

Abstract
Smooth muscle contraction can be divided into two phases: the initial contraction determines the amount of developed force and the second phase determines how well the force is maintained. The initial phase is primarily due to activation of actomyosin interaction and is relatively well understood, whereas the second phase remains poorly understood. Force maintenance in the sustained phase can be disrupted by strains applied to the muscle; the strain causes actomyosin cross-bridges to detach and also the cytoskeletal structure to disassemble in a process known as fluidization, for which the underlying mechanism is largely unknown. In the present study we investigated the ability of airway smooth muscle to maintain force after the initial phase of contraction. Specifically, we examined the roles of Rho-kinase and protein kinase C (PKC) in force maintenance. We found that for the same degree of initial force inhibition, Rho-kinase substantially reduced the muscle's ability to sustain force under static conditions, whereas inhibition of PKC had a minimal effect on sustaining force. Under oscillatory strain, Rho-kinase inhibition caused further decline in force, but again, PKC inhibition had a minimal effect. We also found that Rho-kinase inhibition led to a decrease in the myosin filament mass in the muscle cells, suggesting that one of the functions of Rho-kinase is to stabilize myosin filaments. The results also suggest that dissolution of myosin filaments may be one of the mechanisms underlying the phenomenon of fluidization. These findings can shed light on the mechanism underlying deep inspiration induced bronchodilation.
Keywords: muscle mechanics, asthma, bronchoprotection, ultrastructure
asthma attacks are associated with excessive constriction of airways due to airway smooth muscle (ASM) contraction (1). However, it is not clear whether a change in the intrinsic ASM contractility underlies the exacerbation because other factors such as airway remodeling (e.g., increased ASM mass or thickness of the submucosal layer) could lead to exaggerated narrowing of the airway lumen without an increase in ASM contractility (18). A recent study from our laboratory (4) showed that oscillatory strain induced a smaller decrease in isometric force in ASM from asthmatic subjects compared with that from nonasthmatic subjects, even though the ASM strips from both groups generated the same stress and shortening velocity. In the same study a greater force recovery after oscillatory strain was also observed in asthmatic ASM. This suggests that there is a difference between asthmatic and nonasthmatic ASM in terms of their mechanical properties, even though the difference is not in the isometric stress, shortening velocity, or other commonly used parameters for assessing muscle contractility. The difference appears to be in the ability of the muscle to resist mechanical perturbation and maintain force.
The mechanism underlying smooth muscle's ability to maintain force in a mechanically dynamic environment such as the lung is not clear. Swärd et al. (23) have demonstrated that Rho-kinase activation is important in the tonic phase of contraction in ileum muscle, suggesting a key role for Rho-kinase in force maintenance. It is not known whether Rho-kinase plays a similar role in airway smooth muscle. Although disruption of cross-bridge attachment may account for some of the force decrease observed in smooth muscle subjected to oscillatory strain (7), the slow rate and incomplete force recovery suggest that other mechanisms may be involved, such as “fluidization” as described by Fredberg and colleagues (3, 12). Kuo et al. (14) showed that in airway smooth muscle decrease in force induced by oscillatory strain accompanied a similar degree of decrease in the mass of myosin filaments, suggesting that fluidization may involve depolymerization of myosin filaments. The present study was undertaken to investigate the relationship between Rho-kinase activity and myosin filament formation and the role they play in maintaining isometric force in airway smooth muscle under static and dynamic conditions.
MATERIALS AND METHODS
Tissue preparation and equilibration.
Sheep tracheas were obtained from a local abattoir. All experimental procedures were approved by the Animal Care Committee and Bio-safety Committee of the University of British Columbia and conformed to the guidelines set out by the Canadian Council on Animal Care. Tracheas were transported and stored in Krebs solution (pH 7.4; 118 mM NaCl, 4 mM KC, 1.2 mM NaH2PO4, 22.5 mM NaHCO3, 2 mM CaCl2 and 2 g/l dextrose) at 4°C. The experiments were performed within 4 days of obtaining the tissue. The in situ muscle length was determined from a tracheal segment by measuring the distance between the insertion points of the muscle bundles to the C-shaped cartilage. Caution was taken to ensure that the muscle was relaxed before the length was measured. A wrinkled epithelial layer indicates contraction of the smooth muscle layer. If this was observed, the trachea was immersed in zero-calcium Krebs until the wrinkles disappeared before the length measurement was taken. The in situ length was used as a reference length (Lref) for normalization of all length measurements. After removal of the cartilage, the muscle bundles were held at Lref and were dissected free of adventitial connective tissue, epithelium, and submucosal layer. Muscle strips measuring 1–1.5 mm wide, 0.2–0.3 mm thick, and ∼6 mm in length were removed and attached to aluminum foil clips at each end. The muscle strips were then vertically mounted onto a custom-designed myograph capable of measuring tension and length changes. The resonant frequency of the force transducer is ∼500 Hz and the muscle lever is capable of making a 1-mm step in ∼1 ms. While attached to the myograph, the muscle was entirely submerged in a tissue bath filled with prewarmed Krebs solution (37°C). An outer water-jacket maintained the temperature of the tissue bath at 37°C. The muscle strips held at Lref were initially stimulated for 10 s with electrical field stimulation (EFS) at 5-min intervals during a 1.5-h period of equilibration. EFS consisted of a 60-Hz alternating current at a voltage of 15–20 V, which resulted in maximal force generation. EFS-induced force was mediated by acetylcholine (ACh) released from nerve endings within the muscle preparation, and this force could be completely inhibited by 10−6 M of atropine. Krebs solution was aerated with 5% CO2 and 95% O2 to maintain a pH of 7.4 and was changed in the tissue bath at 5-min intervals. The tissue was considered equilibrated when the muscle generated consistent maximal isometric force (Fmax) with minimal resting tension (less than 1 mN or ∼3% of Fmax).
Overview of experiments.
Force maintenance under static and oscillatory conditions was quantified by monitoring changes in force during a 100-s period after the onset of stimulation by ACh in the presence and absence of various enzyme inhibitors. For the oscillatory condition, force recovery after cessation of oscillation was monitored for an additional 100 s. A schematic is shown in Fig. 1 to illustrate the protocol for determining force maintenance under oscillatory conditions. The inhibitors were H1152 (Rho-kinase) from Torcris Bioscience, Y27632 (Rho-kinase) from Abcam, GF109203x (protein kinase C, PKC) from Enzo Biochem, and ML-7 (myosin light-chain kinase, MLCK) from Sigma-Aldrich. The number of myosin filaments per cell cross-section was quantified in ACh-activated ASM in the presence and absence of Rho-kinase inhibitors and PKC inhibitor.
