

Background: PTH1–34 alleviates radiation-induced osteoporosis in a preclinical radiotherapy model.
Results: PTH1–34 attenuates radiation damage on osteoblasts by increasing Ku70 amount, accelerating DNA repair, and suppressing osteoblast apoptosis. Pathway analyses identify canonical Wnt pathway as an important mediator.
Conclusion: PTH1–34 blocks radiation-induced osteoblast apoptosis through activation of β-catenin.
Significance: PTH1–34 or Wnt agonist could be possible therapy for radiation-induced osteoporosis.
Keywords: Apoptosis, DNA Repair, Osteoblast, Parathyroid Hormone, Wnt Pathway, Ionizing Radiation
Abstract
Focal radiotherapy for cancer patients has detrimental effects on bones within the radiation field and the primary clinical signs of bone damage include the loss of functional osteoblasts. We reported previously that daily injection of parathyroid hormone (PTH, 1–34) alleviates radiation-induced osteopenia in a preclinical radiotherapy model by improving osteoblast survival. To elucidate the molecular mechanisms, we irradiated osteoblastic UMR 106-01 cells and calvarial organ culture and demonstrated an anti-apoptosis effect of PTH1–34 on these cultures. Inhibitor assay indicated that PTH exerts its radioprotective action mainly through protein kinase A/β-catenin pathway. γ-H2AX foci staining and comet assay revealed that PTH efficiently promotes the repair of DNA double strand breaks (DSBs) in irradiated osteoblasts via activating the β-catenin pathway. Interestingly, Wnt3a alone also blocked cell death and accelerated DNA repair in primary osteoprogenitors, osteoblastic and osteocytic cells after radiation through the canonical signaling. Further investigations revealed that both Wnt3a and PTH increase the amount of Ku70, a core protein for initiating the assembly of DSB repair machinery, in osteoblasts after radiation. Moreover, down-regulation of Ku70 by siRNA abrogated the prosurvival effect of PTH and Wnt3a on irradiated osteoblasts. In summary, our results identify a novel role of PTH and canonical Wnt signaling in regulating DSB repair machinery and apoptosis in osteoblasts and shed light on using PTH1–34 or Wnt agonist as possible therapy for radiation-induced osteoporosis.
Introduction
Radiotherapy is one of the major treatments for tumor malignancy and is accompanied by adverse late effects, including its damage to the skeleton within the radiation field (1). Such damage leads to a spectrum of bone changes from mild osteopenia to osteoradionecrosis with an increased risk of in-field fractures. These fractures are common and clinically significant, particularly in patients receiving radiotherapy in the pelvic region (2–5), and pelvic fractures are a substantial cause of morbidity and mortality in the elderly (6, 7). Understanding the mechanisms behind these adverse effects of radiation on bone and identifying new treatments for such bone disorders is imperative to improve the quality of life for these patients.
Bone is a dynamic organ that undergoes constant remodeling, which requires a balance between osteoblast and osteoclast activities. Preclinical and cell culture studies indicate that radiation damages bone formation by decreasing the number of osteoblasts, arresting their cell cycle progression, altering their differentiation ability, and sensitizing them toward apoptosis signals (8–11). It is generally accepted that ionizing radiation induces programmed cell death through its genotoxic actions. Radiation exposure rapidly generates a large amount of DNA lesions, among which DNA double strand break (DSB)4 is the most deleterious one that causes cell death (12). Cells respond by initiating a set of highly spatiotemporally orchestrated events to repair damage and restrict cell cycle progression. In the meantime, DNA damage triggers both death and survival signals. If lesions are not resolved in a timely manner, cell death signals may prevail. We expect that radiation will cause DNA damage and apoptosis in mature osteoblasts in a similar manner. Therefore, signaling pathways that improve DNA repair and prevent apoptosis in osteoblasts would be promising therapeutic targets for the treatment of radiation-induced bone loss.
Intermittent injection of parathyroid hormone (PTH, 1–34) is the only Food and Drug Administration-approved treatment for osteoporosis that greatly stimulates bone formation. In bone, the PTH receptor (PTH1R), a seven-transmembrane domain receptor coupled to G-proteins, is highly expressed in osteoblasts and osteocytes (13). Binding of PTH to its receptor immediately activates two well defined signal transduction pathways in osteoblasts: the PKA pathway (14) in which Gαs stimulates production of cAMP and activation of PKA, and PKC pathway (15) in which Gαq activates phospholipase Cβ with subsequent formation of diacylglycerol and 1,4,5-inositol trisphosphate and PKC activation. We and others (16) have demonstrated that PKA pathway is the major route responsible for the anabolic action of PTH on bone in rodents. Several cellular mechanisms have been proposed to explain the potent stimulatory effect of PTH on new bone formation (17, 18). One of these mechanisms is through its suppressive action on the apoptosis of mature osteoblasts. Previous studies have shown that PTH treatment attenuates the apoptosis of mature osteoblasts lining trabecular bone surface in rodents under normal (19, 20) and pathological conditions (21, 22).
We recently established a clinically relevant radiation model using skeletally mature rats and a newly available small animal radiation research platform that replicates focal clinical radiotherapy in rodents. Using this model, we reported that daily injections of PTH prevent local trabecular bone loss in rat proximal tibia after a therapeutic dose of radiation by protecting the mature osteoblasts and osteocytes from radiation-induced apoptosis (23). However, the molecular mechanisms have not been investigated. In the studies described herein, we adopted in vitro and ex vivo approaches to examine the survival effect of PTH on osteoblasts after radiation and explore the downstream pathways. We found that PTH prolongs the survival of osteoblasts via activation of PKA followed by Wnt/β-catenin pathway. The canonical Wnt pathway is one of the most important signaling pathways governing bone homeostasis (24). Whereas its osteogenic differentiation activity is well studied, its anti-apoptosis action on osteoblasts has been largely ignored. Our present study revealed a novel role of both PTH1R and Wnt/β-catenin pathways in stimulating DNA repair machinery after radiation. Together with their similar effects on regulating apoptosis, these two pathways are promising therapeutic targets for preserving or elevating bone formation and hence bone strength after radiotherapy.
EXPERIMENTAL PROCEDURES
Chemicals
Recombinant human PTH1–34, human PTH1–31, and bovine PTH3–34 was purchased from Bachem. U0126, wortmannin, and H89 were purchased from Calbiochem. IWR-endo was a product of Cayman Chemicals. 8-Bromoadenosine-3′,5′-cyclic monophosphate, ethidium bromide (EB), and acridine orange (AO) were purchased from Sigma-Aldrich. Phorbol 12-myristate 13-acetate was purchased from EMD Millipore. 2′,7′-Dichlorodihydrofluorescein diacetate was purchased from Molecular Probes®.
