

Background:LukGH is a member of the family of two-component bacterial toxins of Staphylococcus aureus that lyse human phagocytic cells.
Results:The crystal structure of LukGH and mutagenesis revealed the molecular basis for heterodimer formation in solution.
Conclusion:LukGH differs from other two-component leukocidins that interact only upon cell contact.
Significance:These data might assist with development of therapeutics that counteract Staphylococcus aureus pathogenesis.
Keywords: Bacterial Toxin, Crystal Structure, Crystallography, Microbial Pathogenesis, Mutagenesis, Staphylococcus aureus (S. aureus)
Abstract
The bi-component leukocidins of Staphylococcus aureus are important virulence factors that lyse human phagocytic cells and contribute to immune evasion. The γ-hemolysins (HlgAB and HlgCB) and Panton-Valentine leukocidin (PVL or LukSF) were shown to assemble from soluble subunits into membrane-bound oligomers on the surface of target cells, creating barrel-like pore structures that lead to cell lysis. LukGH is the most distantly related member of this toxin family, sharing only 30–40% amino acid sequence identity with the others. We observed that, unlike other leukocidin subunits, recombinant LukH and LukG had low solubility and were unable to bind to target cells, unless both components were present. Using biolayer interferometry and intrinsic tryptophan fluorescence we detected binding of LukH to LukG in solution with an affinity in the low nanomolar range and dynamic light scattering measurements confirmed formation of a heterodimer. We elucidated the structure of LukGH by x-ray crystallography at 2.8-Å resolution. This revealed an octameric structure that strongly resembles that reported for HlgAB, but with important structural differences. Structure guided mutagenesis studies demonstrated that three salt bridges, not found in other bi-component leukocidins, are essential for dimer formation in solution and receptor binding. We detected weak binding of LukH, but not LukG, to the cellular receptor CD11b by biolayer interferometry, suggesting that in common with other members of this toxin family, the S-component has the primary contact role with the receptor. These new insights provide the basis for novel strategies to counteract this powerful toxin and Staphylococcus aureus pathogenesis.
Introduction
Staphylococcus aureus is a versatile microbe equipped with numerous virulence mechanisms that can transform this harmless colonizer into a powerful human pathogen causing a broad spectrum of diseases ranging from skin infections to severe deep tissue infections, pneumonia, bacteremia, and sepsis. The hallmark of S. aureus pathogenesis is the ability to avoid and survive the most important innate immune defense: phagocytic killing (1, 2).
The bi-component leukocidins of S. aureus are important virulence factors that attack human phagocytic cells, greatly contributing to immune evasion. A single S. aureus strain can express up to five different bi-component leukocidins: γ-hemolysins HlgAB and HlgCB, Panton-Valentine leukocidin (PVL or LukSF), LukED and LukGH (3–5). These leukocidins belong to the family of structurally related pore-forming toxins (6). The mechanism of pore formation, mostly studied using γ-hemolysin and LukSF, is thought to be similar for all bi-component toxins and involves two separately secreted polypeptides, the S- and F-components, which are distinct in sequence and have a molecular mass between 32 and 38 kDa. Binding studies revealed that the S-component binds first and recruits the F-component to the surface of phagocytic cells, which is followed by oligomerization (7). This step induces structural changes in the stem domains of both subunits that lead to membrane insertion and the formation of a β-barrel pore in the cell membrane, resulting in osmotic lysis (8). Biochemical and biophysical studies of LukSF and HlgAB (including the HlgAB crystal structure) revealed octameric structures formed by four alternating S- and four F-components (9–11). By analogy, the other two leukocidins, LukED and LukGH are also thought to form similar structures.
In the last two years, the cellular receptors of most leukocidins have been identified (reviewed in Refs. 4, 5, and 12). Importantly, all of these molecules play a significant role in immune regulation. LukSF binds complement receptors C5aR and C5LR, LukGH interacts with the C3R (Mac-1 complex, CD11b component), and LukED recognizes chemokine receptors CXCR1 and -2 and CCR5 on phagocytic cells and lymphocytes, respectively (13–16). Based on competition studies, it is likely that HlgC uses the same receptor as LukS (17).
LukGH is the most recently discovered member of the bi-component leukocidin family. It is more distantly related to the other S. aureus leukocidins with only 30–40% amino acid sequence homology, whereas the other leukocidins have greater similarity to each other (60–80%).
Most importantly, we and others discovered that LukGH is fundamentally different from the other bi-component toxins due to heterodimer formation in solution (18). Therefore, strategies aimed at inhibiting the interaction of S- and F-components (for example, with monocomponent specific antibodies) are unlikely to be successful in preventing the formation of active toxin. Instead, steric interference with the binding of the LukGH complex to cells or oligomerization of dimers into pore complexes seem to be more promising strategies. For this, it is critical to determine at the molecular level how LukG and LukH heterodimerize. Here we describe the x-ray crystal structure of LukGH for the first time and determine the amino acid residues crucial for dimer formation, oligomerization, and cytolytic activity.
EXPERIMENTAL PROCEDURES
Bacterial Strains
The S. aureus USA300 strain TCH1516 (ATCC® BAA-1717TM) was obtained from ATCC (Manassas, VA). An isogenic mutant of this strain lacking hla, hlgABC, lukED, and lukSF was generated using gene-specific primers and the shuttle vector described previously (19).
Generation of Recombinant Toxins
Four different LukH and LukG proteins were expressed based on genome sequences of the S. aureus TCH1516 (USA300; CC8, ST8), MRSA252 (EMRSA16, CC30, ST36), MSHR1132 (CC75, ST1850), and H19 (livestock MRSA, CC10) strains. Genes for LukS and LukF were derived from the TCH1516 strain. Toxin genes were codon optimized for Escherichia coli expression, generated by gene synthesis (GenScript) and cloned into expression vectors as follows: NusA tagged LukS, LukF, and LukG_TCH1516 in pET44a (Novagen) at the PshAI/BamHI sites, untagged LukG_TCH1516 and LukH_TCH1516 in pET200D/TOPO (Invitrogen) at the NdeI/XhoI sites, and LukH and LukG proteins from MSHR1132, MRSA252, and H19 strains in pET24a (Novagen) at the NdeI/BamHI sites. The toxins were produced in the Tuner DE3 expression strain without signal peptide sequences (determined by the SignalP 4.1 program), except for LukH_TCH1516 where the PrediSi program was used. Protein expression was induced with isopropyl β-d-1-thiogalactopyranoside for 20 h at 20 °C for LukS, LukF, and the LukGH complexes or for 4 h at 37 °C for the individual LukG and LukH proteins.
Mutant LukG and LukH (LukG1, LukG2, LukH1, and LukH2) proteins were generated by QuikChange mutagenesis (Stratagene) using the lukG_TCH1516 and lukH_TCH1516 templates and the primers in Table 1. Correct sequences for both strands of all DNA constructs were confirmed.
TABLE 1.
