

Background: Ten mitochondrial sites of superoxide/H2O2 generation are known, but their contributions in vivo are undefined.
Results: We assessed their rates ex vivo in conditions mimicking rest and exercise.
Conclusion: Sites IQ and IIF generated half the signal at rest. During exercise, rates were lower, and site IF dominated.
Significance: Contributing sites ex vivo probably reflect those in vivo.
Keywords: Exercise, Hydrogen Peroxide, Mitochondria, Mitochondrial Respiratory Chain Complex, Reactive Oxygen Species (ROS), Rat Skeletal Muscle, Superoxide
Abstract
The sites and rates of mitochondrial production of superoxide and H2O2 in vivo are not yet defined. At least 10 different mitochondrial sites can generate these species. Each site has a different maximum capacity (e.g. the outer quinol site in complex III (site IIIQo) has a very high capacity in rat skeletal muscle mitochondria, whereas the flavin site in complex I (site IF) has a very low capacity). The maximum capacities can greatly exceed the actual rates observed in the absence of electron transport chain inhibitors, so maximum capacities are a poor guide to actual rates. Here, we use new approaches to measure the rates at which different mitochondrial sites produce superoxide/H2O2 using isolated muscle mitochondria incubated in media mimicking the cytoplasmic substrate and effector mix of skeletal muscle during rest and exercise. We find that four or five sites dominate during rest in this ex vivo system. Remarkably, the quinol site in complex I (site IQ) and the flavin site in complex II (site IIF) each account for about a quarter of the total measured rate of H2O2 production. Site IF, site IIIQo, and perhaps site EF in the β-oxidation pathway account for most of the remainder. Under conditions mimicking mild and intense aerobic exercise, total production is much less, and the low capacity site IF dominates. These results give novel insights into which mitochondrial sites may produce superoxide/H2O2 in vivo.
Introduction
Mitochondrial generation of superoxide and hydrogen peroxide was discovered in the 1960s and 1970s (1, 2) and has been well studied (3–15). It is not a single process; the signal obtained is the sum of rates from different sites that prematurely reduce oxygen to superoxide or H2O2. Each site has its own unique properties. The sites can be distinguished and studied in situ by providing electrons from appropriate substrates and using specific electron transport inhibitors to isolate them pharmacologically. In this way, at least 10 distinct sites have been characterized in rat skeletal muscle mitochondria (16). These sites are represented as red circles in Fig. 1. In order of their maximum capacities, they are as follows: IIIQo4 in complex III (17); IQ (18, 19) and IIF (20) in complexes I and II; OF, PF, and BF in the 2-oxoglutarate, pyruvate, and branched-chain 2-oxoacid dehydrogenase complexes (16); GQ in mitochondrial glycerol phosphate dehydrogenase (21); IF in complex I (16); EF in ETF/ETF:Q oxidoreductase (22); and DQ in dihydroorotate dehydrogenase (23).
FIGURE 1.

Sites of superoxide and H2O2 production during electron flow from different blocks of metabolites through the mitochondrial electron transport chain. Metabolic substrates are grouped into five blocks colored to match Table 1: ketone bodies, amino acids, tricarboxylic acid (TCA) cycle, glycerol 3-phosphate (GP) shuttle, and β-oxidation. Unfilled boxes represent intermediate metabolites not added in the media. Electrons from the oxidation of these reduced substrates enter the mitochondrial electron transport chain through different isopotential groups of redox centers, denoted by the two planes, each operating at about the same redox potential (Eh): NADH/NAD+ at Eh ∼−280 mV and QH2/Q at Eh ∼+20 mV (59). The flow of electrons through NADH to the Q pool at complex I and from the Q pool to cytochrome c (cyt c) at complex III is indicated by the large green arrows dropping down through the isopotential planes. Enzymes that feed electrons into each isopotential group are represented as ovals, electron transport inhibitors are drawn with red blunted arrows, and CN-POBS, a suppressor of electron leak at site IQ that does not inhibit electron transport, is drawn with a green blunted arrow. Electrons from NAD-linked substrates enter the NADH/NAD+ pool through appropriate NAD-linked dehydrogenases (DH), including those for branched-chain 2-oxoacids (BCOADH), pyruvate (PDH), 2-oxoglutarate (OGDH), and others that are grouped together because there is no evidence that they produce superoxide/H2O2. Electrons from NADH flow into complex I (site IF) and then drop down via site IQ to QH2/Q in the next isopotential pool, providing the energy to generate protonmotive force (pmf). Q oxidoreductases, including complex II, mitochondrial glycerol 3-phosphate dehydrogenase (mGPDH), ETF:QOR, and dihydroorotate dehydrogenase (DHODH), can also pass electrons into the Q pool. Electrons flow from QH2 through complex III to cytochrome c and finally to oxygen (not shown), again pumping protons and generating pmf. The redox state of NADH (outlined in blue) reports the redox state of the first isopotential group. The redox state of cytochrome b566 (outlined in blue) reports the redox state of the second isopotential group (24). Red circles indicate sites of superoxide/H2O2 production: the flavin/lipoate of the dehydrogenases for branched-chain 2-oxoacids (BF), pyruvate (PF), and 2-oxoglutarate (OF), the complex I flavin (IF) and Q-binding site (IQ), the flavin site of complex II (IIF), the quinone site of mitochondrial glycerol 3-phosphate dehydrogenase (GQ), the flavin site of ETF:QOR (EF), the quinone site of dihydroorotate dehydrogenase (DQ), and the outer quinol-binding site of complex III (IIIQo).
The rates of superoxide/H2O2 production under native conditions (i.e. in the absence of added inhibitors) are much less than these maximum capacities (24). Therefore, the maximum capacities of the sites are not necessarily related to the actual engagement and rate of each site under native conditions. To establish the native rate from each site, we devised novel methods based on measurements of the redox states of endogenous reporters in isolated mitochondria oxidizing conventional substrates (24, 25). These studies led to two striking conclusions. First, the overall rates of H2O2 production differed 5-fold between different conventional substrates; they were much higher with succinate than with glutamate plus malate as substrate. Second, the relative contribution of each site was completely dependent on the substrate being oxidized. With succinate as substrate, site IQ was dominant; with glutamate plus malate, sites IF, IIIQo, and OF all contributed; with palmitoylcarnitine, site IIF was also important; and with glycerol 3-phosphate, five sites contributed significantly, including GQ (25). Thus, the relative and absolute contribution of each specific site to the production of superoxide/H2O2 in isolated mitochondria depends very strongly on which conventional substrate is being oxidized.
