

Background: Chromatin structure and histone modifications regulate transcription in eukaryotes.
Results: Activation of the early growth response gene 1 involves sliding and/or eviction of nucleosomes around the transcription start site and nucleosome-specific, time-dependent changes in histone modifications.
Conclusion: Remodeling mechanisms and histone modifications are specific for each nucleosome.
Significance: Mononucleosomal level studies give unique information on chromatin functions.
Keywords: Chromatin Remodeling, Histone Modification, Liver, Mouse, Small Interfering RNA (siRNA), Transcription Regulation
Abstract
Histone post-translational modifications and nucleosome remodeling are coordinate events involved in eukaryotic transcriptional regulation. There are relatively few data on the time course with which these events occur in individual nucleosomes. As a contribution to fill this gap, we first describe the nature and time course of structural changes in the nucleosomes −2, −1, and +1 of the murine Egr1 gene upon induction. To initiate the transient activation of the gene, we used the stimulation of MLP29 cells with phorbol esters and the in vivo activation after partial hepatectomy. In both models, nucleosomes −1 and +1 are partially evicted, whereas nucleosomes +1 and −2 slide downstream during transcription. The sliding of the latter nucleosome allows the EGR1 protein to bind its site, resulting in the repression of the gene. To decide whether EGR1 is involved in the sliding of nucleosome −2, Egr1 was knocked down. In the absence of detectable EGR1, the nucleosome still slides and remains downstream longer than in control cells, suggesting that the product of the gene may be rather involved in the returning of the nucleosome to the basal position. Moreover, the presence of eight epigenetic histone marks has been determined at a mononucleosomal level in that chromatin region. H3S10phK14ac, H3K4me3, H3K9me3, and H3K27me3 are characteristic of nucleosome +1, and H3K9ac and H4K16ac are mainly found in nucleosome −1, and H3K27ac predominates in nucleosomes −2 and −1. The temporal changes in these marks suggest distinct functions for some of them, although changes in H3K4me3 may result from histone turnover.
Introduction
It is widely accepted that any approach to understanding the mechanisms of transcriptional regulation in eukaryotes has to take into account that nuclear DNA is organized in a complex structure called chromatin. The nucleosome core, formed by 147 bp of DNA wrapping an octamer of two copies each of the H2A, H2B, H3, and H4 histones, is the basic structural motif of chromatin, highly conserved through evolution of eukaryotes (1). The structures of the histone octamer (2, 3) and that of the nucleosome core (4) are known in great detail.
When going from an inactive to an actively transcribing gene, its chromatin structure has to be remodeled to allow the binding of the basal transcriptional machinery and other factors to their proper DNA sequences, often hindered by the presence of nucleosomes. Two main remodeling mechanisms are involved in these processes, namely nucleosome sliding and partial or total histone eviction (5–9). For RNA polymerase processivity, nucleosomes also represent an obstacle, and they also have to be remodeled, frequently with the aid of histone chaperones (10–13).
Along with nucleosome remodeling, histones acquire precise and definite epigenetic marks, especially acetylation of lysine side chains, methylation of lysines and/or arginines, and phosphorylation of serines, to allow transcription of a gene (14). These two events, remodeling and epigenetic modification, are not mutually exclusive, as many times the latter serves as a coded mark to elicit the former.
After the original Loidl's proposal that the versatility of histone acetylation may be related to a signaling effect in which the cell machinery would be able to distinguish among some of the different combinations of acetylated lysines in the four histones (15), a plethora of data on acetylation and other histone modifications are accessible, and nowadays more than 140 different modifications have been described (16). Strahl and Allis (17) proposed the histone code hypothesis, which assumed that histone modifications may act in a sequential or combinatorial fashion to specify different functions. Great effort has been dedicated to interpret the functional meaning of many of these modifications. For instance, from a transcriptional point of view, acetylation is normally associated with activation, although methylation may be regarded either as a repressive or as an activating mark. In the recent years, genome-wide studies have provided us with much data on the usage of the different histone modifications (18–24), although, as pointed out by Rando and Chang (25), they raised some doubts as to the functional significance of the combination of histone marks.
Nevertheless, most of these studies do not give information on the dynamics of histone modifications and are seldom informative on their sequential mode of action. Moreover, most often the ChIP methodology to study specific modification of histones is applied to sonicated chromatin. In this way, albeit short amplicons are used to analyze the results by PCR, the ascription of a given epigenetic mark to a particular nucleosome leads to imprecise results.
We are interested in examining the sequence in which the different modifications of histones are acquired in the different nucleosomes, as well as to study the structural outcome of remodeling of individual nucleosomes during transcription. To do this we have selected the murine Egr1 gene, an immediate-early gene (26), which codes for a zinc finger transcriptional factor. We have previously found that this gene is rapidly activated in response to the treatment of MLP29 mouse progenitor hepatocyte cells with phorbol esters, and we have determined the time course of factor assembly in its promoter. It is also known that the mediator complex assembles on the proximal promoter to stimulate productive transcription by RNA polymerase II (27), but nothing is known about the influence of chromatin structure on the binding of the complex and of the transcription factors. Moreover, as the transcription of the gene, which passes a maximum of 30 min after phorbol ester addition, declines by 180 min (28), Egr1 represents a convenient model to study the sequence of events in both the activating and repressing phases of the expression peak. In this study, we describe the time-dependent changes in the nucleosome occupancy at the promoter (nucleosomes −1 and −2) and proximal coding region (nucleosome +1) upon activation of the gene both in MLP29 cells induced with phorbol esters and in vivo following induction by partial hepatectomy. As the product of the gene has a definite role in its repression (28), the effects of functional knocking down of the gene on the transcriptional rate and on the chromatin structure have been studied. Finally, the time course of epigenetic changes in the three nucleosomes mentioned has also been studied.
EXPERIMENTAL PROCEDURES
Biological Materials
Cells from mouse progenitor hepatocyte cell line MLP29 were cultured and treated with 12-O-tetradecanoylphorbol-13-acetate (TPA)6 as described previously (28). For in vivo experiments, adult mice (8–10 weeks) of the CD1 strain were held at 22 °C with a 12-h light/12-h dark cycle and fed ad libitum with free access to water. Animals were cared for and handled in conformance with European Union guidelines. Liver regeneration was triggered by partial hepatectomy, by removing the median and left lobes after ligation under light isoflurane anesthesia, essentially following the method of Higgins and Anderson (29). The remnant livers were removed at various times. Sham-operated control animals were treated in a similar way, but the ligations and lobe removal were omitted. All the procedures involving live animals were performed in a properly equipped operating room. The study was approved by the Ethical Research Committee of the University of Valencia.
Isolation of Mononucleosomes
To isolate mononucleosomes from MLP29 cells, they were fixed with 1% formaldehyde in PBS by shaking gently at room temperature for 5 min. Cells were collected, washed, and resuspended in cell lysis buffer (100 mm NaCl, 3 mm MgCl2, 30 mm sucrose, 10 mm EDTA, 0.5% (v/v) Nonidet P-40, 10 mm Tris-HCl, pH 7.5) supplemented with 2 μl/ml protease inhibitors (Sigma). Cells were lysed in a manually operated Potter homogenizer, and they were allowed to stand at 0 °C for 15 min. The 500 × g sediment (5 min) was resuspended in washing buffer (15 mm NaCl, 3 mm MgCl2, 60 mm KCl, 20% (v/v) glycerol, 15 mm Tris-HCl, pH 7.5). The nuclear fraction was digested with 25 units of micrococcal nuclease (Roche Applied Science) per A260 unit to give a nuclease concentration of 500 units per ml of a washing buffer supplemented with 3 mm CaCl2. Digestion was carried out for 15 min at 37 °C, and it was stopped by adding cold EDTA to 10 mm. In our hands, the above procedure yielded a preparation of mononucleosomes, without visible traces of dinucleosomes or degradation products. To isolate mononucleosomes from regenerating liver, the remaining lobes of the hepatectomized animals were removed and immediately treated for 15 min at room temperature with 1% formaldehyde in PBS with gentle shaking. After stopping the reaction with 0.125 m glycine in PBS for 5 min, the liver fragments were rinsed and suspended in 10 ml of PBS with 2 μl/ml protease inhibitor mixture (Sigma). Fixed livers were homogenized in a Potter-Elvehjem tissue grinder. The homogenate was filtered through gauze and centrifuged at 3000 × g for 5 min. The nuclear fraction was suspended in cell lysis buffer and processed as above.
Quantitative RT-PCR and Real Time PCR
RNA was isolated by the TRIzol method (30). Quantitative RT-PCR and real time PCR was performed in a thermocycler Bio-Rad C1000 using SYBR Green Platinum (Invitrogen). The manufacturer's instructions were followed at every step. The appropriate pairs of primers for each case are given in Table 1. Results were analyzed with CFX Manager 2.1 software (Bio-Rad). It was routinely checked that the designed oligonucleotide primers gave a single amplification product by recording the melting profiles with genomic DNA as template.
