

Background: Although the solute-sodium symporter (SSS) vSGLT and the neurotransmitter-sodium symporter (NSS) LeuT have similar structural folds, their crystallographically identified substrate sites diverge in location and composition.
Results: We identified second substrate sites in two SSSs that align with the crystallographically identified site in LeuT.
Conclusion: Substrate transport by SSSs involves two substrate sites.
Significance: NSS and SSS share common mechanistic features.
Keywords: Amino Acid Transport, Computational Biology, Galactose, Membrane Transport, Molecular Dynamics, Transporter, Equilibrium Binding, Scintillation Proximity Assay, Solute-Sodium Symporter, Structural Alignment
Abstract
The structure of the sodium/galactose transporter (vSGLT), a solute-sodium symporter (SSS) from Vibrio parahaemolyticus, shares a common structural fold with LeuT of the neurotransmitter-sodium symporter family. Structural alignments between LeuT and vSGLT reveal that the crystallographically identified galactose-binding site in vSGLT is located in a more extracellular location relative to the central substrate-binding site (S1) in LeuT. Our computational analyses suggest the existence of an additional galactose-binding site in vSGLT that aligns to the S1 site of LeuT. Radiolabeled galactose saturation binding experiments indicate that, like LeuT, vSGLT can simultaneously bind two substrate molecules under equilibrium conditions. Mutating key residues in the individual substrate-binding sites reduced the molar substrate-to-protein binding stoichiometry to ∼1. In addition, the related and more experimentally tractable SSS member PutP (the Na+/proline transporter) also exhibits a binding stoichiometry of 2. Targeting residues in the proposed sites with mutations results in the reduction of the binding stoichiometry and is accompanied by severely impaired translocation of proline. Our data suggest that substrate transport by SSS members requires both substrate-binding sites, thereby implying that SSSs and neurotransmitter-sodium symporters share common mechanistic elements in substrate transport.
Introduction
Secondary active transport proteins harness electrochemical ion gradients to fulfill fundamental processes in all living cells, including excretion of toxic compounds, termination of neurotransmission, ion homoeostasis, and nutrient uptake. Interestingly, the structural fold of LeuT (1), a prokaryotic member of the neurotransmitter-sodium symporter family, is shared among several sodium (Na+) or proton-coupled transporter families that only show insignificant sequence homology (2), including the solute-sodium symporter (SSS)3 (3, 4), the nucleobase:cation symporter-1 (5, 6), the betaine/carnitine/choline transporter (7–9), and the amino acid/polyamine/organocation transporter (10) families. This common structural fold is characterized by two 5-transmembrane (TM) segment inverted repeats and suggests similar features and requirements in the conformational transitions associated with their transport mechanisms (11, 12).
In this study, we focus on the SSS family that encompasses Na+-coupled symporters from bacteria to man, including SGLT1 (the Na+/glucose transporter 1) and the Na+/I− symporter, which have been directly implicated in metabolic disorders (13, 14). Crystal structures of the SSS member vSGLT, the Na+/galactose transporter from Vibrio parahaemolyticus, were resolved in inward-occluded (4) and inward-open (3) states. Such an inward-facing preference is in contrast to that of LeuT, which has been readily crystallized in outward-facing conformations (1, 15–18), whereas several mutations in conjunction with conformation-specific antibody fragments were required to stabilize an inward-open LeuT structure (18). Based on these vSGLT structures and in combination with experimental investigations, computational modeling and analysis studies have revealed important mechanistic insights regarding Na+ coupling (19, 20), the dynamics of residues at the intracellular gate of the substrate-binding site (3, 21, 22), and the conformational transition to inward-open state in vSGLT (3) that is analogous to that in LeuT (18, 23).
Curiously, the substrate-binding sites identified in the vSGLT and LeuT structures have distinct TM compositions. In the substrate-bound vSGLT structure (PDB code 3DH4) (4), the substrate galactose was found to interact with TM1′, -2′, -6′, -7′, and -10′ (to facilitate the comparison between vSGLT and LeuT, based on the alignment to the two inverted repeats of LeuT, TM2–11 in vSGLT are renumbered to TM1′–10′ (12)), whereas the central occluded substrate-binding site (S1 site) in LeuT (24) is formed by residues located in TM1, -3, -6, and -8 (1). Interestingly, the galactose-binding site in the vSGLT crystal structure is positioned between the S1 site and the more extracellular second substrate binding (S2) site in LeuT, the latter of which was initially identified by computational modeling and subsequently confirmed with spectroscopic methods and tracer flux and binding studies (24–27). The existence of two sites in LeuT, however, has been challenged because the S2 substrate binding has not yet been captured crystallographically (28). Note that detergents used in crystallography can bind to the S2 site and also likely stabilize LeuT in conformations that are not optimal to bind the S2 substrate (26, 29, 30).
The different location and configuration of the crystallographically identified substrate-binding sites in vSGLT and LeuT raised the question whether the constitution of these substrate sites depends on particular inward- or outward-facing conformational states captured under energetically favorable crystallization conditions (26, 29). Thus, we hypothesized that members of the SSS family in distinct states may bind substrate in additional binding sites, which may not be readily identified with crystallographic approaches. To this end, by using a combined approach encompassing computational modeling and different substrate binding assays, we show that two SSSs, vSGLT and the homologous Na+/proline transporter (PutP), can simultaneously bind two substrate molecules under equilibrium conditions in the presence of Na+, like LeuT of the neurotransmitter-sodium symporter family (24). Mutations that target key residues in the individual sites of vSGLT and PutP reduce the molar binding stoichiometry. In PutP, an experimentally tractable SSS system for which the assessment of different modes of transport has been established (31), these mutations result in severe reduction of proline translocation.
EXPERIMENTAL PROCEDURES
Structure-Structure Alignment between LeuT and vSGLT
The structure-structure alignment of LeuT (PDB code 2A65) (1) and vSGLT (PDB code 3DH4) (4) was performed using Matt (32) with default parameters. The segments considered in the alignment are the two conserved 5-TM inverted repeats, TM1–10 in LeuT and TM1′–10′ in vSGLT (Fig. 1). The resulting sequence alignment was further manually refined by removing the gaps in helical regions and by local adjustments to align the positions in the two structures that face the permeation pathways. The final alignment is shown in Fig. 1. According to this alignment, the superposition of the structures shown in Fig. 2 is based on the middle portions of the more conserved TM-1/1′, -3/3′, -6/6′, and -8/8′, which do not have drastic rearrangements between outward-facing and inward-facing conformations, specifically residues 23–26, 106–111, 251–254, and 351–358 of LeuT versus residues 65–68, 140–145, 261–264, and 361–368 of vSGLT.
FIGURE 1.

Sequence alignment of vSGLT, PutP, and LeuT. The alignment is based on a structure-structure alignment using Matt (32), followed by manual adjustments to remove gaps in helical regions. TM boundaries for LeuT and vSGLT are defined according to those of Yamshita et al. (1) and Faham et al. (4), respectively. The unwound regions in the TM1/1′ and TM6/6′ are underlined. Substrate-binding site residues identified in the crystal structures of LeuT and vSGLT (PDB codes 2A65 and 3DH4, respectively) are highlighted with cyan and red, respectively; the S1 residues of LeuT are from Ref. 52, and the Se residues of vSGLT are defined as having closest heavy atom distance <4.5 Å to the substrate galactose.
FIGURE 2.

Crystallographically identified substrate-binding site of vSGLT is located more extracellularly than that of LeuT. A and B show the superimposed orientations of a LeuT model bound with two substrates Leu (24) and the vSGLT structure (PDB code 3DH4), respectively, based on a structure-structure alignment (see text). The TMs directly involved in substrate binding in either LeuT or vSGLT are in non-white colors and in the same color codes for both panels. In particular, the equivalent TMs in the two 5-TM inverted repeats of LeuT structural fold, e.g. TM1 and TM6, are in the same color. The substrate-binding site in the vSGLT structure, termed Se here, is enclosed by TM1′, -2′, -6′, -7′, and -10′, whereas for LeuT the crystallographically identified S1 site is formed by residues from TM1, -3, -6, and -8, and the computationally identified S2 site is formed by residues from TM 1, -3, -10, and EL4. C and D are the zoom-in views of the substrate-binding sites in LeuT and vSGLT, respectively. The cavity below Tyr-263 of vSGLT may potentially form a substrate-binding site. Thus Phe-253 of LeuT and Tyr-263 of vSGLT may play similar roles as the gating residues to separate more centrally located substrate-binding sites from the extracellular milieu. The Cα-Cβ bonds of substrate binding residues are shown as sticks (cyan for the S1/Sc site and green for the S2/Se sites) with the Cβ atoms represented as small spheres. D, residues Ser-365, Ser-368, and Ser-372 of vSGLT are aligned to Thr-341, Cys-344, and Val-348 of PutP, which have been found to be critical for proline uptake (42).
Induced-fit Docking (IFD) of Substrate to the Se Site
By using the IFD protocol (33), in the presence of a substrate galactose bound in the crystallographically identified substrate site (Se site), a second galactose was docked in the putative Sc cavity (see under “Results”) of our vSGLT model, which was described previously (20). Specifically, we targeted four sub-pockets as follows: (i) immediately below the gate residue, Tyr-263; (ii) between Tyr-263 and Ser-368; (iii) near the Na+ site identified in the vSGLT structure; and (iv) at the midpoint of i and iii. The docking poses from individual trials are pooled together for the clustering analysis. Three representative poses with the highest IFD scores were selected for extensive MD simulations.
Similarly, starting from an equilibrated PutP model based on the crystal structure of vSGLT (PDB code 3DH4) and with a substrate proline bound in the Se site (20), we docked a second proline in its Sc cavity, according to the equilibrated poses of the Sc substrate in vSGLT (see “Results”). Three representative poses with the highest IFD scores were selected for the following MD simulations.