In complex systems, such as intact cells, different substrates are metabolized simultaneously. The main groups of substrates oxidized by mitochondria are represented in Fig. 1 and Table 1 by colored boxes. In skeletal muscle, oxidation of ketone bodies, amino acids, tricarboxylic acid cycle intermediates, glycerol 3-phosphate, and fatty acids feeds electrons into multiple sites in the matrix dehydrogenases and electron transport chain. Therefore, in vivo, it is likely that several sites produce superoxide/H2O2 simultaneously at different rates.
TABLE 1.
Concentrations of substrates and effectors during rest, mild aerobic exercise, and intense aerobic exercise in rat skeletal muscle cytosol and compositions of the three media mimicking these conditions ex vivo
Data are for all metabolites assumed to be relevant, from the literature for rat skeletal muscle as indicated (values for carnitine and acetylcarnitine include some human data; values for free Ca2+ include some mouse data). Where appropriate, calculations used Equation 3 of Ref. 60 and assumed that muscle wet weight is 77% water (60–64) and 81% intracellular (60–62, 64–66) or that 18% of muscle wet weight is protein (66, 67). Published values were corrected for plasma concentrations where appropriate and for the distribution of intracellular metabolites between cytosol and mitochondria, using the following matrix/cytosol ratios from heart and liver (or the assumed values for metabolites in parentheses): glycerol 3-phosphate, glutamine (dihydroxyacetone phosphate, taurine): 0 (68, 69); citrate, isocitrate: 10 (69–71); malate (succinate): 4 (69, 70, 72, 73); 2-oxoglutarate: 7 (69–73); pyruvate (acetoacetate, 3-hydroxybutyrate, arginine, lysine): 2 (70, 73); aspartate (neutral amino acids, acylcarnitines): 1 (69, 70, 72, 74); glutamate: 3.5 (69, 70, 72, 74); carnitine: 0.74 (75); ATP: 0.25 (69); phosphate: 5 (76). Mitochondrial volume was assumed to be 10% of intracellular (77–82). Values were rounded to convenient whole numbers. Total Mg2+ and Ca2+ concentrations to give the targeted free values were calculated using the software MaxChelator. Targeted free Ca2+ concentrations were 0.05 μm (rest), 1 μm (mild aerobic exercise), and 1 μm (intense aerobic exercise) (83–86). Targeted free Mg2+ concentrations were 600 μm (rest), 600 μm (mild aerobic exercise), and 1000 μm (intense aerobic exercise) (40, 87–89). Targeted Na+ concentrations were 16 mm for all media (65, 89). Media had K+ and Cl− adjusted to give an osmolarity of 290 mosm (90–92). Total K+ concentrations were 80 mm (rest), 77 mm (mild aerobic exercise), and 74 mm (intense aerobic exercise). The “basic media” had the compositions shown; the “complex substrate mixes” contained all of the substrates in the colored boxes. *, no data found, so values are assumed; **, maximum value to avoid mitochondrial uncoupling in vitro.
The conditions experienced by muscle mitochondria within cells differ substantially between rest and exercise. In particular, ADP supply, free Ca2+, cytosolic pH, and the availability of different substrates are very different, and this will have profound effects on the mitochondrial production of superoxide and H2O2. During exercise, the levels of both reactive oxygen species and reactive nitrogen species are increased (26). The production of such species is crucial for the beneficial effects of exercise (27–29) and for force development (30). However, excess production is detrimental for skeletal muscle performance (30, 31). Mitochondria are leading candidates for the increased production of these reactive species during exercise (30, 32), although others argue for non-mitochondrial sources (26, 32).
Several different probes can be used in intact cells to report changes in reactive oxygen species (33, 34), but they cannot reliably distinguish the mitochondrial sites active under particular physiological or pathological conditions. This is because pharmacological or genetic manipulation of particular mitochondrial sites invariably alters the redox states of other sites, changing their contributions to overall production of superoxide/H2O2 and making interpretation unreliable. Also, these probes invariably compete with the endogenous antioxidant defenses and measure steady state levels rather than rates of radical production. On the other hand, the rate from each site can now be quantified using isolated mitochondria (24, 25). However, given the strong substrate dependence of their superoxide/H2O2 production, isolated mitochondria oxidizing conventional substrates are not sufficiently physiologically relevant.
To improve the physiological relevance of the more amenable mitochondrial system, in the present work, we develop an ex vivo system in which isolated muscle mitochondria are incubated acutely in novel complex media. These media contain the measured physiological concentrations of all metabolites and effectors assumed to be relevant in skeletal muscle cytosol at rest and during mild and intense aerobic exercise. Using this system, we quantify the contribution of each mitochondrial site to total H2O2 production to gain novel insights into the rates and sites of mitochondrial superoxide/H2O2 production in skeletal muscle during rest and exercise.
EXPERIMENTAL PROCEDURES
Animals, Mitochondria, and Reagents
Female Wistar rats were from Charles River Laboratories, age 5–10 weeks, and fed chow ad libitum with free access to water. Mitochondria were isolated from hind limb skeletal muscle at 4 °C in Chappell-Perry buffer (CP1; 100 mm KCl, 50 mm Tris, 2 mm EGTA, pH 7.4, at 4 °C) by standard procedures (35) and kept on ice during the assays (<5 h). Protein was measured by the biuret method. The animal protocol was approved by the Buck Institute Animal Care and Use Committee in accordance with IACUC standards. Reagents were from Sigma except for Amplex UltraRed, from Invitrogen.
