

Background: STAT3 promotes UVB-induced keratinocyte cell proliferation.
Results: TC-PTP inhibits STAT3-mediated mouse keratinocyte skin cell proliferation and survival following UVB irradiation.
Conclusion: TC-PTP plays an important role in the skin cell response to UVB radiation by regulating cell proliferation and survival.
Significance: TC-PTP shows tumor-suppressive capabilities in skin.
Keywords: Cell Proliferation, Keratinocyte, Skin, STAT3, Tyrosine-protein Phosphatase (Tyrosine Phosphatase), UVB
Abstract
Chronic exposure to UV radiation can contribute to the development of skin cancer by promoting protein-tyrosine kinase (PTK) signaling. Studies show that exposure to UV radiation increases the ligand-independent activation of PTKs and induces protein-tyrosine phosphatase (PTP) inactivation. In the present work, we report that T-cell PTP (TC-PTP) activity is stimulated during the initial response to UVB irradiation, which leads to suppression of keratinocyte cell survival and proliferation via the down-regulation of STAT3 signaling. Our results show that TC-PTP-deficient keratinocyte cell lines expressed a significantly increased level of phosphorylated STAT3 after exposure to low dose UVB. This increase corresponded with increased cell proliferation in TC-PTP-deficient keratinocytes following UVB irradiation. Loss of TC-PTP also reduced UVB-induced apoptosis. Corroborating with these results, overexpression of TC-PTP in keratinocyte cell lines yielded a decrease in phosphorylated STAT3 levels, which corresponded with a significant decrease in cell proliferation in response to low dose UVB. We demonstrate that TC-PTP activity was increased upon UVB exposure, and overexpression of TC-PTP in keratinocyte cell lines further increased its activity in the presence of UVB. Treatment of TC-PTP-deficient keratinocytes with the STAT3 inhibitor STA21 significantly reduced cell viability following UVB exposure in comparison with untreated TC-PTP-deficient keratinocytes, confirming that the effect of TC-PTP on cell viability is mediated by STAT3 dephosphorylation. Combined, our results indicate that UVB-mediated activation of TC-PTP plays an important role in the STAT3-dependent regulation of keratinocyte cell proliferation and survival. Furthermore, these results suggest that TC-PTP may be a novel potential target for the prevention of UVB-induced skin cancer.
Introduction
Phosphotyrosine-based signaling is one essential mechanism in cellular homeostasis. The phosphorylation of proteins at specific tyrosine residues is catalyzed by protein tyrosine kinases (PTKs)2 (1–3). PTKs regulate various cellular processes, including growth, differentiation, metabolism, and motility. Genetic mutations of PTKs and/or aberrant activation of their signaling pathways can lead to an array of human diseases, including cancer (4, 5). PTKs are regulated by endogenous negative feedback mechanisms involving protein-tyrosine phosphatases (PTPs), which negatively regulate the rate and duration of phosphotyrosine signaling (4, 6–8). One substrate of PTKs and PTPs is STAT3 (signal transducer and activator of transcription 3), a member of a family of cytoplasmic proteins that functions as a transcription factor in normal cellular responses to cytokines and growth factors (9–12). STAT3 modulates various physiological functions, including apoptosis, cell cycle regulation, and tumor angiogenesis, through regulation of gene expression, and its constitutive activation is associated with a number of human epithelial cancers (11, 13). Studies have shown that constitutive activation of STAT3 is found in a variety of human tumors and cancer cell lines, including human epithelial cancers, and its inhibition can suppress the growth of cancer cells, suggesting that STAT3 plays a critical role in cancer cell proliferation (9, 10, 14, 15). More importantly, recent studies have shown that activation of STAT3 might be a predominant factor in the development of skin tumors induced by UVB or chemical carcinogen (16–23).
T-cell protein tyrosine phosphatase (TC-PTP; encoded by PTPN2) was one of the first members of the PTP gene family to be identified. TC-PTP is one of 17 intracellular, non-receptor PTPs that is ubiquitously expressed in embryonic and adult tissues, although it is highly expressed in hematopoietic tissues (24, 25). There are two forms of TC-PTP that are generated by alternative splicing at the 3′ end of the gene: TC45 (TC-PTPa) and TC48 (TC-PTPb). TC45 (45 kDa) is the major form of TC-PTP in most species, including humans and mice. TC45 contains a bipartite nuclear localization signal and is found primarily in the nucleus, whereas TC48 (48 kDa) contains a hydrophobic C terminus and is localized to the endoplasmic reticulum. In mouse tissue, almost all TC-PTP mRNA encodes TC45; TC48 mRNA is not detectable by Northern blot analysis (26–28). Substrates targeted by TC-PTP include JAK1, JAK3, STAT1, STAT3, and STAT5, suggesting that it negatively regulates the JAK/STAT signaling pathway (29, 30). Recent studies showed that STAT3 is dephosphorylated in keratinocytes in response to UVB irradiation, and treatment with sodium vanadate (a pan-PTP inhibitor) recovered the level of phosphorylated STAT3 (22). Furthermore, studies demonstrated that TC-PTP, SHP1, and SHP2 cooperatively dephosphorylate STAT3 following UVB irradiation in skin (31).
Based on previous work (22, 31), we suggest that exposure to UVB radiation triggers PTP activation in skin, which attenuates STAT3 signaling by dephosphorylating STAT3. PTP activation plays an important role in the initial skin cell response to UVB that protects cells from the increase in phosphorylated STAT3 that could lead to hyperproliferation and cancer. Skin cells are more susceptible to UVB-induced cellular proliferation and survival without the activation of PTP. In the present study, we examined the impact of activation of PTP, especially TC-PTP, in the regulation of UVB-induced cell proliferation and survival using mouse keratinocyte cell lines.
MATERIALS AND METHODS
Cell Culture and UVB Irradiation
3PC, an immortalized mouse keratinocyte cell line (32), was cultured in keratinocyte growth medium containing 1% fetal bovine serum to 80–85% confluence, at which time cells were washed with DPBS. A small volume of DPBS was added to coat the attached cells with a thin layer of DPBS, and then keratinocyte cell lines were irradiated with UVB. Immediately after irradiation, DPBS was removed, and prewarmed medium was added to cells for additional culture before harvest.
RNA Interference (RNAi)
3PC cells were grown overnight to ∼40% confluence and transiently transfected with ON-TARGETplusTM mouse TC-PTP (PTPN2), SHP1 (PTPN6), or SHP2 (PTPN11) short interfering RNA (siRNA) and ON-TARGETplusTM siCONTROL® non-targeting pool siRNA as control (Thermo Scientific Dharmacon, Lafayette, CO). Transfection was performed with Lipofectamine® RNAiMAX (Invitrogen) according to the manufacturer's instructions.