Fig. 1.
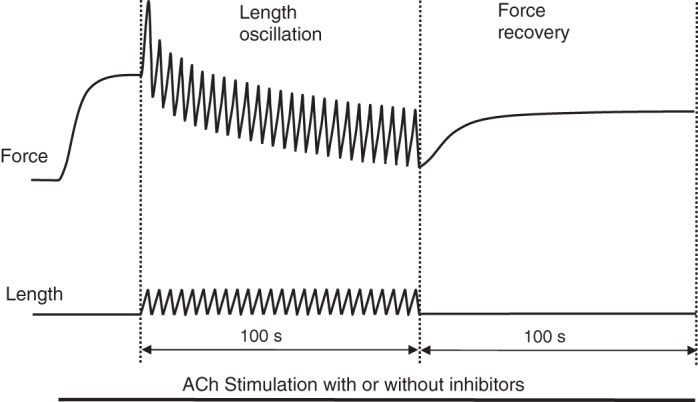
Protocol for determining maintenance of peak force (during stretch) and force recovery after length oscillation. In experiments where enzyme inhibitors were used, the muscle was preincubated with an inhibitor for a predetermined period of time (see text) before commencement of this protocol.
Stimulation with ACh and length oscillations.
Each ASM strip in the inhibitor group was preincubated with a selected inhibitor and stimulated with ACh (10−4 M) in the presence of the inhibitor. To facilitate comparison, we matched the force among different inhibitor groups by choosing a dose of each inhibitor which targets a value of 50% of Fmax (the isometric force induced by 10−4 M ACh). The final concentration and conditions used for each inhibitor was H1152, 3 μM with 30-min incubation time; Y27632, 3 μM with 90-min incubation time; GF109203x, 15 μM with 30-min incubation time; and ML-7, 5 μM with 60-min incubation time. The ACh concentration for the control group (without inhibition) was determined in the same way. The concentrations of ACh used for the control group to match 50% Fmax were within the range of 10−7 and 10−5 M. The matched force was defined as F50. To determine the muscle's ability to maintain active force after mechanical perturbation, oscillatory strain was applied to the muscle strip at the force plateau. Length oscillation (asymmetrical triangular wave) was applied at a frequency of 0.25 Hz for a 100-s period with an amplitude of 25% Lref (3 s for isovelocity stretch and 1 s for isovelocity release). Data were analyzed by examining the resulting peak forces in each cycle of the oscillation.
Electron microscopy.
The protocol for EM fixation has been described previously (25). Briefly, after reaching the force plateau the muscle was fixed while it was still attached to the myograph so that the muscle was minimally disturbed and the force could be continuously monitored. The primary fixative was a cocktail consisting of 1.5% paraformaldehyde, 1.5% glutaraldehyde, and 0.5% tannic acid in 100 mM sodium cacodylate solution warmed to 37°C. After initial fixation the muscle was cut into 6–8 blocks and put in the same primary fixing solution on a shaker at 4°C for 2 h. The blocks were then washed once with 100 mM sodium cacodylate and twice with 1.25% sodium bicarbonate for 10 min each. The blocks were processed in a secondary fixative of 1% OsO4-1.25% NaHCO3 for 1.5 h, followed by three washes in distilled water (10 min each). En bloc staining was accomplished by placing the blocks in 1% uranyl acetate for 1 h, followed by three washes with distilled water. The blocks were then dehydrated using increasing concentrations of ethanol (50, 70, 80, 90, 95%, 10 min each; 100%, 3 × 15 min each) followed by three washes with propylene oxide (10 min each). The blocks were gradually infiltrated overnight with TAAB 812 resin mix and then embedded in molds and placed in an oven at 60°C overnight. The embedded blocks were sectioned on a microtome with a diamond knife and placed on copper mesh grids. The section thickness was 50 nm. After sectioning the grids were stained with 1% uranyl acetate followed by Reynolds lead citrate for 4 and 3 min, respectively. The images were obtained with an electron microscope (Philips/FEI Tecnai 12 TEM).
Measurement of myosin filament density.
Electron micrograph images were taken from transverse sections of the tissue. For measurements of thick filament density, images of single cells were randomly taken from the sections and a blinded evaluator manually counted myosin filaments and measured the cross-sectional area of the cell by manual tracing using Image Pro Plus 5 (Media Cybernetics, Bethesda, MD). The myosin filament content within the cell visible to EM was quantified by counting the filaments in cell cross-sections. The filament content or mass of a cell is the product of the number of filaments in the cell and the average length of the filaments, as explained previously (13). The number of filaments per unit area as a measurement does not provide information about the length or number of the filaments, but the mass. An increase in filament density therefore could be due to an increase in filament number or length, or both. The measurements were carried out under two conditions: control vs. Rho-kinase inhibition (Y27632), and control vs. PKC inhibition (GF109203x). Three groups (a pair per group) of fixed tissues per condition (from 3 repeats of experiments) were analyzed with 15 cell images taken from each tissue, that is, 90 images per condition.
Mathematical model.
We employ the computational model of Donovan (6) to test the hypothesis that alterations to the myosin filament length distribution could be responsible for changes in the force response to oscillatory strain with Rho-kinase inhibition. This model is based on the cross-bridge models developed by many authors [e.g., Huxley (9), Hai and Murphy (8), and Fredberg et al. (7)], but further employs the idea that shorter myosin filaments are more likely to be lost from functional contractile units when strain is imposed. Thus it is an ideal tool for testing the underlying hypothesis here.
We first simulate the experimental protocol using the parameters as in the original paper for the control situation (6), before altering the myosin filament length distribution to mimic Rho-kinase inhibition. The underlying model fits the myosin filament length distribution in terms of the dimer length as a (discrete) geometric distribution, which is described by a single parameter denoted p. For the control situation we employ p = 0.45 as in Donovan (6) [which was fitted to the data of Liu et al. (15)] and assume p = 0.8 to represent generically a distribution biased toward shorter filaments. In the computational model, only the distribution of filament lengths was altered between the two cases and all other elements remain identical.
Statistical analysis.
Data are presented as means ± SE; n stands for the number of tracheas from different sheep. Two-way ANOVA test and repeated-measures Bonferroni post tests were used for all oscillation data analyses. One-way ANOVA followed by Student-Newman-Keuls test was used for myosin density and force matching analyses. SigmaPlot 11 and Prism Graphpad were used for statistical analyses.
RESULTS
Force matching.
