria. Together, these findings support the concept of a unique cellular phenotype resulting from expression of hSP-CI73T, which represents an acquired cellular quality-control disorder, whereby the toxic gain of function is induced indirectly by mistrafficking of an aberrant gene product.
MATERIALS AND METHODS
Reagents.
The p-enhanced green fluorescent protein (pEGFP)-C1 and red fluorescent protein from the gene dsRed (pDsRed)-C1 monomer plasmids were purchased from Clontech (Mountain View, CA). The EGFP-ABCA3 expression plasmid cloned in our laboratory has been described previously (5, 9). Tissue culture medium was produced by the Cell Center Facility, University of Pennsylvania. Except where noted, all other reagents were electrophoresis, tissue culture, or analytical grade and were purchased from Sigma Chemical (St. Louis, MO) or Bio-Rad (Hercules, CA).
Cell lines and treatment.
The embryonic kidney epithelial (HEK293) (HEK) cell line was originally obtained through the American Type Culture Collection (ATCC, Manassas, VA). HEK cells were grown as monolayers at 37°C, in a 5% CO2-supplemented incubator. cDNAs encoding for human wild-type (hSP-CWT) or mutant (hSP-CI73T) full-length proSP-C (197 amino acids) were inserted into either pEGFP-C1 or pDsRed-C1 vector as described previously (71, 72). Stable HEK cells lines containing EGFP- and DsRed-tagged hSP-CWT or hSP-CI73T were generated using CaPO4 transfection described previously with subsequent selection steps taken to isolate individual clones using 1 mg/ml G418 (13). Stable cell lines were maintained in DMEM supplemented with 500 μg/ml G418, 2 mM glutamine, 1% penicillin/streptomycin, and 10% fetal bovine serum.
Parental or stably expressing HEK cell lines grown to 75–85% confluence in 35-mm plastic dishes were subjected to reporter transfection or pharmacological treatments. Transient transfection with either empty vector (pcDNA3), a GFP-LC3 plasmid (gift of Dr. Ravi Amaravadi, University of Pennsylvania), or EGFP-tagged huntingtin exon 1 constructs containing poly Q25 or poly Q72 originally developed by Dr. Alan Tobin, University of California, Los Angeles, CA (a gift from Dr. Randy Pittman, University of Pennsylvania, Philadelphia, PA) were performed using CaPO4 as previously published (45) at doses of 1–2 μg/dish. Media were replaced after 18 h, and cells were cultured for an additional 24 h before harvest or fixation.
Where designated, cells were treated with MG132 (Sigma-Aldrich), bafilomycin A1 (BafA1; Sigma-Aldrich), Torin (Tocris Bioscience, Bristol, UK), or rapamycin (LC Laboratories, Woburn, MA) at indicated concentrations and duration.
Cytosolic and mitochondria-enriched organellar fractions were prepared as described (25). Briefly, cells harvested by scraping were washed in PBS, and protein concentration was determined using the Lowry method (Bio-Rad) with BSA as the standard. The cells were lysed mechanically in isolation medium (250 mM sucrose, 1 mM EDTA, 10 mM Tris, pH 7.4, supplemented with protease and phosphatase inhibitors). After nuclei were removed by centrifugation (1,500 g), the organelle fraction was pelleted at 11,800 g for 10 min. The pellet was washed and repelleted before resuspension in isolation medium (organelles). A fixed percentage of the recovered mitochondrial-enriched organellar pellet along with equal amounts of the cytosolic fraction (supernatant) were analyzed by SDS-PAGE. The first postnuclear supernatant (cytosol) was subjected to one additional low-speed centrifugation (1,500 g) before analysis and contained no detectable amounts of mitochondrial proteins by Western analysis for MTCO2.
Case summary and biopsy from a patient with hSP-CI73T missense mutation.
A 13-mo-old preterm female with a history of eczema and failure to thrive was admitted to the hospital for increased work of breathing associated with a viral upper respiratory infection. A prolonged oxygen requirement and tachypnea after resolution of her viral syndrome led to a pulmonary consultation. Family history was insignificant. A chest radiograph revealed marked hyperinflation and diffuse bilateral pulmonary opacities. A noncontrasted CT scan of the chest revealed diffuse ground glass bilateral pulmonary opacities, concerning for interstitial lung disease. A wedge biopsy was performed, showing interstitial expansion by epithelioid cells without collagen, fibroblasts, or hemorrhage, with focally prominent type II cell hyperplasia. Some alveoli showed filling by foamy macrophages, cholesterol clefts, and/or paplike proteinaceous fluid. No viral cytopathic effect was noted, and periodic acid-Schiff staining was negative; a diagnosis of chronic pneumonitis of infancy was assigned. Portions of this biopsy were also fixed for transmission electron microscopy as part of a standard protocol for evaluation of pediatric lung disease. Genetic testing revealed a heterozygous c.218 T>C substitution in one allele of SFTPC resulting in the described I73T missense mutation in the proSP-C sequence. The Institutional Review Boards at both the University of Pennsylvania and University of North Carolina have classified the clinical data and pathology specimens as an exempt study (not human subject research).
Immunoblotting and antibodies.
Cells collected from 35-mm culture dish by scraping and quantitated by hemocytometer counting were solubilized in sample loading buffer (93.75 mM Tris·HCl, pH 6.8, 3% SDS, 15% glycerol, and 0.015% bromophenol blue) supplemented with 5% dithiothreitol. Samples were sonicated on ice in 10-s bursts twice at 50 W and heated to 95°C for 5 min, and proteins were separated by SDS/PAGE and transferred to PVDF membranes. Immunoblotting of transferred samples was performed using incubations with primary anti-EGFP (Clontech), 1:2,000; anti-β-actin (Sigma), 1:5,000; anti-LC3B (Cell Signaling, Danvers, MA), 1:1,000; anti-p62 (Cell Signaling), 1:1,000; anti-MTCO2 (Abcam, Cambridge, MA), 1:1,000; anti-parkin (Cell Signaling), 1:1,000; anti-DsRed (Clontech), 1:2,000; anti-Rab7 (Cell Signaling), 1:1,000, or anti-Lamp1 (Abcam) 1:1,000; and horseradish peroxidase-conjugated secondary antibodies as previously described (72). Bands were visualized by enhanced chemiluminescence using a commercially available kit, ECL Western Blotting Detection Reagents (Amersham, Arlington Heights, IL). Chemiluminescence images were produced either by exposure to film or by direct acquisition using a CCD camera system (Kodak 440 Imaging System; Kodak, New Haven, CT).
Immunocytochemistry.
Colocalization studies were performed by immunostaining plated cells that were fixed by immersion of coverslips in 4% paraformaldehyde. Following permeabilization, cells were immunolabeled with primary antibodies for 3 h at room temperature at the following dilutions: anti-CD63 (Sanquin, Amsterdam, the Netherlands), 1:2,500; anti-EEA1 (BD Biosciences, San Jose, CA), 1:200; and anti-epithelial cell adhesion molecule (Ep-CAM) (Santa Cruz Biotechnology, Santa Cruz, CA), 1:100. FITC-conjugated secondary goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) at 1:400 dilution and Texas Red-conjugated secondary goat anti-mouse IgG monoclonal or secondary goat anti-rabbit IgG polyclonal antibodies (Jackson ImmunoResearch Laboratories) at 1:200 dilution were used for visualization.
Fluorescence microscopy.
Fluorescence images of either live cells in 35-mm culture dishes or cells that were on air-dried and Mowiol-mounted slides were viewed on an Olympus I-70 inverted fluorescent microscope (Olympus, Melville, NY) with filter packages High Q fluorescein isothiocyanate for EGFP (excitation at 480 nm, emission at 535/550 nm), and High Q TR for DsRed or mitotracker Red (excitation at 579/599 nm, emission at 645/675 nm) obtained from Chroma Technology (Brattleboro, VT). Fluorescence and phase images were captured using a Hamamatsu 12-bit coupled-charged device camera (Hamamatsu, Hamamatsu, Japan). Image acquisition, processing, and overlay analysis were performed using Metamorph 7.5 software (Molecular Devices, Sunnyvale, CA). For confocal microscopy, cells were examined using the TE300 Nikon coupled Radiance 2000 imaging system (Carl Zeiss, Thornwood, NY). All images were subsequently transformed from 24-bit program files to 8-bit TIFF files for assembly and labeling.
Vital imaging of mitochondrial function.
The presence of mitochondria with an intact transmembrane potential (ΔΨ) was determined by incubation of live cells with Mitotracker Red (Molecular Probes, Eugene, OR). Conditions were as follows: CMXRos (Mitotracker Red) stock solution prepared in DMSO was dissolved in prewarmed low-serum DMEM (1% penicillin/streptomycin and 5% fetal bovine serum) to a final concentration of 25 nM. Cell monolayers were incubated with CMXRos-DMEM for 15 min at 37°C with 5% CO2, washed in fresh media, and imaged. Cells were examined for fluorescence signals using an Olympus X-40 inverted microscope and captured with a Hamamatsu 12-bit coupled-charged device camera. Image acquisition, fluorescence quantitation, processing, and analysis were performed using the Metamorph 7.5 software (Molecular Devices) by utilizing identical imaging criteria within experimental groups including exposure time, image scaling, and image γ.