Establishment of TC-PTP-deficient Stable Cell Lines
TC-PTP-deficient 3PC cell lines were established by lentiviral transduction with MISSION® shRNA constructs specific for mouse TC-PTP (PTPN2) (Sigma-Aldrich). In brief, 293T packaging cells were co-transfected with lentiviral shRNA constructs and lentiviral helper vectors (gag/pol packaging vector and VSV-G envelope vector). After a 48 h incubation, the viral supernatant fraction was collected and filter-sterilized. 3PC cells were infected with lentiviral particles for 24 h and selected with puromycin over a 3-day period.
Generation of TC-PTP-expressing Adenoviral Vector and Transient Expression
Mouse keratinocyte cell lines overexpressing TC-PTP (referred to as Ad/TC-PTP) were established using the AdEasyTM adenoviral vector system (Agilent Technologies, Santa Clara, CA). Briefly, wild-type mouse TC-PTP (45 kDa) cDNA from pcDNA4/myc-His-TC45-WT (31) was amplified by PCR and cloned into the pCMV6/AN/HA vector (Origene, Rockville, MD). TC-PTP cDNA containing an N-terminal HA tag (HA.TC-PTP) was amplified by PCR and cloned into the pShuttle-CMV vector (Addgene, Cambridge, MA). Then the linearized recombinant plasmid was co-transformed with the pAdEasy-1 vector into Escherichia coli BJ5183 cells to generate the final adenovirus plasmid containing the HA.TC-PTP insert. Next, AD-293 packaging cells were transfected with the recombinant TC-PTP adenoviral vector or control vector. Finally, 3PC cells were transduced with the adenoviral particles for 24 h, followed by selection with puromycin.
Establishment of TC-PTP-overexpressing Stable Cell Lines
Stable mouse 3PC keratinocyte cell lines overexpressing TC-PTP (referred to as pEF/mTC45) were established by lentiviral transduction with a modified lentiviral system based on Gateway® cloning technology (Invitrogen) (33). Wild-type mouse TC-PTP (45 kDa) cDNA from pcDNA4/myc-His-TC45-WT (31) was amplified by PCR and cloned into the lentivirus entry vector pEF-1α/pENTR B (Addgene). Gateway® LR Clonase® enzyme mix (Invitrogen) was used to catalyze an LR recombination reaction between the pEF-1α/pENTR B/TC-PTP and pLenti X1 Puro DEST (Addgene) destination vector to generate the final lentiviral expression clone for TC-PTP. Finally, 3PC cells were infected with lentiviral particles from the TC-PTP lentiviral expression clone as described above.
Western Blot Analysis
Cells were resuspended with lysis buffer containing 40 mm Tris, pH 7.4, 120 mm NaCl, 10 mm EDTA, 0.1% (v/v) Nonidet P-40, protease inhibitor mixture, and phosphatase inhibitor mixtures 1 and 2 (Sigma-Aldrich). Equal amounts of total protein were resolved using SDS-PAGE and transferred to PVDF membrane (GE Healthcare). The membrane was incubated overnight at 4 °C with primary antibody, followed by incubation with a horseradish peroxidase-conjugated secondary antibody. Chemiluminescent detection reagents (GE Healthcare) were used to detect immunoreactive protein. The following antibodies were used: anti-phospho-STAT3 (Tyr-705), anti-STAT3, anti-SHP1, anti-SHP2; anti-cyclin D1; anti-PARP; anti-cleaved caspase-3 (Cell Signaling Technology, Inc., Beverly, MA), anti-TC-PTP (R&D Systems Inc., Minneapolis, MN), and anti-β-actin (Sigma-Aldrich). For densitometric analysis, ImageJ software (version 1.49e, National Institutes of Health) was used to measure the integrated optical density (IOD). Relative protein expression was calculated as IODexperimental/IODcontrol.
Caspase-3 Activity Assay
Caspase-3 activity was measured by the caspase-3/CPP32 colorimetric assay kit according to the manufacturer's instructions (Biovision Inc., Milpitas, CA). Briefly, after UVB irradiation, 3PC cells were resuspended with lysis buffer and incubated on ice at 4 °C for 10 min, followed by centrifugation at 12,000 × g for 10 min. The clarified cell lysate protein was mixed with DEVD-p-nitroanilide caspase-3-specific substrate in a 96-well plate and then incubated at 37 °C for 2 h. Absorbance at 405 nm was measured on the SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA).
STAT3 Activation Assay
STAT3 activation was measured using the colorimetric TransAM STAT3 transcription factor assay kit according to the manufacturer's instructions (Active Motif, Carlsbad, CA). Briefly, 3PC cell nuclear protein extract was incubated for 1 h in a 96-well plate coated with an oligonucleotide containing the STAT consensus binding site. STAT3 was detected by incubation with anti-STAT3 primary antibody followed by incubation with a horseradish peroxidase-conjugated secondary antibody. Absorbance at 450 nm was measured, and data were normalized to untreated control. Nuclear protein fractions were prepared from 3PC keratinocyte cell lines using NE-PER® nuclear and cytoplasmic extraction reagents according to the manufacturer's instructions (Pierce).
TC-PTP Activity Assay
TC-PTP activity was measured using the Duoset® IC Human/Mouse/Rat Active TC-PTP activity assay kit (R&D Systems) according to the manufacturer's instructions. Briefly, protein extract was incubated in a 96-well plate coated with a capture antibody specific for TC-PTP at 4 °C for 3 h. After washing twice, tyrosine phosphatase substrates (200 μm) were added for the dephosphorylation reaction catalyzed by TC-PTP. The level of free phosphate was determined by a sensitive dye-binding assay using malachite green and molybdic acid, followed by measurement of the absorbance at 620 nm.
Cell Proliferation Assay
3PC keratinocyte cell proliferation was assessed using a 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-1) (Dojindo Laboratories, Kumamoto, Japan) assay. Briefly, 3PC cells were plated at a density of 5 × 103 cells/well in 96-well cell culture plates and incubated overnight. Then cells were irradiated with UVB as described above and recultured for 24–72 h. After cultivation, a 1:100 dilution of WST-1 solution was added to each well and incubated for 2 h. Absorbance at 450–690 nm was measured.
Lactate dehydrogenase Cytotoxicity Assay
Cytotoxicity of STA-21, a STAT3-specific inhibitor, on 3PC cells was assessed using the Pierce lactate dehydrogenase cytotoxicity assay kit (Thermo Scientific). Briefly, 3PC cells were plated at a density of 5 × 103 cells/well in a 96-well cell culture plate, and then the cells were treated with 0–50 μm STA-21 for 18 h. Lactate dehydrogenase activity in the medium was quantified by measuring the absorbance at 490 and 680 nm.