To gain insight into the mechanisms regulating force maintenance in ASM, we employed four different inhibitors including two Rho-kinase inhibitors (H1152 and Y27632), PKC inhibitor GF109203x, and MLCK inhibitor ML-7. After the muscle strips equilibrated and the EFS-induced isometric force reached a maximal steady state, 10−4 M ACh was used to induce maximal contraction (Fmax). Then through trial and error, a dose of ACh (within the range of 10−7 to 10−5 M) was obtained and used to stimulate the muscle to reach 50% Fmax. Isometric contraction was recorded for 100 s after the muscle reached a force plateau to monitor the ability of the muscle to maintain the contraction. By preincubating a muscle strip in a predetermined dose (also through trial and error) of inhibitor the force generation of 50% Fmax by 10−4 M ACh in the presence of the inhibitor was obtained to match the control group. Force matched data after normalization by Fmax (F50) are shown in Fig. 2; the results are F50 = 0.515 ± 0.022 Fmax for the control group, 0.516 ± 0.029 for the H1152 group, 0.508 ± 0.031 for the Y27632 group, 0.510 ± 0.029 for the GF109203x group, and 0.509 ± 0.037 for the ML-7 group. There is no statistical difference among all 5 groups (1-way ANOVA, P = 0.992).
Fig. 2.
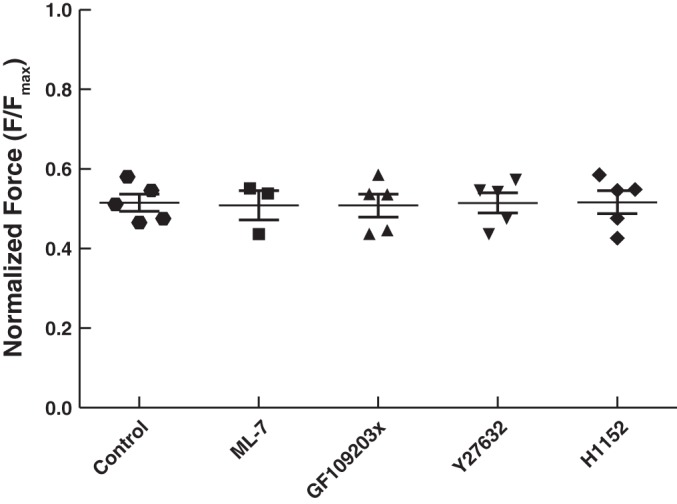
Force generation from all groups matched to 50% Fmax (maximal contraction induced by ACh 10−4 M). For the control group, a lower dose of Ach (within the range of 10−7 to 10−5 M) was used to stimulate the muscle to match 50% Fmax; for the 4 groups with inhibitors, the force generation was induced by 10−4 M Ach with the inhibitors. There is no statistically significant difference among the groups (1-way ANOVA, P = 0.992). The average Fmax values are 215.5 ± 37.4 kPa (n = 5) for the control group, 207.9 ± 39.3 kPa (n = 3) for the ML-7 group, 232.5 ± 23.7 kPa (n = 5) for the GF109023x group, 189.7 ± 17.6 kPa (n = 4) for the Y27632 group, and 198.1 ± 23.6 kPa (n = 5) for the H1152 group.
Force maintenance without oscillatory strain.
One example of normalized force trace recorded during ACh stimulation is shown in Fig. 3A: the force generated with H1152 declined after the muscle reached its maximal contraction, whereas in the absence of the inhibitor the force plateau was maintained over 100 s. To quantify the force decline over the 100-s period, force values were plotted at 4-s intervals as shown in Fig. 3B. Force from the control group decreased to 97.9 ± 1.3% F50 in 100 s without oscillation while the ML-7 group and GF109203x group decreased to 96 ± 1.8% F50 and 96.1 ± 3.5% F50, respectively; ML-7 had no effect on the force maintenance (2-way ANOVA, P = 0.259) while PKC inhibitor GF109203x showed a small but significant effect on force maintenance (2-way ANOVA, P < 0.05). Although the effect of GF109203 is statistically significant, it is doubtful that this effect is physiologically significant. The force records for ML-7 and GF109203x were not different from each other (2-way ANOVA, P = 0.683). Force generated in the presence of Y27632 and H1152 100 s after stimulation were 89.6 ± 2.7% F50 and 89 ± 2.9% F50, respectively, representing a significant decline (2-way ANOVA, P < 0.0001); both records with Rho-kinase inhibitors are significantly different from the control record and records with ML-7 and GF109203x (2-way ANOVA, P < 0.0001), but with no difference between the two (2-way ANOVA, P = 0.1846).
Fig. 3.
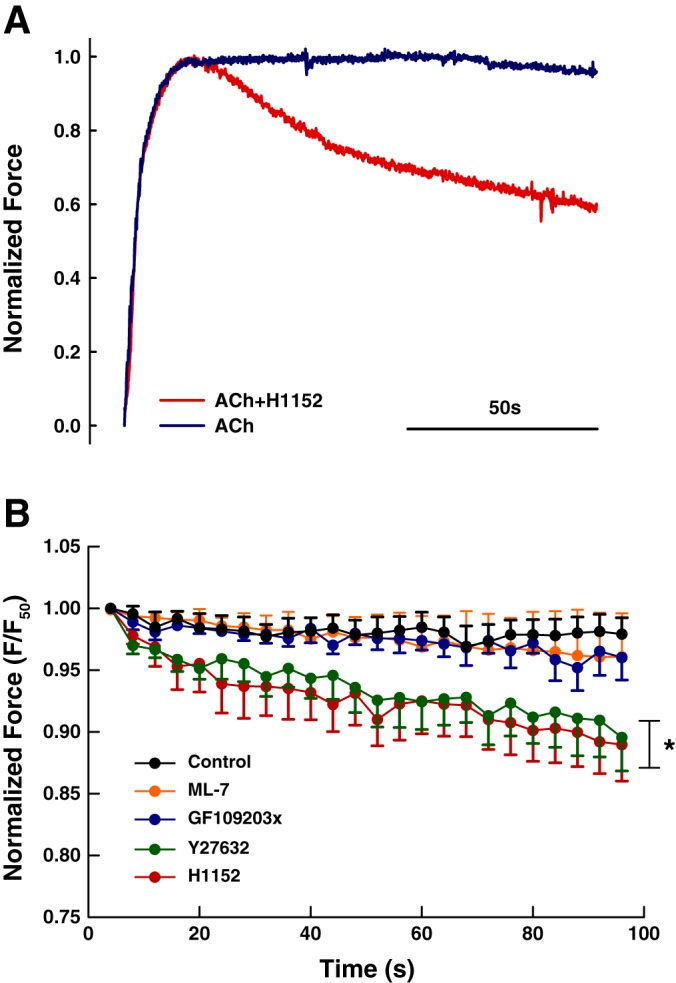
A: representative force records for ACh (10−6 M) stimulation with and without H1152 (3 μM). B: force maintenance over a 100-s period. Both H1152 and Y27632 groups are significantly different compared with control, ML-7, and GF109203x groups (2-way ANOVA, *P < 0.0001). The two Rho-kinase inhibitors are not different from each other (2-way ANOVA, P = 0.1846.)