Transmission electron microscopy.
Transmission electron microscopy of HEK cells was performed in the Imaging Core Facility in the Institute for Environmental Medicine at the University of Pennsylvania School of Medicine using the method of Hayat and modified as we have previously published (26, 28). Cell lines grown on plastic dishes were rinsed in PBS and then fixed overnight at 4°C in 2% glutaraldehyde with 0.1 M sodium cacodylate buffer, pH 7.4. Cells harvested by scraping were pelleted by centrifugation and osmicated for 60 min at 4°C in 2% osmium tetra oxide in 0.1 M sodium cacodylate buffer. Following the washing with Na cacodylate buffer to remove osmium, samples were stained en bloc with 2% aqueous uranyl acetate for 30 min. Following dehydration in graded alcohol, pellets were embedded in epon 812 and cured at 70°C for 48 h. Seventy-nanometer-thick sections were generated with a Leica Ultracut S ultramicrotome and collected on 200 mesh copper grids, stained in 50% alcoholic uranyl acetate, and counter stained with bismuth subnitrite. Air-dried grids of both fixed cell lines and clinical biopsy samples were subsequently examined on a JEOL JEM 1010 electron microscope. Images were collected using a Hamamatsu CCD camera.
Samples from patient with hSP-CI73T were fixed in 2.5% glutaraldehyde, buffer rinsed in sodium phosphate buffer, postfixed in osmic acid, rinsed in water, dehydrated in ascending percentage of alcohols, cleared in propylene oxide, infiltrated in propylene oxide and epon, and then embedded in fresh epon. Thick sections (1 μm) were taken, placed on a glass slide, and then stained in toluidine blue. The slides were then reviewed for specific areas for thin sections. Thin sections (90 nm) were taken, placed on a copper grid, stained with uranyl acetate and lead citrate, and then examined in a Zeiss LEO 910 transmission electron microscope.
Statistical analysis.
Experimental data were analyzed by one-way ANOVA with the Tukey-Kramer post hoc test using GraphPad InStat software, version 3.0 for Windows (GraphPad Software, San Diego, CA). All values are means ± SE unless otherwise indicated. Significance was accepted at P < 0.05.
RESULTS
Expression of hSP-CI73T results in the deposition of aberrantly processed mutant protein in plasma membrane and endosomal compartments.
Because of well-appreciated technical issues with generating stable cell lines from primary AT2 cells, we chose to characterize the influence of chronic expression of the SFTPC I73T missense substitution on cellular homeostasis by generation of several HEK293 cell lines stably expressing fluorescently tagged hSP-C isoforms. Using transiently transfected cell lines (such as HEK293, A549, and primary AT2 cells), we and others have previously reported that, whereas wild-type proSP-C is directly routed via LE/multivesicular bodies (MVB) to LROs and LBs, respectively (6, 7, 33, 70, 72), hSP-CI73T expression is concentrated in the plasma membrane and early endosomes (4). Consistent with those findings, in HEK cells stably expressing EGFP/hSP-CI73T, the subcellular distribution of the expressed hSP-CI73T fusion products included the plasma membrane (as confirmed by costaining for Ep-CAM) and EEA-1-positive cytosolic vesicles (Fig. 1A). These findings are consistent with recent immunogold data in another hSP-CI73T cell line demonstrating significant expression of hSPCI73T on the plasma membrane (61). Furthermore, whereas hSP-CI73T was significantly excluded from LROs as identified by the use of an ABCA3/EGFP reporter construct (Fig. 1B), we observed a subset of hSP-CI73T expression within the lumen of cytosolic structures staining positive for Rab7, a small GTP-binding protein used as a LE marker antigen (Fig. 1C). In addition, some of the mutant isoform also coexpressed with CD63 (a marker antigen predominantly expressed in LE/MVB and lysosomes) (53) (Fig. 1D). In contrast to mutant hSP-CI73T, stably expressed hSP-CWT fusion protein was highly colocalized to LROs (ABCA3/EGFP-positive vesicles) (Fig. 1B) and a subset of smaller punctate CD63-positive vesicles (Fig. 1D) but was excluded from EEA-1, Rab7, and plasma membrane compartments (Fig. 1, A and C). When combined with prior studies utilizing transient expression, these results are consistent with a model whereby the I73T missense substitution results in the diversion of proSP-CI73T away from LROs, and its mistrafficking to the plasma membrane is followed by subsequent retrograde deposition within early endosomes and eventually in Rab7/CD63-positive LE/MVB.
Fig. 1.

Stably expressed human surfactant protein C with the mutation for threonine for isoleucine missense substitution at residue 73 (hSP-CI73T) is localized to the plasma membrane and endosomal compartments and undergoes aberrant processing. A: representative confocal images of human embryonic kidney epithelial (HEK293) (HEK) cells stably expressing enhanced green fluorescent protein (EGFP)/wild-type hSP-C (hSP-CWT) or EGFP/hSP-CI73T immunostained for Texas Red-conjugated early endosomal antigen 1 (EEA1) (top) or epithelial cell adhesion molecule (Ep-CAM) (bottom) antibodies. Solid boxes from the merged images are magnified to illustrate colocalization or noncolocalization (right). EGFP/hSP-CI73T was highly colocalized with the plasma membrane marker Ep-Cam and early endosomes (EEA-1). Bar = 5 μm. B, left: representative fluorescent images of HEK cells stably expressing red fluorescent protein from the gene dsRed (DsRed)/hSP-CWT (top) or DsRed/hSP-CI73T (bottom) 48 h following expression of EGFP-human ATP binding cassette class A3 (hABCA3) using CaPO4 transfection method. Solid boxes from the merged images are magnified to illustrate colocalization or lack of colocalization. Right: counts of vesicle coexpressing hABCA3/EGFP and DsRed/hSP-C isoforms. Data were obtained from 2 separate experiments, each with counts averaging 50–100 vesicles per cell in at least 20 transfected cells. *P < 0.05. C: representative fluorescent images of HEK cells stably expressing DsRed/hSP-CWT (top) or DsRed/hSP-CI73T (bottom) immunostained with Alexa Fluor 488-conjugated anti-Rab7. Solid boxes from the merged images are magnified to illustrate colocalization of hSP-CI73T but not DsRed/hSP-CWT with Rab7. White arrowheads highlight expression of DsRed/hSP-CI73T within large Rab7 vesicles. Bar = 5 μm. D: representative confocal images of HEK cells stably expressing EGFP/hSP-CWT or EGFP/hSP-CI73T immunostained with Texas Red-conjugated CD63 antibodies. Solid boxes from the merged images are magnified to illustrate colocalization (right). EGFP/hSP-CWT and EGFP/hSP-CI73T each partially colocalized with CD63, which is highly expressed in late endosomes and lysosomes. Bar = 5 μm. E: representative anti-GFP immunoblot bands (left) of HEK whole cell lysate samples stably expressing either EGFP/hSP-CWT (middle lane) or EGFP/hSP-CI73T (right lane) are displayed to demonstrate equal expression of SP-C isotypes. PC, parental cell line. β-Actin was used to normalize for equal loading and calculation of relative band intensity (right).
Mistrafficking of hSP-CI73T in these in vitro models was associated with aberrant posttranslational processing of the mutant proSP-C primary translation product. Western blotting of cells stably expressing EGFP/hSP-CI73T or EGFP/hSP-CWT using antisera against the NH2-terminally placed EGFP tag demonstrated the presence of multiple mutant hSP-CI73T intermediate forms that differed distinctly from the banding pattern seen for EGFP/hSP-CWT, which is characteristic of a normal post-Golgi proteolytic processing en route to LROs (Fig. 1E). Aberrant processing was also detected in another stable cell line expressing HEK-DsRed/hSP-CI73T (data not shown) and was consistent with processing profiles previously observed in prior reports using transiently transfected cell lines (4, 76).
Stable expression of hSP-CI73T induces increases in autophagic vacuole number and size.
On the basis of the expression data shown in Fig. 1 coupled with prior data showing that expression of mutant hSP-CI73T was associated with an impairment in uptake and degradation of exogenously added surfactant phospholipids (4), we hypothesized that the cellular dysfunction observed in the hSP-CI73T phenotype represents an acquired disruption in the endosomal-lysosomal pathway and its dependent cellular functions such as cellular quality control, including macroautophagy (37). We examined the influence of chronic hSP-CI73T expression on autophagy using a variety of independent static and dynamic measures. GFP-tagged LC3, a mammalian homolog of yeast Atg8, represents a specific marker for developing autophagosomes. HEK cells stably expressing DsRed-tagged hSP-CWT or hSP-CI73T were transiently transfected with GFP/LC3. DsRed/SP-CWT cells displayed a primarily diffuse subcellular distribution of the GFP/LC3 fusion in with occasional small cytosolic puncta, indicative of low basal numbers of autophagosomes (Fig. 2A, top). In contrast, DsRed/SP-CI73T-expressing cells exhibited numerous large GFP/LC3-labeled cytosolic punctae with some containing intraluminal DsRed/SP-CI73T (Fig. 2A, bottom). Quantitative assessment by counting vesicles revealed that chronic SP-CI73T expression induces significant increases in both size and number of LC3-containing autophagosomes (Fig. 2B).