RESULTS
TC-PTP Has a Major Effect on STAT3 Regulation in Skin Keratinocytes
In our previous work, we demonstrated that PTPs are required to regulate STAT3 activation in mouse 3PC keratinocyte cells. Specifically, three PTPs (TC-PTP and the Src homology 2 domain-containing PTPs, SHP1 and SHP2) were shown to cooperate in the dephosphorylation of STAT3 that occurs following exposure to UVB radiation (31); therefore, we further investigated the role(s) of TC-PTP, SHP1, and SHP2 in the regulation of STAT3 signaling in 3PC cell lines. As shown in Fig. 1A (see also supplemental Fig. 1 and 2), the level of phosphorylated STAT3 in keratinocyte cells transiently transfected with TC-PTP (PTPN2), SHP1 (PTPN6), or SHP2 (PTPN11) siRNA was increased compared with cells transiently transfected with scrambled siRNA. Interestingly, the level of phosphorylated STAT3 in TC-PTP knockdown was higher relative to SHP1 knockdown or SHP2 knockdown (Fig. 1A), which implies that TC-PTP has a greater effect on STAT3 dephosphorylation than the other two PTPs. Knockdown of TC-PTP by transient siRNA transfection steadily decreased TC-PTP expression 1–5 days after transfection, and then expression levels recovered due to loss of siRNAs by regular cell division. The level of phosphorylated STAT3 expression increased with the decrease in TC-PTP expression and then decreased with the recovery of TC-PTP expression at a later time. Similarly, the level of the cell cycle regulator cyclin D1, a downstream target of STAT3, also increased and then decreased in correspondence with TC-PTP levels (Fig. 1B), suggesting that TC-PTP can play a critical role in the regulation of STAT3 signaling in keratinocyte. To further investigate whether TC-PTP has a major effect on STAT3 dephosphorylation in response to UVB irradiation, 3PC cells were transiently transfected with PTP-specific siRNA or scrambled control siRNA and then treated with UVB (80 mJ/cm2) (Fig. 1C and supplemental Fig. 3). Following UVB irradiation, STAT3 was rapidly dephosphorylated; however, knockdown of TC-PTP significantly inhibited this event. Knockdown of SHP1 or SHP2 had little to no effect (Fig. 1C and supplemental Fig. 4). These results demonstrate for the first time that of three PTPs that were identified to participate in STAT3 dephosphorylation in mouse keratinocytes, TC-PTP has a major impact on the regulation of STAT3 signaling in the presence or absence of UVB radiation.
FIGURE 1.

TC-PTP is a major PTP in UVB-induced STAT3 dephosphorylation in keratinocytes among three PTPs. A, 3PC keratinocytes were cultured and transfected with siRNA specific for TC-PTP, SHP1, or SHP2. Cells were collected after 48 h of siRNA transfection. Total cell lysates were resolved by SDS-PAGE and immunoblotted with antibody specific for phosphorylated STAT3 (PY-STAT3). B, 3PC keratinocytes were cultured and transfected with siRNA specific for TC-PTP. Cells were collected at the indicated time. C, 3PC keratinocytes were cultured and transfected with siRNA specific for TC-PTP, SHP1, or SHP2. Cells were irradiated with UVB at a dose of 80 mJ/cm2 and then collected 1 h after irradiation.
Loss of TC-PTP in Keratinocytes Inhibits the Dephosphorylation of STAT3 That Occurs in Response to Low Dose UVB Irradiation
High dose UVB irradiation triggered the rapid dephosphorylation of STAT3 by PTPs, especially TC-PTP, in mouse keratinocyte cell lines (see Fig. 1C) (31). In mouse epidermis, the level of phosphorylated STAT3 initially decreased upon exposure to UVB, and then it increased significantly at later time points (31), so we utilized low dose UVB irradiation to investigate the long term effect of TC-PTP on the STAT3 response to UVB irradiation. After exposure to low dose UVB light, the level of STAT3 phosphorylation in 3PC cells also quickly decreased, and then it was restored 6 h after UVB irradiation (Fig. 2A), which is similar to previous in vivo results in mouse epidermis (31). However, transient knockdown of TC-PTP with siRNA allowed for a more rapid recovery of STAT3 phosphorylation 6–24 h after UVB exposure compared with control cells. In particular, expression of phosphorylated STAT3 is higher in TC-PTP-deficient cells 24 h after UVB irradiation, at which time expression increased almost 8-fold in comparison with untreated control cells (Fig. 2B). The results suggest that TC-PTP plays a critical role in the regulation of the STAT3 response to UVB irradiation.
FIGURE 2.

TC-PTP deficiency on STAT3 dephosphorylation in keratinocytes following low dose of UVB irradiation. A, representative Western blot analysis of phosphorylated STAT3 (PY-STAT3) in total cell lysates. 3PC keratinocytes were cultured and transfected with control or TC-PTP siRNA. Cells were collected at the indicated time after UVB irradiation (10 mJ/cm2). B, relative levels of phosphorylated STAT3 were quantified by densitometry. Results are the mean ± S.D. (error bars) from three independent experiments. *, p < 0.05 by t test for equality of means.
TC-PTP Deficiency Induces Keratinocyte Proliferation following Low Dose UVB Exposure
Studies have shown that STAT3 contributes to UVB-induced skin tumorigenesis through its role in keratinocyte cell survival and proliferation (19). Given that TC-PTP has the ability to negatively regulate STAT3 through dephosphorylation following low dose UVB, we studied the impact of TC-PTP on mouse keratinocyte proliferation and survival in the presence or absence of low dose UVB radiation. In the absence of UVB, transient knockdown of TC-PTP in 3PC cells using siRNA resulted in a significant increase in cell proliferation compared with cells transiently transfected with scrambled control siRNA after 48 h of culturing (Fig. 3A). In the presence of low dose UVB radiation, we observed an initial decrease in cell number in both control and TC-PTP-deficient cells due to UVB damage (Fig. 3, B–D). Upon exposure to 5 or 10 mJ/cm2 of UVB, initial UVB-induced damage was severe, so keratinocyte cell growth did not recover until 48 h after exposure (Fig. 3, C and D), yet knockdown of TC-PTP considerably increased cell proliferation following UVB irradiation (Fig. 3, B–D). After irradiation with 2 mJ/cm2 of UVB, the number of TC-PTP-deficient cells was significantly higher than control cells 48 and 72 h after exposure (Fig. 3B). The number of TC-PTP-deficient cells was significantly higher than control cells 24, 48, and 72 h after a dose of 5 mJ/cm2 UVB (Fig. 3C). Cell proliferation significantly increased in TC-PTP-deficient cells compared with control cells 72 h after irradiation with 10 mJ/cm2 of UVB (Fig. 3D).
FIGURE 3.

Knockdown of TC-PTP on the regulation of keratinocyte proliferation after low dose of UVB irradiation. A–D, 3PC keratinocytes were cultured and transfected with control or TC-PTP siRNA for 24 h. Cell viability was measured using the WST-1 assay. Results are the mean ± S.D. (error bars) from three independent experiments. *, p < 0.05 by Student's t test for equality of means. A, cells were collected at 24 and 48 h after transfection. B–D, cells were irradiated with UVB and then collected 24, 48, and 72 h after irradiation. B, 2 mJ/cm2; C, 5 mJ/cm2; D, 10 mJ/cm2. E, cells were transfected with siRNA specific for TC-PTP, SHP1, or SHP2. Cells were collected at 24, 48, and 72 h after UVB irradiation at a dose of 2 mJ/cm2.