Primers used for mutagenesis
| Primer name | Interface | Primer sequence |
|---|---|---|
| LukG_R23A_fwd | 1 | gatacgaaaatgtacacggccacggcaaccacgagcg |
| LukG_R23A_rev | 1 | cgctcgtggttgccgtggccgtgtacattttcgtatc |
| LukG_K217A_fwd | 1 | cttcacgccgaaagacgcaatgccggtcaccgtg |
| LukG_K217A_rev | 1 | cacggtgaccggcattgcgtctttcggcgtgaag |
| LukG_E171A_fwd | 2 | cgttggttggaaagtcgcagcgcacctgatcaataac |
| LukG_E171A_rev | 2 | gttattgatcaggtgcgctgcgactttccaaccaacg |
| LukG_D189A_D191A_fwd | 2 | gtcaactgacgaatgctagcgccaaccgcacgaaatc |
| LukG_D189A_D191A_rev | 2 | gatttcgtgcggttggcgctagcattcgtcagttgac |
| LukH_D75A_fwd | 1 | cgatgacccgacctacgccaaaaatgtgctgctgg |
| LukH_D75A_rev | 1 | ccagcagcacatttttggcgtaggtcgggtcatcg |
| LukH_D197A_fwd | 1 | ggtctgtcattgctaacgctctgaaatatggcggtg |
| LukH_D197A_rev | 1 | caccgccatatttcagagcgttagcaatgacagacc |
| LukH_R215A_fwd | 2 | cgaactgctgttttacgctaatacccgcatcgcgac |
| LukH_R215A_rev | 2 | gtcgcgatgcgggtattagcgtaaaacagcagttcg |
| LukH_R234A_fwd | 2 | cgcgtcgaaatatgcttacccggccctggtg |
| LukH_R234A_rev | 2 | caccagggccgggtaagcatatttcgacgcg |
| LukH_R240A_fwd | 2 | cccggccctggtggcctccggttttaatcc |
| LukH_R240A_rev | 2 | ggattaaaaccggaggccaccagggccggg |
The LukH and LukG proteins were expressed without tags, isolated from inclusion bodies solubilized in 8 m urea (Fisher BioReagents), refolded, and purified in two steps. The refolded LukH proteins were purified by size exclusion chromatography on a Superdex 75 16/60 column (GE Healthcare) equilibrated in 50 mm sodium phosphate, pH 7.5, with 500 mm NaCl, followed by cation exchange chromatography on SulfoPropyl (SP)-Sepharose Fast Flow (FF) column (HiTrap, 5 ml, GE Healthcare) equilibrated in 20 mm sodium phosphate, pH 7.2, with 50 mm NaCl, and eluted with a linear gradient of 0.05–1 m NaCl in the same buffer. The denatured LukG proteins were purified by cation exchange chromatography on the SP FF column equilibrated in 50 mm acetate, pH 4.0, with 8 m urea, and eluted with a linear gradient of 0.05–1 m NaCl in the same buffer, followed by refolding in 20 mm CAPS3, pH 10.2 (pH 11.0 for LukG_MSHR1132), with 150 mm NaCl. The refolded LukG proteins were further purified by anion exchange chromatography on a Q-Sepharose FF column (HiTrap, 5 ml, GE Healthcare) equilibrated in 20 mm CAPS, pH 10.2 (pH 11.0 for LukG_MSHR1132), with 75 mm NaCl and eluted with a linear gradient of 0.075–0.5 m NaCl in the same buffer.
LukS and LukF were expressed in soluble form with an N-terminal NusA/His6 tag, which was removed proteolytically after the first purification step. Purification typically involved three chromatographic steps: 1) immobilized metal affinity column (IMAC), 2) cation exchange or IMAC, and 3) size exclusion chromatography. The clarified cell extract was loaded onto an IMAC (HiTrap, 5 ml, GE Healthcare) and the fusion protein was eluted with 500 mm imidazole. Following buffer exchange into cleavage buffer (20 mm Tris, pH 7.5, 200 mm NaCl, 2 mm CaCl2) and digestion with enterokinase (New England Biolabs), the mature protein was separated from the NusA/His6 tag by metal ion affinity (IMAC for LukF) or cation exchange (SP FF for LukS) chromatography. The final purification step on the gel filtration column equilibrated in 50 mm sodium phosphate, pH 7.5, with 300 mm NaCl, yielded pure proteins (>95%) based on SDS-PAGE analysis.
The LukGH complexes were generated by co-transformation of E. coli with two plasmids containing different antibiotic resistance markers, carrying either the lukH or lukG genes. LukG was expressed as a fusion protein with NusA/His6 at the N terminus to allow metal ion affinity purification of the complex, whereas LukH was expressed in un-tagged form. The two proteins were found in the soluble fraction and were co-purified by IMAC. The NusA/His6 tag was removed proteolytically with enterokinase giving the un-tagged, mature LukGH complex, which was further purified by cation exchange chromatography using an SP FF column (HiTrap, 1 ml, GE Healthcare) equilibrated in 20 mm sodium phosphate, pH 7.4, with 75 mm NaCl and the complex eluted with 150–200 mm NaCl in the same buffer.
The proteins were assayed for purity by SDS-PAGE, monomeric state by size exclusion chromatography, and for functionality in in vitro toxin potency assays. Biotinylated proteins were generated with the amino reactive reagent Sulfo-NHS-LC biotin (Thermo Scientific), according to the manufacturer's instructions, yielding biotinylation levels of 1–2.5 biotin/protein.
Purification of the I-domain of Human and Mouse CD11b
The I-domains of human and mouse CD11b (amino acids 111–321 for each) were cloned from CD11b cDNAs (OriGene) into pET28a (Novagen) cleaved with NcoI and BamHI. The expression of the proteins was induced at 20 °C with 0.4 mm isopropyl β-d-1-thiogalactopyranoside, and the proteins were purified by cation exchange and size exclusion chromatography. Biotinylation was performed using the same method as described for the recombinant toxins.
Binding Analysis with Biolayer Interferometry (BLI)
Binding of LukG (wild-type and mutants) to biotinylated LukH and LukH2 was performed by immobilizing the biotinylated toxin on streptavidin sensors (fortéBio, Pall Life Sciences) and monitoring the association of LukG (1–16 μg/ml) to the preloaded sensors for 10 min in PBS containing 1% BSA. Biotinylated HlgC was used as a negative control to determine unspecific binding. Binding of LukGH and LukH variants to the I domain of human CD11b was performed by immobilizing the biotinylated receptor on streptavidin sensors and monitoring the association of LukGH or LukH (0.2–10 and 0.1–5 μg/ml, respectively) to the preloaded sensors for 10 min to 1 h and the dissociation for 300 s in PBS buffer containing 1% BSA and 1 mm MgCl2. The Kd values were determined using the Data Analysis 7 software (fortéBio, Pall Life Sciences).
Dynamic Light Scattering (DLS) Analysis
Complex formation in solution was investigated by analyzing LukG_TCH1516, LukH_TCH1516, and their mixture, or the LukGH co-expressed complexes derived from the TCH1516, MRSA252, and H19 strains by DLS, using a DynaPro NanoStar (Wyatt) instrument equipped with a static light scattering detector. The molecular weights of the proteins (MW-S) were determined from the static light scattering measurements, whereas the abundance of the monomeric forms (% Mass) and the molar masses estimated from the protein conformation (MW-R) were determined from the DLS measurements.
Tryptophan Fluorescence Measurements
The fluorescence measurements detecting intrinsic tryptophan fluorescence were performed on a Hitachi F-7000 fluorimeter (Hitachi High Technologies) in 50 mm sodium phosphate, pH 7.5, with 150 mm NaCl, using an excitation wavelength of 280 nm, and monitoring emission in the 300–450 nm range with emission and excitation slits of 2.5 and 1.5 nm, respectively. Different LukH concentrations (0–0.5 μm) were titrated into a cuvette containing either LukG at a fixed concentration (0.25 μm) or buffer only (as reference), and difference spectra were calculated by subtracting the reference spectra.
Circular Dichroism (CD) Analysis
Far UV (195–250 nm) CD spectra were recorded on a Chirascan (Applied Photophysics) spectrometer at protein concentrations of 0.1–0.3 mg/ml in 20 mm sodium phosphate, pH 7.5, with 200 mm NaCl for the LukGH and 50 mm sodium phosphate, pH 7.5, with 500 mm NaCl for the LukH variants.
Chemical Cross-linking
Cross-linking of mixtures of LukG and LukH variants (35 μg/ml each) was performed by incubation with glutaraldehyde (1 mm) at 37 °C in 20 mm HEPES, pH 7.5, with 50 mm NaCl for up to 2 min and the reaction was stopped by adding 0.1 m Tris, pH 8.0. The samples were analyzed by SDS-PAGE using 4–12% BisTris gels (Novex, Invitrogen).