Oxygen Consumption and Superoxide/H2O2 Production
Skeletal muscle mitochondria (0.3 mg of protein·ml−1) were incubated at 37 °C for 4–5 min in the appropriate “basic medium” (Table 1) mimicking the cytosol of skeletal muscle during rest (basic “rest” medium plus oligomycin), mild aerobic exercise (basic “mild aerobic exercise” medium with no further additions), or intense aerobic exercise (basic “intense aerobic exercise” medium plus glucose and sufficient hexokinase after titration (about 0.08 units·ml−1) to give 90% of the maximum state 3 oxygen consumption rate). ATP (6 mm) was injected into the chamber, and then after 1 min, the appropriate “complex substrate mix” was added (Table 1). Oxygen consumption rates were measured using a Clark-type electrode fitted in a water-jacketed chamber (Rank Brothers, Bottisham, UK). Rates of superoxide/H2O2 production were measured collectively as rates of H2O2 production as two superoxide molecules are dismutated by endogenous or exogenous superoxide dismutase to yield one H2O2. H2O2 was detected using 5 units·ml−1 horseradish peroxidase and 50 μm Amplex UltraRed in the presence of 25 units·ml−1 superoxide dismutase (36) in a Varian Cary Eclipse spectrofluorometer (λexcitation = 560 nm, λemission = 590 nm) with constant stirring and calibrated with known amounts of H2O2 in the presence of all relevant additions because some of them quenched fluorescence (36).
NAD(P)H and Cytochrome b566 Redox State
The reduction state of endogenous NAD(P)H was determined by autofluorescence in mitochondria incubated as described for H2O2 production (most of the signal is from bound NADH in the matrix, and NADPH hardly changes in the present experiments, but for transparency we call it “NAD(P)H”) using a Shimadzu RF5301-PC dual wavelength spectrophotometer at λexcitation = 365 nm, λemission = 450 nm (18, 24). NAD(P)H was assumed to be 0% reduced after 5 min without added substrate. 100% reduction was established internally by adding saturating conventional substrate (e.g. 5 mm malate plus 5 mm glutamate) and 4 μm rotenone at the end of each run. Intermediate values of NAD(P)H reduction were measured at 3–4 min after the addition of the appropriate mix of substrates and were determined as percentage of reduced NAD(P)H relative to the 0 and 100% values. The reduction state of endogenous cytochrome b566 was measured using 1.5 mg of mitochondrial protein·ml−1 with constant stirring at 37 °C in an Olis DW 2 dual wavelength spectrophotometer at A566 nm–A575 nm (17). This signal reports ∼75% cytochrome b566 and ∼25% cytochrome b562 (17, 37). Cytochrome b566 was assumed to be 0% reduced after 5 min without added substrate. 100% reduction was established in separate cuvettes with 5 mm succinate and 2 μm antimycin A. Intermediate values of cytochrome b566 reduction were measured over 45 s, ∼20 s after the addition of the appropriate complex substrate mix, and were determined as percentage of reduced cytochrome b566 relative to the 0 and 100% values.
Correction for Matrix Peroxidase Activity
H2O2 production rates in Figs. 3 and 12 (but not in other figures) were corrected for losses of H2O2 caused by peroxidase activity in the matrix to give a better estimate of actual superoxide/H2O2 production rates (38). Rates were mathematically corrected to those that would have been observed after pretreatment with 1-chloro-2,4-dinitrobenzene (CDNB) to deplete glutathione and decrease glutathione peroxidase and peroxiredoxin activity, using an empirical equation,
where ν is the rate of H2O2 production in pmol of H2O2·min−1·mg protein−1.
FIGURE 3.

Maximum capacities for superoxide/H2O2 production of the 10 characterized sites in the mitochondrial electron transport chain and matrix compared with the native ex vivo rates using isolated mitochondria incubated in the absence of inhibitors in media mimicking the cytosol of skeletal muscle during rest, mild aerobic exercise, or intense aerobic exercise. The sites in the NADH/NAD+ isopotential group (Fig. 1) are OF (flavin site of the 2-oxoglutarate dehydrogenase complex), PF (flavin site of the pyruvate dehydrogenase complex); BF (flavin site of the branched-chain 2-oxoacid dehydrogenase complex); and IF (flavin site of complex I). Site IQ of complex I is between the two isopotential groups. The sites in the QH2/Q isopotential groups are IIIQo (outer ubiquinone binding site of complex III); IIF (flavin site of complex II); GQ (quinone site of site mitochondrial glycerol 3-phosphate dehydrogenase); EF (flavin site of the electron-transferring flavoprotein/ETF:ubiquinone oxidoreductase system), and DQ (quinone site of dihydroorotate dehydrogenase). The first nine bars are replotted from Ref. 16, and the DQ bar is replotted from Ref. 23. Ex vivo native rates are from Fig. 2D. All rates were corrected for matrix peroxidases using Equation 1. Values are means ± S.E. (error bars) (n = 3–20).
FIGURE 12.