We showed that knockdown of TC-PTP in mouse keratinocytes had a greater inhibitory effect on STAT3 dephosphorylation following UVB irradiation than knockdown of either SHP1 or SHP2 (Fig. 1C). To determine whether the same effect applied to cell proliferation following UVB exposure, 3PC cells were transiently transfected with PTP-specific siRNA or scrambled siRNA and then treated with low dose UVB (Fig. 3E). Although knockdown of TC-PTP showed a significant increase in cell proliferation following UVB, knockdown of either SHP1 or SHP2 had no effect on keratinocyte cell proliferation compared with control cells (Fig. 3E), which provides further evidence that TC-PTP is the major PTP involved in the regulation of STAT3 signaling in response to UVB radiation.
Use of stable 3PC keratinocyte cell lines that are deficient in TC-PTP also demonstrated that loss of TC-PTP significantly increases cell proliferation in the presence or absence of UVB radiation (Fig. 4). We were able to generate two keratinocyte cell lines by lentiviral transduction of shRNA (hereafter referred to as TC-PTP shRNA 1 and TC-PTP shRNA 2); TC-PTP shRNA 1 cells showed a partial silencing of TC-PTP expression, and TC-PTP shRNA 2 cells showed a complete loss of TC-PTP expression (Fig. 4A). The level of phosphorylated STAT3 was increased in TC-PTP shRNA 1 cells and TC-PTP shRNA 2 cells in comparison with control. Furthermore, the level of phosphorylated STAT3 was higher in TC-PTP shRNA 2 cells than TC-PTP shRNA 1 cells (Fig. 4, A and B). The levels of cyclin D1 and c-Myc were increased in correlation with decreased TC-PTP and increased STAT3 in TC-PTP shRNA 1 and 2 cells (Fig. 4A). Consistent with this observation, cell proliferation was significantly increased in both TC-PTP-deficient cell lines compared with control cells after 48 h of culturing, and cell proliferation was higher in TC-PTP shRNA 2 cells than in TC-PTP shRNA 1 cells (Fig. 4C). Upon exposure to low dose UVB, cell proliferation increased in both TC-PTP-deficient cell lines 24–72 h after irradiation, and again, cell proliferation was higher in TC-PTP shRNA 2 cells in comparison with TC-PTP shRNA 1 cells (Fig. 4D). In particular, the complete loss of TC-PTP in TC-PTP shRNA 2 cells significantly minimized the reduction in cell number due to UVB-induced DNA damage at 24 h. Together, the results indicate that TC-PTP is required to negatively regulate keratinocyte cell proliferation.
FIGURE 4.

Stable knockdown of TC-PTP with shRNA on the regulation of keratinocyte survival and proliferation after low dose of UVB irradiation. A, generation of TC-PTP-deficient 3PC cell lines with mouse TC-PTP shRNA. B, relative levels of phosphorylated STAT3 were quantified by densitometry. Results are the mean ± S.D. (error bars) from three independent experiments. *, p < 0.05 by Student's t test for equality of means. C and D, control (Scrambled) or TC-PTP-deficient cells were cultured. Cell viability was measured using the WST-1 assay. Results are the mean ± S.D. from three independent experiments. *, p < 0.05 by Student's t test for equality of means. C, cells were collected at 24 and 48 h without UVB irradiation. D, cells were irradiated with UVB at a dose of 2 mJ/cm2 and then collected 24, 48, and 72 h after irradiation.
TC-PTP Overexpression Inhibits Keratinocyte Cell Proliferation following Low Dose UVB Exposure
To further investigate the potential role of TC-PTP in mouse keratinocyte cell proliferation, TC-PTP was overexpressed in 3PC cells by either transient or stable expression (Fig. 5). Transient overexpression of TC-PTP in 3PC cells using adenovirus significantly reduced the level of phosphorylated STAT3 compared with control cells (Fig. 5A). Keratinocyte cells overexpressing TC-PTP showed a significant decrease in cell proliferation relative to control cells after 24 and 48 h of culturing (Fig. 5B). After irradiation with 10 mJ/cm2 UVB, UVB-induced apoptosis reduced the number of cells in both control and transient TC-PTP-overexpressing 3PC cells 72 h after UVB exposure; however, cells overexpressing TC-PTP were more sensitive to UVB-induced apoptosis, so cell proliferation was significantly decreased compared with control (Fig. 5C). A stable 3PC keratinocyte cell line that overexpresses TC-PTP also was generated by lentiviral transduction. Like transient TC-PTP overexpression, the level of phosphorylated STAT3 was significantly lower in stable TC-PTP-overexpressing 3PC cells than in control cells (Fig. 5D). Consistent with this observation, stable TC-PTP overexpression in keratinoxcytes significantly decreased cell proliferation after 24 and 48 h of culturing (Fig. 5E). Following exposure to 2 mJ/cm2 of UVB, the number of TC-PTP-overexpressing cells was significantly lower than control cells 48 and 72 h after UVB (Fig. 5F). The results confirm that overexpression of TC-PTP inhibits mouse keratinocyte cell proliferation and further demonstrate that TC-PTP is critical to the regulation of keratinocyte cell proliferation and survival.
FIGURE 5.

Overexpression of TC-PTP on the regulation of keratinocyte survival and proliferation after low dose of UVB irradiation. A–C, ectopic overexpression of TC-PTP in keratinocytes. 3PC keratinocytes were infected with an adenoviral vector expressing TC-PTP tagged with HA or empty vector as an infection control. A, Western blot analysis of TC-PTP and phosphorylated STAT3 (PY-STAT3). B and C, cell viability was evaluated in the absence or presence of UVB irradiation using the WST-1 assay. B, cell viability was measured 24 and 48 h after adenoviral infection without UVB irradiation. C, cells were irradiated with UVB (10 mJ/cm2) after infection. Cell viability was measured 24, 48, and 72 h after UVB irradiation. Results are the mean ± S.D. (error bars) from three independent experiments. *, p < 0.05 by Student's t test for equality of means. D–F, stable overexpression of TC-PTP in keratinocytes. D, generation of stably TC-PTP-overexpressing 3PC cell lines with pEF/mTC45 lentiviral vector. Empty pEF lentiviral vector was used as control. E and F, cell viability was measured using the WST-1 assay. E, cells were collected at 24 and 48 h without UVB irradiation. F, cells were irradiated with UVB at a dose of 2 mJ/cm2 and then collected 24, 48, and 72 h after irradiation.