Cell Culture, Radiation, and Apoptosis Assay
UMR106-01 cells were maintained in minimum essential medium (MEM) supplemented with 5% fetal bovine serum (FBS), 100 international units/ml penicillin and 100 μg/ml streptomycin. Rat primary calvarial osteoprogenitors were obtained from neonatal calvariae by sequential collagenase and trypsin digestion as described previously (25) and cultured in the growth medium (10% MEM, 1% nonessential amino acids, 100 international units/ml penicillin and 100 μg/ml streptomycin). Osteocyte cell line, OCY454 (kindly provided by Dr. Paola Divieti-Pajevic), was cultured in α-MEM containing 10% FBS, 100 international units/ml penicillin, and 100 μg/ml streptomycin. For radiation experiments, cells were seeded in 12-well plates at 13,000 cells/cm2 for 2 days. Radiation by X-Rad 320i was then applied to the cells at a rate of 1.04 Gy/min to a final dosage of 8 Gy for UMR and Ocy454 cells and 24 Gy for primary osteoprogenitors. These dosages, which correspond to the dosage used in our previous animal study (23), were determined by our pilot experiments that radiation caused significant cell death in those cells at those doses. PTH1–34 (10 nm) was added to the medium 30 min later. To treat cells with Wnt3a, conditioned medium (CM) was collected from Wnt3a-overexpressing L cells that were cultured in serum-depleted DMEM for 1 day and applied at 50% (v/v) to cells at 30 min after radiation. CM from non-overexpressing L cells was used as control. Two days later, EB/AO staining (26) was performed to detect apoptotic cells. Apoptotic cells were identified as cells having condensed chromatin (green at early apoptotic stage and red at late apoptotic stage). Living cells had a characteristic green chromatin staining with a normal morphology of the nucleus. For TUNEL staining to detect apoptosis, cells were stained with Apoptag in situ peroxidase kit (Millipore).
siRNA Knockdown Studies
Cells at 50–60% confluency were transfected with 10 nm siRNAs for β-catenin (RNC.RNAI.N053357.12.1,), Ku70 (RNC.RNAI.N139080.12.1 and RNC.RNAI.N139080.12.3), or NC1-control duplex (TriFECTa siRNA kit, Integrated DNA technologies) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. One day later, cells were radiated and treated with either PTH1–34 (10 nm) or Wnt3a CM.
Immunofluorescence
After radiation, cells were fixed with 4% paraformaldehyde, permeabilized, and incubated with antibodies against γ-H2AX (Millipore), β-catenin (BD Biosciences), Ku70 (Abcam), or cleaved caspase-3 (Cell Signaling) at room temperature for 1 h, followed by Alexa Fluor-conjugated fluorescent secondary antibodies and DAPI staining. The number of γ-H2AX foci per cell and percentages of nuclear β-catenin+ and cleaved caspase-3+ cells were quantified under a fluorescence microscope (Nikon Eclipse 90i) using ImageJ software. For Ku70, fluorescence intensity in individual cells was quantified. At least 200 cells per experiment point were examined.
Comet Assay
Cells were analyzed with Trevigen Comet Assay® kit under neutral conditions. Images were uniformly adjusted to subtract the camera background. For each experiment point, at least 100 cells were measured. Using a comet assay plugin from ImageJ that allows a mathematical calculation of DNA contents in the head and tail of a comet, DNA damage was quantified as the percentage of tail DNA.
Immunoblotting
Cells were lysed in a modified RIPA buffer (1× RIPA buffer, 2 mm EDTA, 50 mm β-glycerophosphate, 1 mm Na3VO4, 1 mm PMSF, and protease inhibitor mixture). Fifteen μg of protein from each sample was separated on 10% SDS-PAGE gels and then transferred to polyvinylidene difluoride membranes (Bio-Rad). Protein amount was measured by immunoblotting with the following antibodies: anti-γ-H2AX (Millipore), anti-Ku70 (Abcam), anti-β-catenin (BD Biosciences), and anti-β-actin (Santa Cruz Biotechnology). The bands were visualized using corresponding secondary antibodies conjugated with horseradish peroxidase followed by chemiluminescence (Amersham Biosciences ECLTM, Western blotting detection reagent, GE Healthcare). Bands were quantified from three individual gels normalized against β-actin, which was used as loading control.
Culture and Radiation of Calvariae
All animal work performed in this report was approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania. Calvariae harvested from neonatal C57BL/6 mouse pups or Sprague-Dawley rat pups (postnatal days 3–5) were cultured as relatively intact tissue and processed for paraffin sections as described previously (26, 27). Briefly, calvariae, cleaned free of surrounding soft tissue, were cut into two equal halves at the sagittal suture. Each hemi-calvaria was placed on a metallic mesh, in DMEM with 10% FBS, 100 international units/ml penicillin, and 100 μg/ml streptomycin. One day later, calvariae were irradiated with X-Rad 320i at a rate of 1.04 Gy/min to a final dosage of 8 Gy. Control calvariae were placed in the irradiator for the same period without radiation. Then, calvariae were immediately pretreated with either dimethyl sulfoxide or IWR-endo (10 μm) for 30 min, followed by addition of PTH1–34 (10 nm) or Wnt3a CM.
Immunohistology of Calvariae
Calvariae were fixed in 10% neutralized buffered formalin overnight, decalcified in 10% EDTA for 2 days, processed, and embedded in paraffin. Five μm-thick sections were cut parallel to the plane of the sagittal suture, and comparisons were made at equivalent positions in each sample. Sections of mouse calvariae were stained with Apoptag in situ peroxidase kit (Millipore), anti-γ-H2AX (Cell Signaling), and anti-cleaved caspase-3 (Cell Signaling) antibodies. Sections of rat calvariae were stained with anti-Ku70 (Abcam) antibody, which does not recognize mouse Ku70. Target immunogens were detected either by secondary antibodies followed by DAB detection (VECTASTAIN® ABC kit, for TUNEL assay and cleaved caspase-3 staining) or by a secondary goat antibody conjugated with Alexa Fluor 594 (for γ-H2AX foci staining) or Alexa Fluor 488 (for Ku70 staining). Images were acquired under the microscope at 200× magnification, and the numbers of total, apoptotic, and cleaved caspase-3+ osteoblasts, as well as γ-H2AX foci and Ku70 nuclear staining in osteoblasts were quantified using ImageJ software.
Statistics
All results are expressed as mean ± S.E. of triplicate measurements. All experiments were repeated independently at least three times with a similar conclusion, and only data from a representative experiment were presented. Unpaired Student's t test was used to evaluate the statistical difference between control and treated groups. In cases of multiple groups, differences were first analyzed by one-way analysis of variance followed by Bonferroni's post test. Values of p < 0.05 were considered significant.
RESULTS
PTH Alleviates Radiation-induced Apoptosis in Osteoblasts via PKA/β-catenin Pathway
Our previous studies demonstrated that PTH1–34 daily injections are able to protect mature osteoblasts lining trabecular bone surfaces from cell death induced by small animal radiation research platform focal radiation (23). To extend these in vivo findings and to investigate the underlying mechanisms, we irradiated osteoblast-like UMR106–01 cells at 8 Gy and treated them with 10 nm PTH1–34 30 min later. Cell apoptosis assay using EB/AO staining showed that radiation strongly increased the percentage of apoptotic cells from 1.0 to 12.6% and that PTH1–34 greatly diminished the apoptotic cells to 2.6% (Fig. 1A). Another apoptotic assay (TUNEL staining) generated the same results (Fig. 1B). Next, we used specific inhibitors for pathway analysis. After binding to its receptor PTH1R, PTH1–34 stimulates both PKA and PKC pathways (18). In line with our previous report that the major signaling pathway mediating the anabolic action of PTH is the cAMP/PKA pathway (16), the PKA inhibitor, H89, completely abrogated the protective effect of PTH (Fig. 1A). To further confirm this, truncated PTH fragments that specifically activate individual pathways and pathway-specific activators were tested (Fig. 1C). Only PTH1–31, which activates PKA, and PKA activator (8-bromoadenosine-3′,5′-cyclic monophosphate) rescued cells from radiation-induced apoptosis, whereas PTH3–34, which activates PKC, and PKC activator (phorbol 12-myristate 13-acetate) had no such effect. Other inhibitors, such as MEK inhibitor (U0126) and PI3K inhibitor (wortmannin), did not alter the survival effect of PTH on irradiated UMR cells (Fig. 1A). Interestingly, IWR-endo, a Wnt pathway inhibitor that acts by stabilizing the destruction complex for β-catenin, also abolished the protective effect of PTH (Fig. 1A), implying that the canonical Wnt/β-catenin pathway contributes to the PTH action on irradiated osteoblasts. Neither PKA nor Wnt inhibitors affected osteoblastic cell survival under non-radiated and radiated conditions without PTH treatment (data not shown).