Construction of the Simulation Systems and MD Simulations
The vSGLT models with the additional Sc substrate were reinserted back to the equilibrated explicit water-bilayer-water environment by aligning them to the reference vSGLT model in the original simulation system (20). In this system, the water phase includes Na+ and Cl− ions corresponding to a concentration of 150 mm NaCl; the entire system includes ∼73,400 atoms, including 171 palmitoyloleoylphosphatidylcholine molecules and ∼14,000 waters.
Three MD simulations with different initial poses of the Sc substrate were carried out for both vSGLT and PutP (Table 1), using Desmond (version 3.0) (34) with the OPLS2005 (35) all-atom force field. Periodic boundary condition was employed. The relaxation protocol modified from that developed by Schrödinger Inc. (Desmond 3.0; Schrodinger, Inc., New York, NY), including minimization, heating, and solvation of buried cavities, and equilibrations were carried out before the production runs. First, two steepest descent minimizations with and without harmonic constraints of 50 kcal/mol on the solute heavy atoms with a maximum of 2000 steps were performed, followed by a 60-ps MD simulation raising the temperature from 10 to 310 K in the canonical ensemble (NVT), during which the solute heavy atoms were restrained. The system was then equilibrated in two successive steps with a Martyna-Tobias-Klein isothermal-isobaric ensemble (NPT) (36), where a harmonic constraint with a force constant of 10 kcal/mol on the solute heavy atoms was applied at the first step and removed afterward. In the production runs, the time step was 1 fs, and a constant surface tension of 4000 bar·Å was added (NPγT). Electrostatic interactions were calculated with particle mesh Ewald (PME) (37) throughout the simulations, and the short range cutoff was set to 10.0 Å.
TABLE 1.
vSGLT and PutP MD trajectories
The frames from the last 100 ns of each trajectory are used for the analysis. The root mean square deviations (r.m.s.d.) were calculated against the starting model for each simulation.
| Transporter | Starting Sc binding mode | Length of the trajectories | r.m.s.d. of the Cα in TMs | Movement of substrate Sc |
|---|---|---|---|---|
| ns | Å | |||
| vSGLT | Sub-pocket 1, near Na2 | 250 | 1.3 ± 0.1 | Moves within sub-pocket 1 |
| Sub-pocket 2, between Tyr-263 and Ser-368 | 238 | 1.2 ± 0.1 | Moves within sub-pockets 2 and 3 | |
| Sub-pocket 3, right below Tyr-263 | 213 | 1.6 ± 0.2 | Moves within sub-pockets 2 and 3 | |
| PutP | Sub-pocket 1, near Na2 | 118 | 2.8 ± 0.1 | Moves within sub-pocket 1 |
| Sub-pocket 2, between Tyr-248 and Cys-344 | 108 | 2.0 ± 0.1 | Moves within sub-pocket 2 | |
| Sub-pocket 3, right below Tyr-248 | 500 | 2.9 ± 0.1 | Moves within sub-pocket 3 |
Clustering of the Substrate Binding Poses in the Sc Cavity and MM/GBSA Calculations
After each MD frame was superimposed on the crystal structure (PDB code 3DH4) based on the putative Sc site residues (Table 2), the substrate poses in the Sc cavity were clustered according to the their center-of-mass (COM) positions with respect to the bound Na+ in the crystal structure of vSGLT, in a spherical coordinate system (see the definition in Fig. 3). As the θ values of the poses vary in a narrow range (within a range of 30°), we clustered the poses according to the distribution of the r and ϕ values, and we considered a densely populated region as a cluster (Fig. 4, A and C). For each frame, the MM/GBSA receptor-ligand energy was calculated using Prime (version 3.1; Schrödinger).
TABLE 2.
Composition of the Sc and Se binding sites of vSGLT and PutP
The binding site residues in vSGLT and PutP are based on the clusters with averagely more favored MM/GBSA binding energy, i.e., clusters 2 and 3 for vSGLT and clusters 1 and 3 for PutP. Residues within 4.5 Å of the ligand in >50% of the frames in each cluster are listed here.
* The S1 residues of LeuT are highlighted in cyan (see Fig. 1).
FIGURE 3.
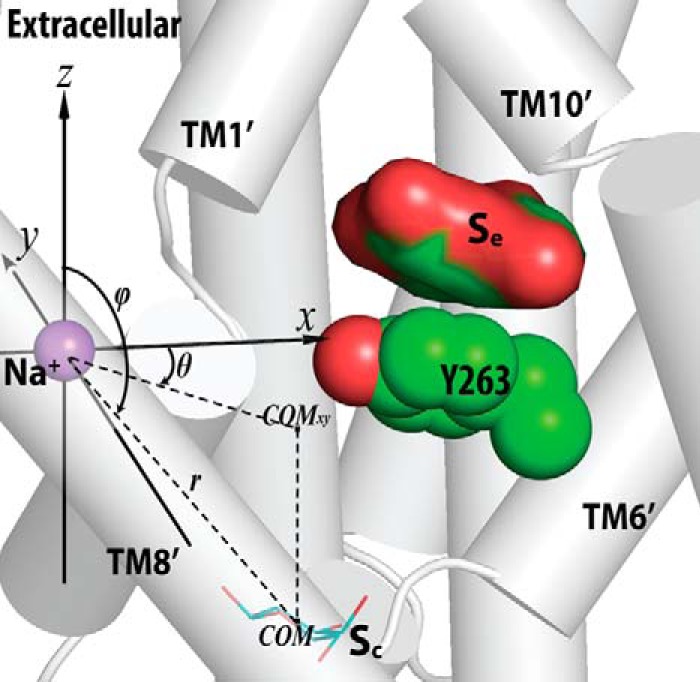
Definition of the spherical coordinate system. This system is used to describe the center-of-mass (COM) position of the Sc substrate with respect to the Na2 in vSGLT. Thus, the Na2 position is defined as the reference point (0,0,0). The x,y,z directions shown are based on the reoriented coordinates of PDB code 3DH4 from the Orientations of Proteins in Membranes (OPM) database (53), which have the z axis perpendicular to the membrane, and the x axis largely pointing from Na2 to the side chain of Tyr-263. The projection of the COM on the xy plane is shown as the point COMxy. “r” is the distance between Na2 and COM; “θ” is the angle between the x axis and the line connecting Na2-COMxy, and “ϕ” is the angle between the z axis and the line connecting Na2-COM.
FIGURE 4.

Substrate binding poses in the Sc site of vSGLT and PutP. A, distribution of the probabilities of finding the galactose molecule at a given position in the Sc site of vSGLT (expressed in a spherical coordinate (r, θ, ϕ) and using the coordinate to Na2 as the reference point, see Fig. 3). The galactose binding poses are divided into three clusters based on this distribution. B, average MM/GBSA energies of each the galactose pose cluster in the Sc site. Corresponding results for PutP are shown in C and D. The substrate binding poses in the Sc sites of vSGLT (E) and PutP (F) are represented by vectors (from the center of the ring pointing toward the carbons outside the rings for both galactose and proline). In addition to the gating residues Tyr-263 of vSGLT and Tyr-248 of PutP, residues that were experimentally tested are shown as sticks. The Se-bound substrates are in sphere representations.
Protein Expression and Purification
vSGLT mutations were performed using the QuikChange methodology (Agilent Technologies) with the vSGLT gene in pJexpress as template. All mutants were verified by DNA sequencing, and vSGLT variants were expressed in Escherichia coli XL1-Blue cells. Briefly, an overnight starter culture in Terrific Broth supplemented with 30 μg/ml kanamycin was inoculated into 1-liter flasks of the same media to a final absorbance at 600 nm of 0.05 and cultivated aerobically at 37 °C. Protein expression was induced at an absorbance at 600 nm of 1.8 with 0.75 μm isopropyl 1-thio-β-d-galactopyranoside for 3 h at 33 °C. Cells were collected at 7000 × g for 10 min and frozen at −20 °C prior to the preparation of membrane vesicles with an EmulsiFlex-C3. Membrane vesicles were solubilized by the addition of 2% n-dodecyl β-d-maltopyranoside (Anatrace). vSGLT was bound to a nickel-nitrilotriacetic acid column (GE Healthcare) in 70 mm Tris-Cl, pH 8.0, 150 mm NaCl, 4 mm Na3-citrate, 20 mm imidazole, 6% glycerol, 5 mm 2-mercaptoethanol, 0.0174% n-dodecyl β-d-maltopyranoside and eluted in the same buffer with 200 mm imidazole. Peak fractions were concentrated using an Amicon Ultra 50 kDa (Millipore) and loaded onto an analytical Superdex 200 column (GE Healthcare) equilibrated in 25 mm Tris-Cl, pH 7.4, 150 mm NaCl, 2 mm 2-mercaptoethanol, 0.0174% n-dodecyl β-d-maltopyranoside. Only the center of the elution peak consisting of 0.5–1 mg of total protein was pooled and used for the equilibrium dialysis assays.
Recombinant PutP wild-type (WT) and variants (containing a C-terminal His6 tag) (38) were produced in E. coli WG170 and purified as described (39). Mutations of Tyr-248 to Gly, Pro-252 to Cys, Cys-344 to Ala, and Leu-398 to Ser were performed with PCR-based site-directed mutagenesis and confirmed by sequencing. Protein was assayed with the Amido Black method (40).