TABLE 1.
Primers used for PCR
| Amplicon | Primer sequence |
Size | |
|---|---|---|---|
| Forward | Reverse | ||
| bp | |||
| +405 | TACCCCAAACTGGAGGAGATGAT | GCAGCACCGAGGAACTGG | 62 |
| +362 | GATCTCTGACCCGTTCGGC | CATCATCTCCTCCAGTTTGGG | 73 |
| +305 | CCACCCAACATCAGTTCTCC | GTGGGTGAGTGAGGAAAGGA | 119 |
| +241 | GTGTGCCCTCAGTAGCTTCG | GCGAGCTGGAGAACTGATGT | 64 |
| +171 | GGGGCCCACCTACACTCC | GTTGGCCGGGTTACATGC | 58 |
| +60 | CGAGAGATCCCAGCGCGCAG | TGGTGGACGCAGGGCTGCC | 119 |
| +38 | CGAGAGATCCCAGCGCGCAG | TCTTGCGGCGGCGGAAGCTG | 77 |
| −69 | GCCGGTCCTTCCATATTAGG | TCAAGGGTCTGGAACAGCAC | 90 |
| −126 | GTGCCCACCACTCTTGGAT | GCAGGAAGCCCTAATATGGA | 80 |
| −178 | GCTTTCCAGGAGCCTGAGC | AGCCCTCCCATCCAAGAGT | 81 |
| −240 | GGCAAGCTGGGAACTCCA | AGGCTCCTGGAAAGCCTAGT | 75 |
| −307 | CGCCTTATATGGAGTGGCCC | CTGGAGTTCCCAGCTTGCC | 96 |
| −342 | CGCCGGAACAGACCTTATTT | CTCCCAGAGCCGGAAGC | 77 |
| −373 | GACCCGGAAACGCCATATAAG | CCATATTGGGCCACTCCATATAAG | 91 |
| −425 | CTCCCAGTTGGGAACCAAG | GGGATCCTTCCTGCTCCTTA | 88 |
| −494 | CCGCGCGCCCAGCTCTAC | CCTCCTTGGTTCCCAACTG | 93 |
| −550 | GCCTGGGCTTCCCTAGC | CCAGGCGCGTAGAGCTG | 73 |
| −600 | CCAGGATGACGGCTGTAGAA | CGAGCTGGGCTAGGGAAG | 78 |
| −666 | TCTGCCCTAGCCCGCCCTGCC | TTCTACAGCCGTCATCCT | 94 |
| −746 | GCCTCAGTTTTCCCGGTGACA | AGGGCGGGCTAGGGCAGAGGC | 102 |
| −787 | TGCTAGCTAGGCAGTGTCCCAA | GCCCTGACCTGAGCCTCTACTAAT | 108 |
| p | TTCCCTAGCCCAGCTCGCAC | GAACAGCAGCAGTGGCCTCC | 352 |
| α-actin | CACCTGACCACAGGGCATCC | AACTGGCTCCAAGGCTCACG | 227 |
Nucleosome Occupancy
To investigate nucleosome occupancy of the promoter and proximal coding region of the Egr1 gene, a micrococcal nuclease protection assay was performed. DNA isolated from mononucleosomes was used as a template for quantitative PCR. Primers were designed to amplify tiled amplicons of about 60–120 bp along the promoter and the proximal coding region. The amplicons were identified by the position of their center relative to the TSS, and the corresponding primers and amplicon sizes are given in Table 1. To correct for the different efficiency of the primers, the results were normalized using the data obtained with genomic DNA, sonicated to an average size of 300 bp in a Sonic Vibra Cell VCX-500 instrument. The normalized data were plotted against the position of the amplicon center.
Egr1 Knockdown
MLP29 cells were cultured in 100-mm plates to 50–60% confluence and treated with 40 μl if Lipofectamine RNAi Max (Invitrogen) per plate and 25 nm of either scrambled siRNA (Ambion 4390843), Egr1 siRNA 1 (Ambion 4390771, ID: S65378), or Egr1 siRNA 2 (Ambion 4390771, ID: S65380) following the instructions of the manufacturer. The cells were then incubated in CO2 atmosphere for 8–10 h at 37 °C, and the medium was replaced by DMEM with 0.5% bovine fetal serum. After 20 h, the cells were treated with TPA and processed for micrococcal nuclease digestion as above.
Other Procedures
ChIP and mononucleosomal immunoprecipitation were carried out as described elsewhere (31). To study the binding of chromatin remodelers, conventional ChIP analysis and semiquantitative PCR were carried out as described (28). The antibodies used were as follows: anti-H3K9ac (Abcam, ab-4441); anti-H3K9me3 (Abcam, ab-8898); anti-H3K27ac (Abcam, ab-4729); anti-H3K27me3 (Millipore, 07-449); anti-H3K4me3 (Abcam, ab-8580); anti-H3S10phK14ac (Millipore, 07-081); anti-H3K14ac (Millipore, 07-353); anti-H4K16ac (Millipore, 07329); anti-EGR1 (Santa Cruz Biotechnology, sc-110); anti-H3 (Abcam, ab-1791); anti-BRM (Santa Cruz Biotechnology, sc-28710); anti-BRG1 (Santa Cruz Biotechnology, sc-10768); and anti-β-actin (Sigma, A5441).
RESULTS
Nucleosome Occupancy at Egr1 Promoter and Proximal Coding Region in MLP29 Cells
The analysis of micrococcal nuclease protection along 1200 bp (between −800 and +400), covering the promoter and proximal coding region, revealed the presence of three protected areas, two in the promoter and the third one at the beginning of the coding region. Taking into account the data of Fig. 1A and the length of the amplicons, the size of the nuclease-protected areas in the promoter can be estimated between a minimum of 265 bp and a maximum of 380 bp for the upstream area and between 140 and 230 for the downstream one. In other words, there is room enough for a nucleosome in both areas. Therefore, we henceforward assumed that they respectively correspond to nucleosomes −2 and −1. The protected area at the beginning of the coding region is less definite. It is wider than the other areas, and its profile is somewhat blurred. It can be assumed that this region is occupied by nucleosome +1.
FIGURE 1.
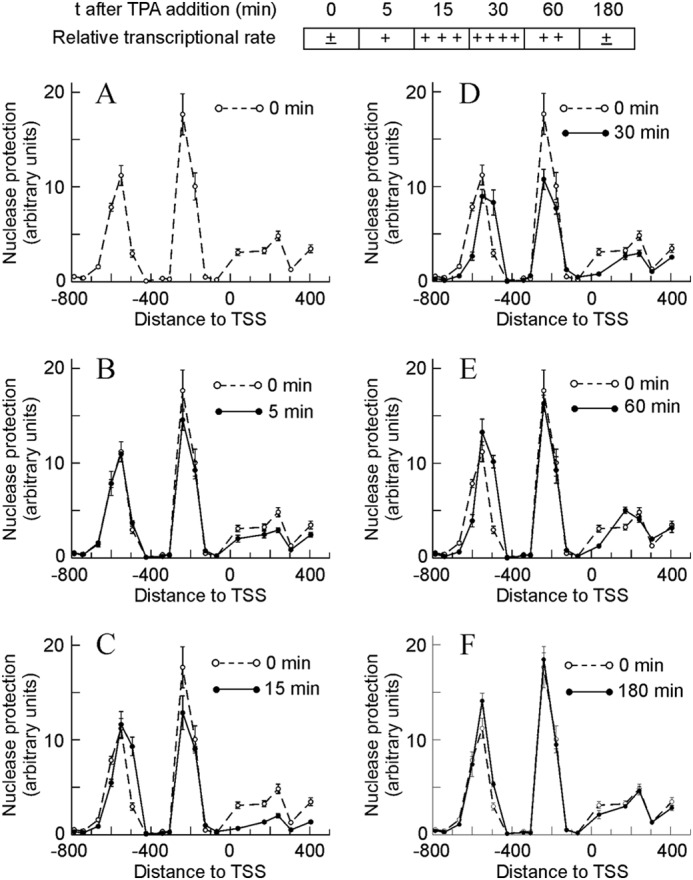
Time-dependent changes in micrococcal nuclease protection in the promoter and proximal coding region of the Egr1 gene during TPA induction. To facilitate the interpretation of the results, a semiquantitative diagram showing the relative transcriptional rate is included at the top. The protection against nuclease digestion at different times after adding TPA, as shown in A–F, was determined and plotted against the distance to TSS. For comparison purposes, the nuclease protection at t = 0 min was shown in all the panels. The plotted experimental points correspond to the means ± S.E. of three determinations.