FIGURE 1.

PTH protects osteoblasts from radiation-induced apoptosis via PKA and β-catenin pathways. A, analysis of apoptotic UMR106-01 cells by EB/AO staining at 2 days after radiation with or without 10 nm PTH treatment. For inhibitor assay, control (con, DMSO) and inhibitors (10 μm H89, 10 μm IWR-endo (IWR), 20 μm U0126, and 1.5 μm wortmannin (WMN)) were added to the culture 30 min before PTH. &, p < 0.001 radiated (R) versus non-radiated cells (NR); %, p < 0.01; δ, p < 0.001 PTH versus vehicle (veh). B, analysis of apoptotic UMR cells by TUNEL staining at 2 days after radiation with or without 10 nm PTH treatment. &, p < 0.001 versus NR; δ, p < 0.001 PTH versus vehicle. C, PTH stimulation of cell survival is PKA-dependent. UMR cells were treated with various PTH peptides (1–34, 1–31, and 3–34, 10 nm) or pathway-specific activators (0.5 mm 8-bromoadenosine-3′,5′-cyclic monophosphate (8BA) and 3 μm phorbol 12-myristate 13-acetate (PMA)). &, p < 0.001 versus NR; δ, p < 0.001 versus R and vehicle. D and E, immunofluorescence staining of cleaved caspase-3 (casp3) in UMR cells at 8 h after radiation with and without PTH and inhibitor treatments. The percentages of cells with cleaved casp3 nuclear staining were quantified (E). $, p < 0.01 versus NR; %, p < 0.01 versus R; ^, p < 0.01 versus R, PTH, and control. F–I, calvariae harvested from neonatal mouse pups were irradiated and treated with 10 nm PTH1–34 in growth medium. Control (con, DMSO) and 10 μm IWR-endo were added to the culture 30 min before PTH. Two days later, calvariae were fixed for paraffin sections followed by TUNEL (F) and cleaved casp3 (H) staining. The percentages of apoptotic (G) and cleaved casp-3+ (I) osteoblasts (OB) in calvariae were quantified. &, p < 0.001 versus NR; δ, p < 0.001 versus R; ^, p < 0.01; Σ, p < 0.001 versus R, PTH, and control.
Caspase-3 is the major effector caspase that provokes cellular destruction by cleaving several hundreds of cellular proteins, and therefore, cleaved (active) caspase-3 is a hallmark of apoptosis (28). Immunofluorescence staining revealed that radiation strikingly increased the amount of cleaved caspase-3 in the nucleus (Fig. 1, D and E). PTH1–34 treatment greatly reduced their nuclear amount, and the remaining protein was mostly located in the cytoplasm. Moreover, consistent with EB/AO apoptosis staining results, H89 and IWR-endo each abrogated this PTH effect.
To confirm above in vitro data, we harvested calvariae from newborn mouse pups and cultured them ex vivo. This calvarial organ culture preserves much of the skeletal structure and offers greater physiological relevance than osteoblastic cell lines. We irradiated these calvariae at 8 Gy followed by addition of 10 nm PTH1–34. After 2 days, both TUNEL and cleaved caspase-3 staining (Fig. 1, F–I) showed that radiation remarkably increased the percentage of apoptotic osteoblasts from 10% to ∼50% and that PTH treatment almost completely blocked the increase in cell death. Similar to cell culture results, IWR-endo reversed the anti-apoptotic effect of PTH, substantiating our conclusion that PTH treatment attenuated radiation-induced osteoblastic cell death in a canonical β-catenin-dependent manner.
Previous reports have demonstrated that PTH regulates β-catenin activity through multiple mechanisms such as promoting Lrp6 phosphorylation (29), recruiting the adapter protein of Wnt receptor Dishevelled (30), and activating Smad3 (31), etc. We found that neither radiation nor PTH altered the mRNA expression of β-catenin in osteoblastic cells (data not shown). However, radiation slightly increased its protein amount (Fig. 2A) and stimulated its nuclear translocation (Fig. 2, B and C), a step required for β-catenin to act as a co-transcriptional factor to activate Wnt target genes. Compared with vehicle, PTH treatment further enhanced β-catenin amount and its nuclear translocation in osteoblasts after radiation. Pretreating cells with inhibitors H89 or IWR-endo not only decreased β-catenin amount but also completely abolished its nuclear translocation after radiation and PTH treatment, indicating that activation of β-catenin by PTH is through the PKA pathway (Fig. 2, B and C). To further validate the role of β-catenin in PTH action, we used siRNA against β-catenin to eliminate PTH-induced nuclear translocation of β-catenin after radiation (Fig. 2D). Consistent with the inhibitor results, β-catenin siRNA also abrogated the prosurvival effect of PTH on radiated cells (Fig. 2E). Taken together, our data clearly demonstrate that PTH1–34 activates PKA followed by β-catenin pathway to alleviate radiation-induced osteoblastic cell death.
FIGURE 2.

PTH activates β-catenin in osteoblasts after radiation. A, immunoblotting of β-catenin in UMR cells after 6 h of radiation in the absence and in the presence of PTH and pathway-specific inhibitors. B, immunofluorescence of β-catenin in UMR cells treated with the same conditions as in A. Arrows point to nuclear β-catenin-positive cells. C, the numbers of cells with β-catenin nuclear staining were quantified. &, p < 0.001 versus non-radiated cells (NR); %, p < 0.01 versus radiated cells (R); Σ, p < 0.001 versus R, PTH, and control (DMSO) for inhibitors. D, the numbers of cells with β-catenin nuclear staining in UMR cells pretreated with either 10 nm mock or β-catenin siRNA were quantified at 6 h after radiation. &, p < 0.001 versus NR; δ, p < 0.001 versus R and mock siRNA; Σ, p < 0.001 versus R, PTH, and mock siRNA. E, the percentages of apoptotic UMR cells were quantified at 2 days after radiation. &, p < 0.001 versus NR; δ, p < 0.001 versus R and mock siRNA; Σ, p < 0.001 versus R, PTH, and mock siRNA.
PTH1–34 Accelerates DNA Repair after Radiation
Generation of reactive oxygen species (ROS) is one of the immediate actions for radiation to damage cells and induce cell death. To study whether PTH affects this step, we irradiated UMR cells, treated them with vehicle or PTH1–34, and then measured ROS levels in the cells using 2′,7′-dichlorodihydrofluorescein diacetate, a ROS indicator. Quantification of its oxidized form by fluorometer revealed that PTH did not affect the generation of intracellular ROS amount caused by radiation (Fig. 3A).
FIGURE 3.