Binding Studies
Equilibrium dialysis was performed (29) with the HTD96b dialysis 96-well apparatus and 12,000–14,000 molecular weight cutoff membranes in binding buffer (150 mm Tris/Mes, pH 7.5, 50 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine, 20% glycerol, 0.1% (w/v) n-dodecyl β,d-maltopyranoside) for 16 h at 4 °C with 250 pmol of vSGLT (or variants thereof) or 50 pmol of PutP. d-[1-3H]Galactose (20 Ci/mmol; American Radiolabeled Chemicals, Inc.) or l-[2,3,4,5-3H]proline (90 Ci/mmol; American Radiolabeled Chemicals) was used at the indicated concentration range. 20-μl samples were taken from each dialysis compartment separated by the dialysis membrane (total volume per compartment was 50 μl) and incubated with scintillation mixture for 24 h prior to photo-multiplier tube-based decays/min) counting. To capture protein bound to the dialysis membrane, the exposed membrane section was excised and subjected to scintillation counting. Nonspecific radiotracer binding to the membrane was determined by performing the assay in the absence of protein, and data were corrected accordingly. Decays/min were transformed into picomoles using known amounts of [3H]galactose or [3H]proline. Scintillation proximity assay (SPA)-based binding of [3H]proline to purified PutP variants was performed as described (39) with 0.92 pmol of purified protein per assay in binding buffer.
[3H]Proline Transport Measurements
Na+-coupled [3H]proline uptake into intact E. coli WG170 harboring plasmids coding for given PutP variants or a control plasmid was performed in 50 mm Tris/Mes, pH 6.0, 50 mm NaCl as described (31). Facilitated diffusion down the proline concentration gradient was performed either in the same buffer or in 100 mm Tris/Mes, pH 6.0 (Na+-free condition) with de-energized WG170 harboring indicated PutP variants. Briefly, 200-μl cell suspensions at a protein concentration of 0.07 mg/ml were incubated for 5 min prior to the start of the uptake reaction in the presence of 5 μm carbonyl cyanide m-chlorophenylhydrazone (CCCP) and 5 μm monensin to dissipate the transmembrane electrochemical Na+ and H+ gradients (31) prior to the addition of 20 μm [3H]proline. For counterflow experiments, cells were incubated in 50 mm Tris/Mes, pH 6.0, in the presence of 1 mm Pro and 10 mm NaCl for 2 h at 4 °C prior to washing the cells three times in 50 mm Tris/Mes, pH 7.5, 1 mm Pro, 5 μm CCCP, 5 μm monensin. Concentrated cell stocks were diluted ∼200-fold in buffer composed of 50 mm Tris/Mes, pH 7.5, 5 μm CCCP, 5 μm monensin plus 1 μm [3H]Pro to obtain a final total cell protein concentration of 0.03 mg/ml and incubated for the indicated periods of time. Reactions were quenched with ice-cold 100 mm KPi, pH 6.0, 100 mm LiCl followed by filtration through a 0.75-μm GF/F glass fiber filter (Advantec MFS). The captured radioactivity on the filter was determined by liquid scintillation counting. Transformation of decays/min to picomoles was performed using standards of known amounts of [3H]proline.
Data Analysis
Data fits of kinetic analyses were performed using nonlinear regression algorithms in Prism (GraphPad), and errors represent the standard error of the fit.
RESULTS
Crystallographically Identified Substrate-binding Sites in vSGLT and LeuT Are At Different Locations
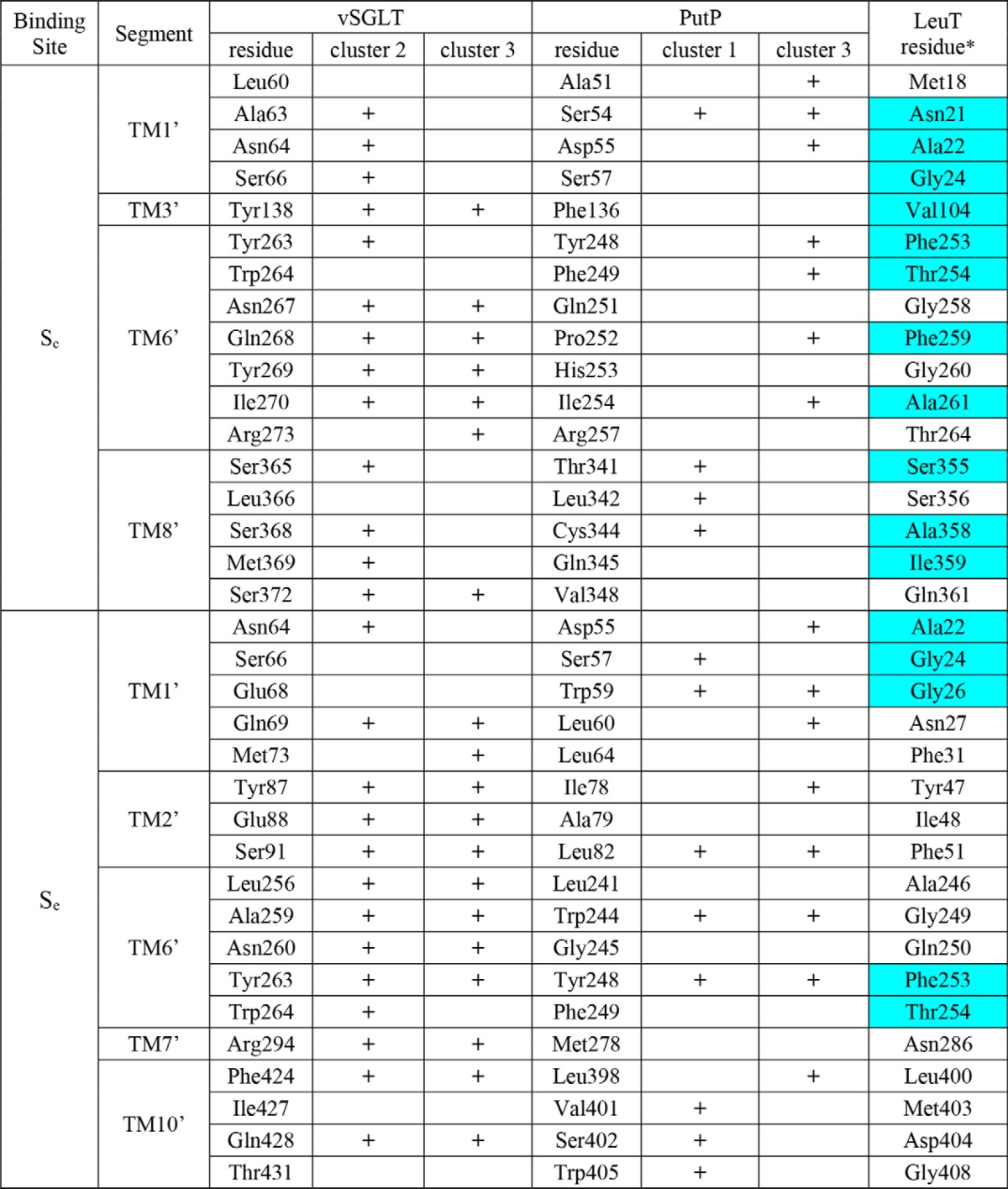
To identify common and unique key structural elements of LeuT and vSGLT, we performed a structure-structure alignment between the crystal structures of LeuT and vSGLT with Matt (32). When we superimposed LeuT and vSGLT with the core TM regions (TM1–10 of LeuT and TM1′–10′ of vSGLT; Fig. 1), we found that the substrate-binding site of vSGLT, which is formed by residues from TM1′, -2′, -6′, -7′, and -10′, is situated more extracellularly than the central occluded substrate (S1) site in LeuT, which is enclosed by TM1, -3, -6, and -8 (Fig. 2). Based on the superposition shown in Fig. 2, the bound substrates, galactose in vSGLT and leucine in LeuT, have a COM distance of 8.0 Å. In fact, the vSGLT substrate-binding site is at a location between the S1 site and the extracellular second substrate (S2) site of LeuT, the latter of which was identified using computational modeling and simulations, as well as radiotracer and flux measurements (24). Whereas the S2 site in LeuT is located in the middle of the so-called extracellular vestibule (EV) that is composed of residues shown to participate in binding tricyclic antidepressants (15) and selective serotonin reuptake inhibitors (41) in outward-facing conformations, the substrate-binding site of vSGLT is at the intracellular end of the collapsed EV in the inward-facing conformation. To account for these differences, we refer to the substrate-binding site revealed by the crystal structure of vSGLT (PDB code 3DH4) as the extracellular substrate (Se) site.
In particular, Tyr-263 from TM6′ of vSGLT at the intracellular end of the galactose-binding site is similarly located as Phe-253 from TM6 of LeuT, although there are subtle divergences between the unwound regions of TM6′ in vSGLT and TM6 in LeuT in terms of length and orientation; Tyr-263 of vSGLT and Phe-253 of LeuT are both at the bottom of the EV and above the cavity formed by TM1′/1, -3′/3, -6′/6, and -8′/8, i.e. the S1 site in LeuT. Thus, although the vSGLT substrate galactose is located right above (extracellular to) Tyr-263, the S1 substrate of LeuT is positioned right below (intracellular to) Phe-253, which was found to act as the gate at the extracellular side of the S1 site; it separates the S1 site from the EV (1, 24). On the extracellular side of the Se site of vSGLT, Phe-424 and Gln-428 from TM10′ can be aligned precisely to Leu-400 and Asp-404 of LeuT, residues that have been found to be involved in S2 substrate binding (24–26).
Our structure-structure alignment of vSGLT and LeuT also aligns the deduced Na+-binding site of vSGLT (4) precisely to the Na2 site of LeuT (1). Curiously, near this Na+-binding site and below Tyr-263 of vSGLT, there is an empty pocket formed by TM1′, -3′, -6′, and -8′, which are aligned to the TMs that enclose the S1 site of LeuT. In the inward-facing conformation of vSGLT, this pocket is highly exposed to the intracellular water phase (3, 4). Although the carboxyl group of the S1 substrate in LeuT directly coordinates the bound Na+ in the Na1 site and is in close vicinity to Na2 (with a COM distance of ∼6.5 Å), the substrate bound in the Se site in vSGLT is significantly further away from the crystallographically identified single Na+ site (with a COM distance of ∼14.5 Å). Thus, we hypothesize that in another conformational state the pocket below Tyr-263 in vSGLT may form a central substrate-binding site that is close to that Na+ site. In the following, we termed this pocket of vSGLT as the central substrate (Sc) site, which structurally aligns with the S1 site of LeuT.