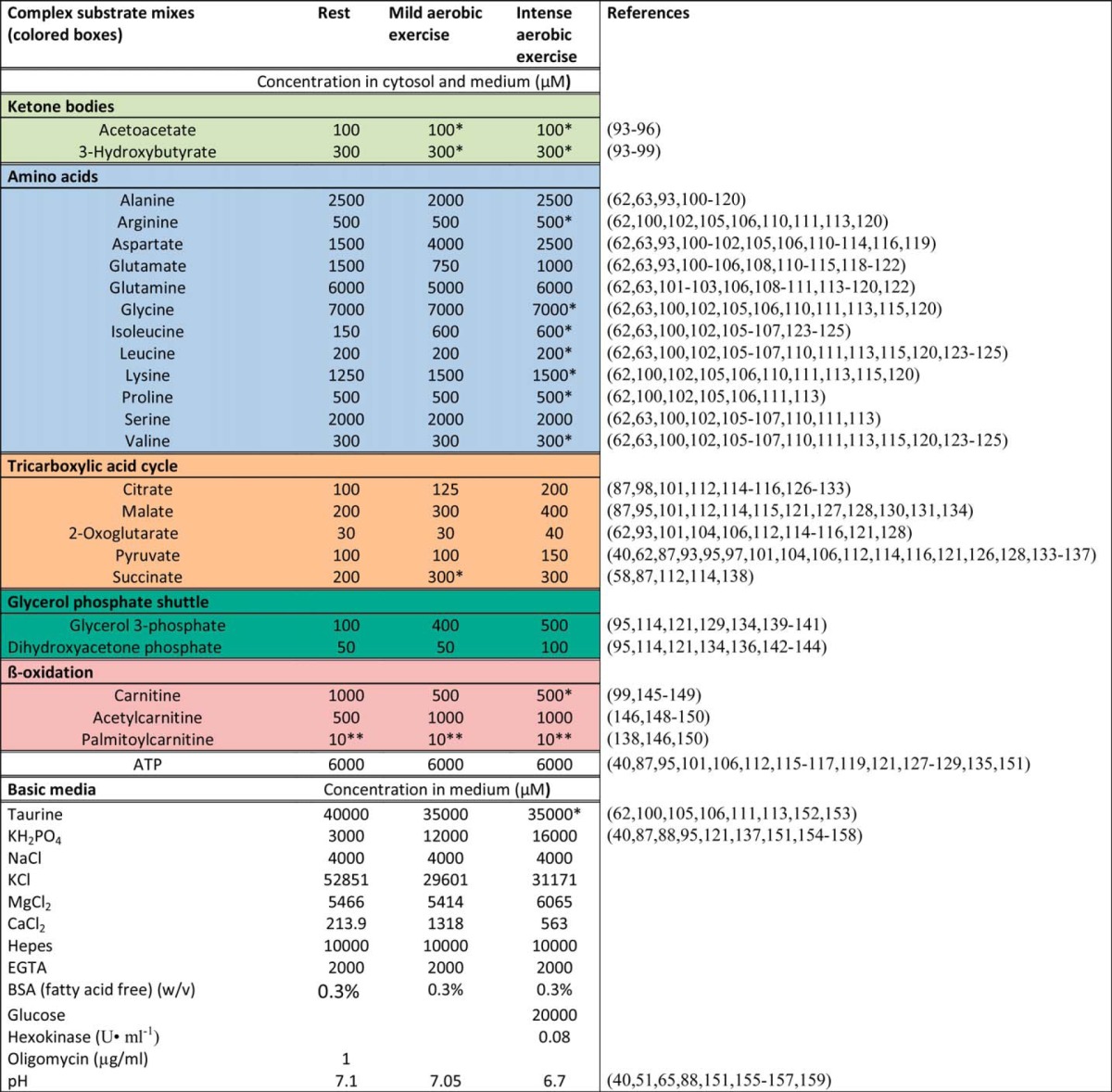
Contributions of different sites to superoxide and H2O2 production by isolated skeletal muscle mitochondria ex vivo in media mimicking rest, mild aerobic exercise, and intense aerobic exercise. Positive values taken from Figs. 8D, 9, and 11 were corrected for the losses of H2O2 caused by the activity of mitochondrial matrix peroxidases using Equation 1. Because of their topology, only 50% of the signal assessed for sites IIIQo and GQ was corrected, slightly diminishing their contributions relative to other sites. Inverted error bars indicate the propagated errors for each site, and conventional error bars indicate the propagated sum of these errors. Values are means ± S.E. (error bars) (n = 3–20).
Due to the non-linearity of the curve, the correction is different at different overall rates. Therefore, the total H2O2 production rates were first corrected using Equation 1. The corrected rate from each site (and its S.E.) was then back-calculated based on its relative contribution in a given condition (Table 3). All sites produce superoxide/H2O2 exclusively into the mitochondrial matrix, except for IIIQo and GQ, which generate ∼50% of the superoxide/H2O2 to the cytosol (24). For these sites, only 50% of the rate was corrected.
TABLE 3.
Corrections applied for changes in redox state of NAD(P)H and cytochrome b566, and rate of superoxide/H2O2 production from each site during “rest,” “mild aerobic exercise,” and “intense aerobic exercise”
Δ control values in columns B and C were used to correct the observed changes in H2O2 production rate in column A for changes in the redox states of NAD(P)H and cytochrome b566, as shown in the last column.
a 2.5 μm CN-POBS decreased IQ superoxide production by only 65%; therefore, values were scaled to account for CN-POBS potency.
b 10 μm CN-POBS reduced IQ superoxide production by only 75%; therefore, values were corrected for CN-POBS potency.
c The relative contribution was calculated based on the CDNB corrected rates for each site. The S.E. was scaled according to the internal error for each site. DHAP, dihydroxyacetone phosphate; nd, not determined.
Curve Fitting
Data were fit by exponential functions in Figs. 5C, 6C, and 10 (C and F) to yield the parameter values in Equations 2–5, respectively,
where νH2O2 is the rate of H2O2 production.
FIGURE 5.
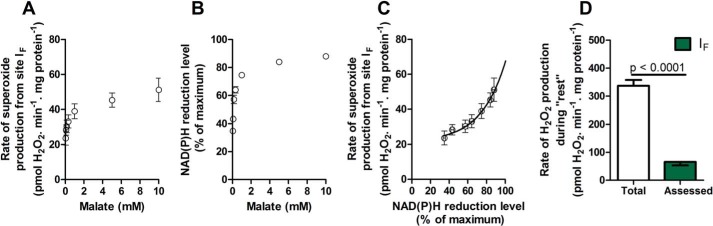
Contribution of site IF at “rest.” Mitochondria were incubated in medium mimicking rest (Table 1). A, rate of superoxide production from site IF, defined by the presence of 4 μm rotenone, 1.5 mm aspartate, and 2.5 mm ATP, at different malate concentrations. B, dependence of NAD(P)H reduction level on malate concentration under the same conditions (100% reduction was established by adding 5 mm malate plus 5 mm glutamate). C, calibration curve obtained by combining the y axes from A and B, showing the dependence of the rate of superoxide production from site IF on the NAD(P)H reduction level. D, total rate of H2O2 production at “rest” (from Fig. 2D) and the contribution of site IF assessed from the reduction state of NAD(P)H in Fig. 4 and the calibration curve in C (green; the inverted error bar indicates the propagated error for site IF). Values are means ± S.E. (error bars) (n = 3–20). p < 0.0001 by Welch's t test.