Knockdown of TC-PTP in Keratinocytes Suppresses UVB-induced Apoptosis
Skin exposure to UVB radiation yields cellular DNA damage, which can trigger cell death unless it is either repaired or tolerated. We showed that constitutive STAT3 activation prevents UVB-induced apoptosis, whereas STAT3 deficiency sensitizes mouse keratinocyte cells to UVB-induced apoptosis (19, 22). To examine the role of TC-PTP in UVB-induced cell apoptosis, 3PC cells transiently transfected with either scrambled or TC-PTP siRNA were treated with UVB and analyzed by Western blot with two apoptotic markers, PARP and caspase-3. Levels of cleaved PARP and cleaved caspase-3 were increased in both control and TC-PTP-deficient cells 12–48 h after UVB treatment, most likely as the result of DNA damage (Fig. 6A). However, the levels of cleaved PARP and cleaved caspase-3 were significantly lower in TC-PTP-deficient cells 12 and 24 h after UVB irradiation compared with control cells (Fig. 6, A–C). In agreement with these results, caspase-3 activity was also significantly decreased in TC-PTP-deficient cells after UVB irradiation when compared with control (Fig. 6D). These results suggest that TC-PTP is required to promote apoptosis induced by low dose UVB.
FIGURE 6.

Knockdown of TC-PTP in keratinocytes on the regulation of UVB-induced apoptosis. A, representative Western blot analysis of apoptotic proteins in total cell lysates. 3PC keratinocytes were cultured and transfected with control or TC-PTP siRNA. Cells were collected at the indicated time after UVB irradiation (5 mJ/cm2). B, relative levels of PARP were quantified by densitometry. Results are the mean ± S.D. (error bars) from three independent experiments. *, p < 0.05 by Student's t test for equality of means. C, relative levels of cleaved caspase-3 were quantified by densitometry. D, caspase-3 activity was measured at 24 h after UVB irradiation.
UVB Exposure Activates TC-PTP in Skin Keratinocytes
Previous studies suggested that UVB radiation inactivates PTPs by oxidizing the cysteine residue within the conserved active site of the PTP catalytic domain (34–36). Our studies suggest for the first time that TC-PTP is activated by UVB exposure, so it can contribute to the dephosphorylation of STAT3 in mouse 3PC keratinocyte cells. To determine whether or not TC-PTP is activated by UVB radiation, 3PC cells were treated with UVB, and TC-PTP activity was quantified by colorimetric assay. TC-PTP activity steadily increased until activation peaked at 3 h after treatment with 10 mJ/cm2 UVB. TC-PTP activity then decreased slightly, yet TC-PTP activity was still significantly higher in UVB-treated cells when compared with untreated control cells at 0 h (Fig. 7A). Higher doses of UVB light also triggered a significant increase in TC-PTP activity (Fig. 7B), which is reasonable, given that TC-PTP contributes to the rapid dephosphorylation of STAT3 induced by high dose (100 mJ/cm2) UVB exposure (31).
FIGURE 7.

UVB irradiation activates TC-PTP in keratinocytes. A, TC-PTP activity was assessed at the indicated time after UVB irradiation (10 mJ/cm2) in 3PC keratinocytes. Results are the mean ± S.D. (error bars) from three independent experiments. *, p < 0.05 by Student's t test for equality of means. B, TC-PTP activity was assessed 3 h after different doses of UVB irradiation in keratinocytes. C, TC-PTP activity was measured at the indicated times after UVB irradiation (10 mJ/cm2) in control (empty vector) or TC-PTP-overexpressing keratinocytes.
To verify that TC-PTP is activated by UVB exposure, we examined TC-PTP activity in a stable 3PC cell line overexpressing TC-PTP. Overexpression of TC-PTP in 3PC keratinocytes enhanced the increase in TC-PTP activity following UVB irradiation relative to control keratinocytes (Fig. 7C). Together, the results imply that, in response to UVB exposure, TC-PTP is activated in mouse keratinocytes, allowing TC-PTP to inhibit cell proliferation and promote UVB-induced apoptosis.
Recovery of TC-PTP-deficient Cell Proliferation after UVB Exposure Is Dependent on STAT3
To demonstrate that the increase in cell proliferation of TC-PTP-deficient mouse keratinocyte cells in response to UVB irradiation is dependent on STAT3 signaling, we directly quantified STAT3 transcriptional activity by colorimetric assay in 3PC cells transiently transfected with TC-PTP siRNA or scrambled control siRNA (Fig. 8A). STAT3 transcriptional activity was higher in TC-PTP-deficient cells compared with control cells in the absence of UVB. After irradiation with low dose UVB, STAT3 activity was significantly increased in TC-PTP-deficient keratinocytes compared with control keratinocytes (Fig. 8A), which is in agreement with our Western blot data. We also utilized STA-21, a STAT3-specific inhibitor, to determine the impact of STAT3 on TC-PTP-deficient cell proliferation (37). STA-21 blocks STAT3 dimerization and DNA binding (37), and it did not show cytotoxicity in 3PC cells even at a concentration as high as 50 μm (Fig. 8B). Cell proliferation was significantly increased in TC-PTP-deficient cells compared with control cells following UVB irradiation. However, pretreatment of TC-PTP-deficient and control cells with STA-21 before UVB exposure almost completely suppressed cell proliferation in both control and TC-PTP-deficient cells after UVB treatment (Fig. 8C). The results demonstrate that TC-PTP-mediated regulation of keratinocyte cell proliferation in response to UVB is dependent on STAT3 signaling.
FIGURE 8.

TC-PTP regulates keratinocyte survival and proliferation after UVB irradiation by inhibiting STAT3 signaling. A, STAT3 transcriptional activity was assessed after UVB irradiation (5 and 10 mJ/cm2) in 3PC keratinocytes. Keratinocytes were cultured and transfected with control or TC-PTP siRNA. Cells were collected 24 h after UVB irradiation. Results are the mean ± S.D. (error bars) from three independent experiments. *, p < 0.05 by Student's t test for equality of means. B, analysis of STA-21 cytotoxicity on 3PC keratinocytes. Keratinocytes were plated and treated with different concentrations of STA-21 for 18 h. Cytotoxicity was determined by lactate dehydrogenase release in medium. C, keratinocytes were cultured and transfected with control or TC-PTP siRNA individually and irradiated with UVB at a dose of 2 mJ/cm2. STA-21 or DMSO as a control was pretreated 1 h before UVB treatment. Cell viability was evaluated 48 h after UVB treatment. Results are the mean ± S.D. from three independent experiments. *, p < 0.05 by Student's t test for equality of means.
DISCUSSION
Nonmelanoma skin cancer is the most common form of cancer in the United States, and data show that the incidence of skin cancer is increasing at a substantial rate. Among several risk factors for skin cancer, excessive exposure to UV radiation from the sun is the major cause (38). In the current study, we show that TC-PTP is involved in the mouse keratinocyte cell response to an assault by UVB radiation. TC-PTP can function as a tumor suppressor by attenuating STAT3 signaling to inhibit cell proliferation and by promoting apoptosis for the elimination of UVB-damaged cells. Loss of TC-PTP in keratinocytes increased the level of phosphorylated STAT3 and significantly increased cell proliferation in response to low dose UVB. Overexpression of TC-PTP in keratinocytes significantly decreased cell proliferation in response to low dose UVB, which correlated with a decreased level of phosphorylated STAT3. We show for the first time that TC-PTP activity is stimulated following UVB exposure, and we demonstrate that inhibition of STAT3 by its specific inhibitor completely reverses the enhanced cell proliferation observed in TC-PTP-deficient keratinocytes following low dose UVB irradiation. Combined, these results suggest that TC-PTP-mediated signaling in keratinocytes may serve as part of an initial protective mechanism against UVB-induced skin carcinogenesis (Fig. 9).