A recent study of PutP using the substituted cysteine-accessibility method (SCAM) supports the presence of the Sc site (42). In this study, labeling of the individual thiol group at residue positions 344, 347, 348, and 351 was reduced in the presence of the substrate proline but not by Na+ alone (42). Interestingly, the aligned residues at these positions in vSGLT face the Sc site, but are not involved in forming the crystallographically identified Se site in vSGLT (Fig. 2D).
Potential Substrate-binding Residues in the Sc Site of vSGLT Align to the S1 Residues of LeuT
To specifically identify the residues in the Sc site that are critical for substrate binding, using the comprehensive vSGLT model constructed and equilibrated previously (20), we first performed induced-fit docking to position a second substrate galactose in the Sc site to identify its optimal poses in the presence of the galactose bound in the Se site.
The resulting poses of galactose in the Sc site were clustered into three groups (Table 1). A representative Sc pose from each group was selected based on the MM/GASA binding energy (see “Experimental Procedures”). After reinsertion of these vSGLT models bound with galactose at both the Sc and Se sites back into the explicit water-membrane simulation system, they were further relaxed and characterized with MD simulations. The MD simulations were carried out until the root mean square deviations of the TM regions plateaued for at least 100 ns (Table 1).
Whereas the Se substrate stayed in the pose identified in the crystal structure in all simulations, the substrate in the Sc site fluctuated around the original docked location but remained within this site. We clustered the Sc substrate poses from all MD trajectories into three clusters (Fig. 4A). Strikingly, for the two clusters with more favored binding energies (clusters 2 and 3 in Fig. 4, B and E), most of the substrate-interacting residues are aligned to the S1 residues of LeuT (Table 2). Specifically, in the MD frame with the most favored galactose binding energy, among the 10 galactose-interacting residues, seven can be aligned to the S1 substrate-binding residues in LeuT. In this frame, the hydroxyl groups of Sc galactose appear to optimally form H-bonds with the side chains of Ser-66, Arg-273, Ser-365, and Ser-368, and the backbone of Tyr-269 (Fig. 5). Interestingly, the aligned Na2 binding residues Ser-365 in vSGLT and Ser-355 in LeuT are similarly able to both coordinate the Na+ ion and be in close proximity to the substrate. In contrast, among the residues forming the Se site, only the gating residues Asn-64 and Tyr-263 and the nearby Trp-264 can be aligned to the S1 residues of LeuT (Table 2).
FIGURE 5.
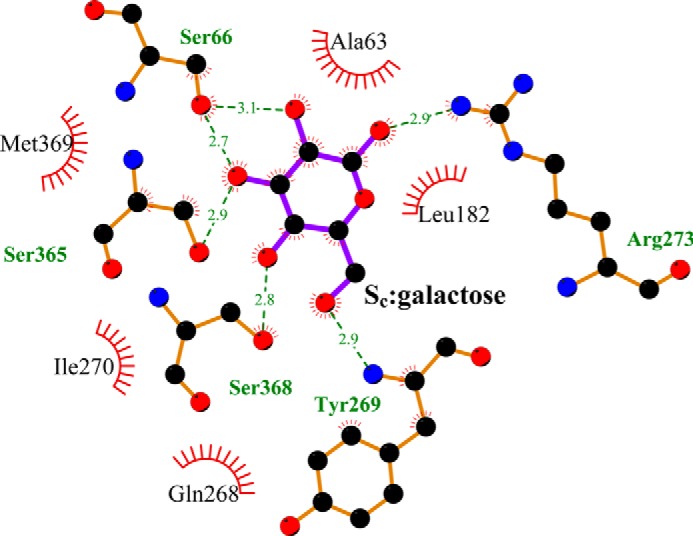
Residues interacting with the galactose bound in the Sc site of vSGLT. The MD frame shown is the frame with the most favored galactose binding energy. The two-dimensional representation was generated using the Ligplot+ diagram (54). The H-bonds are shown as green dotted lines with the distances indicated in Å. The spoked arcs represent hydrophobic interactions.
vSGLT Has a Molar Binding Stoichiometry of 2
To experimentally test the computational findings, we performed [3H]galactose saturation binding using equilibrium dialysis with known amounts of purified vSGLT to deduce the galactose-to-vSGLT molar binding stoichiometry (MBS). This method allowed for the assessment of the binding activity without relying on a potential bias introduced by the calculations of counting efficiency in different detection modes, as all samples and calibration standards were subjected to the same scintillation counting format (29). The MBS of [3H]galactose binding to vSGLT in the presence of 50 mm Na+ was concentration-dependent and reached a maximum of ∼2 (Fig. 6A). Subjecting the data of three independent experiments to a single-site global fitting analysis (GraphPad Prism 5) yielded a Kd (concentration at half-maximum binding) of 179.2 ± 19.9 μm and a Bmax of 2.07 ± 0.07 molecules of galactose bound per molecule of vSGLT.
FIGURE 6.

Substrate binding in vSGLT. Na+-dependent equilibrium dialysis-based binding was measured with [3H]galactose (0.02 Ci/mmol) and purified vSGLT-WT (A), vSGLT-E88A (B), vSGLT-F424S (C), or vSGLT-S368A (D). Data of ≥3 independent experiments were subjected to global fitting in GraphPad Prism 5 using single-site models.
Mutations of the Sc or Se Site Residues of vSGLT Reduce the MBS to One
To assess the impact of disrupting the substrate binding in the Se site, we targeted Phe-424 and Glu-88 that are on the extracellular side of the Se site and relatively far from the proposed Sc site. Specifically, Phe-424 aligns with Leu-400 in LeuT, a residue that has been shown to participate in the binding of substrate in the S2 site of LeuT (24), whereas Glu-88 has been previously shown to be important for galactose transport (4). By mutating Phe-424 to Ser or Glu-88 to Ala, the MBS of two observed for vSGLT-WT was reduced to ∼1. Fitting the data to a one-site model showed small alterations of the Kd values (133.3 ± 12.1 μm and 176.5 ± 20.3 μm for E88A and F424S), whereas the MBSs were reduced to 1.20 ± 0.03 and 1.20 ± 0.04 for E88A (Fig. 6B) and F424S (Fig. 6C), respectively.
To disrupt the proposed Sc site, we substituted Ser-368, an Sc residue relatively far from the Se site and the Na+ site, with Ala. [3H]Galactose binding by vSGLT-S368A yielded an MBS of 1.17 ± 0.03 and a Kd of 111.6 ± 10.8 μm (Fig. 6D). Taken together, the saturation binding data support our hypothesis that under equilibrium conditions vSGLT, like LeuT, can simultaneously bind two substrate molecules, only one of which has been identified in the available crystal structures of vSGLT (4).
Homologous PutP Can Bind Two Substrate Molecules
To test whether other SSS members also exhibit an MBS larger than unity, we performed equilibrium dialysis-based saturation binding of [3H]proline to purified PutP, a well characterized and experimentally tractable SSS (31, 38, 43–46), which has a sequence homology of 42% (sequence identity of 20%) to vSGLT in the TM1′–10′ (Fig. 1). Consistent with the simultaneous occupation of PutP by two substrate molecules under equilibrium conditions, the proline-to-PutP MBS was ∼2 (Fig. 7A). However, a plateau in the binding curve (between 5 and 15 μm [3H]proline) was noticeable for PutP (Fig. 7A, inset), which prevented reliable fitting with one-site or two-site models. Therefore, the observed two phases were fit independently with one-site models. Fitting the data ranging between 0 and 15 μm [3H]proline yielded a Kd of 1.53 ± 0.32 μm, whereas fitting the data points between 15 and 250 μm [3H]proline yielded a Kd of 24.3 ± 1.66 μm. The Bmax was calculated to be 2.15 ± 0.12 molecules of proline bound per molecule of PutP (proline-to-PutP) (Fig. 7A).
FIGURE 7.

Substrate binding by PutP variants. A, equilibrium dialysis-based binding of [3H]proline (0.5 Ci/mmol) by purified PutP. B–E, SPA-based saturation binding of [3H]proline (1 Ci/mmol) using purified PutP-WT (B), PutP-L398S (C), PutP-C344A (D), PutP-Y248G (E), or PutP-P252C (F). Data in B–F represent the mean ± S.E. of triplicate determinations of representative experiments, and data were subjected to nonlinear regression fitting in GraphPad Prism 5. See text for the dissociation constants (Kd) and proline-to-PutP molar binding stoichiometries. Data in A are from five independent experiments and were subjected to global fitting in GraphPad Prism 5.
In addition, we performed proline binding studies with PutP by means of the scintillation proximity assay (SPA), a rapid and sensitive method to assess the detailed binding kinetics (Fig. 7, B–F) (24, 39). Consistent with the equilibrium binding data (Fig. 7A), SPA-based [3H]proline binding under equilibrium conditions revealed a biphasic saturation binding curve for PutP-WT with a plateau between 8 and 18 μm [3H]proline. This curve was also best fit with two separate one-site models (Fig. 7B), yielding a Bmax of 1.92 ± 0.15, a Kd of 1.30 ± 0.29 μm for the first phase (data points between 0 and 15 μm), and a Kd of 28.72 ± 2.18 μm for the second phase (data points between 15 and 70 μm). Note that due to the relatively low affinity of galactose for vSGLT and observed low signal-to-noise ratio, SPA-based [3H]galactose binding to vSGLT could not be assessed in a reliable and reproducible fashion.