Force maintenance with oscillatory strain.
To test the muscle's ability to maintain active force against mechanical perturbation, length oscillation in the form of an asymmetric triangular wave was applied at a frequency of 0.25 Hz for 100 s with an amplitude of 25% Lref. To examine the specific effect of oscillatory strain on force maintenance, we subtracted the baseline decline in force (under static condition, Fig. 3B) from the averaged peak force response obtained during length oscillation. The peak force was normalized to F50 and plotted in Fig. 4. Rho-kinase inhibition by both H1152 and Y27632 significantly depressed force maintenance compared with the other three groups (control, GF109203x, and ML-7) (2-way ANOVA, P < 0.0001). There was a small but significant difference between the H1152 and Y27632 groups (2-way, ANOVA, P < 0.05). The difference is mostly due to an approximate 10% shift in normalized force with the time course for the force decline virtually unchanged (Fig. 4). This suggests that H1152 and Y27632 acted on the same target (Rho-kinase) and the difference could be abolished by adjusting the dosage of the inhibitors. There was no difference between the GF109203x and the control group and between the ML-7 and the control group (2-way ANOVA, P = 0.294 and P = 0.828, respectively), and there was no difference either between the GF109203x and the ML-7 group (Two-way ANOVA, P = 0.1019).
Fig. 4.

Decline of peak force due to oscillation. Force record in the absence of oscillation (Fig. 2B) was subtracted from force record with the same inhibitor in the presence of oscillation. Note that in the presence of oscillatory strain only the peak force during each stretch was recorded. The Y27632 and the H1152 groups are significantly different from control, ML-7, and GF109203x (2-way ANOVA, *P < 0.0001). Incubation with GF109203x and ML-7 had no significant effect on force response during length oscillation compared with control (2-way ANOVA, P = 0.294 and P = 0.8280, respectively).
Force recovery after length oscillation.
Redevelopment of force after length oscillation was plotted in Fig. 5. No statistically significant difference was found in the force measured immediately after the oscillation. For the control group, force recovered from 7.4 ± 1% F50 to 54.2 ± 2.6% F50; force recovered from 7.9 ± 1.6% F50 to 57.2 ± 1.7% F50 for the ML-7 group, and this was not statistically different from the control group (2-way ANOVA, P = 0.12). GF109203x, H1152, and Y27632 reduced the ability of the muscle to recover from oscillatory strain significantly compared with both the control and the ML-7 group (2-way ANOVA, P < 0.0001); no difference in force redevelopment was found between GF109203x and H1152 and between GF109203x and Y27632 (2-way ANOVA, P = 0.13 and P = 0.85, respectively). The force recovered from 4.5 ± 0.9% F50 to 42.2 ± 2% F50 in the GF109203x group. No significant difference was found between the H1152 and the Y27632 group in force recovery after oscillation (2-way ANOVA, P = 0.27). Force redeveloped from 3.7 ± 0.6% F50 to 37.5 ± 3% F50 in the H1152 group; and 4.7 ± 0.7% F50 to 41.1 ± 4.6% F50 in the Y27632 group.
Fig. 5.
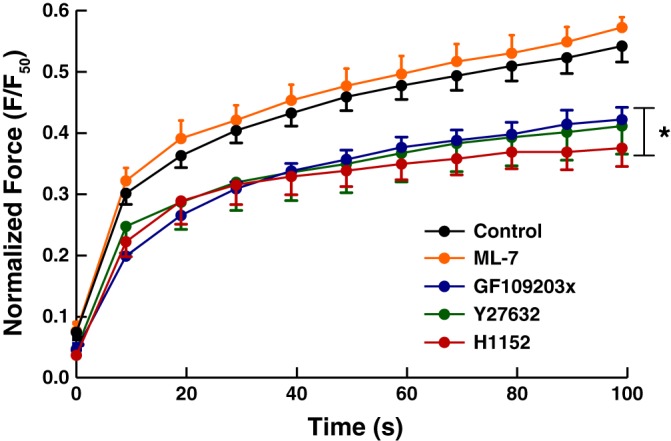
Force recovery after length oscillation. The ML-7 and the control group are not significantly different (2-way ANOVA, P = 0.1245). The H1152, Y27632, and GF109203x groups are significantly different from control and ML-7 group (2-way ANOVA, *P < 0.0001), The GF109203x group is not different from the H1152 and Y27632 groups (2-way ANOVA, P = 0.1305 and P = 0.8451, respectively), and the two Rho-kinase inhibitors are not different from each other (2-way ANOVA, P = 0.2708). Force measured at time points 60, 70, 80, 90, and 100 s in the H1152 group are significantly different compared with the control; for the Y27632 group, only force measured at 100 s is significantly different from the control (Bonferroni post tests, P < 0.001).
Quantification of myosin filament mass.
Figure 6 shows examples of electron micrographs of transverse sections of tracheal smooth muscle fixed under different conditions. Because myosin filament density has been shown to be related to strain-induced force decrease (fluidization) (14), we examined the filament density under conditions where the muscle's ability to maintain force was compromised due to fluidization. In fully activated airway smooth muscle (10−4 M ACh), addition of Y27632 (3 μM) resulted in a ∼23% decrease in force (Fig. 7A). The same degree of force reduction was matched by reducing the ACh concentration (without Y27632) (Fig. 7A). In the force-matched pair, it was found that the myosin filament density was ∼63% lower (P < 0.01) in the Y27632 group (Fig. 7B). We also inhibited PKC with compound GF109203x (15 μM). In fully activated muscle (10−4 M ACh), incubation with GF109203x resulted in a ∼30% decrease in force (Fig. 8A). The same degree of force reduction was matched using lower concentrations of ACh (without GF109203x). In the force-matched pair, no difference was found in the filament density (Fig. 8B, 1-way ANOVA, P = 0.92).
Fig. 6.