Fig. 2.

Stable expression of DsRed/hSP-CI73T results in an increased number and size of autophagosomes, which contain mutant hSP-CI73T protein. A: representative confocal images of DsRed/hSP-CWT and DsRed/hSP-CI73T stable cell lines that were transiently transfected with an EGFP/microtubule-associated protein 1 light-chain 3 (LC3) plasmid for 48 h. Regions delineated by white boxes were enlarged to provide additional resolution (right). Bar = 5 μm. B, left: percentage (%) of EGFP/LC3-expressing cells containing puncta. At least 150 cells per group were counted from at least 3 separate experiments. *P < 0.001. Right: count of EGFP/LC3-positive puncta per cell. At least 50 cells per group from 3 separate experiments were counted. *P < 0.001.
Consistent with these findings, the ultrastructure of hSP-CI73T cells was notable for the presence of a significant number of abnormally large electron-dense bodies (Fig. 3A, bottom, arrowheads) containing cellular and organellar debris surrounded by limiting double membranes, a hallmark of autophagic vacuoles. Qualitatively, the presence of such electron-dense structures was conspicuously absent in hSP-CWT-expressing cells (Fig. 3A, top). Quantitatively, both the number and size of autophagic vacuoles were increased in response to the expression of hSP-CI73T (Fig. 3B).
Fig. 3.
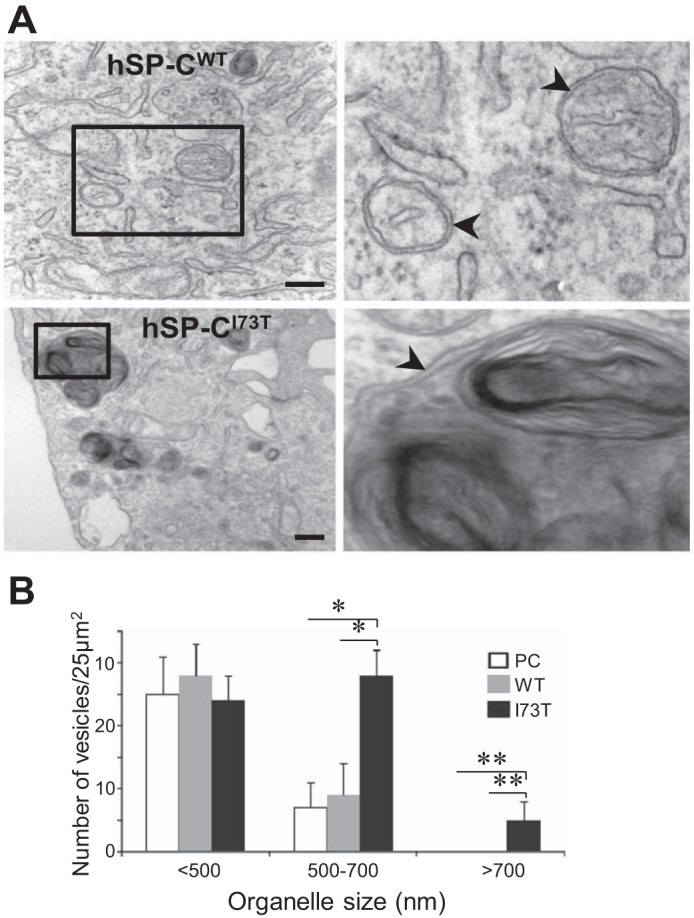
Chronic expression of hSP-CI73T leads to increases in enlarged autophagic vacuoles. A: representative electron microscopy (EM) images of HEK cells stably expressing DsRed/hSP-CWT (top) and DsRed/hSP-CI73T (bottom) showing normal (top) and enlarged (bottom) double-membraned (arrowheads) autophagosome-like vesicles. Solid boxes are magnified on left for better resolution (right). Bar = 500 nm. B: subcellular quantification of vesicles according to size. EM images from at least 20 cells per group were counted. *P < 0.01, **P < 0.001.
Autophagic inclusions are induced by hSP-CI73T in vivo.
Histological examination by light microscopy of a lung biopsy from a patient heterozygous for hSP-CI73T mutation showed interstitial expansion, prominent AT2 hyperplasia, and accumulation of foamy alveolar macrophages (Fig. 4, top). Moreover, ultrastructural evaluation of the biopsy specimen revealed AT2 cells with highly enlarged and structured double membrane-limited organelles containing various amorphous debris and organellar fragments (Fig. 4, middle and bottom). Thus the ultrastructural changes observed in vitro using stable cell lines phenocopied changes induced by hSP-CI73T in vivo.
Fig. 4.

The ultrastructural phenotype in lung epithelia from a SP-C I73T patient. A and B: histological examination of a lung biopsy from a patient heterozygous for hSP-CI73T mutation showing hematoxylin and eosin-stained sections with ×10 (left) and ×20 (right) magnifications. Histopathological features include hyperplasia of AT2 cells and accumulation of foamy macrophages (arrows) in the alveolar lumen. C and E: representative transmission EM images of lung biopsy from the same patient showing large, electron-dense vesicles containing various organellar debris. D and F: solid boxes in C and E are magnified for better resolution to show the double-membrane-encased autophagic vacuoles and cellular debris (arrowheads).
Cells expressing hSP-CI73T demonstrate increased expression of autophagy and LE components.
The influence of hSP-CI73T expression on autophagy was further assessed by biochemical profiling for key molecular components associated with the generation and turnover of autophagosomes. DsRed/hSP-C stable cell lines were transfected with a GFP/LC3 reporter construct, and both endogenous and the transfected GFP fusion forms of LC3 were assessed. The conversion of LC3-I to a lipidated LC3-II form by its coupling to phosphatidylethanolamine marks the activation of LC3 and its localization to the autophagosomal membrane. DsRed/hSP-CWT cells expressed minimal amounts of either endogenous LC3 isoform (Fig. 5A, lane 1). Overnight incubation with the proteasome inhibitor MG132 (previously shown to stimulate autophagy in some systems) (17) resulted in a mild to moderate increase in both LC3-I expression and conversion to LC3-II using either endogenous LC3 or the transfected GFP-tagged forms of LC3 as readouts (Fig. 5A, lane 2). In contrast, hSP-CI73T-expressing cells demonstrated robust LC3-I expression and conversion of both endogenous and exogenous fusion forms to LC3-II at baseline (Fig. 5A, lane 3) that was minimally enhanced by MG132 treatment (Fig. 5A, lane 5).
Fig. 5.

hSP-CI73T induces accumulation of autophagic substrates and Rab7 biomass while inhibiting autophagic flux. A: representative immunoblot of cell lysates from stable DsRed/hSP-CWT and DsRed/hSP-CI73T cell lines 48 h following transfection of GFP/LC3. MG132 (1 μM) or Rapamycin (Rapa) (1 μM) were added during the final 18 h of incubation. Results shown are from probing of a single membrane, which was later cut to remove additional lanes. Large increases in endogenous and exogenous LC3-I and LC3-II are apparent in cells expressing DsRed/hSP-CI73T. To normalize for loading, the same blots were reprobed for β-actin. B: representative immunoblots of the autophagy substrate protein, p62, in total lysates prepared from stable expressing HEK-DsRed/hSP-CWT and HEK-DsRed/hSP-CI73T cells showing p62 protein levels increased in cells expressing hSP-CI73T. β-Actin was used to normalize for equal loading. C: 2 clones each of stable DsRed/hSP-CWT and DsRed/hSP-CI73T cell lines probed for Rab7 or Lamp1 as indicated. β-Actin was used to normalize for equal loading. D: densitometric quantitation of Rab7 band intensity normalized to β-actin from 2 separate experiments. Data are normalized to actin and expressed as fold change over WT hSP-C values. *P < 0.05 vs. hSP-CWT. No differences were observed for Lamp-1 bands. E: representative immunoblots from stable HEK whole cell lysates showing time-dependent changes in LC3 species by 1 μM Rapa only in cells expressing DsRed/hSP-CWT. F: densitometric analysis of LC3-II/actin ratio E and 2 additional independent experiments. All data were normalized to WT 0 h (1) in each experiment. Time-dependent changes in LC3-II levels induced by rapamycin occurred in cells expressing DsRed/hSP-CWT but not DsRed/hSP-CI73T. *P < 0.05 vs. each time point after rapamycin addition.
To differentiate between enhanced autophagic induction and reduced autophagosomal turnover, we examined levels of p62 (SQSTM1). P62 is an autophagic substrate that facilitates delivery of polyubiquitinated substrates to the autophagosome through direct interaction with LC3 during autophagosome biogenesis (11, 50, 66), serving as a linker between ubiquitinated proteins and autophagy-mediated degradation. Compared with hSP-CWT cells, significant increases in p62 levels were observed in cells expressing hSP-CI73T (Fig. 5B). Because, in the course of normal autophagic flux, p62 itself is selectively and continually degraded along with its cargo and LC3 (50), the accumulation of p62 in the presence of an enhanced LC3-II signal is supportive of a late block in autophagy in cells expressing hSP-CI73T.