Mutations of the Sc or Se Residues in PutP Reduce the Proline-to-PutP MBS
To gain insight into the determinants of proline binding by PutP at the atomistic level, based on the fact that PutP shares significant sequence homology with vSGLT and can similarly bind two substrate molecules simultaneously, we carried out computational analyses with vSGLT-based PutP homology models. Previously, we built and equilibrated a PutP model with a proline bound in the Se site (20), the equilibrated state of which has a root mean square deviation of ∼2 Å to the vSGLT structure. As proline is smaller than galactose, we found the Se site of PutP is formed by residues from TM1′, -2′, -6′, and -10′ but does not include any TM7′ residue (Table 2). Starting from this model, we docked a second proline in the sub-pockets of the Sc site that align to those in vSGLT (Table 1), and we selected three representative Sc poses for the following MD simulations to explore the potential Sc binding residues. We carried out clustering analysis of the resulting Sc proline binding poses from the MD simulations, and we calculated the MM/GBSA energies to identify the poses with more favored binding energies (Fig. 4, C, D, and F). Similar to the situation in vSGLT, we found many Sc residues of PutP from TM1′, -3′, -6′, and -8′ are aligned to the S1 residues in LeuT (Table 2).
To experimentally validate the computationally identified substrate-binding modes in PutP, we created mutants of the residues located at the intracellular end of the proposed Sc site and the extracellular end of the Se site, i.e. mutating Cys-344 to Ala and Leu-398 to Ser, respectively. These residues are aligned to Ser-368 and Phe-424 of vSGLT (see above). Furthermore, we also mutated Tyr-248, a residue located between the Se and Sc sites, which can potentially interact with the bound substrates from both sites (Table 2), and Pro-252, another residue facing the proposed Sc site and located more intracellularly than Tyr-248 in TM6′. The binding data observed for PutP-L398S was best fit by a single-site model with a Kd of 6.1 ± 2.1 μm and an MBS of 1.0 ± 0.1 proline-to-PutP (Fig. 7C). Proline binding by PutP-C344A resulted in a complex binding isotherm that saturated at an MBS of 1.05 ± 0.05 proline-to-PutP and was fit best with two separate single-site models with a Kd of 4.57 ± 0.64 μm for data points between 0 and 10 μm and a Kd of 18.3 ± 3.08 μm for data points between 15 and 70 μm (Fig. 7D). The reduction of the MBS to ∼1 was also detected when Tyr-248 was replaced with Gly (Fig. 7E) because fitting the data with a single-site model yielded a Bmax of 1.09 ± 0.05 proline-to-PutP and a Kd of 11.53 ± 1.48 μm. Replacing Pro-252 with Cys yielded an MBS of 1.58 ± 0.21 proline-to-PutP, and fitting two individual curves for data points between 0 and 20 μm and 20 and 70 μm, respectively, yielded Kd values of 12.3 ± 1.36 and 23.5 ± 0.01 μm (Fig. 7F).
Mutations of Residues in the Sc or Se Site in PutP Disrupt Proline Transport
When we measured the time course of 2 μm [3H]proline transport in the presence of saturating NaCl (10 mm) (31), we found the initial transport rates of PutP-L398S and -C344A to be reduced to 13.0 ± 0.2 and 28.8 ± 0.5% that of PutP-WT, whereas their steady-state level of accumulation was 13.3 ± 0.3 and 23.0 ± 0.8%, respectively, of the WT value (Fig. 8A). The initial rate and steady state level of Na+-coupled proline transport by PutP-Y248G were 6.4 ± 0.5 and 10.7 ± 2.1% compared with those of PutP-WT. PutP-P252C exhibited 43.7 ± 2.3 and 71.6 ± 2.7% of the initial rate of transport and steady state level of proline accumulation, respectively, of the rates measured for WT.
FIGURE 8.

PutP-mediated transport. A, time course of 2 μm [3H]proline (1 Ci/mmol) uptake by E. coli WG170 harboring PutP-WT (■), PutP-C344A (∇), PutP-L398S (▴), PutP-Y248G (Δ), and PutP-P252C (□) or a control plasmid (○) in the presence of 10 mm NaCl. B–F, kinetics of Na+-coupled proline transport. Initial rates of transport were determined at [3H]proline concentrations ranging between 0.1 and 100 μm for 10 s (PutP-WT and PutP-P252C) or 30 s (PutP-Y248G, PutP-C344A, or PutP-L398S). Data were normalized to the activity of PutP-WT tested in parallel to account for the batch-specific variations in the expression levels and fit to the Michaelis-Menten equation. Data are shown as mean ± S.E. of triplicate determinations of a representative experiment, and kinetic constants represent the mean ± the S.E. of the fit. The Km values are listed in the corresponding graphs. B, PutP-WT; C, PutP-L398S; D, PutP-C344A; E, PutP-P252C; F, PutP-Y248G. G, time course of proline counterflow. Concentrated cell suspensions of WG170 producing the indicated PutP variant or nontransformed control cells were pre-loaded with 1 mm Pro for 2 h prior to a rapid 200-fold dilution into buffer containing 1 μm [3H]Pro. H, facilitated diffusion of 20 μm [3H]proline by WG170 harboring the indicated PutP variants after treatment of the cells with 5 μm CCCP and 5 μm monensin. Transport was assayed in the presence (solid bar) or absence (open bar) of 10 mm NaCl, and data were normalized with regard to the initial rate of the proline accumulation measured after 10 s for PutP-WT in the presence of NaCl (7.6 ± 0.2 nmol × mg−1 × min−1). Data are normalized with regard to the 1-min uptake activity observed in PutP-WT.
The alterations in the time-dependent proline uptake are reflected in the kinetics of active transport. Compared with PutP-WT, all mutants exhibited reduced maximum transport velocities (Vmax) in the following order: PutP-WT (24.6 ± 0.2 nmol × mg−1 × min−1) > PutP-P252C (52.1 ± 0.5% of the PutP-WT value) > PutP-C344A (24.4 ± 2.4%) > PutP-Y248G (16.4 ± 0.6%) > PutP-L398S (10.2 ± 0.5%). The affinity for Na+-coupled proline transport (Km, proline concentration at 50% Vmax) by the mutants were only marginally affected (Fig. 8, B–F).
We then evaluated the impact of the mutations on proline counterflow (Fig. 8G). Here, the rapid dilution of cells pre-loaded with 1 mm proline into buffer containing 1 μm [3H]proline leads to the exchange of radiolabeled and nonlabeled substrate until equilibrium is reached. In similar fashion to Na+-coupled transport, proline counterflow by the mutants exhibited reduced initial rates when compared with PutP-WT (154 ± 17.9 pmol × min−1 × mg cell protein−1) before reaching equilibrium after about 1 h of incubation as follows: PutP-P252C (51.6 ± 2.1%) > PutP-Y248G (19.8 ± 0.6%) > PutP-C344A (14.1 ± 1.2%) > PutP-L398S (8.5 ± 0.3%). As determined with immunoblotting, the reduced transport activity observed for the protein mutants could not be attributed to different amounts of proteins in the bacterial membrane.
To assess whether the reduced transport activity of the tested PutP mutants could be specifically attributed to disrupted proline but not the coupling of Na+ and proline translocation, we measured the facilitated diffusion of 20 μm [3H]proline in the presence of the uncouplers monensin and CCCP (Fig. 8H). This established approach (31) allows for the assessment of the uncoupled substrate flux down its concentration gradient after dissipating the electrochemical Na+ and H+ gradients (ΔμNa+ and ΔμH+). Furthermore, to test whether the mutations affect proline transport through defects in Na+ binding, we performed the facilitated diffusion of proline also in the virtual absence of Na+ (in presence of 100 mm Tris/Mes, pH 6.0). Consistent with Na+-coupled proline transport and proline counterflow (Fig. 8, A and G), the initial rates of facilitated diffusion of [3H]proline down its concentration gradient by PutP-L398S, PutP-C344A, and PutP-Y248G were reduced by ∼90% compared with PutP-WT, whereas PutP-P252C showed an intermediate effect (∼50% reduction), regardless of whether the assay was performed in the presence or absence of Na+.
DISCUSSION
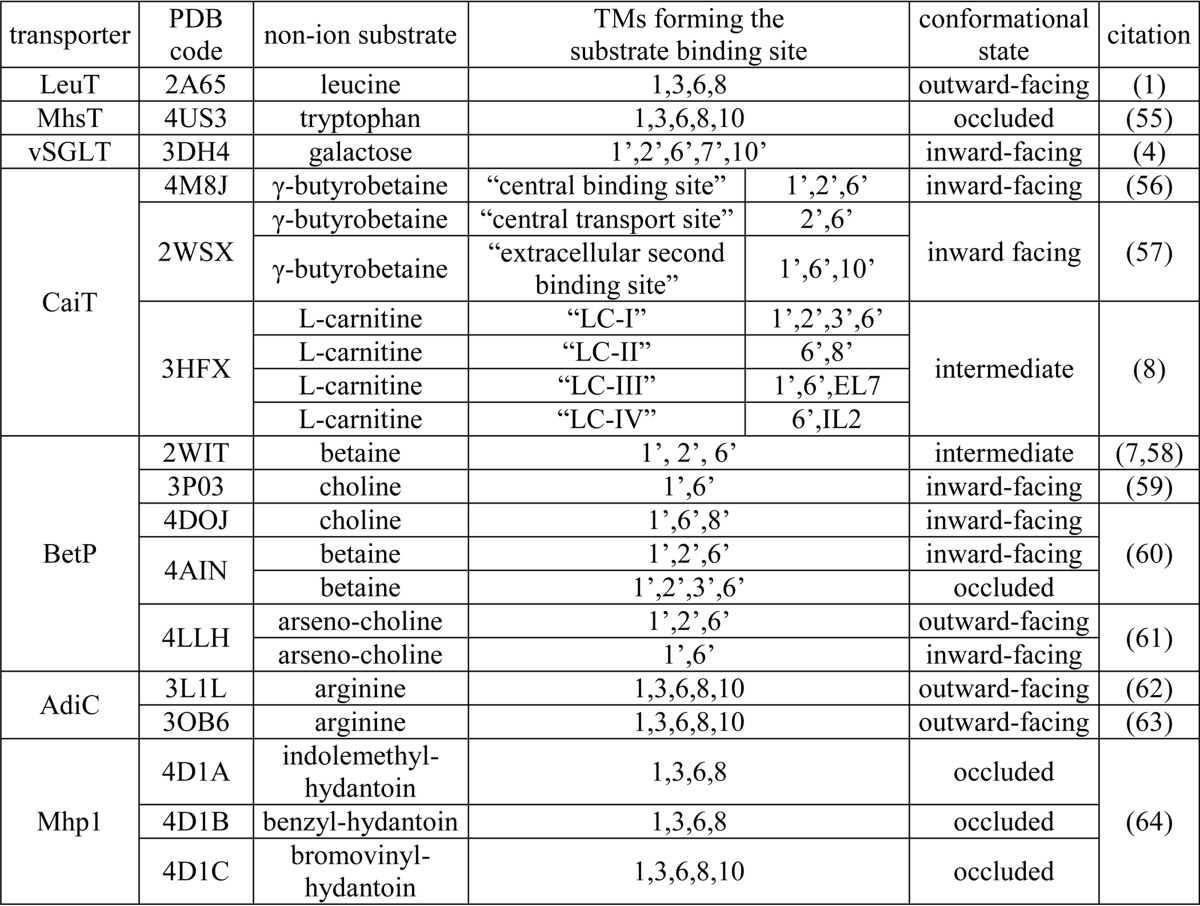
Among the crystal structures of transporters with the LeuT-like structural fold, the number and location(s) of the substrate-binding site(s) in each transporter family are not necessarily identical (Table 3). This raises the question whether such a divergence reflects unique mechanistic features in each family or whether previously unknown commonalities are yet to be revealed, as only certain conformational states for each transporter are readily amenable to crystallography. In this study, we evaluate whether under equilibrium conditions the SSS members vSGLT and PutP can bind substrate in an additional computationally identified site at a location similar to the S1 site of LeuT that is distinct from the galactose binding (Se) site identified in the crystal structure of vSGLT. Our modeling and simulation results indicate that the pocket below the conserved and functionally important tyrosine residue (Tyr-263 in vSGLT (3, 21) and Tyr-248 in PutP (45)) of TM6′, which is enclosed by TM1′, -3′, -6′, and -8′, may form such an additional substrate-binding site, termed the Sc site.