Protein Crystallization, Data Collection, Structure Determination and Refinement
Co-purified LukGH_TCH1516 was dialyzed against 20 mm HEPES buffer, pH 7.5, concentrated to ∼10 mg/ml, and supplemented with 10% glycerol. Diffraction quality crystals were obtained using sitting drop vapor diffusion at 20 °C. 100 nl of protein and 100 nl of reservoir solutions were mixed, and crystals were obtained after 7 days from condition A8 of the Morpheus screen (20) containing 30 mm MgCl2, 30 mm CaCl2, 0.1 m Na-HEPES/MOPS buffer, pH 7.5, and 12.5% (v/v) 2-methyl-2,4-pentanediol (MPD), 12.5% (w/v) PEG 1000, and 12.5% (w/v) PEG 3350. The crystals were harvested directly from their crystallization drops due to the high content of MPD, which functioned as cryoprotectant.
Data were collected at station I911-3 of the MAX-II synchrotron, Lund, Sweden (21), equipped with a marMosaic 225 CCD detector. Data were indexed and integrated using XDS (22) and scaled and merged using SCALA (23) from the CCP4 package (24).
The structure was solved by molecular replacement using Phaser (25) and unmodified PDB entry 3B07 (the octameric pore form of γ-hemolysin from S. aureus) as a search model. Phaser produced unique solutions with two octamers in the asymmetric unit. After refinement in Refmac5 (26), the R-factor Rmodel for the all-atom model was 47.2% and Rfree (calculated on 5% of the data, or 11411 reflections) was 47.9%. Chains A and B of Protein Data Bank code 3B07 were processed using Chainsaw (27), and two full octamers were then generated by superimposing them on the 8 dimers in the octameric pores. Refinement of this model with 8-fold non-crystallographic symmetry (NCS) restraints between chains resulted in Rmodel = 47.2%, Rfree = 47.6%. Removal of the NCS restraints gave Rmodel = 46.9, Rfree = 47.5%, thus no NCS restraints were used in further refinement. Chains A and B were rebuilt using Coot (28) and the full octamers once more regenerated by NCS. This procedure was repeated several times, resulting in Rmodel = 27.0, Rfree = 30.9%, before starting to rebuild the chains individually. After many cycles of optimization of Ramachandran and rotamer outliers, the final values for Rmodel and Rfree were 24.1 and 29.2%, respectively. Water molecules were placed where the density and bonding were unambiguous. Finally the size of the Rfree set was reduced to 1% (2308 reflections) and the structure refined for a final cycle. The structure quality was analyzed using MolProbity (29). Protein interfaces were analyzed with PISA (30) and the protein figures were generated with PyMOL (Schrödinger LLC).
PMN Purification and Differentiation of HL-60 Cells
Cell based assays were performed using either human polymorphonuclear neutrophils (PMNs) or “neutrophil-like” differentiated HL-60 cells. Neutrophils were isolated from heparinized fresh human whole blood, either obtained from the Austrian Red Cross or from healthy volunteers. To aggregate erythrocytes 1 part HetaSep solution (Stem Cell Technologies) was added to 5 parts of blood, mixed, and incubated at 37 °C, 5% CO2 until the plasma/erythrocyte interphase was at ∼50% of the total volume. The leukocyte-enriched plasma layer was carefully layered on a 2-step Percoll gradient (GE Healthcare) (73 and 63% Percoll Plus diluted in Hanks' balanced salt solution) and centrifuged at 680 × g, at room temperature for 30 min, no braking to decelerate. The first and second layers of the post-spin gradient (mainly serum and monocytes) were removed by aspiration. Neutrophils were harvested from the second opaque layer and washed twice in 50 ml of Hanks' balanced salt solution (Invitrogen) + 10 mm glucose. The number of viable cells was counted using trypan blue dye exclusion in a hemocytometer. The described isolation method usually yielded 1–5 × 108 neutrophils/50 ml of whole blood with a viability >95%. For cytotoxicity assays, cells were resuspended in RPMI 1640 culture medium (PAA Laboratories) supplemented with 10% FCS, l-glutamine, and penicillin/streptomycin (=neutrophil medium). HL-60 cells (ATCC CCL-240TM) were cultured in neutrophil medium and differentiated with N,N-dimethylformamide at 100 mm for 5 days, as described previously (31). Differentiation was determined by a significant reduction in CD71 and increase in CD11b expression based on staining intensity with phycoerythrin-conjugated anti-CD71 (clone OKT9, eBioscience) and Brilliant Violet 421-conjugated anti-CD11b (clone ICRF44, BioLegend) monoclonal antibodies.
Cytotoxicity Assays
Activity of bi-component toxins toward neutrophils and differentiated HL-60 cells was assessed by measuring ATP levels of intoxicated cells. Briefly, an equimolar mixture of the F- and S-components or the co-expressed LukGH complex was serially diluted in neutrophil medium and used for intoxication of 25,000 cells for 4 h at 37 °C at 5% CO2. Cell viability was then measured using the Cell Titer-Glo® Luminescent Cell Viability Assay Kit (Promega) according to the manufacturer's instructions. Percent viability was calculated relative to mock-treated cells (100% viability).
For LukGH inhibition studies, cells were preincubated with the mutant LukGH dimers or corresponding amounts of the individually expressed single components (12.75 nm) for 15 min at 37 °C, 5% CO2. The preincubated cells were then intoxicated with a serial dilution of the wild-type LukGH dimer as described above.
Detection of Toxin Binding to Target Cells
Biotinylated LukG, LukH, LukS, and LukF (55 nm) were incubated with 1 × 106 differentiated HL-60 cells or freshly isolated human PMNs in Hanks' balanced salt solution containing 0.5% BSA for 30 min on ice. After a washing step in Hanks' balanced salt solution, cell-bound toxins were detected with Alexa 488-labeled streptavidin (Molecular Probes, Invitrogen), and analyzed by flow cytometry. Toxin binding is expressed as median fluorescent intensity and compared with control cells (no toxin, only secondary reagent and non-relevant biotinylated recombinant protein).
RESULTS
LukG and LukH Bind to Target Cells as a Complex Due to Heterodimerization in Solution
We studied the interaction of the S- and F-components of leukocidins with their target cells using biotinylated subunits expressed in recombinant forms in E. coli. In contrast to the other bi-component toxins, we could not detect surface binding of neither LukG nor LukH to human PMNs or differentiated HL-60 cells. Binding to cells was detected only when both components were present (data shown for PMNs in Fig. 1A). In parallel experiments, biotinylated LukS displayed strong surface binding, which was augmented in the presence of LukF (Fig. 1B). This is consistent with previously published data showing that LukS is the cell binding component of LukSF and recruits its F-component to the membrane to form active toxin (7). The cytolytic potencies of labeled and unlabeled toxins were comparable (Fig. 1C), confirming that labeling did not affect the conformation of the monomers and pore assembly. The binding signal was consistently much stronger with LukSF on both PMNs and differentiated HL-60 cells compared with those obtained with LukGH, suggesting markedly lower LukGH receptor density. Despite that, the cytolytic potencies of the two toxins were comparable.
FIGURE 1.

The LukGH toxin binds to target cells as a dimer. Biotinylated LukH or LukG (A) and LukS or LukF (B) was incubated with human PMNs in the absence or presence of the unlabeled toxin pair. Surface bound, biotinylated proteins were detected with Alexa 488-labeled streptavidin and quantified by flow cytometry. Results of three independent experiments are shown (mean ± S.E.). C, biotin-labeled toxin components were tested for functionality in PMN toxicity assays and compared with unlabeled counterparts. Left panel, LukG and LukH; right panel, LukS and LukF. Results represent the mean from three independent experiments ± S.E. D, complex formation by LukG and LukH was investigated by fluorescence spectroscopy detecting changes in intrinsic tryptophan fluorescence upon the addition of increasing amounts of LukH to a constant amount of LukG (250 nm). E, N-terminal NusA/His-tagged LukG and untagged LukH (derived from the S. aureus TCH1516 strain) were co-expressed in E. coli. Proteins isolated from the soluble fraction of E. coli and eluted from the IMAC column were visualized by SDS-PAGE. Lane 1, marker proteins (molecular masses indicated in kDa); lane 2, NusA-LukG and LukH co-eluted from the IMAC column; lane 3, enterokinase digestion of the NusA-LukG and LukH fractions; lane 4, LukGH fraction eluted from the Sulfo-Propyl column. F, cytotoxic potency of the mixture of individually expressed LukH and LukG was compared with the co-expressed complex (derived from the S. aureus TCH1516 strain) using human PMNs or differentiated HL-60 cells. Viability of cells was determined by luminescent measurement of cellular ATP levels. Results of three independent experiments are shown (mean ± S.E.).