Examples of electron micrographs showing cross-sections of trachealis. A: partially ACh-activated trachealis. B: magnified area from A. C: ACh-activated trachealis in the presence Rho-kinase inhibitor (Y27632) and force-matched to A. D: magnified area from C. E: partially ACh-activated trachealis. F: magnified area from E. G: ACh-activated trachealis in the presence PKC inhibitor (GF109203x) and force-matched to E. H: magnified area from G. M, mitochondrion; SR, sarcoplasmic reticulum; DB, dense body; DP, dense plaque; Cav, caveola. Unlabeled arrows in the magnified areas point to myosin filaments surrounded by actin filaments; double arrows point to intermediate filaments. Myosin filaments have irregular cross-sectional profile and an average diameter of 15 nm, larger than that of actin filaments (6 nm) and that of intermediate filaments (10 nm, circular profile), but smaller than dense bodies. Scale bar, 0.5 μm.
Fig. 7.
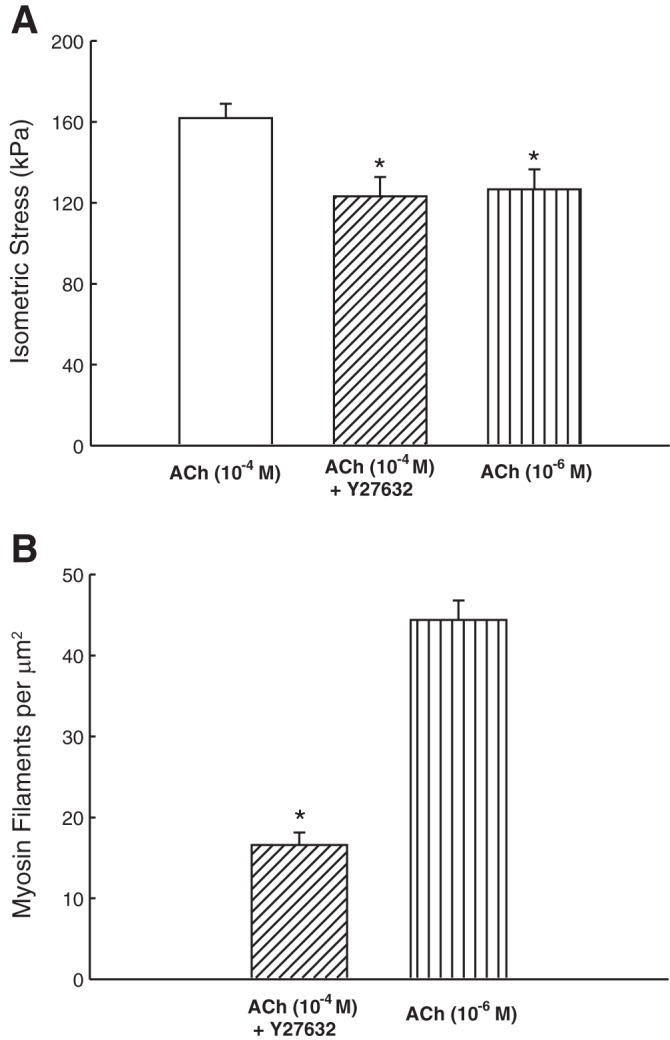
A: maximal (plateau) stress produced by fully activated (ACh, 10−4 M), Y27632 inhibited (10−4 M ACh + 3 μM Y27632), and partially activated (ACh, in the range of 10−6.5–10−6 M) muscles. *Significant difference (1-way ANOVA, P < 0.05) from fully activated force. B: myosin filament density in Y27632 inhibited and partially activated muscles. *Significant difference (1-way ANOVA, P < 0.001).
Fig. 8.
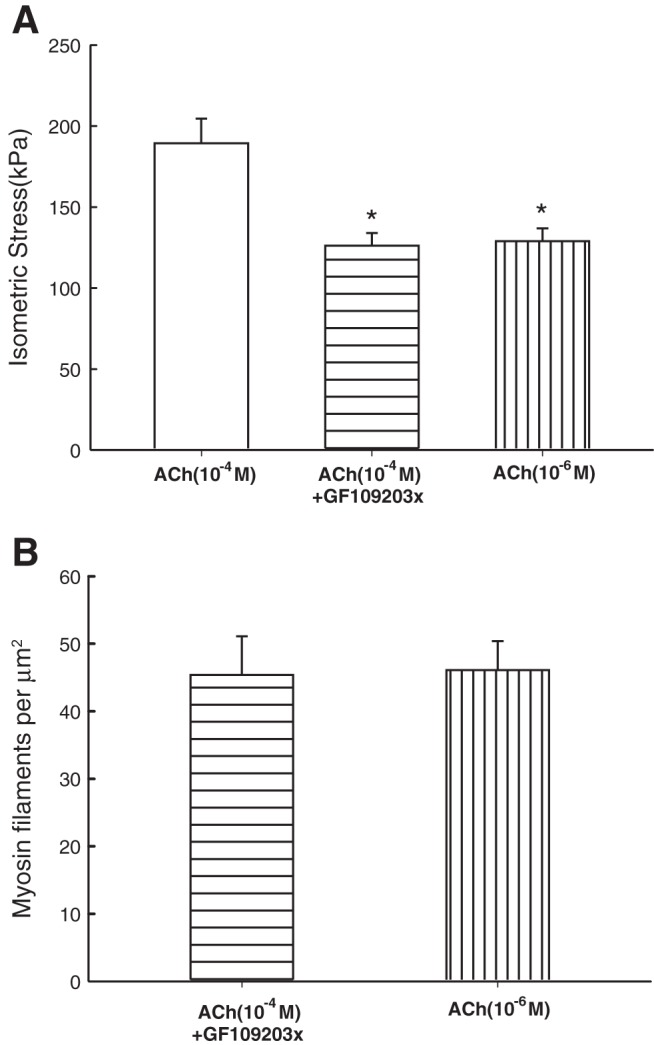
A: maximal (plateau) stress produced by fully activated (ACh, 10−4 M), GF109203x-inhibited (10−4 M ACh + 15 μM GF109203x), and partially activated (ACh, in the range of 10−7–10−5 M) muscles. *Significant difference (1-way ANOVA, P < 0.05) from fully activated force. B: there was no significant difference in the myosin filament densities in GF109203x and its ACh-activated control (1-way ANOVA, P = 0.924).
Mathematical model.
Using the computational model we tested the hypothesis that alterations to the myosin filament length distribution could be responsible for much of the change in force maintenance with oscillatory strain observed in the experiments (due to Rho-kinase inhibition). The simulation results are given in Fig. 9 showing the persistent decrease in force maintenance when the myosin filament distribution was altered to favor shorter lengths; at the peak of the first stretch the difference between the force generated in the two cases was 0.8%, while in the sustained contraction phase the reduction via shorter filaments was 5.2% (at 79 s). The agreement (c.f. Fig. 4) should be interpreted as primarily qualitative, first because inhibition of Rho-kinase has many other roles which are not accounted for here, but also because the altered myosin length distribution is a generic state rather than specifically fitted to the data because inferring the distribution from the myosin filament mass measurements is imprecise.