γ-H2AX assay demonstrates that PTH accelerates DNA repair in osteoblasts after radiation in a β-catenin-dependent manner. A, PTH does not affect ROS generation by radiation. UMR cells were radiated and treated with either vehicle or 10 nm PTH for 2 h. 2′,7′-Dichlorodihydrofluorescein diacetate was added to the culture during the last hour. Cells were then collected for measuring relative fluorescence unit (RFU). &, p < 0.001 versus its respective non-radiated cells (NR). B and C, the time course of immunofluorescence staining of γ-H2AX in UMR cells after radiation with and without PTH treatment. The numbers of γ-H2AX foci per cells were quantified (C). Note that PTH was added at 30 min post-radiation. #, p < 0.05; %, p < 0.01; δ, p < 0.001 versus vehicle (veh). D and E, immunofluorescence of γ-H2AX in calvarial culture after radiation and PTH treatments. The numbers of γ-H2AX foci per calvarial osteoblast (OB) were quantified (E). The arrow points to γ-H2AX staining in calvarial osteoblasts. &, p < 0.001 versus NR; δ, p < 0.001 versus R and vehicle. F and G, the γ-H2AX foci staining experiment was repeated in UMR cells in the absence and presence of PKA (H89) and β-catenin (IWR-endo) inhibitors. The numbers of γ-H2AX foci per cell were quantified (G). &, p < 0.001 versus NR; %, p < 0.01 PTH versus vehicle. H, the γ-H2AX foci/cell was calculated in UMR cells treated with either 10 nm of mock or β-catenin siRNA. &, p < 0.001 versus NR; δ, p < 0.001 versus R and vehicle; ^, p < 0.01 versus R, PTH, and mock siRNA.
Radiation induces highly lethal DNA damage either directly or indirectly through ROS. Among a variety of DNA lesions, DSB is considered to be the major cause of cell death. A sensitive method to detect DSBs is the immunofluorescence staining of γ-H2AX, the phosphorylated histone H2A variant (32). This product, generated rapidly after radiation at the DSB site, spreads hundreds of kilobases along the chromatin from DSBs to form visible foci under microscope and hence, is a DSB marker. As shown in Fig. 3, B and C, the number of γ-H2AX foci per cell increased rapidly within 1 h after radiation, peaked ∼2.5 h, and gradually decreased afterward in vehicle-treated cells. Addition of PTH at 30 min after radiation initially had no effect, but significantly reduced the number of γ-H2AX foci at 2, 4, and 8 h after addition. Radiation causes similar DSB damage in calvarial osteoblasts as revealed by a 16-fold increase in the number of γ-H2AX foci per osteoblast lining the calvarial bone at 6 h postradiation (Fig. 3, D and E). PTH treatment following radiation greatly suppressed such increase, demonstrating that PTH is capable of accelerating DSB repair in osteoblasts after radiation. Next, inhibitor assays showed that the mitigation of γ-H2AX foci by PTH was abolished by pretreating cells with either H89 or IWR-endo (Fig. 3, F and G). Note that H89 and IWR-endo itself had no effect on the production of γ-H2AX under non-radiated condition. Furthermore, β-catenin siRNA strongly blocked the decrease of γ-H2AX foci number induced by PTH1–34 (Fig. 3H).
DSBs in irradiated cells can also be measured by the comet assay (33). During electrophoresis, the broken DNA fragments migrate out of the nucleus to form a comet tail, and thus, the volume of a comet tail correlates with the level of DNA damage in a given cell. We found that radiation induced a 4.2-fold increase in DNA damage at 8 h and that PTH treatment almost completely blocked such increase (Fig. 4, A and B). Pretreatment of cells with either H89 or IWR-endo abolished the effect of PTH in rescuing these DNA damage after radiation. Note that inhibitors themselves modestly but significantly increased DNA damage under non-radiated condition. Moreover, β-catenin siRNA had similar effects as IWR-endo (Fig. 4C). Taken together, the above results demonstrate that PTH accelerates the repair of radiation-induced DNA damage in osteoblasts through a PKA/β-catenin pathway.
FIGURE 4.
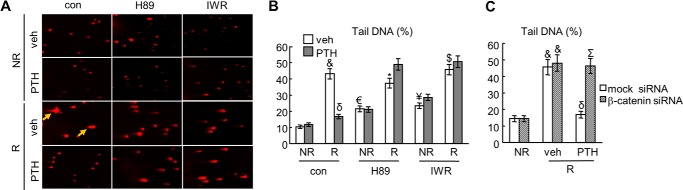
Comet assay confirms that PTH accelerates DNA repair in osteoblasts after radiation. A, representative images of UMR cells subjected to the comet assay and stained with EB. UMR cells were irradiated and treated with or without PTH and pathway-specific inhibitors (H89 and IWR-endo). Arrows point to comet tails. B, the percentages of tail DNA, an indicator of DNA damage, were quantified. *, p < 0.05; $, p < 0.01; &, p < 0.001 versus its respective vehicle (veh) non-radiated cells (NR); €, p < 0.05, ¥, p < 0.01 versus vehicle, NR, and control; δ: p < 0.001 PTH versus vehicle. C, the percentages of tail DNA was calculated in UMR cells treated with either 10 nm mock or β-catenin siRNA. &, p < 0.001 versus NR; δ, p < 0.001 versus veh, radiated (R), and mock siRNA; Σ, p < 0.01 versus R, PTH, and mock siRNA.
Activating Wnt Canonical Pathway Protects Osteoprogenitors, Osteoblasts, and Osteocytes from Radiation-induced Cell Death
Our experiments so far clearly indicate that activating Wnt canonical pathway is a critical step for conveying the survival action of PTH on osteoblasts after radiation. To further explore this mechanism and to investigate whether Wnt signaling itself could facilitate osteoblast survival after radiation, we treated UMR cells with Wnt3a, a canonical Wnt, after radiation. Apoptosis staining revealed that Wnt3a indeed strongly suppressed radiation-induced cell death of UMR osteoblastic cells (Fig. 5A). This survival effect relies on the canonical pathway, as addition of IWR-endo, but not U0126, completely abolished the Wnt3a action on UMR cells. Immunostaining of β-catenin confirmed that Wnt3a enhances the nuclear translocation of β-catenin and thus activates canonical Wnt pathway after radiation (Fig. 5B). β-Catenin siRNA treatment decreased the amount of nuclear β-catenin after Wnt3a and radiation treatment (Fig. 5B) and subsequently abolished the survival effect of Wnt3a on radiated UMR cells (Fig. 5C). Interestingly, Wnt3a not only acted on osteoblastic cells but also exhibited anti-apoptotic effects on other osteoblastic lineage cells after radiation. As shown in Fig. 5D, radiation drastically increased the number of apoptotic cells in both primary osteoprogenitors and osteocytic cells and co-treatment with Wnt3a substantially alleviated the detrimental effect of radiation on these cells.
FIGURE 5.