FIGURE 9.
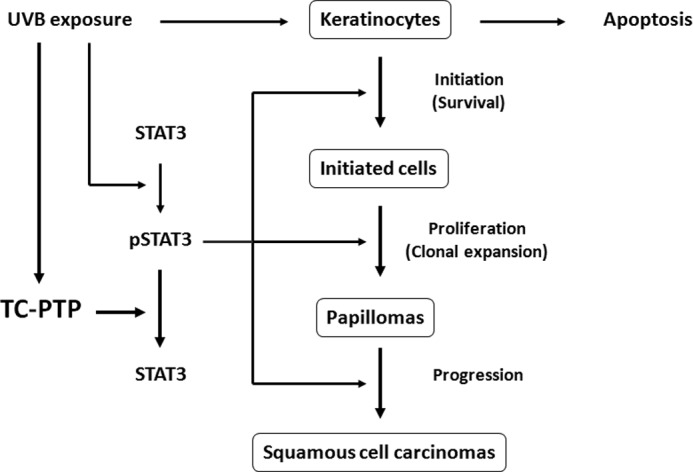
A novel protective mechanism against UVB-induced cellular damage. UVB irradiation triggers STAT3-mediated regulation of epidermal survival and proliferation. In the event that STAT3 becomes activated, this mechanism can lead to enhanced skin cancer formation. As part of an initial protective mechanism, UVB irradiation can also initially activate TC-PTP, which results in the dephosphorylation/inactivation of STAT3, thereby delaying activation of STAT3 signaling to prevent UVB-induced skin carcinogenesis.
In our previous study, we showed that three PTPs, TC-PTP, SHP1, and SHP2, contribute to rapid STAT3 dephosphorylation following UVB irradiation in mouse keratinocytes (31). As shown in this study, TC-PTP is the major PTP among the three PTPs that regulates STAT3 signaling in mouse keratinocytes in the presence or absence of UVB (See Fig. 1). Consistent with this observation, siRNA knockdown of TC-PTP in 3PC cells significantly increased cell proliferation compared with control cells in response to low dose UVB irradiation, whereas siRNA knockdown of either SHP1 or SHP2 had no effect on cell proliferation following exposure to low dose UVB (Fig. 3, A–E). Of the two isoforms of TC-PTP, only the nuclear form of TC-PTP, TC45, is expressed in mouse keratinocytes (31). TC45 has been reported to be primarily localized to the nucleus by its nuclear localization signal (27). Our previous study showed that TC45 is primarily localized in the cytoplasm of keratinocytes, and then it is translocated to the nucleus after UVB exposure. SHP1 and SHP2 are exclusively localized in the cytoplasm in keratinocytes (31). It is possible that TC-PTP has a greater effect on STAT3 dephosphorylation because UVB-induced nuclear translocation of TC-PTP allows it to directly regulate STAT3 before it binds to the consensus sequence of its target gene promoters, which results in reduced cell proliferation after UVB, whereas SHP1 and SHP1 may cooperate to reduce the amount of phosphorylated STAT3 in the cytoplasm so that less STAT3 enters the nucleus. Previous studies (39) and our recent results (data not shown) show that TC-PTP directly interacts with STAT3 in keratinocytes, indicating that STAT3 is a direct substrate of TC-PTP. It is possible that TC-PTP may also be acting upon other cell signaling pathways that regulate cell proliferation. Further studies investigating these potential signaling mechanisms are under way.
STAT3 is known to contribute to cell survival and proliferation by regulating gene expression of specific targets (11). For example, expression of cyclin D1, one critical proliferation marker, is regulated by STAT3 (11). The level of cyclin D1 was increased in correlation with STAT3 activation in mouse skin (19). Cyclin D1 expression is known to increase in UVB-induced skin carcinogenesis (40, 41). Similar to earlier studies, our current study showed that cyclin D1 expression is increased with STAT3 expression in TC-PTP-deficient mouse keratinocyte cell lines (Figs. 1B and 4A). In our previous study, we showed that expression of Bcl-xL, one anti-apoptotic target gene of STAT3, was not decreased with reduced STAT3 phosphorylation after UVB exposure in vitro in mouse keratinocyte cell lines and in vivo in mouse epidermis, suggesting that other signaling mechanisms are involved in the regulation of Bcl-xL expression to compensate for the loss of STAT3-mediated signaling that occurs in response to UVB irradiation. However, our current study showed that UVB-induced apoptosis was significantly lower in TC-PTP-deficient keratinocytes than in control keratinocytes (Fig. 6). There are two possible explanations for these results. First, other anti-apoptotic target genes of STAT3 are involved in the TC-PTP-mediated apoptotic response in mouse keratinocytes. Second, other apoptotic mechanisms regulated by TC-PTP, which are independent of STAT3, are involved in the TC-PTP-mediated apoptotic response in mouse keratinocytes. Further studies investigating these alternative pathways are required.
Genetic mutations of PTKs and/or aberrant activation of their signaling pathways can lead to an array of human diseases, including cancer (4, 5). The aberrant signaling caused by mutation and/or overexpression in 51 of the 90 PTKs has been implicated with various types of cancers (42). Consequently, PTKs are considered to be attractive targets for cancer prevention, and several PTK inhibitors have been developed (2, 4, 43) to treat cancers with high abundance of PTKs (44, 45). Exposure to acute UV irradiation increases the ligand-independent activation of PTKs (46, 47). This event is possible if UV irradiation induces PTP inactivation, and in fact, biochemical studies have revealed that reactive oxygen species (such as H2O2) produced by UV irradiation causes the inactivation of PTPs by oxidizing the cysteine residue within the conserved active site of the PTP catalytic domain (34–36). In our current study, we show that TC-PTP is activated in mouse keratinocytes by low dose UVB (Fig. 7A). It is possible that TC-PTP is activated in skin cells by UVB as part of a novel protective mechanism that protects skin from UVB exposure by initially deactivating oncogenic signaling. Increasing doses of UVB reduced TC-PTP activation 3 h after exposure (Fig. 7B). It is possible that the increase in UVB dose enhances the enzymatic kinetics of TC-PTP, and therefore high doses of UVB exposure result in rapid dephosphorylation of STAT3 (Fig. 1C). Overexpression of TC-PTP in keratinocyte cell lines demonstrated an even greater increase in TC-PTP activity following UVB (Fig. 7C). It is important to note that PTPs are differentially oxidized, although oxidation is a common regulatory mechanism of PTP inactivation (35, 48). PTP oxidation depends on pH and H2O2 concentrations in addition to PTP loop structure (48). It is also reported that PTPs are selectively oxidized by particular physiological stimuli (35). Our previous work also implied that both SHP1 and SHP2 are activated after UVB irradiation. Further studies related to the mechanism of UVB-induced PTP activation will be important to understand the PTP-mediated protective mechanism induced by UVB exposure.