The above assumptions as to the nucleosome occupancy of the studied Egr1 region are, generally speaking, in agreement with a sequence-based prediction of positioning carried out using the NuPoP software tool (32), as shown in Fig. 2A. The differences found will be discussed below. Taken together, all these data allowed us to propose a map of the promoter, which shows the positions of the three nucleosomes studied and those of the different cis elements to facilitate the description of the following results (Fig. 2B). The position proposed for nucleosome +1 is mainly based on the results of in vivo experiments (see below).
FIGURE 2.
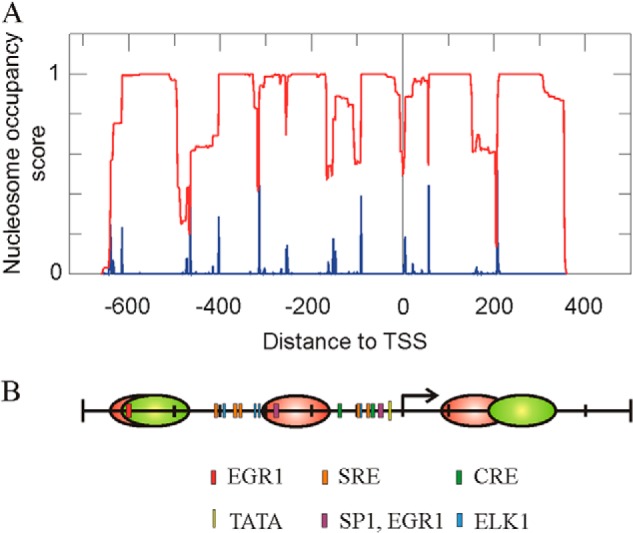
Nucleosome occupancy at the promoter and proximal coding region of the Egr1 gene. A, prediction of the nucleosome occupancy score (red) and of the probability of starting nucleosomes (blue), as determined by the NuPoP program (32). B, map of the Egr1 region under study, showing the location of cis elements and the experimental positions of the nucleosomes in noninduced MLP29 cells (red). The proposed position of nucleosomes −2 and +1 30 min after TPA addition is also shown (green). The proposed positions of nucleosomes are based on the experiments of Figs. 1 and 4.
By using an RNApol-ChIP assay (33), we previously found that transcription of Egr1 can be detected in MLP29 cells as early as 5 min after adding TPA, that it peaks at 30 min, and that at 180 min the activity of the gene is no longer observed (28). Fig. 1, B–F, shows that clear changes occur in the nucleosome occupancy and that the time course of these changes parallels that of transcription. Five min after adding TPA, protection to nuclease slightly diminishes in the region of nucleosomes −1 and +1. This diminution is clearly significant at 15 min, a time point in which a downstream sliding of nucleosome −2 began to be detected. These changes are especially obvious at 30 min, when the transcriptional rate passes a maximum. Once this wave of Egr1 expression has finished, i.e. 180 min after adding TPA, the chromatin structure returns to its initial state (Fig. 1F).
The area under the peak of nucleosome −2, as determined by densitometry of two experiments similar to that of Fig. 1, did not appreciably change with time. This suggests that the nucleosome simply slides downstream. Conversely, the area under the peak of nucleosome −1 dropped to 75% of the control value at 30 min after adding TPA and that of nucleosome +1 dropped to about 65% at 15 min and to about 33% of the control value at 30 min. These results may indicate that nucleosome eviction took place in some of the cells upon induction of the gene. To further explore this question, the promoter occupancy by histone H3 was determined. ChIP experiments with an antibody against the C-terminal domain of histone H3 were carried out with chromatin sonicated to an average DNA length of 500 bp. The presence of H3 was quantified by real time PCR at amplicons −550, −494 (nucleosome −2), −240, −178 (nucleosome −1), +171, and +241 (nucleosome +1), and the results were averaged for each nucleosome. In the region of nucleosome −2, the amount of H3 diminishes only slightly from 0 to 30 min, although the H3 signal significantly decreases in the regions of nucleosomes −1 and +1 (Table 2). These data indicate that, although nucleosome −2 is not appreciably lost on induction of the gene, the histone octamer is evicted from the regions of nucleosomes −1 and +1. Of note, the amount of H3 in the region of nucleosome +1 is roughly 2-fold increased relative to nucleosomes −2 and −1, despite the lower protection to nuclease in that region.
TABLE 2.
Presence of H3 in the Egr1 promoter
The presence of H3 was quantified by RT-PCR at the amplicons indicated, and the results were averaged for each nucleosome.
| Amplicon | Nucleosome | Presence of H3 relative to input |
||
|---|---|---|---|---|
| 0 min | 15 min | 30 min | ||
| −550 | N − 2 | 6.8 ± 1.2 | 6.2 ± 0.7 | 5.9 ± 0.7 |
| −494 | ||||
| −240 | N − 1 | 6.1 ± 0.7 | 4.8 ± 0.7 | 3.4 ± 0.4 |
| −178 | ||||
| +171 | N + 1 | 1.9 ± 0.6 | 6.0 ± 0.2 | 4.2 ± 0.2 |
| +241 | ||||
Two overlapping functional NF-κB sites are present at around −200 in human EGR1 (34, 35). Although no identical sequence is present in mouse Egr1, a somewhat similar motif exists in the region covered by nucleosome −1, and it might be possible that the eviction of the nucleosome had the function of making that motif accessible. Nevertheless, in our hands, no evidence for NF-κB binding was found by ChIP analysis (results not shown).
Presence of Remodeling Complexes in Egr1 Promoter
The finding that promoter nucleosomes are either slid or evicted in connection with Egr1 activation prompted us to study the presence of chromatin remodeling complexes that might be responsible for these alterations. Semiquantitative PCR analysis of a conventional ChIP experiment in an amplicon expanding the promoter from −579 to −228 (amplicon p, Table 1) revealed the presence of the BRM and BRG components of the SWI/SNF complex in the noninduced promoter (Fig. 3). Interestingly, these components left the promoter upon induction, to return when the gene was again silenced. These results agree with those of Sakurai et al. (36), who found that BRM negatively regulates human Egr1. The resolution of the experiment of Fig. 3 did not allow us to decide on which of the nucleosomes the SWI/SNF complex acts, but the region responsive to BRM localizes between −1293 and −502 of the human Egr1 promoter (36). If these data could be extrapolated to the murine model, we might conclude that a BRM-containing complex is related to the sliding of nucleosome −2, as will be discussed below.
FIGURE 3.
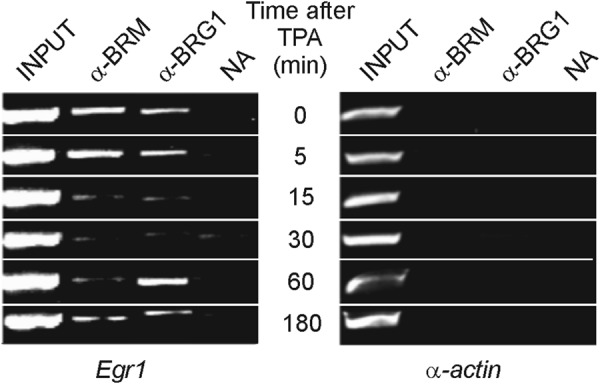
Presence of chromatin remodelers in the Egr1 promoter. A ChIP assay with antibodies against BRG1 and BRM was analyzed by semiquantitative PCR at different times after adding TPA to MLP29 cells. The inactive gene of α-actin was used as control. NA, no antibody added.
Nucleosome Occupancy at Egr1 Promoter and Proximal Coding Region during Hepatic Regeneration after Partial Hepatectomy
We next asked whether the above described mechanism involving the sliding of nucleosome −2 and the partial eviction of nucleosomes −1 and +1 is a peculiarity of the MLP29 cells or constitutes a general way for Egr1 activation at a chromatin level. To address this question, we selected an in vivo model, namely the activation of the gene during hepatic regeneration after partial hepatectomy. Egr1, being an immediate-early gene, becomes activated at the onset of liver regeneration, but the time course of transcription is, as expected, different from that observed in cultured MLP29 cells, and 2 h after partial hepatectomy, the steady level of Egr1 mRNA amounts to 6-fold of the basal level (Fig. 4A). In MLP29 cells, we have previously found that the maximal accumulation of Egr1 mRNA occurs 15 min after the maximal transcription, so we studied the nucleosome occupancy of the Egr1 promoter and proximal coding region 105 min after partial hepatectomy, assuming that at this time the transcriptional rate would be large enough to be reflected in the promoter chromatin structure. When the pattern of nuclease resistance is compared with that obtained in sham-operated animals (Fig. 4B), the same features detected in MLP29 cells are observed. Significant enough differences were found between the samples derived from sham-operated animals and from the hepatectomized ones to conclude that nucleosome −2 slides downstream and the nucleosome −1 is partially evicted when the gene is being actively transcribed. These characteristics were observed in all the six liver regeneration experiments performed. A peculiarity of the in vivo experiments is that the nuclease protection in the region of nucleosome +1 is enhanced relative to MLP29 cells, and this allowed us to map the basal position of this nucleosome roughly within one of the regions predicted by the NuPoP program (Fig. 2B) and to propose that, apart from being partially evicted, nucleosome +1 slides downstream, roughly 30–40 bp, upon induction of the gene.