The canonical Wnt signaling protects osteoblasts from radiation-induced DNA damage and cell death. A, activating canonical Wnt signaling alleviates osteoblast apoptosis after radiation. UMR cells were irradiated, treated with Wnt3a CM, and subjected to EB/AO staining 2 days later. For inhibitor assay, inhibitors (IWR-endo and U0126) were added 30 min before addition of Wnt3a CM. The percentages of apoptotic cells were counted. &, p < 0.001 versus non-radiated cells (NR); δ, p < 0.001 versus radiated cells (R); %: p < 0.01 versus R, Wnt3a, and DMSO control (con). B, the numbers of cells with β-catenin nuclear staining in UMR cells pretreated with either 10 nm mock siRNA or β-catenin siRNA were quantified at 6 h after radiation and Wnt3a treatments. &, p < 0.001 versus NR; δ, p < 0.001 versus R, vehicle, and mock siRNA; Σ, p < 0.001 versus R, Wnt3a, and mock siRNA. C, the percentages of apoptotic UMR cells were quantified 2 days after treatments as described in B. &, p < 0.001 versus NR; δ, p < 0.001 versus R, vehicle, and mock siRNA; Σ, p < 0.001 versus R, Wnt3a, and mock siRNA. D, Wnt3a attenuates cell death in primary osteoprogenitors (left) and osteocytic cells (Ocy454, right) after radiation. Apoptosis assay with EB/AO staining was performed at 2 days after radiation and Wnt3a treatments. $, p < 0.01; &, p < 0.001 versus NR; %, p < 0.01; δ, p < 0.001 versus R and vehicle. E–H, Wnt3a decreases γ-H2AX foci number in radiated osteoblastic (UMR; E and F) and osteocytic (Ocy454; G) cells via activating the canonical β-catenin pathway. DKK1 (500 ng/ml) was added at the same time with Wnt3a CM. In H, cells were transfected with mock or β-catenin siRNA 1 day before radiation. $, p < 0.01; &, p < 0.001 versus NR; %, p < 0.01; δ, p < 0.001 versus R; #, p < 0.05; ^, p < 0.01 versus R, Wnt3a, and control (con); Σ, p < 0.001 versus R, Wnt3a, and mock siRNA. I–K, comet assay shows that Wnt3a reduces the amount of damaged DNA in radiated osteoblastic (UMR) and osteocytic (Ocy454) cells via activating the canonical β-catenin pathway. Representative images in UMR cells are shown in I, and the percentages of tail DNA are quantified in UMR (J) and Ocy454 cells (K). &, p < 0.001 versus NR; δ, p < 0.001 versus R and vehicle Σ, p < 0.001 versus R, Wnt3a, and mock siRNA.
To analyze DNA damage, both γ-H2AX foci staining and comet assay were performed. Identical to PTH1–34, Wnt3a was able to significantly reduce γ-H2AX foci number by 33% in osteoblastic UMR cells (Fig. 5, E and F). The same effect was also observed in an osteocytic cell line, Ocy454 (Fig. 5G). DKK1 is an extracellular antagonist of Wnt co-receptors LRP5 or LRP6, and it mainly suppresses the canonical pathway (34). Addition of either DKK1 or pretransfecting cells with β-catenin siRNA abolished Wnt3a-induced γ-H2AX foci reduction (Fig. 5, F–H), suggesting that Wnt3a regulates γ-H2AX amount through β-catenin. We observed similar results in the comet assay that Wnt3a treatment greatly mitigated the percentages of tail DNA after radiation in UMR cells and osteocytes, and this action of Wnt3a required the canonical β-catenin signaling (Fig. 5, I–K). Similar effects of Wnt3a on DNA damage were also observed in primary osteoprogenitors (data not shown). In summary, we have demonstrated that Wnt canonical pathway is capable of reducing DNA damage caused by radiation and subsequently protecting osteoprogenitors, osteoblasts, and osteocytes from radiation-induced cell death.
Ku70 Mediates DNA Repair and Cell Survival Effects of PTH and Wnt3a after Radiation
The occurrence of DSBs results in decondensation of chromatin at both local and global levels and invokes the activation of DNA damage response and subsequent DSB repair. In mammalian cells, most radiation-induced DSBs are repaired through non-homologous end-joining (NHEJ) pathway (12). The first stage of NHEJ is the binding of the Ku70/80 heterodimer to DNA ends for recruiting the core NHEJ complex. Immunoblots (Fig. 6A) revealed that Ku70 amount increased marginally as a response to radiation-induced DNA damage in UMR cells. Interestingly, we found that Wnt3a treatment significantly increased Ku70 by 5.3- and 7.9-fold at 1 and 4 h, respectively. The up-regulation of Ku70 by Wnt3a requires the canonical pathway as addition of DKK1 partially abolished the increase of Ku70 (Fig. 6B). Immunofluorescence of Ku70 confirmed the elevation of nuclear Ku70 by Wnt3a via the canonical pathway (Fig. 6, C and D). Furthermore, PTH1–34 had similar regulatory effects on nuclear Ku70 amount as shown in Fig. 6E. Again, inhibitor studies revealed that this PTH effect is mediated by PKA and β-catenin pathways. Similar to in vitro results, Wnt3a greatly increased the percentage of nuclear Ku70-positive osteoblasts in the calvarial tissue sections (Fig. 6, F and G). This nuclear localization of Ku70 was abolished by pretreatment with IWR-endo. PTH also stimulates Ku70 amount in the calvarial culture (Fig. 6H).
FIGURE 6.

Ku70 mediates the DNA repair and survival actions of Wnt3a and PTH in osteoblasts after radiation. A, immunoblot delineates the time course of up-regulation of Ku70 amount by Wnt3a in UMR cells. B, stimulation of Ku70 amount by Wnt3a is mediated by the canonical β-catenin pathway. Immunoblot was performed to detect Ku70 protein at 4 h after radiation and Wnt3a treatment in the absence and in the presence of DKK1 (500 ng/ml). The protein amounts shown at the bottom of gels were obtained by densitometric measurement. C, nuclear Ku70 staining in UMR cells is enhanced by Wnt3a at 4 h after radiation as shown by immunofluorescence (IF). D, immunofluorescence confirms that the up-regulation of Ku70 by Wnt3a is mediated by the canonical β-catenin pathway. UMR cells were irradiated and treated with Wnt3a CM in the presence or the absence of DKK1. The fluorescence intensity of Ku70 staining was measured at 4 h after radiation. Moreover, pretransfecting cells with Ku70 siRNA demonstrates the efficiency of this siRNA in knocking down Ku70 amount after Wnt3a treatment. δ, p < 0.001 versus radiated cells (R); %, p < 0.01 versus R, Wnt3a, and control; ^, p < 0.001 versus R, Wnt3a, and mock siRNA. E, PTH1–34 stimulates Ku70 amount through the PKA/β-catenin pathway in UMR cells at 4 h after radiation as shown by immunofluorescence. %, p < 0.01PTH versus vehicle (veh); Σ: p < 0.001 versus R, PTH and control. F, immunohistochemistry of Ku70 in rat calvarial organ culture at 6 h after radiation and Wnt3a treatments with or without IWR-endo. Arrows point to Ku70+ calvarial osteoblasts. G, the percentages of calvarial osteoblasts with nuclear Ku70 staining were quantified. δ, p < 0.001 versus R; Σ, p < 0.001 versus R, Wnt3a, and control. H, PTH treatment increases Ku70 amount in calvarial osteoblasts as shown by immunohistochemistry of calvarial organ sections. δ, p < 0.001 versus R. I–K, knocking down Ku70 by siRNA abolishes the regulatory effects of Wnt3a and PTH on γ-H2AX foci (I), tail DNA (J), and apoptosis (K) in UMR cells. &, p < 0.001 versus NR; δ, p < 0.001 versus R; $, p < 0.01; #, p < 0.001 versus R, Wnt3a (or PTH), and mock siRNA.
To further investigate the role of Ku70 in Wnt and PTH action in DNA repair, we used siRNA against Ku70 to suppress the up-regulation of Ku70 after Wnt3a or PTH1–34 treatment. As shown in Fig. 6D, pretreating cells with Ku70 siRNA efficiently decreased the nuclear Ku70 amount induced by Wnt3a and radiation. Interestingly, γ-H2AX foci measurement and comet assay revealed that accelerated DNA repair caused by either Wnt3a or PTH after radiation was diminished by co-treatment of Ku70 siRNA (Fig. 6, I and J). Moreover, knocking down Ku70 amount clearly abolished the survival effect of Wnt3a and PTH on osteoblasts (Fig. 6K). These findings demonstrate that β-catenin pathway-dependent up-regulation of Ku70, a protein critical for NHEJ DNA repair, could be one of the mechanisms for the accelerated DNA repair by PTH1–34 and Wnt3a.