FIGURE 6.
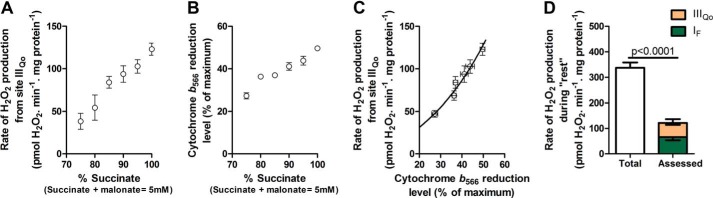
Contribution of site IIIQo at “rest.” Mitochondria were incubated in medium mimicking rest (Table 1). A, rate of superoxide production from site IIIQo, defined as the rate in the presence of 4 μm rotenone sensitive to 4 μm myxothiazol, at different ratios of succinate/malonate (total concentration 5 mm). Data were corrected for changes in the contribution of site IF using changes in the NAD(P)H redox state and the calibration curve in Fig. 5C (Table 3) (24). B, dependence of cytochrome b566 reduction state on the succinate/malonate ratio under the same conditions (100% reduction was established by adding 5 mm succinate plus 2 μm antimycin A). C, calibration curve obtained by combining the y axes from A and B showing the dependence of the rate of superoxide produced from site IIIQo on the cytochrome b566 reduction level. D, total rate of H2O2 production at “rest” (from Fig. 2D) and the contributions of sites IF (from Fig. 5D) and IIIQo (assessed from the reduction state of cytochrome b566 in Fig. 4 and the calibration curve in C). Inverted error bars indicate the propagated errors for each site, and the conventional error bar indicates the propagated sum of these errors. Values are means ± S.E. (error bars) (n = 3–20). p < 0.0001 by Welch's t test.
FIGURE 10.

Calibration of the reporters of sites IF and IIIQo during “intense aerobic exercise.” Mitochondria were incubated in the basic medium mimicking intense aerobic exercise (Table 1). A–C, construction of the calibration curve showing the dependence of the rate of superoxide production from site IF on NAD(P)H reduction level exactly as in Fig. 5, A–C, but using the basic medium mimicking intense aerobic exercise. D–F, construction of the calibration curve showing the dependence of the rate of superoxide production from site IIIQo on the cytochrome b566 reduction level exactly as in Fig. 6, A–C, but using the basic medium mimicking intense aerobic exercise. Values are means ± S.E. (error bars) (n = 3).
Statistics
The calibration curves in Figs. 5C, 6C, and 10 (C and F) were used to calculate the rates of superoxide/H2O2 production at sites IF and IIIQo, both directly when assessing the rates from these sites and indirectly when correcting for the effects on these sites of inhibition at other sites (Table 3). The errors in the calibration curves were taken into account when calculating the S.E. values of the assessed rates. This error was calculated by error propagation as described previously (24).
Because error propagation was used to include the uncertainty from the calibration curve in the assessed rates, the mean ± S.E. values plotted do not represent individual values. Therefore, the statistics were calculated using the averaged values ± S.E. and the number of observations. The significances of differences between the total measured rates and the assessed rates were calculated using Welch's t test. For the regular multiple comparison tests in Figs. 2 and 4, a one-way ANOVA was used, followed by Tukey's post hoc test. When comparing the values with omission of single substrates with the total values, Student's t test was used. p < 0.05 was considered significant.
FIGURE 2.

Oxygen consumption and H2O2 generation by isolated skeletal muscle mitochondria incubated in media mimicking the cytosol of skeletal muscle during rest, mild aerobic exercise, or intense aerobic exercise. A, representative traces of oxygen consumption. The dashed box indicates the interval over which the rates were analyzed. Numbers by the traces indicate mean rates in nmol of O·min−1·mg of protein−1. B, rates of oxygen consumption. The maximum (state 3) rate “Max” was measured in the presence of 20 mm glucose and excess (2.5 units·ml−1) hexokinase. C, representative traces of H2O2 generation. The dashed box indicates the interval over which the rates were analyzed. Numbers by the traces indicate mean rates in pmol of H2O2·min−1·mg of protein−1. D, rates of H2O2 generation. In each panel, mitochondria were incubated in media mimicking rest, mild aerobic exercise, or intense aerobic exercise (Table 1), as indicated. Values are means ± S.E. (error bars) (n = 3–20 biological replicates). *, p < 0.05; ***, p < 0.0001, one-way ANOVA with Tukey's post hoc test.
FIGURE 4.
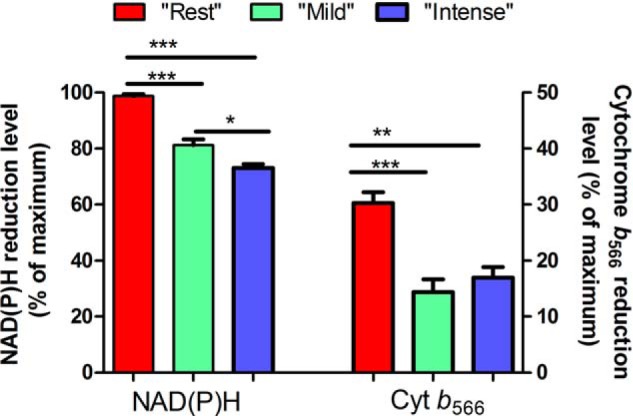
Reduction state of NAD(P)H and cytochrome b566. Mitochondria were incubated in media mimicking rest, mild aerobic exercise, or intense aerobic exercise (Table 1), as indicated. Values are means ± S.E. (error bars) (n = 3–20). *, p < 0.05; **, p < 0.01; ***, p < 0.0001, one-way ANOVA with Tukey's post hoc test.