A functional endosomal network is necessary for the maturation of autophagosomes. Fusion of LE with autophagosomes leads to the formation of an intermediate organelle termed the amphisome, which has been shown to contain subcellular markers of both compartments (10). Western blotting of cell lysates demonstrated a marked enrichment in Rab7, a LE GTPase regulating endosomal/autophagosomal maturation in hSP-CI73T cells (Fig. 5, C and D). Lamp1, a membrane component of lysosomes, was not altered by hSP-CI73T expression. Thus hSP-CI73T leads to increases in biomass of both autophagosomes (LC3, p62) and LE and/or amphisomes (Rab7) consistent with inhibition of late autophagosome maturation to autolysosomes.
Autophagic flux measurements are consistent with a late block in autophagy in hSP-CI73T cells.
To further understand the dynamics underlying the steady-state increases in the levels of autophagy components LC3 and p62 observed in hSP-CI73T cells, autophagic flux studies were performed. First, the response to upstream induction of autophagy was measured by treatment of each cell line with rapamycin, an inhibitor of the mammalian target of rapamycin (mTOR) shown to activate autophagy and alter LC3 turnover (30). Consistent with this, rapamycin treatment resulted in an initial increase in LC3-II followed by a time-dependent overall reduction of LC3 isoforms in hSP-CWT-expressing cells (Fig. 5, E and F) consistent with enhanced utilization and degradation of a rate-limiting cellular pool of LC3-II proteins. By contrast, the high basal levels of LC3 expression in hSP-CI73T cells could not be further enhanced by rapamycin (Fig. 5E, bottom middle and bottom). To guard against overinterpretation of data from use of hSP-CI73T cells with high expression levels of autophagy components, we next analyzed separate clone expressing lower levels of the autophagy-related proteins that matched the basal expression of p62/LC3 observed in hSP-CWT cells (Fig. 6). When treated with BafA1, a v1-ATPase inhibitor shown to disrupt late autophagy events by a variety of mechanisms including autophagosome-lysosome fusion (31, 77), LC3-II and p62 levels could be fully induced by a 5–10-fold lower concentration of BafA1 in cells expressing hSP-CI73T compared with hSP-CWT control (Fig. 6). The shift in the BafA1 dose response curve to the left in hSP-CI73T cells is consistent with a late block in autophagic flux.
Fig. 6.
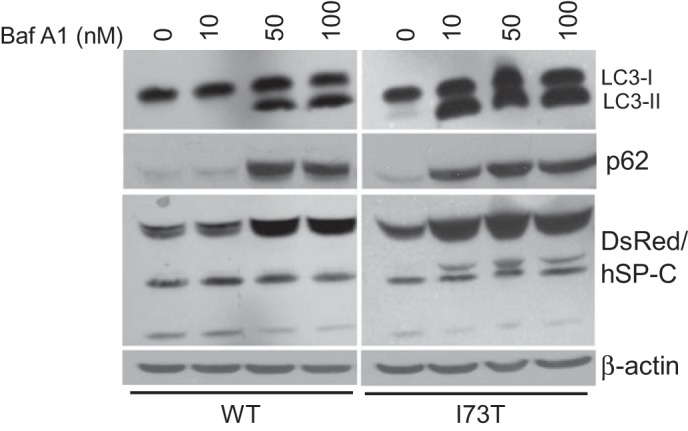
hSP-CI73T-expressing cells are more susceptible to bafilomycin-induced autophagic inhibition. Stable HEK cells expressing DsRed/hSP-CWT and a second DsRed/hSP-CI73T clone expressing similar levels of baseline LC3-I and p62 levels were treated with bafilomycin A1 (BafA1) for 18 h. Induction of LC3-II (top) and p62 (top middle) was evident at a 5-fold lower BafA1 concentration in cells expressing DsRed/hSP-CI73T compared with DsRed/hSP-CWT. Note that DsRed/hSP-C expression (proprotein and processed forms) was similar (bottom middle). β-Actin served as a loading control. Representative of 3 separate experiments.
Chronic expression of hSP-CI73T results in impaired clearance of aggregate-prone protein substrates.
Autophagy represents one of the primary degradation pathways responsible for eliminating abnormal protein aggregates. The accumulation of p62 coupled with the autophagic flux studies suggested such an impairment in autophagic function. Mutant huntingtin proteins (htg) associated with disease contain expanded polyglutamine (PolyQ) tracks (>38 Qs) that render the protein aggregation prone and poor substrates for the proteasome (68) and instead rely on the autophagic pathway for degradation (54, 58). HEK cells stably expressing DsRed-tagged hSP-CWT and hSP-CI73T were transiently transfected to express either EGFP (EGFP-C1) or EGFP-tagged htg-Exon 1 (htg1) containing polyQ repeats either of an aggregation-prone 72-glutamine isoform (EGFP-htg1Q72) or a nonaggregating 25-glutamine (EGFP-htg1Q25) as control. Equal transfection efficiency of each cell line was confirmed by cotransfection of Renilla luciferase (data not shown). Immunoblots from cell lysates harvested 48 h following transfection show the presence of aggregates that migrate with slower electrophoretic mobility at the top of the SDS gels in cells expressing EGFP-htg1Q72 (Fig. 7A). All cells contained both primary translation products as well as smaller processed htg1 isoforms previously described in HEK cells (65). Quantitative analyses by densitometry revealed this to be increased by almost threefold in cells expressing hSP-CI73T compared with hSP-CWT cells (Fig. 7B), indicating impaired autophagic disposal of aggregate-prone protein substrates in hSP-CI73T-expressing cells. To confirm the increase in htg1Q72 aggregation in hSP-CI73T cells, we also utilized fluorescence microscopy to detect the presence of cytosolic EGFP-htg1Q72 (but not EGFP-htg1Q25 or GFP) aggregates. As shown in Fig. 7C, manual counting of over 150 cells per cell line revealed that there was a parallel threefold increase in the percentage of EGFP-htg1Q72-transfected cells containing overt cytosolic aggregates in hSP-CI73T cells vs. hSP-CWT controls. In addition to increases in both p62 and htg1PolyQ aggregates, Western blotting of total cell lysates demonstrated moderate increases in all polyubiquinated proteins in hSP-CI73T cells (Fig. 7D). Taken in total, the data indicate that the late blockade in macroautophagy induced by hSP-CI73T expression results in a functional defect in clearance of ubiquinated protein substrates.
Fig. 7.

Stable hSP-CI73T expression inhibits degradation of aggregate-prone protein substrates. A, top: representative immunoblots of whole cell lysates from stable HEK cells 48 h following transfection of EGFP-C1 vector and EGFP/poly-Q constructs. Increased poly-Q72 aggregates is apparent in cells expressing DsRed/hSP-CI73T compared with DsRed/hSP-CWT. Middle: primary translation products and soluble processed forms of each EGFP-tagged construct demonstrate equal expression across groups. β-Actin was used to normalize for equal loading. NT, Nontransfected. B: quantitative analysis of blots in A. Data are expressed as fold change over WT and are representative of at least 3 independent experiments. *P < 0.05. C: quantification of EGFP/Q72 aggregates in DsRed/hSP-CWT and DsRed/hSP-CI73T cells expressing EGFP/Q72 for 48 h. At least 150 transfected cells per group were counted, *P < 0.05. D: immunoblot analysis with anti-ubiquitin and anti-β-actin in multiple HEK clones expressing HEK-DsRed/hSP-CWT and HEK-DsRed/hSP-CI73T. Representative of 2 separate experiments.
EGFP/hSP-CI73T expression leads to accumulation of dysfunctional mitochondria.
Autophagy plays an additional critical role in cellular homeostasis through the removal and recycling of long-lived and/or dysfunctional organelles. In particular, defective mitochondria are selectively removed and degraded by a regulated autophagic process, mitophagy (29). The consequences of impaired autophagy acquired by hSP-CI73T expression for mitochondrial dynamics and function were assessed. Stable expression of hSP-CI73T led to a 1.5-fold increase in MTCO2, a constituent of mitochondrial membrane, indicative of an increase in overall mitochondrial biomass when compared with hSP-CWT-expressing cells (Fig. 8, A and B). Parkin, a cytosolic protein critical to the autophagy-dependent removal of defective mitochondria, is rapidly translocated from the cytosol to the outer membrane of depolarized (dysfunctional) mitochondria, where it participates in signaling the removal of mitochondria by mitophagy (43, 48, 69). Both cytosolic and mitochondrial (organellar) steady-state levels of parkin were increased in cells expressing hSP-CI73T (Fig. 8, C and D). Analogous to p62 in protein clearance, the accumulation of parkin is consistent with a functional block in the degradation of mitophagy substrates.
Fig. 8.