FIGURE 4.
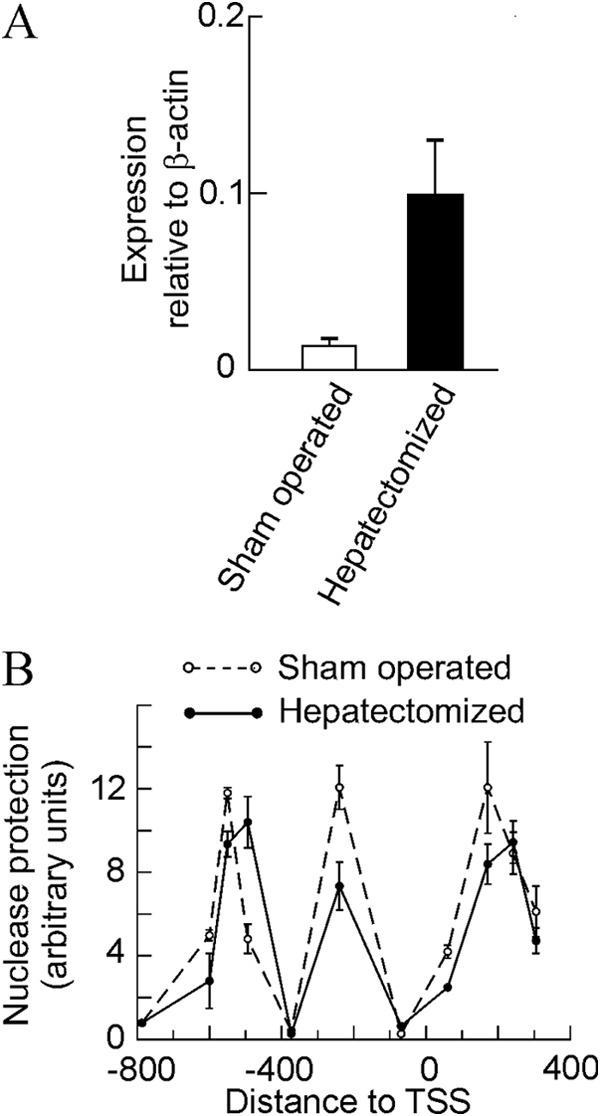
Effects of partial hepatectomy on the expression and chromatin structure of the Egr1 gene. A, quantitative RT-PCR of Egr1 expression in the livers of sham-operated and partially hepatectomized mice (n = 5) 105 min after surgery. B, same livers were used for a nuclease protection experiment as that described in Fig. 1.
Effects of Knocking Down the Expression of Egr1
High resolution ChIP analysis showed that EGR1 protein binds its own gene promoter especially at the −595 site (28). As discussed below, the sliding of nucleosome −2 may facilitate the access of EGR1 to that site. EGR1 recruits NAB1 and NAB2 after 30 min of TPA stimulation of MLP29 cells (28), and this results in the repression of the gene, which is thus only transiently expressed. With these circumstances in mind, we wondered whether the presence of the EGR1 protein causes in some way the sliding of nucleosome −2. To answer this question, we knocked down the expression of Egr1. Two independent experiments, with two different siRNAs each, were carried out. The levels of basal mRNA decreased to 67% relative to scrambled siRNA for siRNA 1 and to 33% for siRNA 2 (Fig. 5A), and in the case of siRNA 2, EGR1 protein was undetectable by Western blotting either in the absence of added TPA or at any time after its addition (Fig. 5B). In both experiments, the time course of mRNA accumulation followed similar patterns, and the full results of one of the experiments, in terms of the TPA-induced increased expression over the basal value, are given in Fig. 6. Despite knocking down the expression of Egr1, after adding TPA the fold increase of mRNA relative to the basal level is higher in cells treated with Egr1 siRNA, especially with siRNA 2, with which the silencing effect is more noticeable. The differences between the results obtained with Egr1 siRNA and with scrambled siRNA are not significant at short times of TPA stimulation. However, 180 min after adding TPA the differences were clearly significant (p < 0.02 for siRNA 1 and p < 0.01 for siRNA 2). These results suggest that the inhibition of Egr1 transcription is delayed in knocked down cells when compared with cells treated with control nucleotides. The differences tend to vanish at longer times.
FIGURE 5.
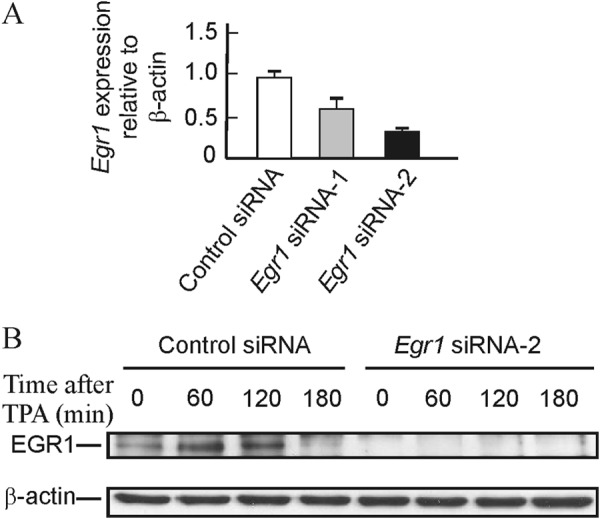
Effects of Egr1 knockdown on the basal level of mRNA and on the content of EGR1 protein. A, basal level of Egr1 mRNA in MLP29 cells treated with scrambled and two Egr1 siRNAs. B, Western blot showing the level of EGR1 protein at different times after TPA addition to MLP29 cells treated with scrambled or siEgr1 2.
FIGURE 6.
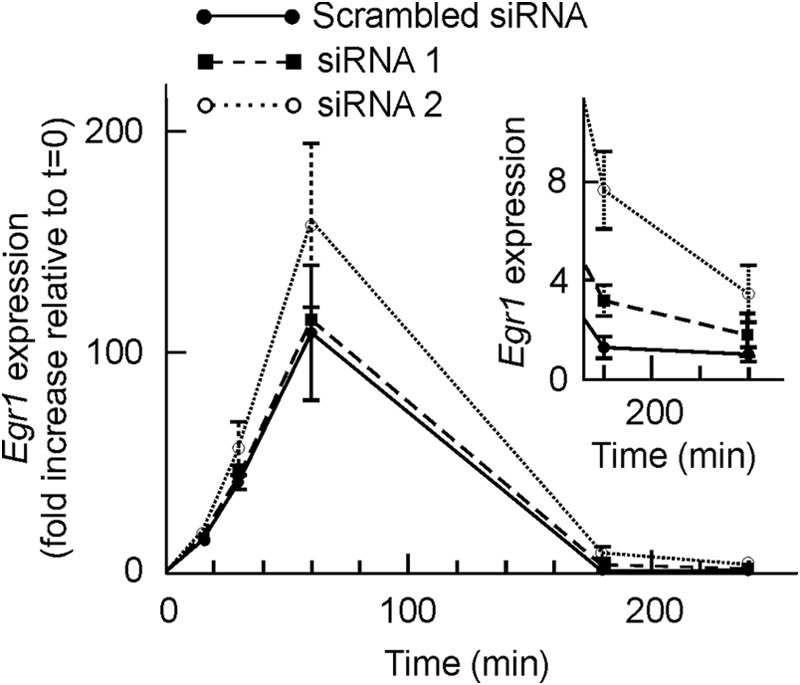
Effects of Egr1 knockdown on the expression of the gene. MLP29 cells were treated with scrambled siRNA, with siEgr1 1, or with siEgr1 2, and the Egr1 expression was determined by quantitative RT-PCR and plotted, as a function of time after induction with TPA, as fold increase relative to t = 0. The inset shows a magnification of the figure for the two longer times. Points correspond to the means ± S.E. of three determinations.