To test the hypothesis that LukG and LukH might form a complex in solution, we performed fluorescence spectroscopy and measured changes in intrinsic tryptophan (Trp) fluorescence. We observed a blue shift (∼4 nm) in the emission spectra at the maximum emission wavelength, indicating that the Trp residues became buried more deeply or the Trp environment became more hydrophobic, an indication for complex formation (data not shown). When LukH was titrated against a constant concentration of LukG, the quenching of Trp fluorescence leveled off at ∼1 LukH eq, indicative of a 1:1 LukG:LukH stoichiometry. The complex formed quantitatively in the concentration range of 50–250 nm, suggesting a dissociation constant in the low nanomolar range (Fig. 1D).
We further investigated the complex formation by co-expressing LukG and LukH in E. coli and were able to co-purify LukH via His6-tagged LukG with a 1:1 molar ratio (Fig. 1E). We also observed different behaviors of co-expressed LukGH compared with individually expressed LukG and LukH. The single components were consistently partitioned to inclusion bodies, whereas the co-expressed LukGH was detected mainly in the soluble fraction of E. coli. The purified LukG and LukH proteins required a buffer with high pH and/or high salt for solubility and stability (to avoid aggregation), whereas co-expressed LukGH complex was soluble at physiological pH and salt concentration (see “Experimental Procedures”). The co-expressed complexes were found to be more active than mixtures of individually expressed LukH and LukG (Fig. 1F).
To determine the size of the LukGH complex in solution we employed DLS measurements. The molecular mass measured with the static light scattering detector (MW-S) was 75-kDa for the co-expressed LukGH complex, which concurs with both the calculated and measured molecular masses of the heterodimer of LukG (36 kDa) and LukH (38 kDa) (Table 2). No significant amounts of higher molecular weight complexes were detected.
TABLE 2.
DLS parameters for LukG, LukH, and the LukGH complex
| LukH | LukG | LukG + LukH | LukGH | |
|---|---|---|---|---|
| Radius (nm) | 2.79 | 2.80 | 3.78 | 3.84 |
| MW-R (kDa) | 37.0 | 38.8 | 75.4 | 78.4 |
We detected several LukGH sequence variants in the public genome databases with as low as 82% amino acid homology to the TCH1516 sequence type we used for our studies (Fig. 2). This indicates that the actual variability is much higher than that typically seen with other S. aureus bi-component toxins that display <5% sequence diversity for most strains. To investigate this further, we expressed additional LukGH complexes representing the most divergent sequence variants derived from the MRSA252, MSHR1132, and H19 strains (alignments shown in Fig. 3). In all cases, co-expressed LukG and LukH co-purified as a 1:1 mixture and formed heterodimers in solution based on DLS analysis (data not shown). In cell viability assays we observed comparable potencies of the four LukGH variants with both human PMNs and differentiated HL-60 cells (Fig. 4).
FIGURE 2.

Conservation of LukG and LukH sequences from different S. aureus strains. Percent amino acid sequence identities between LukH and LukG variants derived from different strains for which annotated genome data are available. Those used in this study are highlighted in red.
FIGURE 3.

Amino acid alignment of the bi-component toxins of S. aureus. Amino acid conservations among S- or F-components of all bi-component toxins of the strain TCH1516 are shown. LukH and LukG sequences from MRSA252, MSHR1132, and H19 strains are included for comparison. Multiple sequence alignment was performed with the ClustalW2 program (36). Amino acids conserved between the LukH or LukG variants are shown in bold, and those involved in contacts in interface 1 and 2 are shown in blue and red, respectively. Residues involved in salt bridges in interface 1 and 2 are highlighted in yellow and cyan, respectively, and those mutated in this study are boxed. Secondary structure elements are indicated; arrows for β-strands and coils for α-helices.
FIGURE 4.
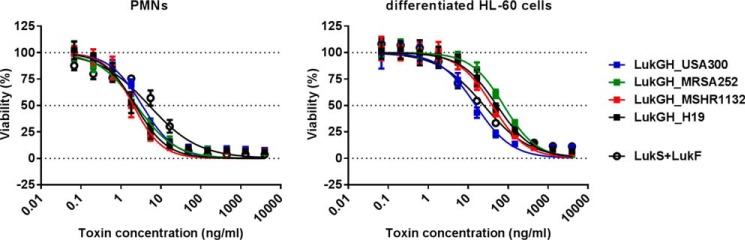
All LukGH variants display comparable toxicity toward human granulocytes. Recombinant LukG and LukH proteins derived from the TCH1516, MRSA252, H19, and MSHR1132 S. aureus strains co-expressed in E. coli cells were incubated with freshly isolated human PMNs or differentiated HL-60 cells at the indicated concentration range. Cell viability was determined by luminescent measurement of cellular ATP levels. Results of three independent experiments (mean ± S.E.) are shown.
X-ray Crystal Structure of LukGH Reveals Differences to Other Leukocidins
The co-purified LukGH_TCH1516 complex was subjected to crystallization. Diffraction quality crystals were obtained only in conditions containing MPD (12.5%). The structure of LukGH was solved at 2.8-Å resolution (Table 3) and revealed an octameric arrangement, with two octamers in the asymmetric unit, composed of four alternating LukG and LukH subunits (Fig. 5A), similar to those obtained with HlgAB (11). With the exception of the N termini, essentially the entire LukH and LukG subunits were visible in the electron density maps, i.e. amino acids Thr35-Glu323 for LukH and Gly16-Asp305 for LukG. The root mean square deviation between LukG and HlgB (chains A) was 1.09 Å, with main differences observed in the loops forming the rim domain (Fig. 5B). The MPD molecule that occupies the phosphocholine binding pocket in HlgB is absent in LukG, where the side chain of the methionine residue at position 178 occupies this pocket. No ordered MPD molecules have been observed in other sites in the structure. The loop at the bottom of the rim domain in HlgB (Trp257-Tyr261) containing the hydrophobic residues Trp257, Phe260, and Tyr261 and shown to be involved in cell binding to human erythrocytes (32), is extended in LukG (Trp262-Tyr270), and contains two polar residues: Arg264 and His265. Polar residues are also found in LukD (Arg258 and His260) and LukF (Asn258 and His260) in the corresponding loops (based on sequence alignments in Fig. 3 and LukF crystal structure, PDB code 1PVL). Tyr72, the other residue shown to be involved in cell binding in HlgB (33), and found in an adjacent loop, is conserved in LukG (Tyr73) (Fig. 5B).
TABLE 3.
Data collection and refinement statistics for structure determination
| Statistics | |
|---|---|
| Data collection | |
| Beamline | I911–3/MAX-II |
| Detector | marMosaic 225 CCD |
| Wavelength (Å) | 1.0000 |
| Space group | P21 |
| Cell dimensions (Å) | a = 135.5, b = 198.6, c = 179.5, β = 103.3° |
| Resolution range (Å)a | 48.19–2.80 (2.95–2.80) |
| Rmerge(I) (%)a | 16.0 (87.1) |
| Mean (I/σ(I)) a | 10.1 (1.7) |
| Completeness (%)a | 97.9 (95.4) |
| Multiplicity a | 3.1 (3.1) |
| No. of observed reflections | 685,808 (96,878) |
| No of unique reflectionsa | 222,018 (31,557) |
| Refinement | |
| Resolution (Å) | 48.19–2.80 |
| Rmodel (F) (%)a | 24.0 (43.0) |
| Rfree (F) (%)a | 29.2 (46.0) |
| No. of unique reflections used | 225,293 |
| No. of non-hydrogen atoms | 2,308 |
| No. of protein atoms | 37,618 |
| Data to parameter ratio | 1.50 |
| No. of water molecules | 205 |
| Average B-factors (Å2) | |
| Protein atoms | 33 |
| Water molecules | 39 |
| Root mean square deviations from ideal geometry | |
| Bond lengths(Å) | 0.014 |
| Bond angles (°) | 1.79 |
| Ramachandran plot | |
| Residues in favored regions | 2,477 (93.3%) |
| Residues in favored plus allowed regions | 2,699 (99.3%) |
a Numbers in parentheses are those for the highest resolution shell.