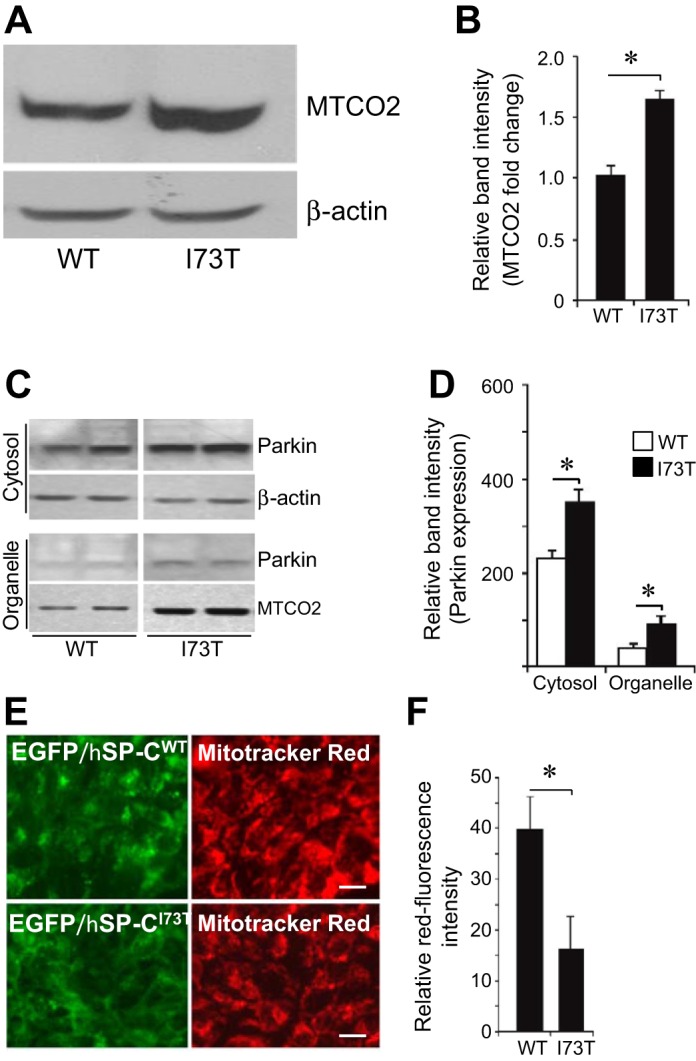
Chronic SP-CI73T protein expression promotes impaired mitophagy and accumulation of functionally impaired mitochondria. A: representative immunoblots probed for the mitochondria marker MTCO2 in total lysates prepared from stable expressing HEK-DsRed/hSP-CWT and HEK-DsRed/hSP-CI73T cells showing increased mitochondrial biomass in cells expressing hSP-CI73T. B: quantitation of MTCO2 content normalized to β-actin from 5 separate experiments. Data are expressed as fold change over WT hSP-C values. *P < 0.05 vs. hSP-CWT. C: representative immunoblots of cytosolic and organelle fractions prepared as described in materials and methods from stable expressing DsRed/hSP-CWT and DsRed/hSP-CI73T cell lysates. Equal amounts of cytosol (as determined by total protein) and an equal percentage of the obtained organellar fractions were loaded. β-Actin served as a loading control for cytosolic fractions. Increased MTCO2 was recovered in organellar fractions consistent with data in A above. Increased levels of parkin were translocated to organelle fractions in cells expressing hSP-CI73T compared with hSP-CWT. D: densitometric quantification of parkin levels from 4 independent experiments. *P < 0.05. E: identification of intact functional mitochondrial using Mitotracker Red showing decreased red fluorescence in live HEK cells expressing EGFP/SP-CI73T (bottom) compared with EGFP/SP-CWT. F: relative red channel fluorescence intensity was quantitatively determined from 3 separate experiments. *P < 0.05.
With impaired mitophagy, increases in mitochondrial mass and parkin expression are accompanied by changes in mitochondria bioenergetics. Mitotracker Red (chloromethyl-X-rosamine) is a cell-permeant mitochondrion-selective fluorescent dye that stains intact mitochondria in live cells, where its partitioning into the mitochondrial matrix is dependent on an intact mitochondrial transmembrane potential (Ψm). When stable cell lines expressing hSP-C isoforms were incubated with Mitotracker Red (Fig. 8, E and F), there was significant reduction in red fluorescence intensity in cells expressing SP-CI73T compared with SP-CWT cells, indicating loss of Ψm in mitochondria in the mutant cell line.
DISCUSSION
Previously, we have demonstrated that the biosynthetic routing of and cellular responses to mutant SFTPC gene products were dependent, in part, on the domain position of the altered amino acid residues within the proSP-C primary sequence. Expression of BRICHOS domain SFTPC mutations, which are highly prone to misfolding, ER retention, and eventual cytosolic aggregation, produces a toxic gain of function including ER stress (46, 47) and apoptosis through multiple UPR signaling pathways (36, 40, 46, 47). In contrast, non-BRICHOS mutations within the proximal COOH propeptide (I73T, E66K) result in very low amounts of ER stress or aggregated SP-C (49, 75) and instead are aberrantly trafficked to the plasma membrane (4, 8, 12, 61) with subsequent internalization and accumulation within the endosomal system, resulting in cellular dysfunction (4, 76). Using hSP-CI73T as a prototype, the present work extends these previous studies to provide molecular mechanisms for cellular dysfunction caused by non-BRICHOS SFTPC mutations by coupling their expression to a striking disruption of normal cellular macroautophagy punctuated by a prominent accumulation of dysmorphic autophagic vacuoles both in vitro and in vivo occurring in association with a diminished functional capacity of the cells to degrade both protein aggregate substrates and dysfunctional mitochondria. Taken together, the hSP-CI73T cellular phenotype is consistent with that of a late block in autophagy, as has been observed in other organellar storage disorders (57, 59).
Whereas aggregation-prone BRICHOS mutations induce canonical UPR signaling (40, 46, 47), mutations within the linker domain such as hSP-CI73T appear to foster its escape from both normal biosynthetic routing and ER quality control sensing, opting for deposition on the plasma membrane (4, 12, 47, 61). The mechanism underlying how clinical mutations involving the luminal linker domain (I73T, E66K) promote the observed mistrafficking remains incompletely defined. It has been suggested that this region serves as a strand for docking the BRICHOS domain to the transmembrane (mature SP-C) domain (74, 75). Despite having highly conserved amino acid residues, the evaluation of the linker domain crystal structure suggests a highly disordered and flexible secondary structure that has been postulated to be capable of disrupting the mature SP-C transmembrane-BRICHOS association of the proprotein (74, 75). Recent evidence suggests that, unlike BRICHOS mutations of SFTPC, hSP-CI73T fails to interact with quality-control chaperones ERDJ4/ERDJ5 that are required for retrotranslocation of aberrantly folded proteins to the proteasome (62). Thus the influence of the I73T substitution on proSP-C trafficking may also be indirect, affecting protein-protein interactions with luminal chaperones or other cofactors with its subsequent misdirection away from LROs or the UPR and with accumulation on the plasma membrane representing a default or constitutive pathway.
Although expression of SFTPC linker domain mutants like hSP-CI73T does not result in significant induction of either ER stress or intrinsic apoptotic cell death pathways, as has been reported for SFTPC BRICHOS mutants (47), the present study has clearly established that chronic expression hSP-CI73T produces a distinct second SFTPC mutant-related cellular phenotype marked by disruption of a key metabolic process, macroautophagy. Perturbation in autophagy was measured by the steady-state number of autophagosomes, which correlates with the autophagosome-associated form of microtubule-associated LC3-II (30). We found accumulation of autophagosomes in the hSP-C I73T mutant cells, as assessed by elevated LC3-II levels and LC3+ vesicles (Figs. 5 and 2, respectively), and autophagic vacuoles analyzed by electron microscopy (Fig. 3). Because both synthesis and degradation affect steady-state LC3-II levels, we determined autophagic flux using a combination of an mTOR inhibitor (rapamycin), which induces autophagy, and BafA1, which prevents lysosomal acidification and accumulates LC3-II by inhibiting its degradation. When treated with rapamycin, hSP-CWT cells demonstrate a normal profile of autophagy induction and LC3 turnover, whereas no further increases in LC3 were observed in hSP-CI73T cells (Fig. 5). Under BafA1-treated conditions, in clones where levels of the autophagy-related proteins were selected to match the basal expression of p62/LC3 observed in hSP-CWT cells, there was enhanced sensitivity to BafA1 in cells expressing hSP-CI73T compared with hSP-CWT control (Fig. 6). Whereas we have documented commensurate alterations in mitochondrial transmembrane potential (ΔΨm) (Fig. 8), we have not found significant differences in total cellular ATP levels (unpublished observation, M. Beers). It is also important to note that BafA1 is a noncompetitive inhibitor of v1-ATPases, and thus its kinetics of inhibition are independent of cytosolic ATP concentration and unlikely to be an explanation for the enhanced BafA1 sensitivity observed in hSP-CI73T cells. Therefore, we conclude that LC3-II accumulation in hSP-CI73T cells is predominantly attributable to decreased autophagosome maturation/degradation (a late block in macroautophagy).