The study of nucleosome occupancy at the Egr1 promoter of MLP29 cells knocked down for Egr1 further supports that idea. Fig. 7 depicts the time course organization of chromatin in the region of nucleosome −2, the one overlapping with the EGR1 site. In cells transfected with both scrambled and Egr1 siRNA 2, the downstream sliding of nucleosome −2 is more noticeable and more durable than in nontransfected MLP29 cells. The maximum of nuclease protection occurs at amplicon −494 instead of −550, and the nucleosome remains slid even at 60 min after TPA addition to cells (compare Figs. 1 and 7). Moreover, subtle but significant differences between control and Egr1-silenced cells were found. Actually, neither the position nor the nuclease protection of nucleosome −2 change in passing from 30 to 60 min after TPA addition in knocked down cells. We have shown that the downstream sliding of nucleosome −2 with the concomitant uncovering of the EGR1 site at −595 is a transcription-associated phenomenon, and therefore, the results of Fig. 7 are compatible with the idea that in Egr1-silenced cells transcription is still active 60 min after TPA addition. More interestingly, the present results show that EGR1 is not involved in the sliding of nucleosome −2 but rather in its return to the basal position. The delay in Egr1 repression may explain the relative increase in the accumulation of mRNA observed in Fig. 6. No significant differences between the behavior of the knocked down cells and those treated with control siRNA were observed in nucleosome −1 (data not shown).
FIGURE 7.
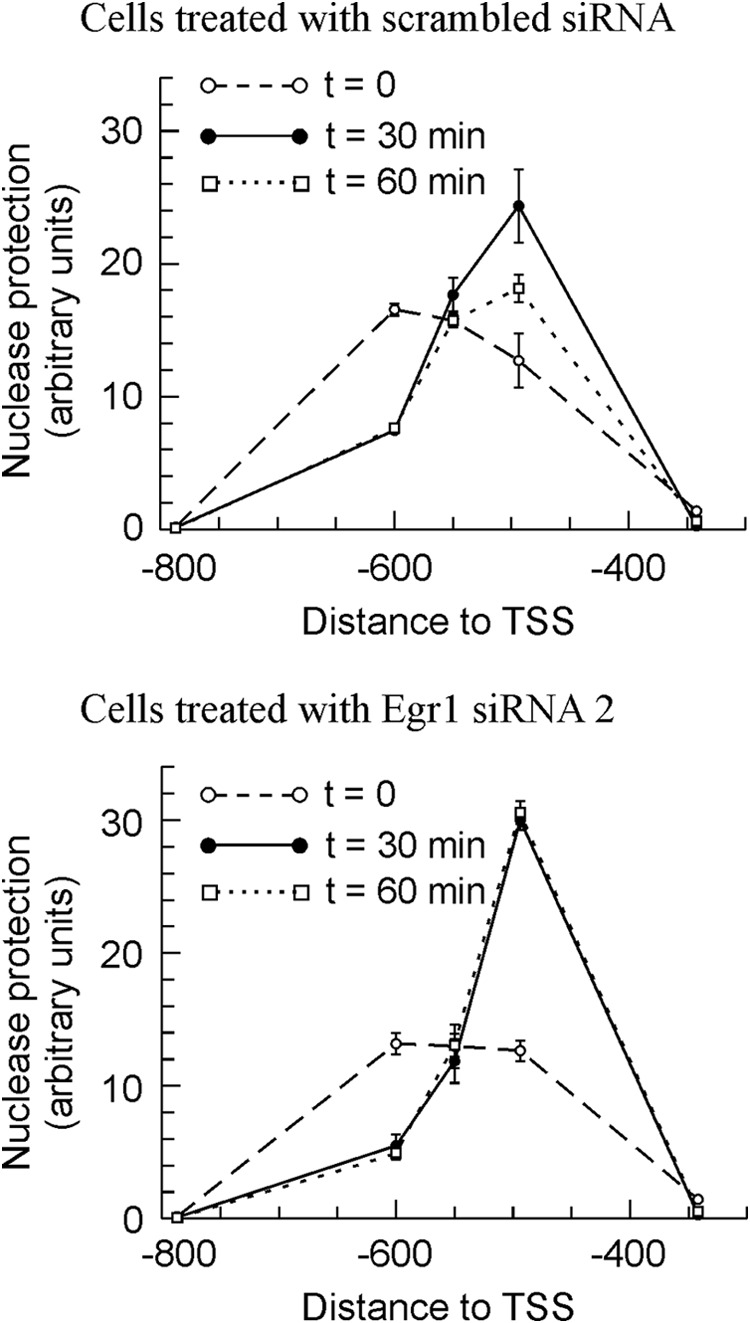
Effects of Egr1 knockdown in MLP 29 cells on the nuclease protection in the region of nucleosome −2. The nuclease protection was measured, as in Fig. 1, at various time points after adding TPA, as indicated. Points correspond to the means ± S.E. of three determinations.
Histone Epigenetic Marks at Egr1 Promoter and Proximal Coding Region
In a previous paper (28), we analyzed the gross epigenetic changes when MLP29 cells are activated by TPA treatment. The analysis was then carried out by conventional ChIP at the whole Egr1 promoter. Results were obtained by semi-quantitative PCR in the repressed gene and after 30 min of TPA treatment. We now describe a more thorough analysis. First, the mononucleosomal immunoprecipitation technique was used, thus allowing us to differentiate the marks in every single nucleosome. To do this, mononucleosomes were immunoprecipitated with the appropriate antibodies, and their DNA was used as template for real time PCR. These determinations were done at two amplicons within each of the nuclease-protected regions corresponding to the nucleosomes. The excellent coincidence between the data obtained from both amplicons (see below) provided an internal control of the results. Moreover, the analysis covered several time points, so the epigenetic changes can be studied from a dynamic point of view at both the activating and repressing phases of gene expression.
The immunoprecipitations and posterior analysis were repeated at least three times, and in every instance the results were consistent. Fig. 8 gives representative results of these experiments. Several general observations on the results can be made. Apart from the close correspondence between the results for both of the amplicons in every nucleosome, the epigenetic marks vary qualitatively and/or quantitatively from one nucleosome to another. Additionally, with only a few exceptions, the intensity of a given mark at 180 min coincides with the basal one. This means that once the gene ceases to be transcribed, the epigenetic state of the nucleosomes returns to its basal value. Finally, the analysis of incompatible epigenetic marks, such as acetylation and methylation of the same lysyl residue, gave opposite results. This obvious finding may also be considered as an internal control of the reliability of the methods used.
FIGURE 8.

Time course of histone modifications in nucleosomes −2, −1, and +1 after adding TPA to MLP29 cells. A–H show the levels of different modifications in a representative experiment, plotted as percent of input, for two amplicons in every nucleosome. Error bars represent the standard deviation of three determinations. The times after adding TPA are given in the inset at bottom right.
The levels of H3S10phK14ac were first determined (Fig. 8A). This double label is a characteristic hallmark of immediate-early genes (37), and we have previously found that it appears in the Egr1 gene, reaching a maximal level 15 min after TPA treatment, and that it returns to the basal level by 30 min. The present results at a mononucleosomal level show that this modification preferentially occurs in nucleosome +1, in which it can be detected as soon as 5 min of activation, and it returns to the basal level by 30 min. This fast and transient modification precedes other modifications (see below) and the transcription itself, which arrives at a maximum at 30 min. H3K14ac was initially recognized as an activating histone modification (38). In our model, the intensity of the mark peaks at 15 min after stimulation with TPA in all the nucleosomes studied, especially in nucleosomes −1 and +1 (Fig. 8B). It seems clear that after 15 min of TPA treatment, the time course of H3S10phK14ac variation in nucleosome +1 is parallel to that of H3K14ac. In that nucleosome, both H3S10phK14ac and H3K14ac return to their basal value 30 min after TPA addition.
Three other of the modifications studied, namely H3K4me3, H3K9me3, and H3K27me3, are specific to nucleosome +1 to a greater or lesser degree (Fig. 8, C–E). Genomic analyses show that H3K4me3 is, in general, present at the start sites of actively transcribed genes (24). This is the case of the Egr1 gene, in which, under basal conditions, H3K4me3 is more abundant in nucleosome +1 than in promoter nucleosomes. In the latter, the mark did not change with induction of the gene, although in nucleosome +1 there is a distinct time-dependent increase of the mark (Fig. 8C). Interestingly, the level of the mark at 180 min, when the transcription has completely ceased instead of returning to the basal value, reached its maximal value. This question will be discussed later. We next determined the distribution of H3K9me3. It was described as a mark of heterochromatin (39) and related to gene repression (40), but it has also been found downstream of TSS in actively transcribed genes (see below). In our case, H3K9me3 is predominantly present in nucleosome +1, and the intensity of the label is maximal between 15 and 60 min after TPA addition (Fig. 8D). The changes in H3K27me3 (Fig. 8E) follow a similar pattern. Nucleosomes −2 and −1 are only scarcely methylated, although nucleosome +1 is clearly modified, with a maximum at 30–60 min after TPA addition. The genome-wide analysis of Barski et al. (18) found that the presence of H3K27me3 is very low in actively transcribing genes and that the modification occurs roughly with similar frequency over the promoters and coding regions of silent genes. Mouse Egr1 does not obey that general pattern, at least in the neighborhood of TSS, as the patterns of the promoter and +1 nucleosomes are different, and the latter is clearly modified.