FIGURE 5.

X-ray structure of the LukG and LukH monomers in the LukGH octamer. A, structure of the LukGH octamer formed by LukG (pink) and LukH (cyan). B, overlay of LukG (pink) and HlgB (orange) (on chains A) with the side chains of the residues involved in membrane binding shown as sticks and MPD and Met178 as orange and pink spheres, respectively. C, overlay of LukH (cyan) and HlgA (green) (on chains B), with LukH residues Thr35-Ile40 and Tyr314-Glu323 colored magenta and red, respectively; the side chains of the residues delineating the loops with a different conformation in LukH are shown as sticks. D, interface 1: LukG and LukH superposed on HlgB and HlgA (chains A and B, respectively, overlay on chains A). E, interface 2: LukH and LukG superposed on HlgA and HlgB (chains B and C, respectively, overlay on chains B). The side chains of the residues involved in electrostatic interactions are shown as sticks and the salt bridges as dotted lines. Electron density map (2Fo − Fc) around residues involved in salt bridges in panel E contoured at 1 σ, is shown as a gray mesh.
The superposition of LukH and HlgA (chains B, root mean square deviation 1.11 Å), also revealed differences in the rim domain, where the loops in LukH (His96-Asn103, Asn196-Asn206, Tyr216-Asn224, and Lys274-Lys290) have a different position and orientation relative to each other compared with those in HlgA (Fig. 5C). Part of the latch domain (Thr35-Ile40) is visible in LukH, but the first 34 amino acids that are specific for LukH are unstructured. The C terminus of LukH, shown to be involved in receptor binding (18), is surface exposed and forms a flexible random coil that lies along the interface between the rim and cap domains in the LukH protomer (Fig. 5C, highlighted in red), with no significant interaction with the LukG monomer. The side chain of Glu323 whose mutation was shown to be sufficient to disrupt toxin activity (18) is not resolved in the structure, and is probably rather flexible but it is clearly exposed to the outer side of the molecule and thus accessible for receptor binding.
Based on this structure, the toxin protomers are involved in two types of interfaces in the octamer: interface 1 between chains A and B and interface 2 between chains B and C (Fig. 5, D and E). The buried surface areas are 2188 and 2461 Å2 for interfaces 1 and 2, respectively. In interface 1, the main interactions occur between the cap domains (Fig. 5D), whereas in interface 2 more interactions were observed between the rim domains of the two monomers (Fig. 5E). Analysis of the contact residues (using 4 Å distance as a threshold) in the LukGH octamer has revealed that interface 1 is stabilized by 34 hydrogen bonds and three electrostatic interactions involving residues Asp39, Asp75, and Asp197 of LukH and Arg23, Lys56, Lys58, and Lys218 of LukG. In HlgAB, only two salt bridges are present in interface 1. In interface 2, there are altogether 56 hydrogen bonds and four electrostatic interactions. The amino acid residues forming the only salt bridge observed in the HlgAB octamer between the cap domains is conserved in the LukGH octamer, formed by Arg49 in LukH and Asp49 in LukG, and also in other S- and F-components (Figs. 3 and 5E). Three additional electrostatic interactions are detected in LukGH, not found in other bi-component toxins, involving residues Arg240 in LukH and Glu171 in LukG between the cap domains, and residues Arg215 and Arg234 in LukH and Asp189 and Asp191 in LukG between the rim domains (Fig. 5E). Importantly, the residues involved in these salt bridges are fully conserved in all LukGH variants (Fig. 3). These data suggest that LukG and LukH are held together with stronger molecular interactions than the other S- and F- components, especially at interface 2.
Charged Residues Forming Salt Bridges Are Involved in Dimer Formation
Based on the crystal structure of the LukGH octamer, we mutated the charged amino acid residues (in TCH1516-derived proteins) involved in electrostatic interactions to test whether these are important for dimer formation. We generated interface 1 mutant proteins by replacing Asp75 and Asp197 in LukH (LukH1) and Arg23 and Lys218 in LukG (LukG1) with Ala. Interface 2 mutants were created by alanine substitution of Arg215, Arg234, and Arg240 in LukH (LukH2) and Asp189, Asp191, and Glu171 in LukG (LukG2) (summarized in Table 4).
TABLE 4.
Overview of mutated residues
| Amino acid mutations | |
|---|---|
| LukH1 | D75A, D197A |
| LukG1 | R23A, K218A |
| LukH2 | R215A, R234A, R240A |
| LukG2 | D189A, D191A, E171A |
We expressed both complexes (co-expression) and the individual monomers in E. coli as described for the wild-type LukGH proteins. The expression levels of the mutant LukGH complexes were comparable, although the amount of soluble complexes varied. The interface 1 mutants LukG1H, LukGH1, and LukG1H1 showed similar solubility to the wild-type complex and were purified using the same procedure as for wild-type LukGH. However, the interface 2 mutant complexes LukG2H, LukGH2, and LukG2H2 were mainly insoluble and therefore not purified. For interface 2 mutants, only the single components, LukG2 and LukH2, were purified. Analysis of the interface 1 mutant LukGH complexes by circular dichroism revealed folding patterns comparable with those of the wild-type complex, indicating that the mutations had no significant effect on the secondary structure of the complex in solution (Fig. 6A).
FIGURE 6.

Interaction of wild-type and mutated LukG with the LukH proteins. A, CD spectra of wild-type and mutant LukGH complexes and LukH normalized to a concentration of 0.5 mg/ml were acquired in 50 mm sodium phosphate, pH 7.5, in the presence of 200 or 500 mm NaCl, respectively. B and C, binding of LukG to biotinylated LukH and LukH2 (B) or binding of LukG, LukG1, and LukG2 to biotinylated LukH (C) was detected by BLI measurements and expressed as response values (obtained after subtracting the values for nonspecific binding when biotinylated HlgC was immobilized on the sensor). D, cross-linking of LukG and LukH. Mixtures of wild-type and mutant LukG and LukH were cross-linked with glutaraldehyde for 2 or 1 min (lanes 2 and 1 in each group, respectively) in the indicated combinations and resolved by SDS-PAGE. Untreated samples are shown in lanes 0.
The mutant monomers LukG1, LukG2, LukH1, and LukH2 expressed as insoluble proteins were purified from inclusion bodies. LukG1, LukG2, and LukH2 purification yields were similar to those observed with LukG and LukH and their CD spectra were also comparable. The LukG monomers had to be kept in highly alkaline formulation buffer, pH 10.2, to prevent aggregation, and under these conditions all LukG proteins showed mainly random coil structures. The LukH1 purification yield was significantly lower than observed with LukH, and the CD spectra revealed a partially unfolded structure (Fig. 6A). Therefore LukH1 was not included in structure-function analyses.
Dimer formation by the individual components was monitored by BLI by immobilizing biotinylated LukH or LukH2 on streptavidin sensors and measuring the binding of LukG to the preloaded sensors. We observed significantly lower binding of LukG to LukH2 compared with LukH (Fig. 6B), suggesting that the mutated charged amino acid residues in LukH are important for dimer formation. The binding strengths of the three LukG mutants to biotinylated LukH also showed significant differences. We detected comparable binding with wild-type LukG and LukG1, whereas LukG2 displayed a very weak interaction (no signal above the nonspecific binding observed when biotinylated HlgC was used as negative control) (Fig. 6C).