TABLE 3.
TMs forming the non-ion substrate binding sites in the representative crystal structures of transporters with LeuT-like structural fold
We consider that a TM is involved in forming a binding site when a heavy atom of any of its residues is within 4 Å of the bound substrate. Note, the nomenclature of the transporter conformational states are not consistently used among different groups. We largely classify the transporter conformations into outward-facing, inward-facing, intermediate, or occluded (“closed”) states.
The majority of the residues forming the Sc site of vSGLT as revealed in our simulations are well aligned to the S1 site residues in LeuT (Table 2). However, because the computational study is based on the inward-facing structure of vSGLT, in which the TMs that form the site are unlikely in their optimal positions to accommodate the Sc substrate, we expect the residue composition of the Sc site to be somewhat different in an inward-occluded state, in which the pocket is prone to binding of the Sc substrate. Such a dynamic formation of a binding site in different conformational states along the transport cycle has been observed previously in LeuT and is likely of mechanistic significance; the significant rearrangements of the intracellular portions of TMs from the inward-close to inward-open states of LeuT (PDB code 2A65 and 3TT3, respectively) (1, 18) lead to the release of substrate from the S1 site during the transition to the inward-open state.
Interestingly, recent simulations from Cheng and Bahar indicate that during LeuT's transitions toward the inward-facing state, the S2 substrate can stably bind right above the gating residue Phe-253 (which aligns to the Tyr-263 of vSGLT and Tyr-248 in PutP) with increased affinity (47). It is thus tempting to speculate that the vSGLT structure (PDB code 3DH4) captured a similar intermediate state with the Se site occupied but with the Sc site not in its optimal configuration to bind the substrate.
To experimentally validate our computational findings, we performed saturation binding experiments with known amounts of protein to determine the MBS. Consistent with our computational predictions, equilibrium dialysis-based saturation [3H]galactose binding confirmed that one molecule of vSGLT can simultaneously bind two galactose molecules under equilibrium conditions. An MBS of two was also reported for Leu and Ala binding to LeuT by us and others (24, 27).
In support of the existence of two substrate sites in vSGLT, individual mutations of residues located in either the Se or the Sc sites reduced the MBS of 2 to ∼1. Obtaining slightly larger than one MBS in those vSGLT mutants may indicate that the mutations did not entirely abolish substrate binding in a particular site but may reflect a severely reduced substrate interaction with the remaining binding residues in that site. From the macroscopic perspective, as the measured stoichiometry is an ensemble average, intermediate stoichiometry may reflect the partial loss of binding to a particular site in a fraction of the protein molecules. Interestingly, in our MD simulations, although the Sc and Se sites in vSGLT are separated by the gating residues Tyr-263 and Asn-64, we observed communication between the two substrate-binding sites. Thus, in the presence of Sc galactose, binding of Se galactose is weakened (with Asn-64, Trp-264, Phe-424, and Gln-428 interacting less frequently), and Se has a tendency to move upward, which demonstrates the dynamics a binding site undergoes during the transport cycle.
We then extended our studies to PutP, an experimentally tractable SSS model system. Similar to vSGLT, our simulations defined Sc- and Se-binding sites in PutP, which are separated by the gating residues Asp-55 and Tyr-248, two residues that have been shown to be functionally important (Table 4) (31, 45). In analogy with binding studies in vSGLT, equilibrium dialysis-based [3H]proline binding by purified PutP revealed an MBS of two as well. Fitting the experimental data to two individual single-site models revealed Kd values that are different by 1 order of magnitude for two sites. Similar to this observation, binding of Ala to LeuT exhibits a complex curve (24, 25) as we observed here for proline binding to PutP. Note that previous binding experiments involving PutP did not exceed radiolabeled proline concentrations of 20 μm, thereby missing the second component of the curve (38, 48). Mutation of Cys-344 or Leu-398, two residues that are located in the Sc and Se sites, respectively, reduced the proline-to-PutP MBS to about one. However, due to the probable complexity of the allosteric interaction of substrate binding to each site, including how binding to one site interferes with the interaction of substrate with the other site, the actual “affinity” of each of the sites may be difficult to determine experimentally.
TABLE 4.
Functional effect of mutations in the Sc and Se sites of SSS proteins
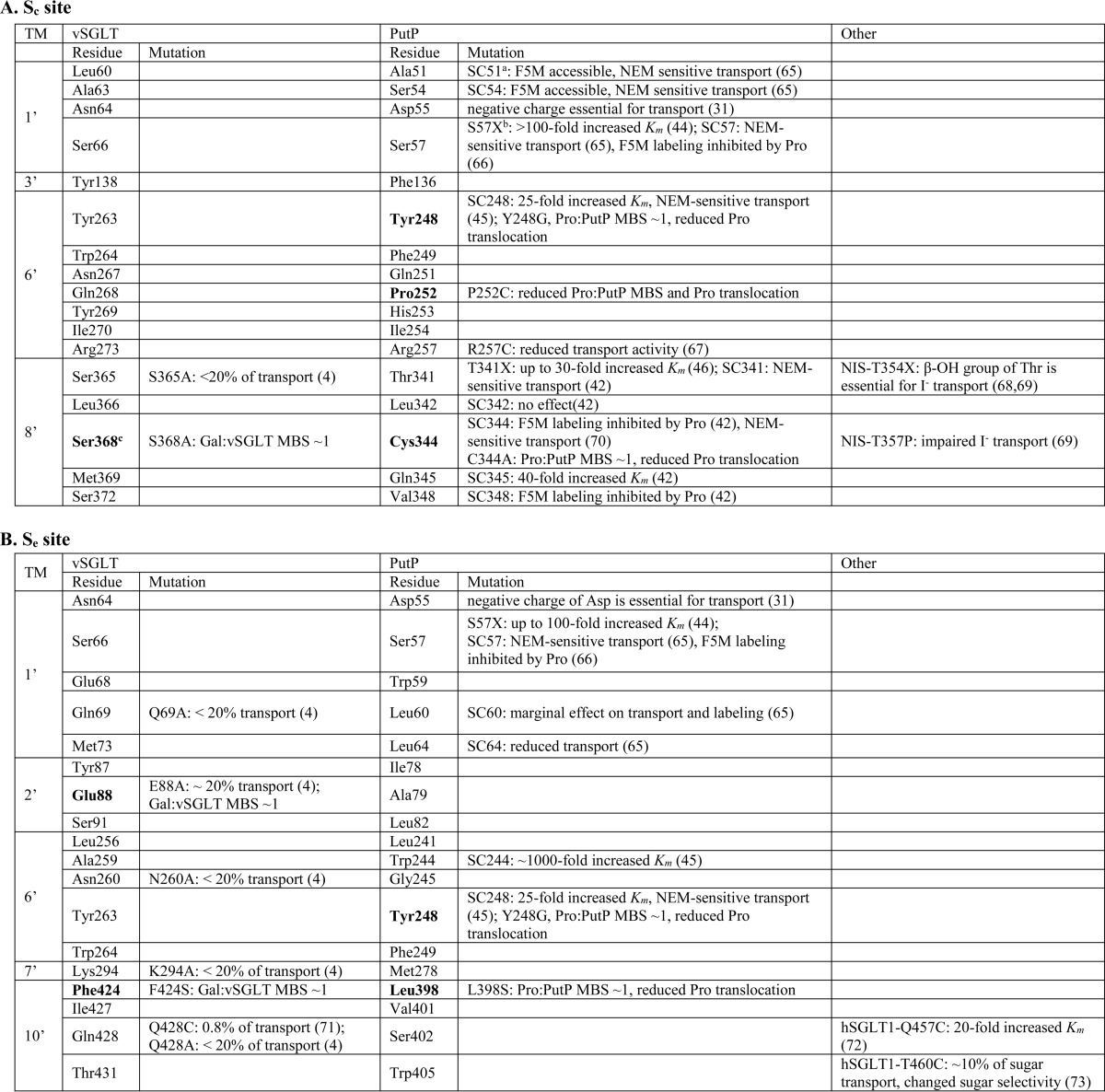
a SC followed by a number indicates the substitution of the native residue at the indicated position with a cysteine in a cysteine-free protein.
b X represents several replacements.
c Boldface type indicates the position studied in this work. Pro, proline; Gal, galactose; NEM, N-ethylmaleimide; F5M, fluorescein 5-maleimide.