Fig. 9.
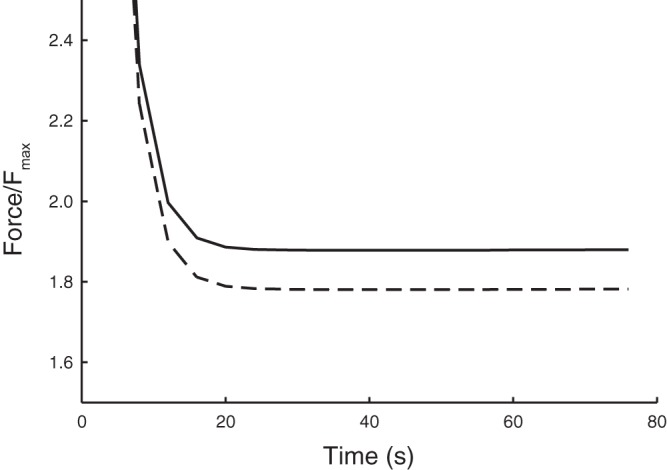
Simulation results using the mathematical model to test the role of myosin filament length distribution on force maintenance in sustained contraction. Here we mimicked the experimental protocol both for a standard filament length distribution (solid line), and an altered distribution with a greater fraction of shorter filaments (dashed line) to observe the alteration in force maintained during the oscillation protocol (see materials and methods). The favorable agreement with the experimental data (c.f. Fig. 3) suggests that changes to the filament length distribution may play an important role in regulating this force response.
DISCUSSION
The most important finding from this study is that in airway smooth muscle Rho-kinase plays an important role in force maintenance after the initial development of force, and that one of the mechanisms by which the Rho-kinase signaling pathway contributes to force maintenance is by stabilizing the myosin filaments. An important implication of the finding is that fluidization observed in smooth muscle (3, 12) is not likely an exclusive phenomenon of the actin cytoskeleton, but involves myosin filament evanescence.
Physiological implications.
It has been documented in vascular smooth muscle that agonist (phenylephrine) stimulation is more effective in maintaining force than stimulation via membrane depolarization by high [K+] (16). This is likely due to agonist activation of G protein-coupled receptor that in turn activates the signaling pathways leading to calcium sensitization, actin cytoskeleton remodeling, contractile filament stabilization, and other events that have yet to be elucidated.
The ability of smooth muscle to maintain force in a sustained contraction has traditionally been attributed to the function of latch-bridges, which are myosin cross-bridges that are dephosphorylated but remain attached to actin filaments (8). Increasing evidence suggests that the latch-bridge hypothesis may not be adequate to explain many smooth muscle behaviors. In a model that incorporated both the normally cycling bridges described by Huxley (9) and the latch-bridges described by Hai and Murphy (8), Fredberg et al. (7) were able to explain relengthening of precontracted muscle due to small-amplitude force oscillation. However, large-amplitude oscillation caused the model to break down. It is now clear that large-amplitude oscillation-induced fluidization in smooth muscle is not likely a phenomenon confined to cross-bridge interaction, but likely involves structural reorganization of the contractile apparatus and cytoskeleton. Brook (2) has recently put forth a model that includes structural disruptions termed “actin-myosin discontinuity” and “parallel-to-series transition” of cross-bridge arrangement, possible elements involved in strain-induced fluidization. With the new model Brook was able to explain features of smooth muscle behavior that could not be explained by the hybridized model of Huxley (9) and Hai and Murphy (8). We have recently found that myosin filaments in smooth muscle are much shorter than previously believed (15), with a possibility that many of them exist as dimers. This imparts an intrinsic instability to the contractile units in smooth muscle in terms of force generation and maintenance under dynamic conditions. This could be the mechanism for the actin-myosin discontinuity and it certainly would facilitate rearrangement of myosin motors in the parallel-to-series (and vice versa) transition, features that appear to be necessary for a model (2) to accommodate data from experiments and to account for at least some aspects of the fluidization phenomenon. A recent model developed by Donovan (6) incorporated a distribution of myosin filament length similar to that described by Liu et al. (15); the model was the first to be able to explain length adaptation (the ability of smooth muscle to recover loss of force due to a length change) over different time scales. We employed this model here to test the hypothesis that changes in the myosin filament length distribution could account for much of the observed change in force maintenance with Rho-kinase inhibition. This hypothesis is supported by the computational modeling, which shows that the degree of alteration which might be expected from changing the myosin filament length distribution alone is comparable to that seen in the experiments (Fig. 4). Even though it would be unreasonable to expect exact quantitative agreement (because of the many roles of Rho kinase in ASM), the good agreement obtained suggests that alteration of the length distribution could play an important role. This is another line of evidence supporting the notion that the intrinsic instability of contractile units in smooth muscle due to myosin filament evanescence is important for normal cellular function and the present finding that Rho-kinase inhibitor affects myosin filament formation indicates that the degree of filament evanescence is regulated at least partially by Rho-kinase.
Disassembly of myosin filaments could occur in at least two ways. One is total dissolution into monomers in which case the muscle's ability to generate force is likely adversely affected. The other way is fragmentation into short filaments, down to dimers. In this case the ability of the muscle to generate isometric force may be unaffected because dimers could act as independent motors, as those described for nonmuscle myosin (24). A myosin dimer lacks the thick backbone of a myosin filament that consists of multiple overlapping helical tails of the myosin molecules and therefore not recognizable as myosin filaments under electron microscope. This could account for the observation made in Fig. 7. That is, even though the isometric force was the same between the control and the test group, the number of observable myosin filaments was significantly less when Rho-kinase was inhibited.
The instability of contractile units manifests itself more prominently under dynamic conditions such as when the muscle is under oscillatory strain. As illustrated in Fig. 10, under static conditions the contractile units may generate the same amount of force because the number of myosin motors is the same (if we define the motor as a myosin dimer whether it is linked to other dimers or not). Under large oscillatory strain, it is conceivable that myosin motors not linked together (thus failing to form filaments) are likely to be lost from the actin filament lattice, especially when both of the motor arms are not overlapped by actin filaments. We propose that this is the mechanism responsible for the greater loss of force observed in Fig. 4 when the Rho-kinase was inhibited, and it is precisely this mechanism that accounts for the force reduction in the mathematical model (Fig. 9). However, this mechanism cannot explain the observations made under static conditions (Fig. 3). The decrease in force in the presence of Rho-kinase inhibitor is likely due to mechanisms related to actin cytoskeleton, especially at the sites of adhesion junction formation where Rho-kinase-dependent phosphorylation of adaptor proteins has been shown to be important in force transmission (26). Note that the force decline shown in Fig. 4 was due exclusively to oscillatory strain because the force loss observed during isometric contraction (Fig. 3) was subtracted from the total force decrease measured during contraction under oscillatory strain.