RESULTS
Media Mimicking Skeletal Muscle Cytosol during Rest, Mild Aerobic Exercise, and Intense Aerobic Exercise ex Vivo
In vivo, the substrates for mitochondrial metabolism come primarily from the catabolism of sugars, proteins, and fats. In the cytosol, they are available to the mitochondria as metabolites that can be categorized into five groups according to their origins: ketone bodies, amino acids, tricarboxylic acid cycle intermediates, glycerol 3-phosphate, and acylcarnitines (Table 1). The major metabolic fates of these substrates are indicated by the colored boxes leading to the mitochondrial dehydrogenases in Fig. 1. These metabolites connect to the 10 sites in skeletal muscle mitochondria known to have significant capacity to produce superoxide or H2O2. In Fig. 1, these sites are grouped in planes reflecting their operating redox potentials. The top plane represents the NADH/NAD+ isopotential group, containing the dehydrogenases that reduce NAD+ and the sites in complex I that oxidize NADH. From complex I, the electrons drop down in energy to a more positive redox potential in the ubiquinone pool. The bottom plane represents the QH2/Q isopotential group, containing the ubiquinone oxidoreductases that reduce ubiquinone and the sites in complex III that oxidize ubiquinol. The electrons are then transferred to cytochrome c and on to the final acceptor, O2, to generate H2O (not shown).
Table 1 lists the consensus concentrations in rat skeletal muscle cytosol of all metabolites and effectors thought to be potentially relevant to mitochondrial electron transport and production of superoxide/H2O2. The values were taken from the extensive literature on rat skeletal muscle obtained mostly by freeze-clamp followed by enzymatic or chromatographic quantitation, both in vivo and in isolated muscle preparations. Where appropriate, they were corrected using consensus values for extracellular contamination and for the estimated compartmentation between the mitochondrial matrix and the cytosol. Values are listed for resting muscle and for two exercise conditions: submaximal stimulation, representing mild aerobic exercise, and extensive stimulation, representing intense aerobic exercise. We assumed that the exact K+ and Cl− concentrations were unimportant and therefore used KCl to bring the osmolarity to the physiological value of 290 mosm.
These literature values enabled us to prepare three different media for mitochondrial incubations ex vivo, containing the relevant metabolites and effectors at the concentrations that would be encountered by mitochondria in vivo during rest and mild and intense aerobic exercise. ATP turnover and hence steady-state ADP concentrations were set based on respiration rates as described below. These media are the first that we know of to be carefully designed to mimic the substrate and effector mix in the cytosol of skeletal muscle cells during “rest,” “mild aerobic exercise,” and “intense aerobic exercise” (Table 1). They enabled us to assess the production of H2O2 from isolated mitochondria in an ex vivo system that closely mimicked the relevant aspects of the cytosol of rat muscle cells in vivo.
Setting ATP Demand during “Rest”, “Mild Aerobic Exercise,” and “Intense Aerobic Exercise”
To deplete endogenous substrates, rat skeletal muscle mitochondria were incubated for 4–5 min in the appropriate basic medium lacking all respiratory substrates (Table 1). ATP was added 1 min before respiration was initiated, to minimize the time available for ATP hydrolysis. To initiate respiration, the appropriate complex mix of substrates mimicking rest or mild or intense aerobic exercise (Table 1) was then added (Fig. 2A).
“Rest”
In resting muscle in vivo, the rates of respiration and ATP synthesis are low (39). To mimic rest ex vivo, a low rate of ATP synthesis by mitochondria incubated in the “rest” medium (Table 1) was achieved by including oligomycin in the medium to fully inhibit the mitochondrial ATP synthase. This was not ideal, because it implies no ATP turnover at rest, but it was necessary because the relatively high contaminating ATPase activity present in the mitochondrial preparation caused an intermediate respiration rate. The rate of oxygen consumption at “rest” was less than 10% of the maximum rate (Fig. 2, A and B, red).
“Mild Aerobic Exercise”
During exercise in vivo, respiratory rates increase due to increased ATP demand and altered substrate supply and concentrations of effectors (pH, Ca2+). Mild exercise induces ≤45% of whole body maximal O2 consumption rate, VO2max (39). To mimic mild aerobic exercise ex vivo, mitochondria were incubated in the basic “mild aerobic exercise” medium in the absence of oligomycin, followed by ATP and then the appropriate complex mix of relevant substrates (Table 1). The initial fast rate of respiration required to rephosphorylate ADP formed from the added ATP (Fig. 2A) was ignored. After this fast phase was complete and ATP/ADP settled to a steady state value, ATP demand and respiration rate ran at an intermediate rate limited by the supply of ADP from contaminating extramitochondrial ATPases. In this phase, the respiratory rate was 22% of the maximum rate (Fig. 2, A and B, green).
“Intense Aerobic Exercise”
Metabolic demand increases with exercise intensity. During intense exercise, respiration is ≥65% of VO2max (39). ADP level increases significantly but is still an order of magnitude lower than the ATP level (40). Cytosolic pH drops to 6.7 due to increased CO2 and lactate production. To mimic intense aerobic exercise ex vivo, mitochondria were incubated in the basic “intense aerobic exercise” medium, followed by ATP and then the appropriate complex mix of relevant substrates. To achieve high respiratory rates but keep ADP levels relatively low, in separate experiments, the O2 consumption rate was titrated to 90% of the maximum rate using hexokinase in the presence of glucose to set the rate of extramitochondrial ATP hydrolysis high but still submaximal in each mitochondrial preparation, and this amount of hexokinase (about 0.08 units·ml−1) was included in the “intense aerobic exercise” medium (Fig. 2, A and B, blue). Maximum phosphorylating respiration was set by adding excess hexokinase (Fig. 2B, white).