DISCUSSION
PTH1–34 is well known for its potent anabolic effects on bone and the intermittent injection of teriparatide has been used as a therapeutic treatment for patients with severe osteoporosis for more than a decade. It greatly stimulates bone mass and improves bone structure and strength at both trabecular and cortical sites. One mechanism to explain this anabolic action is that PTH prolongs the lifespan of osteoblasts (20). Interestingly, this survival effect of PTH seems only restricted to cancellous osteoblasts and does not occur in periosteal osteoblasts, suggesting a site-specific effect (35). Cell culture experiments have shown that PTH prevents osteoblast apoptosis induced by DNA-damaging agents, such as etoposide (20), hydrogen peroxide, and UV irradiation (36). Some DNA damage and repair-related proteins such as PCNA, FOXO3a, and Gadd153 were shown to be regulated by PTH (36). However, the detailed mechanisms, particularly the PTH actions on DNA repair machinery and chromosomal integrity, have not been investigated yet. Our present in vitro and ex vivo studies demonstrate that PTH rescues radiation-induced osteoblast apoptosis by accelerating the repair of DNA DSBs, thus providing mechanistic explanation for our recent in vivo report that PTH1–34 alleviates the loss of local trabecular bone after radiation by preserving bone lining osteoblasts (23). Note that a recent report found that PTH at supraphysiological concentration (100 nm) induces DNA breaks in MC3T3 cells (37). In this work, we did not detect any increases in DNA damage in osteoblastic cells treated with 10 nm PTH under non-radiated conditions.
Our inhibitor assays further reveal that PTH protects osteoblasts from radiation-induced cell death by activating PKA and subsequently β-catenin pathway. Wnt/β-catenin pathway is an important modulator in bone homeostasis (24). Although this pathway is crucial to all bone cell types, it is particularly important for osteoblast lineage cells because it robustly promotes osteogenic differentiation and suppressing chondrogenic and adipogenic differentiation in mesenchymal progenitors. Previous studies also showed that this signaling pathway protects osteoblasts and osteocytes from apoptosis, but the exact mechanism remains elusive (38–41). Pathway analysis has indicated Src/ERK and PI3K/Akt pathways as mediators for Wnt3a survival signal on osteoblasts under serum withdrawal condition (38). However, under our radiation condition, the survival effect of Wnt3a appears to be solely dependent on the canonical β-catenin signaling. This conclusion is also consistent with recent studies showing that PGE2 activates PKA/β-catenin signaling to block glucocorticoid-induced apoptosis (41) and that active β-catenin is required for the radioresistance of mammary epithelial progenitor cells (42). To our knowledge, our study is the first to suggest a novel survival mechanism of the canonical β-catenin pathway in regulating DNA damage repair in osteoblast lineage cells. It is well known that PTH can regulate multiple points upstream of β-catenin, such as Wnt antagonists (sclerostin, secreted frizzled-related proteins, and DKK1), Lrp6, Smad3, and Disheveled, among others (Fei et al. (43)). Further investigation needs to be performed to elucidate the detailed mechanism by which PTH regulates β-catenin to achieve its survival action.
Our studies indicate that PTH and Wnt promotes the repair of radiation-induced DNA damage, particularly, DSBs, as determined by γ-H2AX staining and the comet assay. Radiation causes DNA lesions either directly or indirectly through ROS generation. Repairing these lesions in a timely fashion is essential to help cells avoid the fate of death. Although PTH has no effect on ROS production in osteoblastic culture after radiation, our data demonstrate that both PTH and Wnt are likely to execute their repair action on DSBs by up-regulating the Ku70 protein amounts in the nucleus of osteoblasts, thus possibly promoting the formation of Ku70/80 dimer, a DSB sensor and core component of the NHEJ complex. Ku70 could also regulate apoptosis independent of its DSB repair activity. One study showed that Ku70 sequesters Bax from the mitochondria and mediates Bax deubiquitylation (44). Another study suggested that Ku70 is a deubiquitinase for Mcl1 (45). Both Bax and Mcl1 belong to the Bcl2 family that controls programmed cell death through the intrinsic (mitochondrial) pathway. Despite the importance of Ku70 in repairing DSBs and promoting cell survival, Ku70 knock-out mice were not perinatally lethal and exhibited a significant increase in the basal level of apoptosis only in brain and gastrointestinal tissues (46, 47). They have a shortened lifespan (37 weeks) and show signs of early aging (48). Interestingly, histology revealed a significant reduction of femoral trabecular bone (48), suggesting that Ku70 also participates into bone homeostasis. Further investigations characterizing bone phenotypes of Ku70 knock-out mice and its bone responses toward focal radiation and PTH treatment will provide more mechanistic insight into radiotherapy-induced osteoporosis.
Apart from surgery, radiotherapy is the principal curative treatment for patients with cancer. Focal radiation, normally delivering a much higher radiation dose to tumor sites than whole-body radiation, inevitably causes damage on the neighboring tissues including bone. We initially found that radiation from repetitive in vivo micro-CT scans of young rats at the same proximal tibial site leads to a significant trabecular bone loss within this site and that daily PTH injections rescue such bone loss to a similar level of contralateral non-scanned tibia region (49). To further investigate this observation, we established a clinically relevant focal radiation model in adult rats and demonstrated that 1 month of PTH injections block the loss of trabecular elements and osteoblast apoptosis caused by radiation administered at the beginning of the experiment (days 1 and 3, 8 Gy each) (23). Those data, together with the present mechanistic analyses, suggest that PTH1–34 might be a suitable treatment for radiotherapy-induced osteoporosis and its related fractures. However, due to an increased incidence of osteosarcoma in preclinical rodent studies (50, 51), there are considerable concerns of using PTH in cancer patients with an increased baseline risk of osteosarcoma, including those receiving prior radiation therapy. Another concern of using PTH in cancer patients is the sequence similarity in N terminus between PTH and parathyroid hormone related-protein (PTHrP), a tumor-secreted factor. Both PTH and PTHrP bind to the receptor PTH1R, which is expressed at a much higher level in osteoblasts and osteocytes than non-osseous tumor cells, and PTHrP acts on those osseous cells to stimulate bone resorption for tumor-mediated osteolysis.
Interestingly, our current mechanistic studies reveal that the canonical Wnt pathway has the similar survival effects in protecting osteoblasts from radiation damage and hence present an alternative treatment for radiation-induced osteoporosis. A bone-specific way of activating Wnt/β-catenin can be achieved by injecting sclerostin-neutralizing antibody. Sclerostin is an osteocyte-specific protein that diffuses to bone surface, binds to Lrp5 or -6 in osteoblasts, and then negatively regulates Wnt-mediated osteoblastic bone formation (52). Despite the complex relationship between Wnt signaling and tumorigenesis, sclerostin appears not to be associated with any cancer and therefore targeting it should have less concern. For example, patients with either sclerosteosis, which results from loss-of-function mutations in the sclerostin gene (SOST), or van Buchem disease, which lacks sclerostin due to a deletion of genomic DNA downstream of the SOST, have no increased cancer incidence. SOST knock-out mice with high bone mass phenotype have been studied extensively in many laboratories. These mice have a normal lifespan and exhibit no increased tumor formation during their entire life. The neutralizing antibody against sclerostin is clinically proven to be a potent anabolic treatment for osteoporosis (53) and is currently in a phase 3 clinical trial. We are now testing this antibody treatment in our animal model of focal radiotherapy. We expect this bone-specific anabolic treatment should be more effective and advantageous than other proposed treatments for radiation-induced osteoporosis that do not distinguish between bone and cancer such as antioxidant treatment.