The interface 2 mutants LukH2 and LukG2 were unable to form dimers with LukG and LukH, respectively, based on cross-linking with glutaraldehyde and analysis by SDS-PAGE. In contrast, the interface 1 mutant LukG1-LukH interaction was comparable with wild-type LukG-LukH (Fig. 6D).
Considering all of these data, we concluded that interface 2 is involved in dimer formation, whereas interface 1 forms the dimer-dimer interface in the oligomeric pore structure. Furthermore, the additional three salt bridges in LukGH, not found in other S. aureus bi-component toxins, are crucial for dimerization.
Mutations Disrupting the Salt Bridges Formed by LukGH Monomers and Dimers Abolish Cytolytic Activity
To determine the functional consequences of removing the salt bridges between LukH and LukG and the LukG-LukH dimers, we measured cytolytic activity of the LukGH mutants using differentiated HL-60 cells. We could not detect cytolytic activity with interface 1 or interface 2 mutants, except a weak activity with LukG-LukH2 that was ∼100-fold lower than that observed with wild-type LukG + LukH (Fig. 7A). This indicates that the ionic interactions in both LukG and LukH at the monomer interface and/or dimer-dimer interface (oligomerization) are crucial for formation of functional pores on the target cell membrane.
FIGURE 7.

Cytolytic activity and binding of LukGH is abolished by mutations affecting the electrostatic interactions of LukG and LukH at both interface 1 and 2. A, mixtures of individually expressed LukG and LukH monomers and co-expressed complexes were used in the indicated concentration range to intoxicate differentiated HL-60 cells. Single components were used as controls. Cell viability was determined by luminescent measurement of cellular ATP content. Data are expressed as mean ± S.E. from two independent experiments. B, cell binding of wild-type and mutant proteins to human PMNs and differentiated HL-60 cells was determined using biotinylated toxin components as indicated and quantified by flow cytometry measurement. Results represent the mean ± S.E. of three and two independent experiments with PMNs and differentiated HL-60 cells, respectively. C, differentiated HL-60 cells were preincubated with 14 nm LukGH mutants as indicated for 30 min, followed by intoxication with wild-type LukGH (left panel) and LukSF (right panel) for 4 h in the indicated concentration range. Results represent the mean ± S.E. of three independent experiments.
In subsequent experiments, we tested the same mutants for binding to LukGH target cells. Although we could not detect binding with any of the wild-type or mutated monomers or interface 2 mutant LukGH complexes, interface 1 mutants and wild-type LukGH displayed comparable surface staining on both human PMNs and differentiated HL-60 cells (Fig. 7B).
Next we measured the interaction with CD11b that has been shown to be the cellular receptor for LukGH, more precisely its I-domain (14).4 In BLI-based binding experiments using the recombinantly expressed I-domain of human CD11b encompassing amino acids Gly111-Gly321 (huCD11bI), we detected strong interaction with the wild-type LukGH complex or LukG + LukH mixture with a Kd of ∼3 to 4 nm (Table 5). No binding to the mouse counterpart of the receptor was observed, consistent with previously reported data (18) and with the lack of lysis of mouse PMNs (data not shown). Similar Kd and response values were observed with LukG1H, LukGH1, and LukG1H1 complexes or the LukG1 + LukH mixture in a broad range of concentrations (2.8–140 nm) (Table 5). However, the LukG + LukH2 and LukG2 + LukH mixtures showed a significantly reduced binding to huCD11bI (>25-fold), particularly at lower concentrations, indicating that the mutations in the second interface destabilize the dimer and hence the interaction with the receptor.
TABLE 5.
Binding of wild-type and mutated LukGH and LukH to the human CD11b I-domain
Dissociation constants and BLI response values for LukGH and LukH binding to biotinylated huCD11bI immobilized on streptavidin sensors are shown. Decrease in LukGH cytolytic potency (expressed as increase in EC50 values) with differentiated HL-60 cells in the presence of mutated LukG and LukH proteins is derived from experiments shown in Fig. 7C.
| Toxin (14 nm) | Kd with human CD11bI | Response max | EC50 increase |
|---|---|---|---|
| nm | nm | -fold | |
| LukGH | 2.9 | 2.8 | 1.0 |
| LukG + LukH | 4.0 | 2.4 | |
| LukG1H | 4.3 | 2.4 | 57.5 |
| LukG1 + LukH | 8.4 | 1.9 | 20.8 |
| LukGH1 | 4.4 | 2.4 | 68.7 |
| LukG1H1 | 4.4 | 2.4 | 35.6 |
| LukG + LukH2 | 106 | 0.91 | 0.9 |
| LukG2 + LukH | 146a | 0.37 | 1.2 |
| LukH | 103 | 0.69 | 1.0 |
| LukH2 | 172 | 0.15 | 1.0 |
a Partial fit of the data was used.
The LukG monomer did not show any binding to the human receptor, whereas the LukH interaction was detected and appeared to be ∼25-fold weaker than that of the LukGH dimers (KD = ∼100 nm) (Table 5). These data suggest that LukH is likely to be the primary contact site for the receptor; however, its interaction is weak in the absence of LukG.
We found that preincubation of the target cells with the interface 2 mutants LukG2 + LukH and LukG + LukH2 (0.5 μg/ml, 14 nm) did not influence cell lysis by LukGH (Fig. 7C). This is consistent with our observation that interface 2 mutants are not able to dimerize and bind to either target cells or the cellular LukGH receptor (Table 5). In contrast, all interface 1 mutants significantly reduced the cytolytic activity of wild-type LukGH suggesting competition for the receptor binding site. Notably, preincubation of the same target cells with mutant LukGH proteins had no effect on toxin sensitivity toward LukSF, which binds to different receptor(s) (C5aR and C5L2) (Fig. 7C). Preincubation of the target cells with LukH and LukH2 (up to 5 μg/ml, 140 nm) had no effect on cytolytic activity (data not shown), indicating that the binding of LukH to the receptor on the cell surface is much weaker than suggested by the BLI binding data with the isolated huCD11b I-domain.
LukG and LukH Proteins from Different S. aureus Lineages Can Form Active Toxin Pairs with Different Potencies
To identify which residues might be important for dimerization and oligomerization in addition to those forming salt bridges we tested the formation of active LukGH using three different LukG and LukH variants. LukG and LukH from TCH1516 and MSHR1132 formed non-cognate complexes that had potencies comparable with homologous pairs, whereas the toxin pairs including LukH from MRSA252 were significantly less potent (Fig. 8A). Analysis of the contact residues (defined as being within 4 Å of each other) revealed no significant differences in the LukH_MRSA252 variant that could potentially explain the weaker activity with non-cognate LukG. However, analysis of the amino acids that differ in the LukG and LukH variants and are found close to the two interfaces based on the crystal structure (at a distance <10 Å), revealed some potential contact points where LukG and LukH from the MRSA252 strain might have co-evolved. In interface 1, Trp187 in LukH_TCH1516 is conserved in LukH_MSHR1132 but is replaced by Arg (at position 185) in LukH_MRSA252. Trp187 is in close proximity to Ser30 and Gln31 in LukG_USA300 that are conserved in LukG_MSHR1132 but changed to Thr and Glu (at positions 35 and 36, respectively) in LukG_MRSA252 (Fig. 8B). The Arg185 in LukH_MRSA252 and Glu36 in LukG_MRSA252 could potentially interact electrostatically. In interface 2, Glu51 in LukH_TCH1516 that is conserved in LukH_MSHR1132 is replaced by Val in LukH_MRSA252 (amino acid 46) and is in close proximity to Asn95 and Asn98 in LukG_TCH1516. Although these residues are conserved in LukG_MRSA252 (Asn100 and Asn104), they are further away from each other due to insertion of Ser102 that could provide a closer contact with the hydrophobic side chain of Val46 (Fig. 8C). Gly202 in LukH_TCH1516, conserved in LukH_MSHR1132, is replaced by Asn in LukH_MRSA252, and is in close proximity (∼7 Å) with Leu174 in LukG_TCH1516, which is Ser in LukG_MRSA252. The two polar residues in the MRSA252 variant could potentially form H-bonds. In addition, Lys98 in LukG_MSHR1132, which is an Asn in the other LukG variants, could interact with either Glu46 or Glu48 in LukH_MSHR1132 (these correspond to Glu51 and Val53, respectively in LukH_TCH1516) to form a salt bridge. These data suggest that there are additional contact sites between LukG and LukH that strengthen the binding interaction.