A key consequence produced by the chronic expression of hSP-CI73T used in the current study is the acquisition of a cellular phenotype with impaired cellular quality control (macroautophagy) and is consistent with the ultrastructural phenotype observed in AT2 cells of a patient carrying the hSP-CI73T missense substitution, including intracellular accumulation of autophagosomes containing undegraded macromolecular and organellar debris (Fig. 4). Functionally, this was demonstrated to result in an increase in the cytosolic aggregation of a prototype autophagic reporter substrate, huntingtin (Fig. 7, A–C). Because there is redundancy in the cellular proteostasis system (3), the exact magnitude of the effect of a block in autophagy likely depends on the sum total contributions of both the ubiquitin-proteasome system and all forms of autophagy (macro, micro, and chaperone dependent). Although we also noted increases of baseline endogenous polyubiquinated proteins (Fig. 7D), additional studies utilizing provocative challenges (e.g., under conditions of induced proteolysis) will be required to more completely define the proteostasis system and are in progress.
The most striking feature observed in hSP-CI73T cells is a block in mitophagy. Using a variety of techniques, this study demonstrated that one of the consequences induced by an hSP-CI73T autophagy block is the accumulation of dysfunctional mitochondria. The increased levels of MTCO2 of hSP-CI73T cells at baseline support the case for an increased mitochondrial biomass (Fig. 8A), whereas data using a vital dyes suggest a decrease in the mitochondrial ΔΨm in the mutant cell line, consistent with an enhanced leakiness (Fig. 8, E and F). These findings were accompanied by parallel increases in parkin content in the mitochondrial-enriched organellar fraction (Fig. 8, C and D), and, when combined with the demonstrated block in overall macroautophagy (Figs. 3, 5, and 6), the increased levels of mitochondrial parkin are consistent with a block in mitophagy in which parkin is successfully translocated to the outer membrane of depolarized mitochondria but fails to be processed and degraded along with other mitochondrial components (such as MTCO2/COX4). Although the results presented are most compatible with an accumulation of dysfunctional mitochondria within the autophagosome, there could well be other factors beyond quality control that are contributing to the observed changes in mitochondrial mass and membrane potential. Increases in mitochondrial biogenesis or changes in the bioenergetic efficiency (induced by substrate supply or by fusion/fission events of mitochondrial turnover) respectively cannot be excluded at this point and will require additional experimentation.
In addition to increases in mitochondrial parkin, hSP-CI73T cells also showed upregulation of cytosolic parkin. The mechanism and teleological reasons underlying this finding cannot be answered in the present studies. Upregulation in total parkin content has been described in association with ER stress; however, this is cell type specific, and we have previously shown that hSP-CI73T expression does not enhance markers of ER stress (47). However, parkin has recently been shown to ubiquitinate endogenous BAX, preventing its translocation to mitochondria during stress-induced apoptosis (27). The increased cytosolic availability of parkin at baseline in hSP-CI73T-expressing cells may have antiapoptotic effects and represent an important cellular adaptation to prevent mitochondrial-driven cell death. Supporting this hypothesis, to date, we have not been able to consistently detect significant evidence of either cytochrome c release or caspase 9 activation in this model under baseline conditions (unpublished observations, S. Mulugeta). Additional experiments required to determine the role and function of cytosolic parkin are in progress.
The link between the observed accumulation of hSP-CI73T in plasma membrane/endosomal compartments and the disruption in autophagy is schematically illustrated in Fig. 9. The plasma membrane pool of hSPCI73T can be reinternalized via clathrin-mediated endocytosis and cotransported with transferrin to EEA-1-positive compartments (4). Internalized hSP-CI73T appears to progress from early to LE/MVB identified by colocalization with Rab7 and CD63 (Fig. 1, C and D). Importantly, given its spatial dissociation from hABCA3 expression, it is unlikely that hSPCI73T is transferred to LROs to any high degree (Fig. 1B). During the autophagic process, a cupped-shape structure, also referred to as a phagophore, encloses a portion of cytoplasm, forming an autophagosome. The autophagosome is a double-membrane structure containing undigested cytoplasmic components, including organelles. It is at this level that autophagosomes interact with components of the endocytic pathway (in particular MVBs), generating a hybrid organelle termed amphisomes. The term amphisome is used to describe preautolysosomal compartments containing both autophagic and endocytic material, including marker antigens such as Rab7, M6PR, LC3, and EEA1 (10, 18). Consistent with this, we identified hSP-CI73T in both Rab7 and LC3 vesicles (Figs. 1C and 2A). Finally, by fusion with lysosomes, autophagosomes or amphisomes can form autolysosomes in which intraluminal components and the inner membrane of the autophagosome are degraded. The large autophagic vacuoles visualized on an electron microscope (Figs. 3 and 4), increases in size and number of cytosolic GFP/LC3 vesicles (Fig. 2), increases in autophagy components (p62; LC3) (Fig. 5, A and B), and the dynamic autophagic flux studies (Figs. 5, E and F, and 6) are consistent with an inhibition in autophagosome progression/turnover. Furthermore, the increases in Rab7 biomass seen on Western blot (Fig. 5C) are most compatible with impaired progression of the amphisome to an autolysosome or possibly an impairment in amphisome formation at the level of MVB-autophagosome fusion. Functional MVBs have been shown to be required for autophagic clearance of protein aggregates, such as huntingtin 1 (19), as occurs in our models (Fig. 7).
Fig. 9.
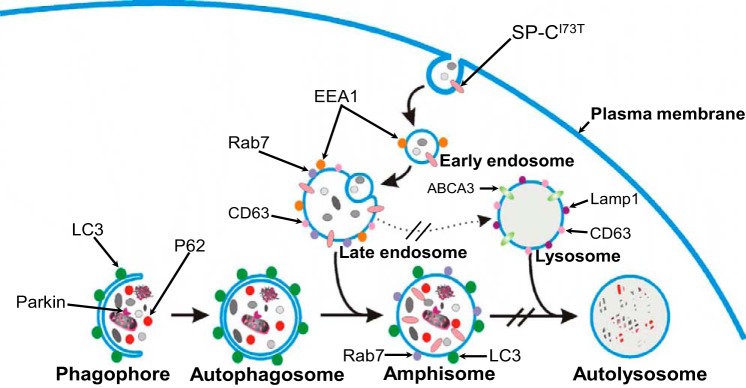
Schematic diagram illustrating model for the cellular phenotype consisting of a late block in macroautophagy induced by expression of the non-BRICHOS SP-CI73T mutant. Details provided in text.
Currently, there are limited data on the role of autophagy or other quality-control pathways in the pathogenesis of parenchymal lung diseases. In data obtained from lung biopsies of adult patients with nonfamilial idiopathic pulmonary fibrosis (IPF), the cellular expression pattern of autophagy biomarkers suggests its overall inhibition in this disease process although it is unclear whether a difference exists between epithelial cells and myofibroblasts (1). Patel et al. (51) have shown using IPF biopsies that overall macroautophagy in the lung appears to be inhibited and that pharmacological enhancement of autophagy can limit fibrosis in murine models. Previously, we have shown in vitro that, in contrast to hSP-CI73T, transient overexpression of BRICHOS mutations produces an inhibition of the proteasome system (46, 47). In preliminary studies, BRICHOS SP-C mutants also initially activate and rely on autophagy to clear protein aggregates, but that inhibition of this process induces apoptosis (39). Thus the body of evidence to date suggests that macroautophagy would be cytoprotective to lung epithelia.
The cellular phenotype induced by hSP-CI73T expression appears similar to the cellular dysfunction described in some storage disorders involving the endosomal-lysosomal network (22, 55, 59). Although storage diseases often result from a deficiency of a single enzyme required for the metabolism and degradation of various macromolecules (i.e., lipids, glycoproteins, and mucopolysaccharides) (35), the eventual accumulation of nondegraded constituents results in the functional disruption of a variety of cellular processes, including endocytosis, lysosomal degradation, and autophagy (20, 21, 55, 59, 64). The increases in autophagosomal structures containing organellar and cellular debris, the impairment in macromolecular housekeeping, and the alterations in mitophagy have been described in a variety of storage disorders, among which include NPC1 (57), Gaucher disease (63), Fabry disease (15), and GM1 gangliosidosis (64). Similar to our findings, the most common theme that emerges from all these studies is the impairment of autophagic flux arising from a late block in autophagy, leading to a secondary accumulation of autophagosomes/amphisomes, polyubiquitinated proteins, aggregated polypeptides, autophagy components (p62, LC3), and/or mitochondria. Whereas direct induction of apoptosis may not play a critical role in the pathogenesis of hSP-CI73T lung disease, there is precedence that disruption of normal organellar homeostasis in the alveolar epithelium can promote apoptosis-independent fibrotic lung remodeling. Using a mouse model of Hermansky-Pudlak syndrome, we recently demonstrated that the accumulation of surfactant lipids and LB enlargement in AT2 cells triggered the release of proinflammatory cytokines, such as monocyte chemoattractant protein-1 contributing to the injury (2). Moreover, in addition to inducing cell death, BRICHOS mutant SFTPC expression has also been shown to trigger release of IL-8 (41), and mutations in the SFTPA2 (SP-A2) gene have resulted in elaboration of TGF-β in vitro (42).