Acknowledgment
We thank Dr. Paola Divieti-Pajevic at Massachusetts General Hospital for providing Ocy454 cells.
This work was supported by National Institutes of Health Grants R01DK095803 (to L. Q.) and R01CA138804 (to B. X.), Penn Center for Musculoskeletal Disorder Grant P30AR050950 (NIAMS, National Institutes of Health), the ASBMR Junior Faculty Osteoporosis Basic Research Award (to L. Q.), and the McCabe Pilot Award (to X. S. L.).
- DSB
- double strand break
- PTH
- parathyroid hormone
- PKA
- protein kinase A
- EB
- ethidium bromide
- AO
- acridine orange
- Gy
- gray
- CM
- conditioned medium
- ROS
- reactive oxygen species
- NHEJ
- non-homologous end-joining
- MEM
- minimum essential medium
- NR
- non-radiated
- R
- radiated.
REFERENCES
- 1. Mitchell M. J., Logan P. M. (1998) Radiation-induced changes in bone. Radiographics 18, 1125–1136; quiz 1242–1123 [DOI] [PubMed] [Google Scholar]
- 2. Baxter N. N., Habermann E. B., Tepper J. E., Durham S. B., Virnig B. A. (2005) Risk of pelvic fractures in older women following pelvic irradiation. JAMA 294, 2587–2593 [DOI] [PubMed] [Google Scholar]
- 3. Moreno A., Clemente J., Crespo C., Martínez A., Navarro M., Fernández L., Minguell J., Vázquez G., Andreu F. J. (1999) Pelvic insufficiency fractures in patients with pelvic irradiation. Int. J. Radiat. Oncol. Biol. Phys. 44, 61–66 [DOI] [PubMed] [Google Scholar]
- 4. Mumber M. P., Greven K. M., Haygood T. M. (1997) Pelvic insufficiency fractures associated with radiation atrophy: clinical recognition and diagnostic evaluation. Skeletal Radiol. 26, 94–99 [DOI] [PubMed] [Google Scholar]
- 5. Uezono H., Tsujino K., Moriki K., Nagano F., Ota Y., Sasaki R., Soejima T. (2013) Pelvic insufficiency fracture after definitive radiotherapy for uterine cervical cancer: retrospective analysis of risk factors. J. Radiat. Res. 54, 1102–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cummings S. R., Melton L. J. (2002) Epidemiology and outcomes of osteoporotic fractures. Lancet 359, 1761–1767 [DOI] [PubMed] [Google Scholar]
- 7. Kanis J. A., Oden A., Johnell O., De Laet C., Jonsson B., Oglesby A. K. (2003) The components of excess mortality after hip fracture. Bone 32, 468–473 [DOI] [PubMed] [Google Scholar]
- 8. Dudziak M. E., Saadeh P. B., Mehrara B. J., Steinbrech D. S., Greenwald J. A., Gittes G. K., Longaker M. T. (2000) The effects of ionizing radiation on osteoblast-like cells in vitro. Plast. Reconstr. Surg. 106, 1049–1061 [DOI] [PubMed] [Google Scholar]
- 9. Gal T. J., Munoz-Antonia T., Muro-Cacho C. A., Klotch D. W. (2000) Radiation effects on osteoblasts in vitro: a potential role in osteoradionecrosis. Arch. Otolaryngol. Head Neck Surg. 126, 1124–1128 [DOI] [PubMed] [Google Scholar]
- 10. Matsumura S., Jikko A., Hiranuma H., Deguchi A., Fuchihata H. (1996) Effect of x-ray irradiation on proliferation and differentiation of osteoblast. Calcif. Tissue Int. 59, 307–308 [DOI] [PubMed] [Google Scholar]
- 11. Szymczyk K. H., Shapiro I. M., Adams C. S. (2004) Ionizing radiation sensitizes bone cells to apoptosis. Bone 34, 148–156 [DOI] [PubMed] [Google Scholar]
- 12. Vignard J., Mirey G., Salles B. (2013) Ionizing-radiation induced DNA double-strand breaks: a direct and indirect lighting up. Radiother. Oncol. 108, 362–369 [DOI] [PubMed] [Google Scholar]
- 13. Jüppner H., Abou-Samra A. B., Freeman M., Kong X. F., Schipani E., Richards J., Kolakowski L. F., Jr., Hock J., Potts J. T., Jr., Kronenberg H. M. (1991) A G protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science 254, 1024–1026 [DOI] [PubMed] [Google Scholar]
- 14. Partridge N. C., Kemp B. E., Veroni M. C., Martin T. J. (1981) Activation of adenosine 3′,5′-monophosphate-dependent protein kinase in normal and malignant bone cells by parathyroid hormone, prostaglandin E2, and prostacyclin. Endocrinology 108, 220–225 [DOI] [PubMed] [Google Scholar]
- 15. Civitelli R., Reid I. R., Westbrook S., Avioli L. V., Hruska K. A. (1988) PTH elevates inositol polyphosphates and diacylglycerol in a rat osteoblast-like cell line. Am. J. Physiol. 255, E660–667 [DOI] [PubMed] [Google Scholar]
- 16. Li X., Liu H., Qin L., Tamasi J., Bergenstock M., Shapses S., Feyen J. H., Notterman D. A., Partridge N. C. (2007) Determination of dual effects of parathyroid hormone on skeletal gene expression in vivo by microarray and network analysis. J. Biol. Chem. 282, 33086–33097 [DOI] [PubMed] [Google Scholar]
- 17. Jilka R. L. (2007) Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone. 40, 1434–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qin L., Partridge N. C. (2012) Parathyroid hormone and parathyroid hormone-related protein: normal function, diseases, and emerging therapeutics in Bone-Metabolic Functions and Modulators (Topics in Bone Biology) (Bronner F., Farach-Carson M. C., Roach H. I., eds.) pp. 1–20, Springer [Google Scholar]
- 19. Bellido T., Ali A. A., Plotkin L. I., Fu Q., Gubrij I., Roberson P. K., Weinstein R. S., O'Brien C. A., Manolagas S. C., Jilka R. L. (2003) Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J. Biol. Chem. 278, 50259–50272 [DOI] [PubMed] [Google Scholar]
- 20. Jilka R. L., Weinstein R. S., Bellido T., Roberson P., Parfitt A. M., Manolagas S. C. (1999) Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Invest. 104, 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Motyl K. J., McCauley L. K., McCabe L. R. (2012) Amelioration of type I diabetes-induced osteoporosis by parathyroid hormone is associated with improved osteoblast survival. J. Cell. Physiol. 227, 1326–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weinstein R. S., Jilka R. L., Almeida M., Roberson P. K., Manolagas S. C. (2010) Intermittent parathyroid hormone administration counteracts the adverse effects of glucocorticoids on osteoblast and osteocyte viability, bone formation, and strength in mice. Endocrinology 151, 2641–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chandra A., Lin T., Tribble M. B., Zhu J., Altman A. R., Tseng W. J., Zhang Y., Akintoye S. O., Cengel K., Liu X. S., Qin L. (2014) PTH1–34 alleviates radiotherapy-induced local bone loss by improving osteoblast and osteocyte survival. Bone 67, 33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baron R., Kneissel M. (2013) WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med. 19, 179–192 [DOI] [PubMed] [Google Scholar]
- 25. Shalhoub V., Conlon D., Tassinari M., Quinn C., Partridge N., Stein G. S., Lian J. B. (1992) Glucocorticoids promote development of the osteoblast phenotype by selectively modulating expression of cell growth and differentiation associated genes. J. Cell. Biochem. 50, 425–440 [DOI] [PubMed] [Google Scholar]