As expected, the pattern of H3K9 (Fig. 8F) and H3K27 (Fig. 8G) acetylation is opposite that of the methylation of the same residues and the most heavily acetylated nucleosomes, i.e. those at the promoter are only scarcely methylated and vice versa. The changes in H3K9ac in nucleosome −1 are particularly interesting, because the peak of this modification, which occurs at 30 min after TPA addition, implies a 5–6-fold increase relative to the basal level (Fig. 8F). The modification, which has been classically related to transcriptional activation (38) and specifically to transcriptional initiation (41, 42), maps close to the initiation site and its extent parallels that of transcription itself. H3K27ac is considered as a mark well correlated with gene expression (24). We previously detected a global increase in H3K27ac upon activation of the gene 30 min after adding TPA (28), but our present results show that the mark is acquired earlier, especially in the promoter nucleosomes (Fig. 8G). In a separate experiment, we detected the increase in this acetylation event in the promoter nucleosomes as early as 5 min after adding TPA (results not shown).
Finally, we examined the time course and nucleosome distribution of the acetylation of H4 lysine 16. The TPA-induced increase in H4K16ac is preferentially found in the promoter nucleosomes, especially in nucleosome −1, where the acquisition of the mark is a very early event in gene activation (Fig. 8H). Five minutes after adding TPA, the label increased to almost 4-fold the basal value, and the maximal level of the mark, at 15 min, reached 6–7-fold the basal value. This maximum precedes those of transcriptional rate and of nucleosome −1 eviction.
DISCUSSION
In this study, we describe an analysis of the chromatin-associated changes accompanying the induction of the murine Egr1 immediate-early gene. Most of the results, centered in the promoter and in the proximal gene body, have been obtained at a mononucleosomal level resolution. The time course of the changes in nucleosome occupancy and in the histone post-translational modifications has been determined, thus allowing us to establish a temporal sequence of the events leading to gene activation.
We first determined the nucleosome occupancy between −800 and +400 relative to the TSS, to find three micrococcal nuclease-protected regions, whose sizes are compatible with bona fide nucleosomes. We ascribed them to nucleosomes −2, −1, and +1. The location of the protected areas reasonably fit the prediction retrieved from the NuPoP program, which is based on experimental data of nucleosome positioning (32). Interestingly, the present data explain why the serum-response factor is constitutively bound to the Egr1 promoter at the two clusters of serum-response elements that are located between −300 and −400 and between −83 and −105 relative to the TSS (28), as both of these clusters are nucleosome-free in nonstimulated cells (Figs. 1A and 2B). The same reasoning is valid to explain the constitutive binding of ELK1, whose binding sites flank nucleosome −1. We have previously observed that CREB binds the Egr1 promoter in the absence of TPA (28). The cAMP-response element sites localize between −50 and −150, i.e. in the nucleosome-free region (Fig. 2B). The binding sites of serum-response factor, ELK1 and CREB, lie in close vicinity (Fig. 2B), and this may result in a cooperative binding, which, according to Segal and Widom (43), can reinforce their competition with nucleosomes in the regions around −400 and the TSS. These would explain the differences found between the NuPoP prediction and the experimental results. The labile binding of SP1 may be weaker than that of the other factors, because, although bound in the absence of TPA, it leaves the promoter as soon as transcription starts (28). In this way, the present data would explain why the SP1 factor prefers a noncanonical CG box at −49 to bind the promoter under basal conditions instead of the canonical site at −277 (28), which lies within nucleosome −1 (Fig. 2B).
The changes in micrococcal nuclease protection after TPA induction (Fig. 1) may be correlated with our data on expression of the gene, which passes a maximum at 30 min (28). As mentioned above, Egr1 transcription is accompanied by a downstream sliding of nucleosome −2 and a partial eviction of nucleosomes −1 and +1.
BRM1 and BRG1, the ATPase subunits of the SWI/SNF-type BAF remodeler (8), are present in the promoter of noninduced Egr1 (Fig. 3). We have already mentioned that BRM negatively regulates the human EGR1 promoter (36) and that it might be assumed that the BRM-containing remodeler is involved in the positioning of nucleosome −2. Fig. 2 shows that the proposed 5′ ends of nucleosome −2 in resting and induced cells coincide with points of maximal probability of starting a nucleosome. If this actually occurs in live cells, the nucleosome would be maintained in the upstream position in the absence of TPA stimulus, and the remodeler complex would be continuously consuming ATP to maintain that position. Upon activation of the gene, the remodeler leaves the promoter (Fig. 3) and the nucleosome shifts toward the downstream position, probably by the action of an unidentified remodeler, with the effect of unblocking the EGR1 site at −595. As a matter of fact, after sliding, nucleosome −2 still lies over the EGR1 site, but the latter is now approximately located in the superhelical positions 1 and 2 of nucleosomal DNA, and their spontaneous unwrapping (44–46) would allow EGR1 to invade its binding site.
The partial eviction of nucleosome −1 does not uncover any critical cis element (Fig. 2), but it may serve the purpose of leaving room for the assembly of the mediator complex. This complex, which stimulates the initiation of transcription by previously bound RNA polymerase II, binds the murine Egr1 promoter mainly between the TSS and −400 (27).
The characteristic patterns of change in the promoter chromatin structure when the Egr1 gene is active, i.e. the downstream sliding of nucleosome −2 and the partial eviction of nucleosomes −1 and +1, also occur in an in vivo model in which the activation of Egr1 during liver regeneration was studied (Fig. 4). It is then interesting to note that both models behave in a similar way. It can be taken for granted that the sliding of nucleosome −2 is required for EGR1 protein to bind its site at −595, which, in turn, leads to the repression of the gene after recruiting NAB1 and NAB2 (28). Nab1 gene is constitutively expressed, but Nab2 is induced by EGR1, thus originating a negative feedback loop (47).
In MLP29 cells, nucleosome +1 is more accessible to nuclease than the other two nucleosomes, despite the higher density of H3 in that region. This suggests that nucleosome +1, apart from being evicted during induction, is already destabilized in noninduced cells. We may assume that this is related to the fact that an immediate-early gene in cultured cells is in some sense permanently poised for transcription. The nuclease protection in the region of nucleosome +1 is much more noticeable in the in vivo experiments than in those using MLP29 cells. This may be related to the reduced transcriptional potential of Egr1 in liver when compared with cultured cells. At any rate, the good definition of the protected area in vivo allows us to conclude that the protection is displaced downstream when the gene is being actively transcribed. We do not know the causes of this sliding, although genome-wide analysis shows that the 5′ border of nucleosome +1 in active genes peaks 30 bp downstream relative to inactive genes (48).
It is worth noting that, after induction, nucleosome −1 returns to its initial state at 60 min, although nucleosome −2 is still altered. This fact agrees with our previous observation that EGR1, NAB1, and NAB2, at that time point, still remain bound to Egr1 promoter (28). The experiments of Egr1 knockdown showed that the sliding of nucleosome −2 is not caused by the presence of the EGR1 protein, which may be rather involved in some way in the returning of the nucleosome to its basal position. The re-assembly of nucleosome −1 at 60 min, when the actual transcription is only residual, probably means that the presence of the mediator complex is no longer required.
The phosphoacetylation of histone H3 (phosphorylation at serine 10 and acetylation at lysine 14) is characteristic of nucleosome +1 (Fig. 8A). The phosphorylation of H3S10, known as the nucleosomal response (49) and catalyzed by MSK1/2, is a primary event in the signal transduction to chromatin, mediated by the ERK and p38 MAPK pathways (50, 51). In our case, the p38 and MEK1/2 pathways are operative, but ERK is not involved in Egr1 activation (28). The subsequent acetylation of lysine 14 is linked to the phosphorylation, and these combined marks seem to be decisive for the recruitment of RNA polymerase II S5ph to the p21 promoter (52). In the case of mouse Egr1, RNA polymerase is present in the promoter in the absence of TPA, although its presence is increased in parallel with the induction of the gene. Inhibition of either p38 or MEK1/2 kinases causes a reduction of RNA polymerase recruitment to the promoter (28). Taken together, all these data suggest that phosphoacetylation of H3 in the +1 nucleosome is decisive for a productive recruitment of RNA polymerase but, in view of the transient pattern of the dual modification H3S10phK14ac (Fig. 8A), this is not required for a maintained transcription.