FIGURE 8.

Active toxin formation by heterologous pairing by LukG and LukH variants. A, human PMNs were treated with mixtures of LukG and LukH monomers derived from TCH1516, MSHR1132, and MRSA252 S. aureus strains in the indicated concentration ranges. Cell viability was determined by luminescent measurement of cellular ATP content. Data are expressed as mean ± S.E. from three independent experiments. B, structure of LukG_TCH1516 (chain A, magenta) and LukH_TCH1516 (chain B, cyan) at interface 1. The side chains of the amino acids that differ in the MRSA252, MSHR1322, and H19 variants are shown as spheres. The side chains of the amino acids that are different in LukH_MRSA252 compared with both LukH_TCH1516 and LukH_MSHR1132 are shown as spheres in pink or yellow if found at the LukGH interface. C, structure of LukH (chain B, cyan) and LukG (chain C, magenta) at interface 2. Representations as described in B.
DISCUSSION
LukGH is one of the most potent of the S. aureus leukocidins toward human phagocytic cells. Its activity is comparable with the potency of LukSF. The lukGH genes are part of the core genome of S. aureus, whereas lukSF is carried by phages and only expressed by ∼5–10% of clinical isolates. Therefore, it is important to understand the mode of action of LukGH to counteract the lysis of human phagocytic cells by S. aureus.
LukGH displays unique features compared with the other bi-component leukocidins. Based on the lower solubility of the subunits of LukGH and lack of binding of LukH or LukG to phagocytic cells, we hypothesized that LukGH forms a complex in solution even before contacting its target cell. Biolayer interferometry and intrinsic tryptophan fluorescence measurements confirmed the interaction of LukH and LukG in solution with an affinity in the low nanomolar range, and based on DLS analysis the size of the complex suggested formation of heterodimers. In addition, we could co-purify LukH with affinity tagged LukG from E. coli by co-expressing the two components with improved solubility. While we were generating these data, DuMont et al. (18) reported co-purification of LukGH from S. aureus culture supernatants and showed that LukG and LukH could be cross-linked by glutaraldehyde.
LukGH displays significant sequence variations in the different S. aureus strains, not observed with other bi-component toxins. The amino acid conservation among the most different forms is in the range of 82–88%. The sequence identity among the other S- and F-components within one strain is up to 81 (between LukS and HlgC) and 82% (between LukF and LukD), respectively. LukSF, LukED, and the γ-hemolysins clearly represent different toxin entities with different species specificity, receptors, and overlapping, but not identical, cell-type specificity (3–5). Therefore, it was important to confirm that all LukGH variants expressed by S. aureus strains showing the most distant evolutionary relationship had comparable toxin activity toward human neutrophils and that LukGH from all strains formed heterodimers in solution.
Our data suggested a different mode of action than described for other bi-component toxins. Therefore, we determined the crystal structure of USA300-type LukGH to compare with those reported for LukSF and HlgAB. Diffraction quality crystals were formed only in the presence of the hydrophobic solvent MPD, also reported for HlgAB (11). The structure of LukGH revealed an octamer arrangement that formed spontaneously under the crystallization conditions, indicating that the octamer is the most stable oligomeric form of LukGH at high protein concentrations in the presence of MPD. This is in good agreement with several previous reports showing that the pore formed by LukSF consists of four molecules of each of the S- and F-components (9–11).
The roles of the two interfaces in the octamer, interface 1 and 2, in dimer formation or oligomerization on the cell surface were explored by site-directed mutagenesis. The three electrostatic interactions (salt bridges) between the rim domains in interface 2 were disrupted by mutating either the negatively charged amino acid residues Glu171, Asp189, and Asp191 in LukG or the positively charged residues Arg215, Arg234, and Arg240 in LukH to Ala. These mutations resulted in lack of binding of LukG to LukH in solution and prevented the potent cytolytic activity seen with wild-type LukGH. The interface 2 mutant LukG2H and LukGH2 complexes were insoluble when co-expressed in E. coli cells, confirming our earlier observations of low solubility of the single components. These data suggest that interface 2 constitutes the binding surface in the dimer. Importantly, the charged residues involved in these interactions in the predicted dimer interface are not conserved between any of the other S- and F-components but are instead fully conserved between the LukGH sequence variants, suggesting that only LukGH has evolved toward heterodimerization in solution.
In contrast, alanine mutations of the two salt bridges in interface 1 formed between the cap domains of LukG (Arg23, Lys217) and LukH (Asp75, Asp197) did not affect formation of soluble complexes in E. coli, dimerization in solution, or binding to target cells, but did eliminate toxin activity. These residues are conserved in all other S- and F-components. We therefore concluded that interface 1 is only required for the oligomerization of LukGH dimers on the cell surface.
The finding that interface 1 mutants were able to form a dimer was further supported by their ability to inhibit LukGH activity, unlike interface 2 mutants. This inhibition is most likely due to competition for the receptor binding sites. Pull-down experiments with biotinylated LukGH and membrane preparations of differentiated HL-60 cells and PMNs identified the CD11b-CD18 complex as binding partner (14).4 DuMont et al. (14) reported direct interaction with CD11b and mapped the LukGH binding site to the CD11b I-domain by showing that a monoclonal antibody specific for this domain inhibited binding of LukGH to target cells. We therefore measured the interaction of the recombinant CD11b I-domain with the wild-type and mutated LukGH complexes. We found that interface 1 mutants bound to the purified I-domain of CD11b with an affinity similar to that of the wild-type complex, whereas interface 2 mutants displayed greatly diminished interaction.
The decrease in LukGH potency in the presence of interface 1 mutant complexes is much greater than expected from a simple competition model. The concentration used for the mutant complex (12.75 nm) was in the range of the Kd value reported for the binding of LukGH to the full-length CD11b-CD18 complex (38 nm) that is ∼10-fold higher than the I domain of CD11b (14). However, the decrease in potency we detected was 10–100-fold. This indicates that toxin activity has a high dependence on receptor concentration, implying that cooperative binding to more than one receptor site is required for the formation of the LukGH pore. This is also supported by the fact that monovalent binding between LukGH and CD11b occurs in the low nanomolar range, whereas LukGH EC50 is at least 10-fold lower (∼100 pm, 10 ng/ml).
Cell binding and receptor identification studies with the bi-component leukocidins suggested that it is the S-component of these toxins (LukS, LukE, and HlgC) that binds to phagocytic cells and their respective receptors, whereas HlgAB seems to contact its main target cells, red blood cells, via the F-component (HlgB) (13–16, 34, 35). An interesting finding from our studies was the weak but detectable binding of LukH to the isolated CD11b I-domain (∼25-fold higher Kd relative to that of the LukGH dimer), not observed with LukG. The lack of binding to target cells by LukH and the absence of inhibition of LukGH cytolytic activity by LukH or LukH2 suggest that although the main contact point between the receptor and LukGH is most likely located in LukH, formation of the LukGH dimer is necessary for a sufficiently strong interaction to initiate oligomerization on the cell surface.
Our data suggest that disruption of complex formation between LukG and LukH is unlikely to be an efficient strategy to prevent LukGH-associated toxicity, because the two components form a complex either inside bacteria or shortly after their secretion. A more promising approach seems to be to inhibit binding of the dimer to the CD11b receptor or oligomerization.
These findings support the development of either small molecule or antibody therapeutics that inhibit LukGH toxin-mediated elimination of phagocytes and dendritic cells, both of which are crucial for effective innate and adaptive immune responses against S. aureus infections.
Acknowledgments
We thank the Campus Support Facility Unit for assisting in DLS and CD measurements. We acknowledge the work by the staff at beamline I911-3 (Lund, Sweden) and crystallization facility at the MAX IV Laboratory (Lund, Sweden). We thank Katharina Havlicek and Manuel Zerbs for technical help.