- 26. Chandra A., Lan S., Zhu J., Siclari V. A., Qin L. (2013) Epidermal growth factor receptor (EGFR) signaling promotes proliferation and survival in osteoprogenitors by increasing early growth response 2 (EGR2) expression. J. Biol. Chem. 288, 20488–20498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mohammad K. S., Chirgwin J. M., Guise T. A. (2008) Assessing new bone formation in neonatal calvarial organ cultures. Methods Mol. Biol. 455, 37–50 [DOI] [PubMed] [Google Scholar]
- 28. Porter A. G., Jänicke R. U. (1999) Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 6, 99–104 [DOI] [PubMed] [Google Scholar]
- 29. Wan M., Yang C., Li J., Wu X., Yuan H., Ma H., He X., Nie S., Chang C., Cao X. (2008) Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev. 22, 2968–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Romero G., Sneddon W. B., Yang Y., Wheeler D., Blair H. C., Friedman P. A. (2010) Parathyroid hormone receptor directly interacts with dishevelled to regulate β-catenin signaling and osteoclastogenesis. J. Biol. Chem. 285, 14756–14763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tobimatsu T., Kaji H., Sowa H., Naito J., Canaff L., Hendy G. N., Sugimoto T., Chihara K. (2006) Parathyroid hormone increases β-catenin levels through Smad3 in mouse osteoblastic cells. Endocrinology 147, 2583–2590 [DOI] [PubMed] [Google Scholar]
- 32. Rothkamm K., Löbrich M. (2003) Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc. Natl. Acad. Sci. U.S.A. 100, 5057–5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Olive P. L., Banáth J. P. (2006) The comet assay: a method to measure DNA damage in individual cells. Nat. Protoc. 1, 23–29 [DOI] [PubMed] [Google Scholar]
- 34. Semënov M. V., Tamai K., Brott B. K., Kühl M., Sokol S., He X. (2001) Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr. Biol. 11, 951–961 [DOI] [PubMed] [Google Scholar]
- 35. Jilka R. L., O'Brien C. A., Ali A. A., Roberson P. K., Weinstein R. S., Manolagas S. C. (2009) Intermittent PTH stimulates periosteal bone formation by actions on post-mitotic preosteoblasts. Bone 44, 275–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schnoke M., Midura S. B., Midura R. J. (2009) Parathyroid hormone suppresses osteoblast apoptosis by augmenting DNA repair. Bone 45, 590–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alves de Oliveira E. C., Szejnfeld V. L., Pereira da Silva N., Coelho Andrade L. E., Heldan de Moura Castro C. (2010) Intermittent PTH1–34 causes DNA and chromosome breaks in osteoblastic and nonosteoblastic cells. Calcif. Tissue Int. 87, 424–436 [DOI] [PubMed] [Google Scholar]
- 38. Almeida M., Han L., Bellido T., Manolagas S. C., Kousteni S. (2005) Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by β-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. J. Biol. Chem. 280, 41342–41351 [DOI] [PubMed] [Google Scholar]
- 39. Bodine P. V., Zhao W., Kharode Y. P., Bex F. J., Lambert A. J., Goad M. B., Gaur T., Stein G. S., Lian J. B., Komm B. S. (2004) The Wnt antagonist secreted frizzled-related protein-1 is a negative regulator of trabecular bone formation in adult mice. Mol. Endocrinol. 18, 1222–1237 [DOI] [PubMed] [Google Scholar]
- 40. Gortazar A. R., Martin-Millan M., Bravo B., Plotkin L. I., Bellido T. (2013) Crosstalk between caveolin-1/extracellular signal-regulated kinase (ERK) and β-catenin survival pathways in osteocyte mechanotransduction. J. Biol. Chem. 288, 8168–8175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kitase Y., Barragan L., Qing H., Kondoh S., Jiang J. X., Johnson M. L., Bonewald L. F. (2010) Mechanical induction of PGE2 in osteocytes blocks glucocorticoid-induced apoptosis through both the β-catenin and PKA pathways. J. Bone Miner Res. 25, 2657–2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Woodward W. A., Chen M. S., Behbod F., Alfaro M. P., Buchholz T. A., Rosen J. M. (2007) WNT/β-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 104, 618–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fei Y., Hurley M. M. (2012) Role of fibroblast growth factor 2 and Wnt signaling in anabolic effects of parathyroid hormone on bone formation. J. Cell. Physiol. 227, 3539–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Amsel A. D., Rathaus M., Kronman N., Cohen H. Y. (2008) Regulation of the proapoptotic factor Bax by Ku70-dependent deubiquitylation. Proc. Natl. Acad. Sci. U.S.A. 105, 5117–5122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang B., Xie M., Li R., Owonikoko T. K., Ramalingam S. S., Khuri F. R., Curran W. J., Wang Y., Deng X. (2014) Role of Ku70 in deubiquitination of Mcl-1 and suppression of apoptosis. Cell Death Differ. 21, 1160–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gu Y., Sekiguchi J., Gao Y., Dikkes P., Frank K., Ferguson D., Hasty P., Chun J., Alt F. W. (2000) Defective embryonic neurogenesis in Ku-deficient but not DNA-dependent protein kinase catalytic subunit-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 97, 2668–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li G. C., Ouyang H., Li X., Nagasawa H., Little J. B., Chen D. J., Ling C. C., Fuks Z., Cordon-Cardo C. (1998) Ku70: a candidate tumor suppressor gene for murine T cell lymphoma. Mol. Cell. 2, 1–8 [DOI] [PubMed] [Google Scholar]
- 48. Li H., Vogel H., Holcomb V. B., Gu Y., Hasty P. (2007) Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Mol. Cell. Biol. 27, 8205–8214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chandra A., Lan S., Zhu J., Lin T., Zhang X., Siclari V. A., Altman A. R., Cengel K. A., Liu X. S., Qin L. (2013) PTH prevents the adverse effects of focal radiation on bone architecture in young rats. Bone 55, 449–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vahle J. L., Long G. G., Sandusky G., Westmore M., Ma Y. L., Sato M. (2004) Bone neoplasms in F344 rats given teriparatide [rhPTH(1–34)] are dependent on duration of treatment and dose. Toxicol. Pathol. 32, 426–438 [DOI] [PubMed] [Google Scholar]
- 51. Vahle J. L., Sato M., Long G. G., Young J. K., Francis P. C., Engelhardt J. A., Westmore M. S., Linda Y., Nold J. B. (2002) Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone (1–34) for 2 years and relevance to human safety. Toxicol. Pathol. 30, 312–321 [DOI] [PubMed] [Google Scholar]
- 52. Moester M. J., Papapoulos S. E., Löwik C. W., van Bezooijen R. L. (2010) Sclerostin: current knowledge and future perspectives. Calcif. Tissue Int. 87, 99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McClung M. R., Grauer A., Boonen S., Bolognese M. A., Brown J. P., Diez-Perez A., Langdahl B. L., Reginster J. Y., Zanchetta J. R., Wasserman S. M., Katz L., Maddox J., Yang Y. C., Libanati C., Bone H. G. (2014) Romosozumab in postmenopausal women with low bone mineral density. N Engl. J. Med. 370, 412–420 [DOI] [PubMed] [Google Scholar]