Mitochondrial H2O2 Production during “Rest,” “Mild Aerobic Exercise,” and “Intense Aerobic Exercise”
The rate of generation of superoxide and H2O2 was measured under the three conditions described above as the rate of extramitochondrial H2O2 production in the presence of exogenous superoxide dismutase to convert any superoxide to H2O2 for assay. Fig. 2C shows that rates were linear for 2–3 min after the addition of the complex substrate mixes and then decreased. Some metabolites were present at very low concentrations (Table 1) and were likely to be consumed quickly, so ideally we would measure initial rates, but because the system was not at steady-state over the first minute in the “exercise” medium (Fig. 2A), we calculated the rates as the pseudolinear rate between 1 and 3 min (dotted boxes in Fig. 2, A and C). Fig. 2D shows that the highest rate of H2O2 production was observed when mitochondria were incubated in the medium mimicking the cytosol of skeletal muscle at rest (336 pmol of H2O2·min−1·mg of protein−1). In the media mimicking mild aerobic exercise and intense aerobic exercise, the rates were significantly lower.
Unphysiological concentrations of conventional substrates and the presence of appropriate electron transport inhibitors favor high rates of mitochondrial superoxide/H2O2 production. These rates can be up to 2% of the total respiration rate of uninhibited resting mitochondria (2, 41). Under more realistic non-inhibited (native) conditions with conventional substrates, the percentage of electron leak to H2O2 is only ∼0.15% (42, 43). Table 2 shows the % electron leak when mitochondria were incubated under conditions mimicking rest, mild aerobic exercise, and intense aerobic exercise. At “rest,” the electron leak was 0.35%. During “exercise,” it was 10–35-fold lower: only 0.03% for “mild aerobic exercise” and 0.01% for “intense aerobic exercise.”
TABLE 2.
Electron leak during “rest,” “mild aerobic exercise,” and “intense aerobic exercise”
Rates were calculated per nmol e− by multiplying the rates of H2O2 production in Fig. 2D in pmol of H2O2·min−1·mg of protein−1 by 0.002 and rates of oxygen consumption in Fig. 2B in nmol of O·min−1·mg of protein−1 by 2. Values are means ± S.E. (n = 3 for oxygen consumption rates; n = 3–20 for H2O2 production rates).
| “Rest” | “Mild aerobic exercise” | “Intense aerobic exercise” | |
|---|---|---|---|
| Rate of H2O2 production (nmol e−·min−1·mg of protein−1) | 0.68 ± 0.04 | 0.16 ± 0.01 | 0.10 ± 0.01 |
| Rate of oxygen consumption (nmol e−·min−1·mg of protein−1) | 196 ± 40 | 492 ± 88 | 2032 ± 73 |
| Percentage electron leak (100·H2O2 production rate/respiration rate) | 0.35% | 0.03% | 0.01% |
The ex vivo data in Fig. 2D and Table 2 suggest that skeletal muscle mitochondria in vivo are unlikely to contribute to the overall increase in reactive oxygen species production observed during exercise, supporting the conclusions of others (26, 32). Instead, the mitochondrial production rate may decrease during exercise, because the redox centers that donate electrons to O2 become more oxidized during exercise (see below).
Sites of Superoxide/H2O2 Production
Ten mitochondrial sites associated with the tricarboxylic acid cycle and electron transport chain are known to produce superoxide or H2O2 (Fig. 1). Their maximum capacities are shown in Fig. 3, on the same scale as the ex vivo rates from Fig. 2D after all of the values have been corrected for consumption of intramitochondrial H2O2 by matrix peroxidases. Clearly, the maximum capacities of several sites are much higher than the ex vivo rates, making it impossible to predict a priori which sites contribute most to the total signal ex vivo or in vivo. The rates of H2O2 production measured under the different conditions in Fig. 2D are the sums of the rates from different sites. Our goal here was to determine the contribution of each site during “rest,” “mild aerobic exercise,” and “intense aerobic exercise” in the tractable ex vivo system because no adequate methods exist to address this question in vivo or in intact cells. We assume that the sites contributing ex vivo in media containing the physiological cytosolic concentrations of all substrates and effectors thought to be relevant will approximate the sites contributing in vivo.
Conventionally, complexes I and III are thought to be the dominant sources of mitochondrial superoxide/H2O2 in isolated mitochondria and cells (10, 13). Complex I produces superoxide at two distinct sites: site IQ, a high capacity site, most obviously active during reverse transfer of electrons from ubiquinol to NADH (18, 19, 44, 45); and site IF (18), now known to have much lower capacity (16). Complex III can produce superoxide at high rates from site IIIQo (2, 17). The rate of superoxide production from site IF depends on the redox state of the complex I flavin, which in turn has a unique relationship to the redox state of mitochondrial NAD(P)H. Similarly, the rate of superoxide production from site IIIQo is related to the redox state of cytochrome b566 (17). The redox states of NAD(P)H and cytochrome b566 were therefore used as endogenous reporters of the rates of superoxide production at sites IF and IIIQo, respectively (24, 25).
Fig. 4 and Table 3 show the reduction levels of NAD(P)H and cytochrome b566 when mitochondria were incubated in the three media. During “rest,” NAD(P)H was nearly 100% reduced, and cytochrome b566 was 30% reduced. During “exercise,” both were significantly more oxidized. To assess the rate of superoxide production by sites IF and IIIQo, calibration curves were built to establish the relationships between superoxide production from these sites and the reduction levels of the endogenous reporters.