In summary, we have shown that the stable expression of an hSP-C missense mutation located within the linker domain of the SFTPC gene associated with fibrotic lung remodeling results in disruption of key cellular quality-control pathways in vitro. The impairment of protein macroautophagy and its related process, mitophagy, represents a second important cellular phenotype for SFTPC-related epithelial cell dysfunction, which is distinct from that produced by aggregation-prone SP-C BRICHOS mutations. These findings, when combined with the role of AT2 cells as progenitor cells in alveolar repair, further support the concept that alveolar epithelial cell susceptibility to second hits and/or death forms the foundational basis for ILD/IPF pathogenesis. Furthermore, detailed understanding of the molecular basis underlying AT2 cell quality-control dysfunction caused by, not only SFTPC mutations, but also other genetic or environmental factors is a key for future development of new therapeutic strategies tailored to specific subcellular targets and pathways for parenchymal lung disease.
GRANTS
This work is supported by the National Institutes of Health (HL119436 to M. Beers, HL090732 to S. Mulugeta, and HL059959 to S. Guttentag), The Department of Veterans Affairs (VA Merit Award BX001176 to M. Beers), The American Heart Association (13GRNT17070104 to M. Beers), and the Cystic Fibrosis Foundation (CF GORALS12L0 to J. Goralski). M. Beers is the Albert M. Rose Established Investigator of the Pulmonary Fibrosis Foundation. A. Hawkins has been supported by NIH 2T32HL007586-26 and NIH T32HL007748-20.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
A.H., S.C., and S.M. performed experiments; A.H., S.M., and M.F.B. analyzed data; A.H., S.H.G., S.M., and M.F.B. interpreted results of experiments; A.H. and S.M. prepared figures; A.H., S.M., and M.F.B. drafted manuscript; A.H., S.H.G., R.D., W.K.F., J.L.G., S.C., S.M., and M.F.B. approved final version of manuscript; S.H.G., S.M., and M.F.B. edited and revised manuscript; S.M. and M.F.B. conception and design of research.
ACKNOWLEDGMENTS
The authors acknowledge the past support and core facilities provided by the Institute for Environmental Medicine at the University of Pennsylvania (Dr. Aron B. Fisher, Director) through NIH PO1 HL19737.
REFERENCES
- 1.Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi M, Kamiya N, Hirano J, Odaka M, Morikawa T, Nishimura SL, Kawabata Y, Hano H, Nakayama K, Kuwano K. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 304: L56–L69, 2013. [DOI] [PubMed] [Google Scholar]
- 2.Atochina-Vasserman EN, Bates SR, Zhang P, Abramova H, Zhang Z, Gonzales L, Tao JQ, Gochuico BR, Gahl W, Guo CJ, Gow AJ, Beers MF, Guttentag S. Early alveolar epithelial dysfunction promotes lung inflammation in a mouse model of Hermansky-Pudlak syndrome. Am J Respir Crit Care Med 184: 449–458, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science 319: 916–919, 2008. [DOI] [PubMed] [Google Scholar]
- 4.Beers MF, Hawkins A, Maguire JA, Kotorashvili A, Zhao M, Newitt JL, Ding W, Russo S, Guttentag S, Gonzales L, Mulugeta S. A nonaggregating surfactant protein C mutant is misdirected to early endosomes and disrupts phospholipid recycling. Traffic 12: 1196–1210, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beers MF, Hawkins A, Shuman H, Zhao M, Newitt JL, Maguire JA, Ding W, Mulugeta S. A novel conserved targeting motif found in ABCA transporters mediates trafficking to early post-Golgi compartments. J Lipid Res 52: 1471–1482, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beers MF, Kim CY, Dodia C, Fisher AB. Localization, synthesis, and processing of surfactant protein SP-C in rat lung analyzed by epitope-specific antipeptide antibodies. J Biol Chem 269: 20318–20328, 1994. [PubMed] [Google Scholar]
- 7.Beers MF, Lomax C. Synthesis and processing of hydrophobic surfactant protein C by isolated rat type II cells. Am J Physiol Lung Cell Mol Physiol 269: L744–L753, 1995. [DOI] [PubMed] [Google Scholar]
- 8.Beers MF, Mulugeta S. Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu Rev Physiol 67: 663–696, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Beers MF, Zhao M, Tomer Y, Russo SJ, Zhang P, Gonzales LW, Guttentag SH, Mulugeta S. Disruption of N-linked glycosylation promotes proteasomal degradation of the human ATP-binding cassette transporter ABCA3. Am J Physiol Lung Cell Mol Physiol 305: L970–L980, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berg TO, Fengsrud M, Stromhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem 273: 21883–21892, 1998. [DOI] [PubMed] [Google Scholar]
- 11.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171: 603–614, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brasch F, Griese M, Tredano M, Johnen G, Ochs M, Rieger C, Mulugeta S, Muller KM, Bahuau M, Beers MF. Interstitial lung disease in a baby with a de novo mutation in the SFTPC gene. Eur Respir J 24: 30–39, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Bridges JP, Xu Y, Na CL, Wong HR, Weaver TE. Adaptation and increased susceptibility to infection associated with constitutive expression of misfolded SP-C. J Cell Biol 172: 395–407, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cameron HS, Somaschini M, Carrera P, Hamvas A, Whitsett JA, Wert SE, Deutsch G, Nogee LM. A common mutation in the surfactant protein C gene associated with lung disease. J Pediatr 146: 370–375, 2005. [DOI] [PubMed] [Google Scholar]
- 15.Chevrier M, Brakch N, Celine L, Genty D, Ramdani Y, Moll S, Djavaheri-Mergny M, Brasse-Lagnel C, Annie Laquerriere AL, Barbey F, Bekri S. Autophagosome maturation is impaired in Fabry disease. Autophagy 6: 589–599, 2010. [DOI] [PubMed] [Google Scholar]
- 16.Crossno PF, Polosukhin VV, Blackwell TS, Johnson JE, Markin C, Moore PE, Worrell JA, Stahlman MT, Phillips JA 3rd, Loyd JE, Cogan JD, Lawson WE. Identification of early interstitial lung disease in an individual with genetic variations in ABCA3 and SFTPC. Chest 137: 969–973, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 171: 513–524, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fader CM, Colombo MI. Autophagy and multivesicular bodies: two closely related partners. Cell Death Differ 16: 70–78, 2009. [DOI] [PubMed] [Google Scholar]
- 19.Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 179: 485–500, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fraldi A, Annunziata F, Lombardi A, Kaiser HJ, Medina DL, Spampanato C, Fedele AO, Polishchuk R, Sorrentino NC, Simons K, Ballabio A. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J 29: 3607–3620, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Fukuda T, Ewan L, Bauer M, Mattaliano RJ, Zaal K, Ralston E, Plotz PH, Raben N. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol 59: 700–708, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol 5: 554–565, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Galetskiy D, Woischnik M, Ripper J, Griese M, Przybylski M. Aberrant processing forms of lung surfactant proteins SP-B and SP-C revealed by high-resolution mass spectrometry. Eur J Mass Spectrom 14: 379–390, 2008. [DOI] [PubMed] [Google Scholar]
- 24.Garmany TH, Wambach JA, Heins HB, Watkins-Torry JM, Wegner DJ, Bennet K, An P, Land G, Saugstad OD, Henderson H, Nogee LM, Cole FS, Hamvas A. Population and disease-based prevalence of the common mutations associated with surfactant deficiency. Pediatr Res 63: 645–649, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 19: 4861–4870, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayat MA. Basic Techniques For Transmission Electron Microscopy. Orlando, FL: Academic, 1986. [Google Scholar]
- 27.Johnson BN, Berger AK, Cortese GP, Lavoie MJ. The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. Proc Natl Acad Sci USA 109: 6283–6288, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabore AF, Wang WJ, Russo SJ, Beers MF. Biosynthesis of surfactant protein C: characterization of aggresome formation by EGFP chimeras containing propeptide mutants lacking conserved cysteine residues. J Cell Sci 114: 293–302, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462: 245–253, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8: 445–544, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 4: 849–850, 2008. [DOI] [PubMed] [Google Scholar]
- 32.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 290: 1717–1721, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kotorashvili A, Russo SJ, Mulugeta S, Guttentag S, Beers MF. Anterograde transport of surfactant protein C proprotein to distal processing compartments requires PPDY-mediated association with Nedd4 ubiquitin ligases. J Biol Chem 284: 16667–16678, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14: 759–774, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Lampe C, Bellettato CM, Karabul N, Scarpa M. Mucopolysaccharidoses and other lysosomal storage diseases. Rheum Dis Clin North Am 39: 431–455, 2013. [DOI] [PubMed] [Google Scholar]