H3K4 methyltransferases associate with the C-terminal domain S5ph form of RNA polymerase II (25), and this explains why H3K4me3 is preferentially associated with the nucleosomes of the proximal coding region (18, 19, 21). However, histone exchange frequently occurs in the “hot” nucleosomes found in regulatory regions and at the beginning of the gene body, and Henikoff (53) proposed that methylation of H3K4 is related to histone turnover. This may explain why H3K4 remained trimethylated when Egr1 transcription had finished (Fig. 8C). H3K27me3 has been classically described as a repressor mark (54), and our results show that both H3K4me3 and H3K27me3 are preferentially associated with the same nucleosome (Fig. 8, C and E), although we do not know whether they reside on the same histone tail. We have also observed the existence of methylated H3K4 and H3K27 in the same nucleosome in the Gas1 gene (31), and the occurrence of these bivalent marks is a somewhat frequent phenomenon. The present results, with their time-dependent analysis of the histone modifications, allow us to propose an explanation for the apparent contradiction of finding both “repressive” and “activating” marks on the same nucleosome. The presence of H3K4me3, according to the Henikoff hypothesis (53, 55), would be a consequence of histone turnover, although that of H3K27me3, which inhibits elongation by RNA polymerase II (56), would be related to the decrease of transcriptional rate from 30 min onward, as this is the moment in which the modification in nucleosome +1 is especially noticeable (Fig. 8E).
Some of our results argue against an unambiguous interpretation of the histone code and of the results obtained at a genome-wide level. For instance, H3K9me3 has been described to be associated with silent genes (24) and to recruit HP1, which was reported as related to heterochromatin and involved in gene silencing (57). Preliminary mononucleosomal immunoprecipitation data from our laboratory,7 indicate that, although the presence of HP1-γ is almost negligible in nucleosome −1, in which the level of H3K9me3 is very low (Fig. 8D), HP1-γ is clearly present in nucleosome +1. These results are in accordance with those of Vakoc et al. (58), who found that the presence of H3K9me3 and HP1-γ is associated with transcriptional elongation. Obviously, these questions deserve further analysis.
The acetylation of H4K16, H3K9, and H3K27 predominates on the promoter nucleosomes (Fig. 8, F--H). Two of these modifications, namely H4K16ac and H3K9ac, are especially intense at nucleosome −1. It is tempting to speculate that they may be associated with the observed eviction of this nucleosome. H4K16ac precedes from a temporal point of view to nucleosome eviction, whereas the profile of H3K9ac coincides with that of −1 eviction. The ChIP-chip analysis of Horikoshi et al. (59) revealed a significant association of the nucleosomes carrying the H4K16ac mark with CREB sites. Interestingly, a functional cAMP-response element, to which CREB is constitutively bound, lies immediately downstream of nucleosome −1 (Fig. 2B), and its maximal acetylation (15 min) coincides with the maximal CREB phosphorylation (28), but we do not know whether a functional relationship exists between these two findings. The time course of H3K9 acetylation (Fig. 8F) coincides with those of nucleosome −1 eviction and transcriptional activity. H3K9 is acetylated by p300 (60), and CREB-binding protein, a related p300 paralog, is present in the Egr1 promoter (28), but we agree with Henikoff and Shilatifard (55) in warning against drawing causal conclusions simply based on the correlation of modification patterns.
Fig. 9 summarizes most of the results given in this paper and some of our previous data (28) to offer a model of the structural changes and histone modifications during the activation and repression of the Egr1 gene. The advantages of carrying out these studies at mononucleosomal resolution may be clearly noticed as the remodeling of chromatin and the acquisition of the activating or repressing marks may act in a distinct nucleosome-specific manner. The ultimate aim of an epigenetic study ought to be the establishment of causal relationships between epigenetic modifications and their downstream effects, but further research would be required to establish these connections.
FIGURE 9.

Summary of the results presented in this paper. The position of nucleosomes −2, −1, and +1 is given at four different times after TPA addition. The partial eviction of nucleosomes is symbolized by increasing the transparency of the ovals. The location of transcription factors serum-response factor, ELK1, SP1, and CREB and the phosphorylation of the latter is based on our previous results (28). The tentative positions of the BRM-containing remodeler and the preinitiation complex (PIC) are also shown. The transcriptional rate is symbolized by placing more or less arrows over the RNA polymerase II (pol II) molecule. The relative level of histone modifications is given according to the color code depicted at the top.
This work was supported in part by FISS Grant FIS PI12/02110 (to G. L.-R.).
A. L. Riffo-Campos, J. Castillo, G. López-Rodas, M. I. Rodrigo, and L. Franco, unpublished results.
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- TSS
- transcription start site
- CREB
- cAMP-response element-binding protein.
REFERENCES
- 1. Kumari S., Swaminathan A., Chatterjee S., Senapati P., Boopathi R., Kundu T. K. (2013) Chromatin organization, epigenetics and differentiation: an evolutionary perspective. Subcell. Biochem. 61, 3–35 [DOI] [PubMed] [Google Scholar]
- 2. Arents G., Burlingame R. W., Wang B.-C., Love W. E., Moudrianakis E. N. (1991) The nucleosomal core histone octamer at 3.1 Å resolution: A tripartite protein assembly and a left-handed superhelix. Proc. Natl. Acad. Sci. U.S.A. 88, 10148–10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arents G., Moudrianakis E. N. (1993) Topography of the histone octamer surface: repeating structural motifs utilized in the docking of nucleosomal DNA. Proc. Natl. Acad. Sci. U.S.A. 90, 10489–10493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 5. Conaway R. C., Conaway J. W. (2009) The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem. Sci. 34, 71–77 [DOI] [PubMed] [Google Scholar]
- 6. Alkhatib S. G., Landry J. W. (2011) The nucleosome remodeling factor. FEBS Lett. 585, 3197–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murawska M., Brehm A. (2011) CHD chromatin remodelers and the transcription cycle. Transcription 2, 244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Euskirchen G., Auerbach R. K., Snyder M. (2012) SWI/SNF chromatin-remodeling factors: multiscale analyses and diverse functions. J. Biol. Chem. 287, 30897–30905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Narlikar G. J., Sundaramoorthy R., Owen-Hughes T. (2013) Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 154, 490–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hondele M., Ladurner A. G. (2013) Catch me if you can: how the histone chaperone FACT capitalizes on nucleosome breathing. Nucleus 4, 443–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kulaeva O. I., Hsieh F. K., Chang H. W., Luse D. S., Studitsky V. M. (2013) Mechanism of transcription through a nucleosome by RNA polymerase II. Biochim. Biophys. Acta 1829, 76–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kulaeva O. I., Hsieh F. K., Studitsky V. M. (2010) RNA polymerase complexes cooperate to relieve the nucleosomal barrier and evict histones. Proc. Natl. Acad. Sci. U.S.A. 107, 11325–11330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsieh F. K., Kulaeva O. I., Patel S. S., Dyer P. N., Luger K., Reinberg D., Studitsky V. M. (2013) Histone chaperone FACT action during transcription through chromatin by RNA polymerase II. Proc. Natl. Acad. Sci. U.S.A. 110, 7654–7659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gardner K. E., Allis C. D., Strahl B. D. (2011) Operating on chromatin, a colorful language where context matters. J. Mol. Biol. 409, 36–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loidl P. (1988) Towards an understanding of the biological function of histone acetylation. FEBS Lett. 227, 91–95 [DOI] [PubMed] [Google Scholar]
- 16. Tan M., Luo H., Lee S., Jin F., Yang J. S., Montellier E., Buchou T., Cheng Z., Rousseaux S., Rajagopal N., Lu Z., Ye Z., Zhu Q., Wysocka J., Ye Y., Khochbin S., Ren B., Zhao Y. (2011) Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Strahl B. D., Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 18. Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. (2007) High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 [DOI] [PubMed] [Google Scholar]
- 19. Guenther M. G., Levine S. S., Boyer L. A., Jaenisch R., Young R. A. (2007) A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130, 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wiencke J. K., Zheng S., Morrison Z., Yeh R. F. (2008) Differentially expressed genes are marked by histone 3 lysine 9 trimethylation in human cancer cells. Oncogene 27, 2412–2421 [DOI] [PubMed] [Google Scholar]