Implicating the functional importance of two substrate-binding sites and their allosteric interaction for substrate transport, the reduction of the MBS from 2 to ∼1 (observed for PutP-Y248G, PutP-C344A, and PutP-L398S) is accompanied by a severe reduction of the proline translocation. This reduction cannot be attributed to defects in Na+ binding, as proline uniport (facilitated diffusion along the proline concentration gradient) in the absence of Na+ shows a similar trend observed with Na+-coupled or Na+-uncoupled transport modes. In contrast, differential effects on uniport in the absence or presence of Na+ have only been observed for mutants that were shown to disrupt Na+ binding directly (31). Replacing Pro-252 with Cys moderately disrupts the binding. Consistently, this mutant shows the smaller effect on all tested transport modes.
Reversibility of active transport has been reported more than 40 years ago (49) and has been generally accepted as a common feature of ion-coupled substrate transport according to which the polarity of transport depends on the direction of the concentration gradient of the transported ions and/or substrates (50). Whereas previous transport measurements of PutP inversely inserted into the membrane of proteoliposomes have revealed its reversibility (51), our counterflow results provide the basis for a detailed analysis of the role of both substrate sites in the reverse transport PutP.
Taken together, our results highlight the functional importance of two substrate-binding sites in vSGLT and PutP, which may be representatives for the other members of the SSS family (Table 4). The location of the Sc site (similar to that of the S1 site in LeuT) brings the substrate and Na+ binding into close proximity, an observation with wide ramifications for future comparative studies that aim to identify the mechanistic elements of Na+-coupled substrate transport by SSS that are either unique or common to other transporters with the LeuT-fold.
Acknowledgments
Computations were performed on Ranger at the Texas Advanced Computing Center (National Science Foundation Grant TG-MCB090022) and the David A. Cofrin computational infrastructure of the Institute for Computational Biomedicine at Weill Cornell Medical College. We thank Drs. Jonathan Javitch and Harel Weinstein for helpful discussions, MiEstrella Miller-Cruz and Trang Nguyen for technical assistance, and Dr. Ernest Wright for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants DA023694 (to L. S.), GM078844 (to J. A.), and DA017293 (to M. Q.). This work was also supported by National Science Foundation Grant MCB1330730 (to M. Q.).
- SSS
- solute-sodium symporter
- vSGLT
- sodium/galactose transporter from V. parahaemolyticus
- PDB
- Protein Data Bank
- CCCP
- carbonyl cyanide p-chlorophenylhydrazone
- MBS
- molar binding stoichiometry
- IFD
- induced-fit docking
- MD
- molecular dynamics
- SPA
- scintillation proximity assay
- TM
- transmembrane
- EV
- extracellular vestibule
- COM
- center-of-mass
- MM/GBSA
- molecular mechanics/generalized born surface area.
REFERENCES
- 1. Yamashita A., Singh S. K., Kawate T., Jin Y., Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 2. Wong F. H., Chen J. S., Reddy V., Day J. L., Shlykov M. A., Wakabayashi S. T., Saier M. H., Jr. (2012) The amino acid-polyamine-organocation superfamily. J. Mol. Microbiol. Biotechnol. 22, 105–113 [DOI] [PubMed] [Google Scholar]
- 3. Watanabe A., Choe S., Chaptal V., Rosenberg J. M., Wright E. M., Grabe M., Abramson J. (2010) The mechanism of sodium and substrate release from the binding pocket of vSGLT. Nature 468, 988–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Faham S., Watanabe A., Besserer G. M., Cascio D., Specht A., Hirayama B. A., Wright E. M., Abramson J. (2008) The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science 321, 810–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weyand S., Shimamura T., Yajima S., Suzuki S., Mirza O., Krusong K., Carpenter E. P., Rutherford N. G., Hadden J. M., O'Reilly J., Ma P., Saidijam M., Patching S. G., Hope R. J., Norbertczak H. T., Roach P. C., Iwata S., Henderson P. J., Cameron A. D. (2008) Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science 322, 709–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shimamura T., Weyand S., Beckstein O., Rutherford N. G., Hadden J. M., Sharples D., Sansom M. S., Iwata S., Henderson P. J., Cameron A. D. (2010) Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science 328, 470–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ressl S., Terwisscha van Scheltinga A. C., Vonrhein C., Ott V., Ziegler C. (2009) Molecular basis of transport and regulation in the Na+/betaine symporter BetP. Nature 458, 47–52 [DOI] [PubMed] [Google Scholar]
- 8. Tang L., Bai L., Wang W. H., Jiang T. (2010) Crystal structure of the carnitine transporter and insights into the antiport mechanism. Nat. Struct. Mol. Biol. 17, 492–496 [DOI] [PubMed] [Google Scholar]
- 9. Schulze J. M., Wang A. Y., Kobor M. S. (2010) Reading chromatin: insights from yeast into YEATS domain structure and function. Epigenetics 5, 573–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shaffer P. L., Goehring A., Shankaranarayanan A., Gouaux E. (2009) Structure and mechanism of a Na+-independent amino acid transporter. Science 325, 1010–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krishnamurthy H., Piscitelli C. L., Gouaux E. (2009) Unlocking the molecular secrets of sodium-coupled transporters. Nature 459, 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abramson J., Wright E. M. (2009) Structure and function of Na+-symporters with inverted repeats. Curr. Opin. Struct. Biol. 19, 425–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wright E. M., Loo D. D., Hirayama B. A. (2011) Biology of human sodium glucose transporters. Physiol. Rev. 91, 733–794 [DOI] [PubMed] [Google Scholar]
- 14. Penheiter A. R., Russell S. J., Carlson S. K. (2012) The sodium iodide symporter (NIS) as an imaging reporter for gene, viral, and cell-based therapies. Curr. Gene Ther. 12, 33–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Singh S. K., Yamashita A., Gouaux E. (2007) Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature 448, 952–956 [DOI] [PubMed] [Google Scholar]
- 16. Singh S. K., Piscitelli C. L., Yamashita A., Gouaux E. (2008) A competitive inhibitor traps LeuT in an open-to-out conformation. Science 322, 1655–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang H., Elferich J., Gouaux E. (2012) Structures of LeuT in bicelles define conformation and substrate binding in a membrane-like context. Nat. Struct. Mol. Biol. 19, 212–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krishnamurthy H., Gouaux E. (2012) X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481, 469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li J., Tajkhorshid E. (2009) Ion-releasing state of a secondary membrane transporter. Biophys. J. 97, L29–L31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mazier S., Quick M., Shi L. (2011) Conserved tyrosine in the first transmembrane segment of solute:sodium symporters is involved in Na+-coupled substrate co-transport. J. Biol. Chem. 286, 29347–29355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zomot E., Bahar I. (2010) The sodium/galactose symporter crystal structure is a dynamic, not so occluded state. Mol. Biosyst. 6, 1040–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li J., Tajkhorshid E. (2012) A gate-free pathway for substrate release from the inward-facing state of the Na+-galactose transporter. Biochim. Biophys. Acta 1818, 263–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao Y., Terry D., Shi L., Weinstein H., Blanchard S. C., Javitch J. A. (2010) Single-molecule dynamics of gating in a neurotransmitter transporter homologue. Nature 465, 188–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shi L., Quick M., Zhao Y., Weinstein H., Javitch J. A. (2008) The mechanism of a neurotransmitter:sodium symporter–inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol. Cell 30, 667–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao Y., Terry D. S., Shi L., Quick M., Weinstein H., Blanchard S. C., Javitch J. A. (2011) Substrate-modulated gating dynamics in a Na+-coupled neurotransmitter transporter homologue. Nature 474, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quick M., Winther A. M., Shi L., Nissen P., Weinstein H., Javitch J. A. (2009) Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor-bound conformation. Proc. Natl. Acad. Sci. U.S.A. 106, 5563–5568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nasr M. L., Singh S. K. (2014) Radioligand binding to nanodisc-reconstituted membrane transporters assessed by the scintillation proximity assay. Biochemistry 53, 4–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Piscitelli C. L., Krishnamurthy H., Gouaux E. (2010) Neurotransmitter/sodium symporter orthologue LeuT has a single high-affinity substrate site. Nature 468, 1129–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Quick M., Shi L., Zehnpfennig B., Weinstein H., Javitch J. A. (2012) Experimental conditions can obscure the second high-affinity site in LeuT. Nat. Struct. Mol. Biol. 19, 207–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Khelashvili G., LeVine M. V., Shi L., Quick M., Javitch J. A., Weinstein H. (2013) The membrane protein LeuT in micellar systems: aggregation dynamics and detergent binding to the S2 site. J. Am. Chem. Soc. 135, 14266–14275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quick M., Jung H. (1997) Aspartate 55 in the Na+/proline permease of Escherichia coli is essential for Na+-coupled proline uptake. Biochemistry 36, 4631–4636 [DOI] [PubMed] [Google Scholar]