Previous studies by Xu et al. (49) showed that acute UV irradiation activates epidermal growth factor receptor (EGFR) by oxidative inhibition of PTPκ in keratinocytes. STAT3 is known to be activated by various cell surface receptors, including EGFR (16, 23). TC-PTP is also known to dephosphorylate EGFR (50). However, our current studies show that UVB-mediated activation of TC-PTP dephosphorylates STAT3, indicating that UVB-induced STAT3 dephosphorylation by TC-PTP is independent of EGFR.
In conclusion, our data demonstrate that TC-PTP is a critical regulator of STAT3 signaling in mouse keratinocytes. TC-PTP activation by low dose UVB irradiation can lead to significant inhibition of keratinocyte cell proliferation and survival by dephosphorylating STAT3. Our previous studies showed that STAT3 has a critical role in UVB skin carcinogenesis (19). Further studies using a TC-PTP-specific transgenic mouse model will be helpful in understanding the role of TC-PTP in UVB-induced skin carcinogenesis. Our preliminary data suggest that TC-PTP activation may be a reversible event that occurs upon each exposure to environmental factors like UVB.3 This implies that repeated STAT3 dephosphorylation by reoccurring activation of TC-PTP could significantly reduce STAT3 signaling and thereby contribute to the prevention of skin cancer by delaying constitutive activation of STAT3. Combined, given the high degree of homology between human and mouse TC-PTP, these studies suggest that TC-PTP may be a novel potential target for both the prevention and treatment of UVB-induced human skin cancer.
This work was supported, in whole or in part, by National Institutes of Health, NIEHS, Grant ES022250 and National Institutes of Health Cancer Center Support Grant P30 CA054174.

This article contains supplemental Figs. 1–4.
H. Lee, L. D. Morales, T. J. Slaga, and D. J. Kim, unpublished data.
- PTK
- protein-tyrosine kinase
- PTP
- protein-tyrosine phosphatase
- TC-PTP
- T-cell protein-tyrosine phosphatase
- DPBS
- Dulbecco's PBS
- PARP
- poly(ADP-ribose) polymerase
- IOD
- integrated optical density
- WST-1
- 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt
- EGFR
- EGF receptor.
REFERENCES
- 1. Lim W. A., Pawson T. (2010) Phosphotyrosine signaling: evolving a new cellular communication system. Cell 142, 661–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hunter T. (2009) Tyrosine phosphorylation: thirty years and counting. Curr. Opin. Cell Biol. 21, 140–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Robinson D. R., Wu Y. M., Lin S. F. (2000) The protein tyrosine kinase family of the human genome. Oncogene 19, 5548–5557 [DOI] [PubMed] [Google Scholar]
- 4. Lemmon M. A., Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Casaletto J. B., McClatchey A. I. (2012) Spatial regulation of receptor tyrosine kinases in development and cancer. Nat. Rev. Cancer 12, 387–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tonks N. K., Neel B. G. (1996) From form to function: signaling by protein tyrosine phosphatases. Cell 87, 365–368 [DOI] [PubMed] [Google Scholar]
- 7. Stoker A. W. (2005) Protein tyrosine phosphatases and signalling. J. Endocrinol. 185, 19–33 [DOI] [PubMed] [Google Scholar]
- 8. Hendriks W. J., Elson A., Harroch S., Pulido R., Stoker A., den Hertog J. (2013) Protein tyrosine phosphatases in health and disease. FEBS J. 280, 708–730 [DOI] [PubMed] [Google Scholar]
- 9. Bowman T., Garcia R., Turkson J., Jove R. (2000) STATs in oncogenesis. Oncogene 19, 2474–2488 [DOI] [PubMed] [Google Scholar]
- 10. Bromberg J. (2002) Stat proteins and oncogenesis. J. Clin. Invest. 109, 1139–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Levy D. E., Darnell J. E., Jr. (2002) Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3, 651–662 [DOI] [PubMed] [Google Scholar]
- 12. Levy D. E., Lee C. K. (2002) What does Stat3 do? J. Clin. Invest. 109, 1143–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Darnell J. E., Jr. (1997) STATs and gene regulation. Science 277, 1630–1635 [DOI] [PubMed] [Google Scholar]
- 14. Bromberg J. F., Wrzeszczynska M. H., Devgan G., Zhao Y., Pestell R. G., Albanese C., Darnell J. E., Jr. (1999) Stat3 as an oncogene. Cell 98, 295–303 [DOI] [PubMed] [Google Scholar]
- 15. Turkson J., Jove R. (2000) STAT proteins: novel molecular targets for cancer drug discovery. Oncogene 19, 6613–6626 [DOI] [PubMed] [Google Scholar]
- 16. Kim D. J., Chan K. S., Sano S., Digiovanni J. (2007) Signal transducer and activator of transcription 3 (Stat3) in epithelial carcinogenesis. Mol. Carcinog. 46, 725–731 [DOI] [PubMed] [Google Scholar]
- 17. Sano S., Chan K. S., DiGiovanni J. (2008) Impact of Stat3 activation upon skin biology: a dichotomy of its role between homeostasis and diseases. J. Dermatol. Sci. 50, 1–14 [DOI] [PubMed] [Google Scholar]
- 18. Kim D. J., Kataoka K., Rao D., Kiguchi K., Cotsarelis G., Digiovanni J. (2009) Targeted disruption of stat3 reveals a major role for follicular stem cells in skin tumor initiation. Cancer Res. 69, 7587–7594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim D. J., Angel J. M., Sano S., DiGiovanni J. (2009) Constitutive activation and targeted disruption of signal transducer and activator of transcription 3 (Stat3) in mouse epidermis reveal its critical role in UVB-induced skin carcinogenesis. Oncogene 28, 950–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chan K. S., Sano S., Kiguchi K., Anders J., Komazawa N., Takeda J., DiGiovanni J. (2004) Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J. Clin. Invest. 114, 720–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kataoka K., Kim D. J., Carbajal S., Clifford J. L., DiGiovanni J. (2008) Stage-specific disruption of Stat3 demonstrates a direct requirement during both the initiation and promotion stages of mouse skin tumorigenesis. Carcinogenesis 29, 1108–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sano S., Chan K. S., Kira M., Kataoka K., Takagi S., Tarutani M., Itami S., Kiguchi K., Yokoi M., Sugasawa K., Mori T., Hanaoka F., Takeda J., DiGiovanni J. (2005) Signal transducer and activator of transcription 3 is a key regulator of keratinocyte survival and proliferation following UV irradiation. Cancer Res. 65, 5720–5729 [DOI] [PubMed] [Google Scholar]