This work was supported by the Forschungsförderungsgesellschaft (FFG) “Basisprogramm” Grants 832915, 837128, 841918, and 845382 from the Austrian Research Promotion Agency (awarded to Arsanis Biosciences). This work was performed by employees of two biotech companies who own stock (A. B., H. R., L. S., D. T. L., I. D., A. T., K. G., B. M., S. W., M. J., and E. N.) and are inventors on patents (A. B., H. R., and E. N.).
The atomic coordinates and structure factors (code 4TW1) have been deposited in the Protein Data Bank (http://wwpdb.org/).
A. Badarau, H. Rouha, S. Malafa, L. Stulik, and E. Nagy, unpublished data.
- CAPS
- N-cyclohexyl-3-aminopropanesulfonic acid
- IMAC
- immobilized metal affinity column
- DLS
- dynamic light scattering
- MPD
- 2-methyl-2,4-pentanediol
- NCS
- non-crystallographic symmetry
- PMN
- polymorphonuclear neutrophil
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- BLI
- biolayer interferometry.
REFERENCES
- 1. Spaan A. N., Surewaard B. G., Nijland R., van Strijp J. A. (2013) Neutrophils versus Staphylococcus aureus: a biological tug of war. Annu. Rev. Microbiol. 67, 629–650 [DOI] [PubMed] [Google Scholar]
- 2. Rigby K. M., DeLeo F. R. (2012) Neutrophils in innate host defense against Staphylococcus aureus infections. Semin. Immunopathol. 34, 237–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vandenesch F., Lina G., Henry T. (2012) Staphylococcus aureus hemolysins, bi-component leukocidins, and cytolytic peptides: a redundant arsenal of membrane-damaging virulence factors? Front. Cell Infect. Microbiol. 2, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alonzo F., 3rd, Torres V. J. (2014) The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol. Mol. Biol. Rev. 78, 199–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aman M. J., Adhikari R. P. (2014) Staphylococcal bicomponent pore-forming toxins: targets for prophylaxis and immunotherapy. Toxins 6, 950–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gouaux E., Hobaugh M., Song L. (1997) α-Hemolysin, gamma-hemolysin, and leukocidin from Staphylococcus aureus: distant in sequence but similar in structure. Protein Sci. 6, 2631–2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Colin D. A., Mazurier I., Sire S., Finck-Barbançon V. (1994) Interaction of the two components of leukocidin from Staphylococcus aureus with human polymorphonuclear leukocyte membranes: sequential binding and subsequent activation. Infect. Immun. 62, 3184–3188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Menestrina G., Dalla Serra M., Comai M., Coraiola M., Viero G., Werner S., Colin D. A., Monteil H., Prévost G. (2003) Ion channels and bacterial infection: the case of beta-barrel pore-forming protein toxins of Staphylococcus aureus. FEBS Lett. 552, 54–60 [DOI] [PubMed] [Google Scholar]
- 9. Miles G., Movileanu L., Bayley H. (2002) Subunit composition of a bicomponent toxin: staphylococcal leukocidin forms an octameric transmembrane pore. Protein Sci. 11, 894–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jayasinghe L., Bayley H. (2005) The Leukocidin pore: evidence for an octamer with four LukF subunits and four LukS subunits alternating around a central axis. Protein. Sci. 14, 2550–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamashita K., Kawai Y., Tanaka Y., Hirano N., Kaneko J., Tomita N., Ohta M., Kamio Y., Yao M., Tanaka I. (2011) Crystal structure of the octameric pore of staphylococcal γ-hemolysin reveals the β-barrel pore formation mechanism by two components. Proc. Natl. Acad. Sci. U.S.A. 108, 17314–17319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DuMont A. L., Torres V. J. (2014) Cell targeting by the Staphylococcus aureus pore-forming toxins: it's not just about lipids. Trends Microbiol. 22, 21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spaan A. N., Henry T., van Rooijen W. J., Perret M., Badiou C., Aerts P. C., Kemmink J., de Haas C. J., van Kessel K. P., Vandenesch F., Lina G., van Strijp J. A. (2013) The staphylococcal toxin Panton-Valentine leukocidin targets human C5a receptors. Cell Host Microbe 13, 584–594 [DOI] [PubMed] [Google Scholar]
- 14. DuMont A. L., Yoong P., Day C. J., Alonzo F., 3rd, McDonald W. H., Jennings M. P., Torres V. J. (2013) Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc. Natl. Acad. Sci. U.S.A. 110, 10794–10799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reyes-Robles T., Alonzo F., 3rd, Kozhaya L., Lacy D. B., Unutmaz D., Torres V. J. (2013) Staphylococcus aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe 14, 453–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alonzo F., 3rd, Kozhaya L., Rawlings S. A., Reyes-Robles T., DuMont A. L., Myszka D. G., Landau N. R., Unutmaz D., Torres V. J. (2013) CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 493, 51–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gauduchon V., Werner S., Prévost G., Monteil H., Colin D. A. (2001) Flow cytometric determination of Panton-Valentine leucocidin S component binding. Infect. Immun. 69, 2390–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DuMont A. L., Yoong P., Liu X., Day C. J., Chumbler N. M., James D. B., Alonzo F., 3rd, Bode N. J., Lacy D. B., Jennings M. P., Torres V. J. (2014) Identification of a crucial residue required for Staphylococcus aureus LukAB cytotoxicity and receptor recognition. Infect. Immun. 82, 1268–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kato F., Sugai M. (2011) A simple method of markerless gene deletion in Staphylococcus aureus. J. Microbiol. Methods 87, 76–81 [DOI] [PubMed] [Google Scholar]
- 20. Gorrec F. (2009) The MORPHEUS protein crystallization screen. J. Appl. Crystallogr. 42, 1035–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ursby T., Unge J., Appio R., Logan D. T., Fredslund F., Svensson C., Larsson K., Labrador A., Thunnissen M. M. (2013) The macromolecular crystallography beamline I911–3 at the MAX IV laboratory. J. Synchrotron Radiat. 20, 648–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kabsch W. (2010) XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Evans P. R. (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 67, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stein N. (2008) CHAINSAW: a program for mutating PDB files used as templates in molecular replacement. J. Appl. Crystallogr. 41, 641–643 [Google Scholar]
- 28. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 31. Romero-Steiner S., Libutti D., Pais L. B., Dykes J., Anderson P., Whitin J. C., Keyserling H. L., Carlone G. M. (1997) Standardization of an opsonophagocytic assay for the measurement of functional antibody activity against Streptococcus pneumoniae using differentiated HL-60 cells. Clin. Diagn. Lab. Immunol. 4, 415–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Monma N., Nguyen V.T., Kaneko J., Higuchi H., Kamio Y. (2004) Essential residues, W177 and R198, of LukF for phosphatidylcholine-binding and pore-formation by staphylococcal γ-hemolysin on human erythrocyte membranes. J. Biochem. 136, 427–431 [DOI] [PubMed] [Google Scholar]
- 33. Yokota K., Kamio Y. (2000) Tyrosine72 residue at the bottom of rim domain in LukF crucial for the sequential binding of the staphylococcal γ-hemolysin to human erythrocytes. Biosci. Biotechnol. Biochem. 64, 2744–2747 [DOI] [PubMed] [Google Scholar]
- 34. Kaneko J., Ozawa T., Tomita T., Kamio Y. (1997) Sequential binding of Staphylococcal γ-hemolysin to human erythrocytes and complex formation of the hemolysin on the cell surface. Biosci. Biotechnol. Biochem. 61, 846–851 [DOI] [PubMed] [Google Scholar]
- 35. Nguyen V. T., Kamio Y., Higuchi H. (2003) Single-molecule imaging of cooperative assembly of γ-hemolysin on erythrocyte membranes. EMBO J. 22, 4968–4979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goujon M., McWilliam H., Li W., Valentin F., Squizzato S., Paern J., Lopez R. (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–W699 [DOI] [PMC free article] [PubMed] [Google Scholar]