Fig. 10.

Schematic illustration of contractile units containing myosin filament fragments. It is assumed that one of the functions of Rho-kinase is to stabilize myosin filaments. With Rho-kinase inhibitor (+), myosin filaments breakdown into shorter fragments; some of the fragments become invisible to EM so that the myosin filament density would appear to decrease (see text for more description).
The signaling pathway downstream of Rho-kinase regulating myosin filament formation is currently unknown. It is known that in solution phosphorylation of the myosin regulatory light chain facilitates myosin filament formation (5, 22). In intact smooth muscle inhibition of myosin light chain kinase leads to a decrease in the observable myosin filaments (19), suggesting that the in vitro mechanism of the light chain phosphorylation (in facilitating myosin filament formation) is also operative in vivo. Interestingly inhibition of PKC had no effect on myosin filament density (Fig. 8), but it has an effect on the force recovery (Fig. 5). It appears that Rho-kinase and PKC do not share the same pathway in regulating myosin filament formation, but the pathways appear to converge in the regulation of force recovery [or resolidification, using the terminology of Krishnan et al. (12) and Chen et al. (3)]. It also appears that the regulation of myosin filament formation is more complicated than just phosphorylation of the myosin light chain; this requires further investigation.
It should be pointed out that inhibition of Rho-kinase and PKC resulted in reduced active force generation in the muscle maximally stimulated by ACh (10−4 M) and that the “control” (Fig. 2) was represented by active force generated by the muscle without inhibitors but at a lower concentration of ACh. In both control and test groups approximately half of the myosin cross-bridges were activated if we use maximal isometric force as an index of the number of activated cross-bridges. The difference between the Rho-kinase-inhibited groups and other groups lies in their ability to maintain isometric force (Fig. 3). Another way to illustrate the effect of Rho-kinase is to induce a contraction by a certain stimulator without activating Rho-kinase and to show that the contraction cannot be maintained, in contrast to a contraction induced by the same stimulator with a Rho-kinase activator. Membrane depolarization by potassium chloride could be such a stimulator, but it has been shown that KCl could partially activate Rho-kinase (10).
Disease relevance.
Airway smooth muscle functions under a mechanically dynamic environment and has likely evolved to work optimally with constant mechanical feedback. This perhaps explains why healthy subjects could reverse bronchoconstriction with a deep inspiration (17) and suppression of deep inspiration over a prolonged period caused healthy subjects to develop asthmalike symptoms (21). Interestingly the bronchodilating effect of deep inspirations is absent in asthmatics (20). Although the mechanism underlying the bronchodilation effect of deep inspiration is not entirely clear and is still under debate, strain on airway smooth muscle due to deep inspiration is one of the possible causes. We have recently shown that oscillatory strain applied to asthmatic airway smooth muscle results in less force reduction compared with the force reduction induced in nonasthmatic airway smooth muscle (4), indicating a greater resistance of asthmatic airway smooth muscle to mechanical perturbation, and suggesting that the intrinsic properties of the muscle may be altered in asthma. Deep inspirations could partially fluidize the cytoskeleton and the contractile apparatus of normal smooth muscle (3, 12) and prevent it from ever achieving its maximal contractile capability. In asthma somehow the break on the muscle's contractility failed. This could be due to a disruption in the transmission of strain to the muscle via extracellular tethers, or it could be due to the muscle's intrinsic refractoriness to mechanical perturbation as suggested by Chin et al. (4). The present finding suggests that abnormally stable myosin filaments due to enhanced signaling from the Rho-kinase pathway could augment the refractoriness of airway smooth muscle to strains that would normally constrain the muscle's contractility to an appropriate level. It is not clear which enzyme within the Rho-kinase pathway is key to the regulation of myosin filament stability. Elucidation of the pathway may provide new targets for intervention in the treatment of asthma.
GRANTS
This work was supported by operating grants from the Canadian Institutes of Health Research (CIHR) (MOP-13271, MOP-37924), Discovery Grant from the National Sciences and Engineering Research Council of Canada, Natural Science Foundation of China (Grant No. 11172340), Training Program for Hundreds of Distinguished Leading Scientists of Chongqing, Specialized Research Fund for the Doctoral Program of Higher Education of China (20120191120032), Chongqing Natural Science Foundation(Project No. cstc-2012jjA0588), Fundamental Research Funds for the Central Universities (CQDXWL-2012-2013, CQDXWL-2013-028), and the National Heart, Lung, and Blood Institute (HL-103405). B. Lan is supported by a scholarship from the China Scholarship Council.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the author(s).
AUTHOR CONTRIBUTIONS
B.L., L.D., P.D.P., C.Y.C. conception and design of research; B.L., L.Y.M.C., H.T.S., J.Z. performed experiments; B.L., C.Y.S., G.M.D. analyzed data; B. L., L.D., G.M.D., L.Y.C., H.T.S., L.W., J.Z., C.D.P., B.A.N., J.C.L., N.E.S., S.M.B., P.D.P., C.Y.S. interpreted results for experiment; L.B., C.Y.S. prepared figures; L.B., C.Y.S. drafted manuscript; B.L., L.D., G.M.D., L.Y.C., H.T.S., L.W., J.Z., C.D.P., B.A.N., J.C.L., N.E.S., S.M.B., P.D.P., C.Y.S. edited and revised manuscript; B. L., L.D., G.M.D., L.Y.C., H.T.S., L.W., J.Z., C.D.P., B.A.N., J.C.L., N.E.S., S.M.B., P.D.P., C.Y.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We give special thanks to Meadow Valley Meats (Pitt Meadows, BC) for the supply of fresh sheep tracheas in kind support for this research project and to D. Solomon for technical assistance.