- 23. Rho O., Kim D. J., Kiguchi K., Digiovanni J. (2011) Growth factor signaling pathways as targets for prevention of epithelial carcinogenesis. Mol. Carcinog. 50, 264–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cool D. E., Tonks N. K., Charbonneau H., Walsh K. A., Fischer E. H., Krebs E. G. (1989) cDNA isolated from a human T-cell library encodes a member of the protein-tyrosine-phosphatase family. Proc. Natl. Acad. Sci. U.S.A. 86, 5257–5261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mosinger B., Jr., Tillmann U., Westphal H., Tremblay M. L. (1992) Cloning and characterization of a mouse cDNA encoding a cytoplasmic protein-tyrosine-phosphatase. Proc. Natl. Acad. Sci. U.S.A. 89, 499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tillmann U., Wagner J., Boerboom D., Westphal H., Tremblay M. L. (1994) Nuclear localization and cell cycle regulation of a murine protein tyrosine phosphatase. Mol. Cell. Biol. 14, 3030–3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kamatkar S., Radha V., Nambirajan S., Reddy R. S., Swarup G. (1996) Two splice variants of a tyrosine phosphatase differ in substrate specificity, DNA binding, and subcellular location. J. Biol. Chem. 271, 26755–26761 [DOI] [PubMed] [Google Scholar]
- 28. Bourdeau A., Dubé N., Tremblay M. L. (2005) Cytoplasmic protein tyrosine phosphatases, regulation and function: the roles of PTP1B and TC-PTP. Curr. Opin Cell Biol. 17, 203–209 [DOI] [PubMed] [Google Scholar]
- 29. Dubé N., Tremblay M. L. (2005) Involvement of the small protein tyrosine phosphatases TC-PTP and PTP1B in signal transduction and diseases: from diabetes, obesity to cell cycle, and cancer. Biochim. Biophys. Acta 1754, 108–117 [DOI] [PubMed] [Google Scholar]
- 30. Xu D., Qu C. K. (2008) Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 13, 4925–4932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim D. J., Tremblay M. L., Digiovanni J. (2010) Protein tyrosine phosphatases, TC-PTP, SHP1, and SHP2, cooperate in rapid dephosphorylation of Stat3 in keratinocytes following UVB irradiation. PLoS One 5, e10290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klann R. C., Fitzgerald D. J., Piccoli C., Slaga T. J., Yamasaki H. (1989) Gap-junctional intercellular communication in epidermal cell lines from selected stages of SENCAR mouse skin carcinogenesis. Cancer Res. 49, 699–705 [PubMed] [Google Scholar]
- 33. Campeau E., Ruhl V. E., Rodier F., Smith C. L., Rahmberg B. L., Fuss J. O., Campisi J., Yaswen P., Cooper P. K., Kaufman P. D. (2009) A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Denu J. M., Tanner K. G. (2002) Redox regulation of protein tyrosine phosphatases by hydrogen peroxide: detecting sulfenic acid intermediates and examining reversible inactivation. Methods Enzymol. 348, 297–305 [DOI] [PubMed] [Google Scholar]
- 35. Tonks N. K. (2005) Redox redux: revisiting PTPs and the control of cell signaling. Cell 121, 667–670 [DOI] [PubMed] [Google Scholar]
- 36. Caselli A., Marzocchini R., Camici G., Manao G., Moneti G., Pieraccini G., Ramponi G. (1998) The inactivation mechanism of low molecular weight phosphotyrosine-protein phosphatase by H2O2. J. Biol. Chem. 273, 32554–32560 [DOI] [PubMed] [Google Scholar]
- 37. Song H., Wang R., Wang S., Lin J. (2005) A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 102, 4700–4705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rogers H. W., Weinstock M. A., Harris A. R., Hinckley M. R., Feldman S. R., Fleischer A. B., Coldiron B. M. (2010) Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch. Dermatol. 146, 283–287 [DOI] [PubMed] [Google Scholar]
- 39. Yamamoto T., Sekine Y., Kashima K., Kubota A., Sato N., Aoki N., Matsuda T. (2002) The nuclear isoform of protein-tyrosine phosphatase TC-PTP regulates interleukin-6-mediated signaling pathway through STAT3 dephosphorylation. Biochem. Biophys. Res. Commun. 297, 811–817 [DOI] [PubMed] [Google Scholar]
- 40. Kim A. L., Athar M., Bickers D. R., Gautier J. (2002) Stage-specific alterations of cyclin expression during UVB-induced murine skin tumor development. Photochem. Photobiol. 75, 58–67 [DOI] [PubMed] [Google Scholar]
- 41. Cooper S. J., MacGowan J., Ranger-Moore J., Young M. R., Colburn N. H., Bowden G. T. (2003) Expression of dominant negative c-jun inhibits ultraviolet B-induced squamous cell carcinoma number and size in an SKH-1 hairless mouse model. Mol. Cancer Res. 1, 848–854 [PubMed] [Google Scholar]
- 42. Blume-Jensen P., Hunter T. (2001) Oncogenic kinase signalling. Nature 411, 355–365 [DOI] [PubMed] [Google Scholar]
- 43. Levitzki A., Mishani E. (2006) Tyrphostins and other tyrosine kinase inhibitors. Annu. Rev. Biochem. 75, 93–109 [DOI] [PubMed] [Google Scholar]
- 44. Shawver L. K., Slamon D., Ullrich A. (2002) Smart drugs: tyrosine kinase inhibitors in cancer therapy. Cancer Cell 1, 117–123 [DOI] [PubMed] [Google Scholar]
- 45. Chow L. Q., Eckhardt S. G. (2007) Sunitinib: from rational design to clinical efficacy. J. Clin. Oncol. 25, 884–896 [DOI] [PubMed] [Google Scholar]
- 46. Sachsenmaier C., Radler-Pohl A., Zinck R., Nordheim A., Herrlich P., Rahmsdorf H. J. (1994) Involvement of growth factor receptors in the mammalian UVC response. Cell 78, 963–972 [DOI] [PubMed] [Google Scholar]
- 47. Coffer P. J., Burgering B. M., Peppelenbosch M. P., Bos J. L., Kruijer W. (1995) UV activation of receptor tyrosine kinase activity. Oncogene 11, 561–569 [PubMed] [Google Scholar]
- 48. Groen A., Lemeer S., van der Wijk T., Overvoorde J., Heck A. J., Ostman A., Barford D., Slijper M., den Hertog J. (2005) Differential oxidation of protein-tyrosine phosphatases. J. Biol. Chem. 280, 10298–10304 [DOI] [PubMed] [Google Scholar]
- 49. Xu Y., Shao Y., Voorhees J. J., Fisher G. J. (2006) Oxidative inhibition of receptor-type protein-tyrosine phosphatase κ by ultraviolet irradiation activates epidermal growth factor receptor in human keratinocytes. J. Biol. Chem. 281, 27389–27397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tiganis T., Bennett A. M., Ravichandran K. S., Tonks N. K. (1998) Epidermal growth factor receptor and the adaptor protein p52Shc are specific substrates of T-cell protein tyrosine phosphatase. Mol. Cell. Biol. 18, 1622–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]