- 21. Nozaki T., Yachie N., Ogawa R., Kratz A., Saito R., Tomita M. (2011) Tight associations between transcription promoter type and epigenetic variation in histone positioning and modification. BMC Genomics 12, 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karmodiya K., Krebs A. R., Oulad-Abdelghani M., Kimura H., Tora L. (2012) H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics 13, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Du Z., Li H., Wei Q., Zhao X., Wang C., Zhu Q., Yi X., Xu W., Liu X. S., Jin W., Su Z. (2013) Genome-wide analysis of histone modifications: H3K4me2, H3K4me3, H3K9ac, and H3K27ac in Oryza sativa L. Japonica. Mol. Plant 6, 1463–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kimura H. (2013) Histone modifications for human epigenome analysis. J. Hum. Genet. 58, 439–445 [DOI] [PubMed] [Google Scholar]
- 25. Rando O. J., Chang H. Y. (2009) Genome-wide views of chromatin structure. Annu. Rev. Biochem. 78, 245–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Janssen-Timmen U., Lemaire P., Mattéi M. G., Revelant O., Charnay P. (1989) Structure, chromosome mapping and regulation of the mouse zinc-finger gene Krox-24; evidence for a common regulatory pathway for immediate-early serum-response genes. Gene 80, 325–336 [DOI] [PubMed] [Google Scholar]
- 27. Wang G., Balamotis M. A., Stevens J. L., Yamaguchi Y., Handa H., Berk A. J. (2005) Mediator requirement for both recruitment and postrecruitment steps in transcription initiation. Mol. Cell 17, 683–694 [DOI] [PubMed] [Google Scholar]
- 28. Tur G., Georgieva E. I., Gagete A., López-Rodas G., Rodríguez J. L., Franco L. (2010) Factor binding and chromatin modification in the promoter of murine Egr1 gene upon induction. Cell. Mol. Life Sci. 67, 4065–4077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Higgins G. M., Anderson R. M. (1931) Experimental pathology of the liver I: restoration of the liver of the white rat following surgical removal. Arch. Path. 12, 186–202 [Google Scholar]
- 30. Chomczynski P., Sacchi N. (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156–159 [DOI] [PubMed] [Google Scholar]
- 31. Sacilotto N., Espert A., Castillo J., Franco L., López-Rodas G. (2011) Epigenetic transcriptional regulation of the growth arrest-specific gene 1 (Gas1) in hepatic cell proliferation at mononucleosomal resolution. PLoS One 6, e23318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xi L., Fondufe-Mittendorf Y., Xia L., Flatow J., Widom J., Wang J. P. (2010) Predicting nucleosome positioning using a duration Hidden Markov Model. BMC Bioinformatics 11, 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sandoval J., Rodríguez J. L., Tur G., Serviddio G., Pereda J., Boukaba A., Sastre J., Torres L., Franco L., López-Rodas G. (2004) RNAPol-ChIP: a novel application of chromatin immunoprecipitation to the analysis of real-time gene transcription. Nucleic Acids Res. 32, e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aicher W. K., Sakamoto K. M., Hack A., Eibel H. (1999) Analysis of functional elements in the human Egr-1 gene promoter. Rheumatol. Int. 18, 207–214 [DOI] [PubMed] [Google Scholar]
- 35. Zheng C., Ren Z., Wang H., Zhang W., Kalvakolanu D. V., Tian Z., Xiao W. (2009) E2F1 Induces tumor cell survival via nuclear factor-κB-dependent induction of EGR1 transcription in prostate cancer cells. Cancer Res. 69, 2324–2331 [DOI] [PubMed] [Google Scholar]
- 36. Sakurai K., Furukawa C., Haraguchi T., Inada K., Shiogama K., Tagawa T., Fujita S., Ueno Y., Ogata A., Ito M., Tsutsumi Y., Iba H. (2011) MicroRNAs miR-199a-5p and −3p target the Brm subunit of SWI/SNF to generate a double-negative feedback loop in a variety of human cancers. Cancer Res. 71, 1680–1689 [DOI] [PubMed] [Google Scholar]
- 37. Nowak S. J., Corces V. G. (2004) Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet. 20, 214–220 [DOI] [PubMed] [Google Scholar]
- 38. Peterson C. L., Laniel M. A. (2004) Histones and histone modifications. Curr. Biol. 14, R546–R551 [DOI] [PubMed] [Google Scholar]
- 39. Peters A. H., Mermoud J. E., O'Carroll D., Pagani M., Schweizer D., Brockdorff N., Jenuwein T. (2002) Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat. Genet. 30, 77–80 [DOI] [PubMed] [Google Scholar]
- 40. Wang Z., Zang C., Rosenfeld J. A., Schones D. E., Barski A., Cuddapah S., Cui K., Roh T. Y., Peng W., Zhang M. Q., Zhao K. (2008) Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 40, 897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liang G., Lin J. C., Wei V., Yoo C., Cheng J. C., Nguyen C. T., Weisenberger D. J., Egger G., Takai D., Gonzales F. A., Jones P. A. (2004) Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc. Natl. Acad. Sci. U.S.A. 101, 7357–7362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bernstein B. E., Kamal M., Lindblad-Toh K., Bekiranov S., Bailey D. K., Huebert D. J., McMahon S., Karlsson E. K., Kulbokas E. J., 3rd, Gingeras T. R., Schreiber S. L., Lander E. S. (2005) Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120, 169–181 [DOI] [PubMed] [Google Scholar]
- 43. Segal E., Widom J. (2009) What controls nucleosome positions? Trends Genet. 25, 335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li G., Levitus M., Bustamante C., Widom J. (2005) Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 12, 46–53 [DOI] [PubMed] [Google Scholar]
- 45. Poirier M. G., Oh E., Tims H. S., Widom J. (2009) Dynamics and function of compact nucleosome arrays. Nat. Struct. Mol. Biol. 16, 938–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Poirier M. G., Bussiek M., Langowski J., Widom J. (2008) Spontaneous access to DNA target sites in folded chromatin fibers. J. Mol. Biol. 379, 772–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kumbrink J., Gerlinger M., Johnson J. P. (2005) Egr-1 induces the expression of its corepressor nab2 by activation of the nab2 promoter thereby establishing a negative feedback loop. J. Biol. Chem. 280, 42785–42793 [DOI] [PubMed] [Google Scholar]
- 48. Schones D. E., Cui K., Cuddapah S., Roh T. Y., Barski A., Wang Z., Wei G., Zhao K. (2008) Dynamic regulation of nucleosome positioning in the human genome. Cell 132, 887–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mahadevan L. C., Willis A. C., Barratt M. J. (1991) Rapid histone H3 phosphorylation in response to growth factors, phorbol esters, okadaic acid, and protein synthesis inhibitors. Cell 65, 775–783 [DOI] [PubMed] [Google Scholar]
- 50. Lee E. R., McCool K. W., Murdoch F. E., Fritsch M. K. (2006) Dynamic changes in histone H3 phosphoacetylation during early embryonic stem cell differentiation are directly mediated by mitogen- and stress-activated protein kinase 1 via activation of MAPK pathways. J. Biol. Chem. 281, 21162–21172 [DOI] [PubMed] [Google Scholar]
- 51. Drobic B., Pérez-Cadahía B., Yu J., Kung S. K., Davie J. R. (2010) Promoter chromatin remodeling of immediate-early genes is mediated through H3 phosphorylation at either serine 28 or 10 by the MSK1 multi-protein complex. Nucleic Acids Res. 38, 3196–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Simboeck E., Sawicka A., Zupkovitz G., Senese S., Winter S., Dequiedt F., Ogris E., Di Croce L., Chiocca S., Seiser C. (2010) A phosphorylation switch regulates the transcriptional activation of cell cycle regulator p21 by histone deacetylase inhibitors. J. Biol. Chem. 285, 41062–41073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Henikoff S. (2008) Nucleosome destabilization in the epigenetic regulation of gene expression. Nat. Rev. Genet. 9, 15–26 [DOI] [PubMed] [Google Scholar]
- 54. Li B., Carey M., Workman J. L. (2007) The role of chromatin during transcription. Cell 128, 707–719 [DOI] [PubMed] [Google Scholar]
- 55. Henikoff S., Shilatifard A. (2011) Histone modification: cause or cog? Trends Genet. 27, 389–396 [DOI] [PubMed] [Google Scholar]
- 56. Chen S., Ma J., Wu F., Xiong L. J., Ma H., Xu W., Lv R., Li X., Villen J., Gygi S. P., Liu X. S., Shi Y. (2012) The histone H3 Lys 27 demethylase JMJD3 regulates gene expression by impacting transcriptional elongation. Genes Dev. 26, 1364–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eissenberg J. C., Elgin S. C. (2000) The HP1 protein family: getting a grip on chromatin. Curr. Opin. Genet. Dev. 10, 204–210 [DOI] [PubMed] [Google Scholar]
- 58. Vakoc C. R., Mandat S. A., Olenchock B. A., Blobel G. A. (2005) Histone H3 lysine 9 methylation and HP1γ are associated with transcription elongation through mammalian chromatin. Mol. Cell 19, 381–391 [DOI] [PubMed] [Google Scholar]
- 59. Horikoshi N., Kumar P., Sharma G. G., Chen M., Hunt C. R., Westover K., Chowdhury S., Pandita T. K. (2013) Genome-wide distribution of histone H4 lysine 16 acetylation sites and their relationship to gene expression. Genome Integr. 4, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Szerlong H. J., Prenni J. E., Nyborg J. K., Hansen J. C. (2010) Activator-dependent p300 acetylation of chromatin in vitro: enhancement of transcription by disruption of repressive nucleosome-nucleosome interactions. J. Biol. Chem. 285, 31954–31964 [DOI] [PMC free article] [PubMed] [Google Scholar]