REFERENCES
- 1.An SS, Bai TR, Bates JH, Black JL, Brown RH, Brusasco V, Chitano P, Deng L, Dowell M, Eidelman DH, Fabry B, Fairbank NJ, Ford LE, Fredberg JJ, Gerthoffer WT, Gilbert SH, Gosens R, Gunst SJ, Halayko AJ, Ingram RH, Irvin CG, James AL, Janssen LJ, King GG, Knight DA, Lauzon AM, Lakser OJ, Ludwig MS, Lutchen KR, Maksym GN, Martin JG, Mauad T, McParland BE, Mijailovich SM, Mitchell HW, Mitchell RW, Mitzner W, Murphy TM, Paré PD, Pellegrino R, Sanderson MJ, Schellenberg RR, Seow CY, Silveira PS, Smith PG, Solway J, Stephens NL, Sterk PJ, Stewart AG, Tang DD, Tepper RS, Tran T, Wang L. Airway smooth muscle dynamics: a common pathway of airway obstruction in asthma. Eur Respir J 29: 834–860, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brook BS. Emergence of airway smooth muscle mechanical behavior through dynamic reorganization of contractile units and force transmission pathways. J Appl Physiol 116: 980–997, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen C, Krishnan R, Zhou E, Ramachandran A, Tambe D, Rajendran K, Adam RM, Deng L, Fredberg JJ. Fluidization and resolidification of the human bladder smooth muscle cell in response to transient stretch. PLos One 5: e12035, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chin LY, Bossé Y, Pascoe C, Hackett TL, Seow CY, Paré PD. Mechanical properties of asthmatic airway smooth muscle. Eur Respir J 40(1): 45–54, 2012. [DOI] [PubMed] [Google Scholar]
- 5.Craig R, Smith R, Kendrick-Jones J. Light-chain phosphorylation controls the conformation of vertebrate non-muscle and smooth muscle myosin molecules. Nature 302: 436–439, 1983. [DOI] [PubMed] [Google Scholar]
- 6.Donovan GM. Modelling airway smooth muscle passive length adaptation via thick filament length distributions. J Theor Biol 333: 102–108, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fredberg JJ, Inouye DS, Mijailovich SM, Butler JP. Perturbed equilibrium of myosin binding in airway smooth muscle and its implications in bronchospasm. Am J Respir Crit Care Med 159: 959–967, 1999. [DOI] [PubMed] [Google Scholar]
- 8.Hai CM, Murphy RA. Cross-bridge phosphorylation and regulation of latch state in smooth muscle. Am J Physiol Cell Physiol 254: C99–C106, 1988. [DOI] [PubMed] [Google Scholar]
- 9.Huxley AF. Muscle structure and theories of contraction. Prog Biophys Biophys Chem 7: 255–318, 1957. [PubMed] [Google Scholar]
- 10.Janssen LJ, Tazzeo T, Zuo J, Pertens E, Keshavjee S. KCl evokes contraction of airway smooth muscle via activation of RhoA and Rho-kinase. Am J Physiol Lung Cell Mol Physiol 287: L852–L858, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Kapsali T, Permutt S, Laube B, Scichilone N, Togias A. Potent bronchoprotective effect of deep inspiration and its absence in asthma. J Appl Physiol 89: 711–720, 2000. [DOI] [PubMed] [Google Scholar]
- 12.Krishnan R, Park CY, Lin YC, Mead J, Jaspers RT, Trepat X, Lenormand G, Tambe D, Smolensky AV, Knoll AH, Butler JP, Fredberg JJ. Reinforcement versus fluidization in cytoskeletal mechanoresponsiveness. PLos One 4: e5486, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuo KH, Herrera AM, Wang L, Paré PD, Ford LE, Stephens NL, Seow CY. Structure-function correlation in airway smooth muscle adapted to different lengths. Am J Physiol Cell Physiol 285: C384–C390, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Kuo KH, Wang L, Paré PD, Ford LE, Seow CY. Myosin thick filament lability induced by mechanical strain in airway smooth muscle. J Appl Physiol 90: 1811–1816, 2001. [DOI] [PubMed] [Google Scholar]
- 15.Liu JC, Rottler J, Wang L, Zhang J, Pascoe CD, Lan B, Norris BA, Herrera AM, Paré PD, Seow CY. Myosin filaments in smooth muscle cells do not have a constant length. J Physiol 591: 5867–5878, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morgan JP, Morgan KG. Stimulus-specific patterns of intracellular calcium levels in smooth muscle of ferret portal vein. J Physiol 351: 155–167, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nadel JA, Tierney DF. Effect of a previous deep inspiration on airway resistance in man. J Appl Physiol 16: 717–719, 1961. [DOI] [PubMed] [Google Scholar]
- 18.Paré PD, Roberts CR, Bai TR, Wiggs BJ. The functional consequences of airway remodeling in asthma. Monaldi Arch Chest Dis 52: 589–596, 1997. [PubMed] [Google Scholar]
- 19.Qi D, Mitchell RW, Burdyga T, Ford LE, Kuo KH, Seow CY. Myosin light chain phosphorylation facilitates in vivo myosin filament reassembly after mechanical perturbation. Am J Physiol Cell Physiol 282: C1298–C1305, 2002. [DOI] [PubMed] [Google Scholar]
- 20.Scichilone N, Kapsali T, Permutt S, Togias A. Deep inspiration-induced bronchoprotection is stronger than bronchodilation. Am J Respir Crit Care Med 162: 910–916, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Skloot G, Permutt S, Togias A. Airway hyperresponsiveness in asthma: a problem of limited smooth muscle relaxation with inspiration. J Clin Invest 96: 2393–2403, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith RC, Cande WZ, Craig R, Tooth PJ, Scholey JM, Kendrick-Jones J. Regulation of myosin filament assembly by light-chain phosphorylation. Philos Trans R Soc Lond B Biol Sci 302: 73–82, 1983. [DOI] [PubMed] [Google Scholar]
- 23.Swärd K, Dreja K, Susnjar M, Hellstrand P, Hartshorne DJ, Walsh MP. Inhibition of Rho-associated kinase blocks agonist-induced Ca2+ sensitization of myosin phosphorylation and force in guinea-pig ileum. J Physiol 522: 33–49, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Umeki N, Jung HS, Sakai T, Sato O, Ikebe R, Ikebe M. Phospholipid-dependent regulation of the motor activity of myosin X. Nat Struct Mol Biol 18: 783–788, 2011. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Herrera AM, Paré PD, Seow CY. Dense-body aggregates as plastic structures supporting tension in smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 299: L631–L638, 2010. [DOI] [PubMed] [Google Scholar]
- 26.Zhang W, Huang Y, Gunst SJ. The small GTPase RhoA regulates the contraction of smooth muscle tissues by catalyzing the assembly of cytoskeletal signaling complexes at membrane adhesion sites. J Biol Chem 287: 33996–34008, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]