- 36.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB, Miller GG, Loyd JE, Blackwell TS. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpes virus infection. Am J Physiol Lung Cell Mol Physiol 294: L1119–L1126, 2008. [DOI] [PubMed] [Google Scholar]
- 37.Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy 8: 719–730, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loyd JE. Pulmonary fibrosis in families. Am J Respir Cell Mol Biol 29, Suppl 3: S47–S50, 2003. [PubMed] [Google Scholar]
- 39.Maguire JA, Malzer E, Marciniak SJ, Beers MF. Autophagy promotes cytoprotection by facilitating removal of aggregation-prone surfactant protein C L188Q BRICHOS mutant in vitro and in vivo [abstract]. Am J Respir Crit Care Med 187: A5182, 2013. [Google Scholar]
- 40.Maguire JA, Mulugeta S, Beers MF. Multiple ways to die: Delineation of the unfolded protein response and apoptosis induced by Surfactant Protein C BRICHOS mutants. Int J Biochem Cell Biol 44: 101–112, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maguire JA, Mulugeta S, Beers MF. Endoplasmic reticulum stress induced by surfactant protein C BRICHOS mutants promotes proinflammatory signaling by epithelial cells. Am J Respir Cell Mol Biol 44: 404–414, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maitra M, Dey M, Yuan WC, Nathanielsz PW, Garcia CK. Lung fibrosis-associated surfactant protein A1 and C variants induce latent transforming growth factor beta1 secretion in lung epithelial cells. J Biol Chem 288: 27159–27171, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Matsuda N, Tanaka K. Uncovering the roles of PINK1 and parkin in mitophagy. Autophagy 6: 952–954, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct 27: 421–429, 2002. [DOI] [PubMed] [Google Scholar]
- 45.Mulugeta S, Beers MF. Processing of surfactant protein C requires a type II transmembrane topology directed by juxtamembrane positively charged residues. J Biol Chem 278: 47979–47986, 2003. [DOI] [PubMed] [Google Scholar]
- 46.Mulugeta S, Maguire JA, Newitt JL, Russo SJ, Kotorashvili A, Beers MF. Misfolded BRICHOS SP-C mutant proteins induce apoptosis via caspase-4- and cytochrome c-related mechanisms. Am J Physiol Lung Cell Mol Physiol 293: L720–L729, 2007. [DOI] [PubMed] [Google Scholar]
- 47.Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol 32: 521–530, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nerelius C, Martin E, Peng S, Gustafsson M, Nordling K, Weaver T, Johansson J. Mutations linked to interstitial lung disease can abrogate anti-amyloid function of prosurfactant protein C. Biochem J 416: 201–209, 2008. [DOI] [PubMed] [Google Scholar]
- 50.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131–24145, 2007. [DOI] [PubMed] [Google Scholar]
- 51.Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, Rosas IO, Morse D. Autophagy in idiopathic pulmonary fibrosis. PLoS One 7: e41394, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Percopo S, Cameron HS, Nogee LM, Pettinato G, Montella S, Santamaria F. Variable phenotype associated with SP-C gene mutations: fatal case with the I73T mutation. Eur Respir J 24: 1072–1073, 2004. [DOI] [PubMed] [Google Scholar]
- 53.Pols MS, Klumperman J. Trafficking and function of the tetraspanin CD63. Exp Cell Res 315: 1584–1592, 2009. [DOI] [PubMed] [Google Scholar]
- 54.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 11: 1107–1117, 2002. [DOI] [PubMed] [Google Scholar]
- 55.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90: 1383–1435, 2010. [DOI] [PubMed] [Google Scholar]
- 56.Sanchez-Pulido L, Devos D, Valencia A. BRICHOS: a conserved domain in proteins associated with dementia, respiratory distress and cancer. Trends Biochem Sci 27: 329–332, 2002. [DOI] [PubMed] [Google Scholar]
- 57.Sarkar S, Carroll B, Buganim Y, Maetzel D, Ng AH, Cassady JP, Cohen MA, Chakraborty S, Wang H, Spooner E, Ploegh H, Gsponer J, Korolchuk VI, Jaenisch R. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep 5: 1302–1315, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sarkar S, Krishna G, Imarisio S, Saiki S, O'Kane CJ, Rubinsztein DC. A rational mechanism for combination treatment of Huntington's disease using lithium and rapamycin. Hum Mol Genet 17: 170–178, 2008. [DOI] [PubMed] [Google Scholar]
- 59.Settembre C, Fraldi A, Jahreiss L, Spampanato C, Venturi C, Medina D, de Pablo R, Tacchetti C, Rubinsztein DC, Ballabio A. A block of autophagy in lysosomal storage disorders. Hum Mol Genet 17: 119–129, 2008. [DOI] [PubMed] [Google Scholar]
- 60.Settembre C, Fraldi A, Rubinsztein DC, Ballabio A. Lysosomal storage diseases as disorders of autophagy. Autophagy 4: 113–114, 2008. [DOI] [PubMed] [Google Scholar]
- 61.Stevens PA, Pettenazzo A, Brasch F, Mulugeta S, Baritussio A, Ochs M, Morrison L, Russo SJ, Beers MF. Nonspecific interstitial pneumonia, alveolar proteinosis, and abnormal proprotein trafficking resulting from a spontaneous mutation in the surfactant protein C gene. Pediatr Res 57: 89–98, 2005. [DOI] [PubMed] [Google Scholar]
- 62.Stewart GA, Ridsdale R, Martin EP, Na CL, Xu Y, Mandapaka K, Weaver TE. 4-Phenylbutyric acid treatment rescues trafficking and processing of a mutant surfactant protein-C. Am J Respir Cell Mol Biol 47: 324–331, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun Y, Grabowski GA. Impaired autophagosomes and lysosomes in neuronopathic Gaucher disease. Autophagy 6: 648–649, 2010. [DOI] [PubMed] [Google Scholar]
- 64.Takamura A, Higaki K, Kajimaki K, Otsuka S, Ninomiya H, Matsuda J, Ohno K, Suzuki Y, Nanba E. Enhanced autophagy and mitochondrial aberrations in murine G(M1)-gangliosidosis. Biochem Biophys Res Commun 367: 616–622, 2008. [DOI] [PubMed] [Google Scholar]
- 65.Tebbenkamp AT, Crosby KW, Siemienski ZB, Brown HH, Golde TE, Borchelt DR. Analysis of proteolytic processes and enzymatic activities in the generation of huntingtin n-terminal fragments in an HEK293 cell model. PLoS One 7: e50750, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vadlamudi RK, Joung I, Strominger JL, Shin J. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem 271: 20235–20237, 1996. [DOI] [PubMed] [Google Scholar]
- 67.van Moorsel CH, van Oosterhout MF, Barlo NP, de Jong PA, van der Vis JJ, Ruven HJ, van Es HW, van den Bosch JM, Grutters JC. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a Dutch cohort. Am J Respir Crit Care Med 182: 1419–1425, 2010. [DOI] [PubMed] [Google Scholar]
- 68.Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell 14: 95–104, 2004. [DOI] [PubMed] [Google Scholar]
- 69.Vives-Bauza C, de Vries RL, Tocilescu M, Przedborski S. PINK1/Parkin direct mitochondria to autophagy. Autophagy 6: 315–316, 2010. [DOI] [PubMed] [Google Scholar]
- 70.Vorbroker DK, Voorhout WF, Weaver TE, Whitsett JA. Posttranslational processing of surfactant protein C in rat type II cells. Am J Physiol Lung Cell Mol Physiol 269: L727–L733, 1995. [DOI] [PubMed] [Google Scholar]
- 71.Wang WJ, Mulugeta S, Russo SJ, Beers MF. Deletion of exon 4 from human surfactant protein C results in aggresome formation and generation of a dominant negative. J Cell Sci 116: 683–692, 2003. [DOI] [PubMed] [Google Scholar]
- 72.Wang WJ, Russo SJ, Mulugeta S, Beers MF. Biosynthesis of surfactant protein C (SP-C). Sorting of SP-C proprotein involves homomeric association via a signal anchor domain. J Biol Chem 277: 19929–19937, 2002. [DOI] [PubMed] [Google Scholar]
- 73.Whitsett JA, Weaver TE. Hydrophobic surfactant proteins in lung function and disease. N Engl J Med 347: 2141–2148, 2002. [DOI] [PubMed] [Google Scholar]
- 74.Willander H, Askarieh G, Landreh M, Westermark P, Nordling K, Keränen H, Hermansson E, Hamvas A, Nogee LM, Bergman T, Saenz A, Casals C, Åqvistg J, Jörnvall H, Berglund H, Presto J, Knight SD, Johansson J. High-resolution structure of a BRICHOS domain and its implications for anti-amyloid chaperone activity on lung surfactant protein C. Proc Natl Acad Sci USA 109: 2325–2329, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Willander H, Hermansson E, Johansson J, Presto J. BRICHOS domain associated with lung fibrosis, dementia and cancer–a chaperone that prevents amyloid fibril formation? FEBS J 278: 3893–3904, 2011. [DOI] [PubMed] [Google Scholar]
- 76.Woischnik M, Sparr C, Kern S, Thurm T, Hector A, Hartl D, Liebisch G, Mulugeta S, Beers MF, Schmitz G, Griese M. A non-BRICHOS surfactant protein c mutation disrupts epithelial cell function and intercellular signaling. BMC Cell Biol 11: 88-2121-11-88, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 23: 33–42, 1998. [DOI] [PubMed] [Google Scholar]