- 32. Menke M., Berger B., Cowen L. (2008) Matt: local flexibility aids protein multiple structure alignment. PLoS Comput. Biol. 4, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sherman W., Day T., Jacobson M. P., Friesner R. A., Farid R. (2006) Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 49, 534–553 [DOI] [PubMed] [Google Scholar]
- 34. Bowers K. J., Chow E., Xu H., Dror R. O., Eastwood M. P., Gregersen B. A., Klepeis J. L., Kolossvary I., Moraes M. A., Sacerdoti F. D., Salmon J. K., Shan Y., Shaw D. E. (2006) in Proceedings of the ACM/IEEE SC 2006 Conference (SC'06), Tampa, FL, Nov. 11–17, 2006, pg. 43, IEEE, New York [Google Scholar]
- 35. Jorgensen W. L., Tirado-Rives J. (2005) Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. U.S.A. 102, 6665–6670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martyna G. J., Tobias D. J., Klein M. L. (1994) Constant pressure molecular dynamics algorithms. J. Chem. Phys. 101, 4177–4189 [Google Scholar]
- 37. Darden T., York D., Pedersen L. (1993) Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 38. Quick M., Jung H. (1998) A conserved aspartate residue, Asp187, is important for Na+-dependent proline binding and transport by the Na+/proline transporter of Escherichia coli. Biochemistry 37, 13800–13806 [DOI] [PubMed] [Google Scholar]
- 39. Quick M., Javitch J. A. (2007) Monitoring the function of membrane transport proteins in detergent-solubilized form. Proc. Natl. Acad. Sci. U.S.A. 104, 3603–3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schaffner W., Weissmann C. (1973) A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal. Biochem. 56, 502–514 [DOI] [PubMed] [Google Scholar]
- 41. Zhou Z., Zhen J., Karpowich N. K., Law C. J., Reith M. E., Wang D. N. (2009) Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat. Struct. Mol. Biol. 16, 652–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Raba M., Baumgartner T., Hilger D., Klempahn K., Härtel T., Jung K., Jung H. (2008) Function of transmembrane domain IX in the Na+/proline transporter PutP. J. Mol. Biol. 382, 884–893 [DOI] [PubMed] [Google Scholar]
- 43. Jung H., Rübenhagen R., Tebbe S., Leifker K., Tholema N., Quick M., Schmid R. (1998) Topology of the Na+/proline transporter of Escherichia coli. J. Biol. Chem. 273, 26400–26407 [DOI] [PubMed] [Google Scholar]
- 44. Quick M., Tebbe S., Jung H. (1996) Ser57 in the Na+/proline permease of Escherichia coli is critical for high-affinity proline uptake. Eur. J. Biochem. 239, 732–736 [DOI] [PubMed] [Google Scholar]
- 45. Olkhova E., Raba M., Bracher S., Hilger D., Jung H. (2011) Homology model of the Na+/proline transporter PutP of Escherichia coli and its functional implications. J. Mol. Biol. 406, 59–74 [DOI] [PubMed] [Google Scholar]
- 46. Hilger D., Böhm M., Hackmann A., Jung H. (2008) Role of Ser-340 and Thr-341 in transmembrane domain IX of the Na+/proline transporter PutP of Escherichia coli in ligand binding and transport. J. Biol. Chem. 283, 4921–4929 [DOI] [PubMed] [Google Scholar]
- 47. Cheng M. H., Bahar I. (2013) Coupled global and local changes direct substrate translocation by neurotransmitter-sodium symporter ortholog LeuT. Biophys. J. 105, 630–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mogi T., Anraku Y. (1984) Mechanism of proline transport in Escherichia coli K12. II. Effect of alkaline cations on binding of proline to a H+/proline symport carrier in cytoplasmic membrane vesicles. J. Biol. Chem. 259, 7797–7801 [PubMed] [Google Scholar]
- 49. Garrahan P. J., Glynn I. M. (1967) The incorporation of inorganic phosphate into adenosine triphosphate by reversal of the sodium pump. J. Physiol. 192, 237–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaback H. R., Sahin-Tóth M., Weinglass A. B. (2001) The kamikaze approach to membrane transport. Nat. Rev. Mol. Cell Biol. 2, 610–620 [DOI] [PubMed] [Google Scholar]
- 51. Jung H., Tebbe S., Schmid R., Jung K. (1998) Unidirectional reconstitution and characterization of purified Na+/proline transporter of Escherichia coli. Biochemistry 37, 11083–11088 [DOI] [PubMed] [Google Scholar]
- 52. Beuming T., Shi L., Javitch J. A., Weinstein H. (2006) A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol. Pharmacol. 70, 1630–1642 [DOI] [PubMed] [Google Scholar]
- 53. Lomize M. A., Lomize A. L., Pogozheva I. D., Mosberg H. I. (2006) OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625 [DOI] [PubMed] [Google Scholar]
- 54. Laskowski R. A., Swindells M. B. (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 [DOI] [PubMed] [Google Scholar]
- 55. Malinauskaite L., Quick M., Reinhard L., Lyons J. A., Yano H., Javitch J. A., Nissen P. (2014) A mechanism for intracellular release of Na+ by neurotransmitter/sodium symporters. Nat. Struct. Mol. Biol. 21, 1006–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kalayil S., Schulze S., Kühlbrandt W. (2013) Arginine oscillation explains Na+ independence in the substrate/product antiporter CaiT. Proc. Natl. Acad. Sci. U.S.A. 110, 17296–17301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schulze S., Köster S., Geldmacher U., Terwisscha van Scheltinga A. C., Kühlbrandt W. (2010) Structural basis of Na+-independent and cooperative substrate/product antiport in CaiT. Nature 467, 233–236 [DOI] [PubMed] [Google Scholar]
- 58. Korkmaz F., Ressl S., Ziegler C., Mäntele W. (2013) K+-induced conformational changes in the trimeric betaine transporter BetP monitored by ATR-FTIR spectroscopy. Biochim. Biophys. Acta 1828, 1181–1191 [DOI] [PubMed] [Google Scholar]
- 59. Perez C., Koshy C., Ressl S., Nicklisch S., Krämer R., Ziegler C. (2011) Substrate specificity and ion coupling in the Na+/betaine symporter BetP. EMBO J. 30, 1221–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Perez C., Koshy C., Yildiz O., Ziegler C. (2012) Alternating-access mechanism in conformationally asymmetric trimers of the betaine transporter BetP. Nature 490, 126–130 [DOI] [PubMed] [Google Scholar]
- 61. Perez C., Faust B., Mehdipour A. R., Francesconi K. A., Forrest L. R., Ziegler C. (2014) Substrate-bound outward-open state of the betaine transporter BetP provides insights into Na+ coupling. Nat. Commun. 5, 4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gao X., Zhou L., Jiao X., Lu F., Yan C., Zeng X., Wang J., Shi Y. (2010) Mechanism of substrate recognition and transport by an amino acid antiporter. Nature 463, 828–832 [DOI] [PubMed] [Google Scholar]
- 63. Kowalczyk L., Ratera M., Paladino A., Bartoccioni P., Errasti-Murugarren E., Valencia E., Portella G., Bial S., Zorzano A., Fita I., Orozco M., Carpena X., Vázquez-Ibar J. L., Palacín M. (2011) Molecular basis of substrate-induced permeation by an amino acid antiporter. Proc. Natl. Acad. Sci. U.S.A. 108, 3935–3940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Simmons K. J., Jackson S. M., Brueckner F., Patching S. G., Beckstein O., Ivanova E., Geng T., Weyand S., Drew D., Lanigan J., Sharples D. J., Sansom M. S., Iwata S., Fishwick C. W., Johnson A. P., Cameron A. D., Henderson P. J. (2014) Molecular mechanism of ligand recognition by membrane transport protein, Mhp1. EMBO J. 33, 1831–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pirch T., Landmeier S., Jung H. (2003) Transmembrane domain II of the Na+/proline transporter PutP of Escherichia coli forms part of a conformationally flexible, cytoplasmic exposed aqueous cavity within the membrane. J. Biol. Chem. 278, 42942–42949 [DOI] [PubMed] [Google Scholar]
- 66. Pirch T., Quick M., Nietschke M., Langkamp M., Jung H. (2002) Sites important for Na+ and substrate binding in the Na+/proline transporter of Escherichia coli, a member of the Na+/solute symporter family. J. Biol. Chem. 277, 8790–8796 [DOI] [PubMed] [Google Scholar]
- 67. Ohsawa M., Mogi T., Yamamoto H., Yamato I., Anraku Y. (1988) Proline carrier mutant of Escherichia coli K-12 with altered cation sensitivity of substrate-binding activity: cloning, biochemical characterization, and identification of the mutation. J. Bacteriol. 170, 5185–5191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pohlenz J., Refetoff S. (1999) Mutations in the sodium/iodide symporter (NIS) gene as a cause for iodide transport defects and congenital hypothyroidism. Biochimie 81, 469–476 [DOI] [PubMed] [Google Scholar]
- 69. De la Vieja A., Reed M. D., Ginter C. S., Carrasco N. (2007) Amino acid residues in transmembrane segment IX of the Na+/I− symporter play a role in its Na+ dependence and are critical for transport activity. J. Biol. Chem. 282, 25290–25298 [DOI] [PubMed] [Google Scholar]
- 70. Yamato I., Anraku Y. (1988) Site-specific alteration of cysteine 281, cysteine 344, and cysteine 349 in the proline carrier of Escherichia coli. J. Biol. Chem. 263, 16055–16057 [PubMed] [Google Scholar]
- 71. Xie Z., Turk E., Wright E. M. (2000) Characterization of the Vibrio parahaemolyticus Na+/glucose cotransporter. A bacterial member of the sodium/glucose transporter (SGLT) family. J. Biol. Chem. 275, 25959–25964 [DOI] [PubMed] [Google Scholar]
- 72. Díez-Sampedro A., Wright E. M., Hirayama B. A. (2001) Residue 457 controls sugar binding and transport in the Na+/glucose cotransporter. J. Biol. Chem. 276, 49188–49194 [DOI] [PubMed] [Google Scholar]
- 73. Hirayama B. A., Loo D. D., Díez-Sampedro A., Leung D. W., Meinild A. K., Lai-Bing M., Turk E., Wright E. M. (2007) Sodium-dependent reorganization of the sugar-binding site of SGLT1. Biochemistry 46, 13391–13406 [DOI] [PubMed] [Google Scholar]