Flavin of Complex I, Site IF, during “Rest”
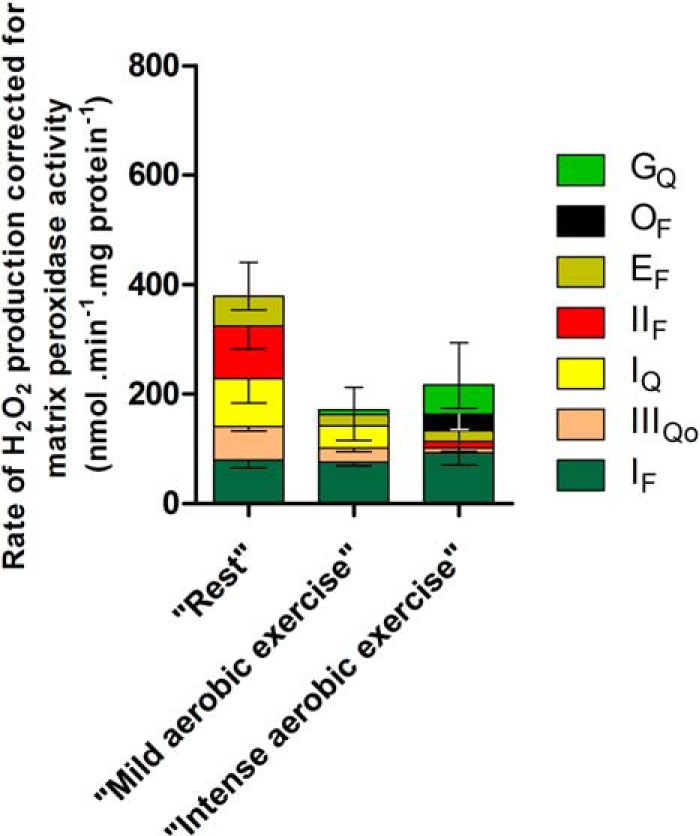
NAD(P)H redox state was calibrated as a reporter of the rate of superoxide production at site IF, as described previously (24). Fig. 5A shows the observed rate of H2O2 production from site IF as a function of the concentration of malate in the presence of rotenone (with added ATP and aspartate to minimize the contribution of site OF (16)). Fig. 5B shows the NAD(P)H reduction state under the same conditions. Fig. 5C replots the data from Fig. 5, A and B, to show the dependence of the measured rate of H2O2 production arising from superoxide production at site IF on the NAD(P)H reduction state. As shown in Fig. 4, NAD(P)H during “rest” was almost 100% reduced. From Fig. 5C, the contribution of site IF during “rest” was assessed to be 65 ± 12 pmol of H2O2·min−1·mg of protein−1 (mean ± S.E.; n = 8). The contributions of site IF and other relevant values developed below are summarized in Table 3.
Site IF accounted for 20 ± 3% of the total rate of H2O2 production measured during “rest” (Fig. 5D). Therefore, other sites also produced superoxide/H2O2 at rest.
Outer Ubiquinone Binding Site of Complex III, Site IIIQo, during “Rest”
Cytochrome b566 redox state was calibrated as a reporter of the rate of superoxide production at site IIIQo as described previously (24). Fig. 6A shows the observed myxothiazol-sensitive rate of H2O2 production from site IIIQo as a function of the ratio of added succinate and malonate in the presence of rotenone. At each point, the rate of H2O2 production from site IF was calculated from the observed NAD(P)H reduction level using Fig. 5C and subtracted from the total observed rate to give the rate specifically from site IIIQo (Table 3). Fig. 6B shows the cytochrome b566 redox state under the same conditions. Fig. 6C replots the data from Fig. 6, A and B, to show the dependence of the rate of H2O2 production arising from superoxide production at site IIIQo on the cytochrome b566 reduction state. Fig. 4 and Table 3 show that cytochrome b566 was 30% reduced during “rest.” Using the calibration curve in Fig. 6C, the contribution of site IIIQo was 55 ± 7 pmol of H2O2·min−1·mg of protein−1 (mean ± S.E.; n = 12) (Fig. 6D).
Site IIIQo accounted for 15 ± 2% of the total rate of H2O2 production measured during “rest” (Fig. 6D and Table 3). After accounting for the contributions of sites IF and IIIQo to the total rate of H2O2 production, there was still a significant difference between the total rates measured and assessed (Fig. 6D). Therefore, other sites also produced superoxide/H2O2 at rest.
Ubiquinone Binding Site of Complex I, Site IQ, during “Rest”
Site IQ has a high capacity for superoxide production during reverse electron transport when succinate is oxidized (19, 43) (Fig. 3) and during forward electron flow from NAD-linked substrates under particular conditions (44). However, the importance of this site in vivo and under semiphysiological conditions (25, 46, 47) is unknown. The contribution of site IQ during “rest” ex vivo was assessed by adding rotenone to inhibit electron transport at this site, followed by correction for consequent changes in the rate of superoxide production from sites IF and IIIQo. Rotenone was added 1 min prior to the substrate mix and after correction significantly decreased the rate of superoxide/H2O2 production at “rest” (Fig. 7A). The rate attributed to site IQ was 79 ± 23 pmol of H2O2·min−1·mg of protein−1 (mean ± S.E.; n = 6).
FIGURE 7.
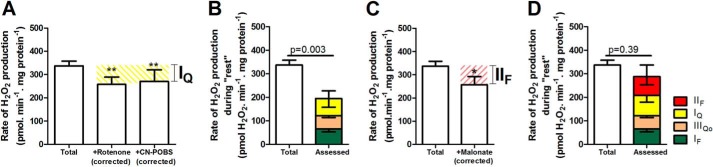
Contributions of sites IQ and IIF at “rest.” Mitochondria were incubated in medium mimicking rest (Table 1). A, total rate of H2O2 production and the rates in the presence of a IQ electron transport inhibitor (4 μm rotenone) or a suppressor of IQ superoxide production (2.5 μm CN-POBS) after correction for changes in the rates of sites IF and IIIQo assessed by changes in NAD(P)H and cytochrome b566 redox state (Table 3). The yellow hatched area represents the contribution of site IQ. B, total rate of H2O2 production at “rest” (from Fig. 2D) and the contributions of sites IF (from Fig. 5D), IIIQo (from Fig. 6D), and IQ (from A). Inverted error bars indicate the propagated errors for each site, and the conventional error bar indicates the propagated sum of these errors. C, total rate of H2O2 production and the rate in the presence of 4 μm malonate to inhibit the flavin site of complex II, after correction for changes in the rates of sites IF, IQ, and IIIQo (Table 3). The red hatched area represents the contribution of site IIF. D, total rate of H2O2 production at “rest” (from Fig. 2D) and the contributions of sites IF (from Fig. 5D), IIIQo (from Fig. 6D), IQ (from A), and IIF (from C). Inverted error bars indicate the propagated errors for each site, and the conventional error bar indicates the propagated sum of these errors. Values are means ± S.E. (error bars) (n = 3–20). **, p < 0.005; ***, p < 0.0001 by one-way ANOVA followed by Tukey's post hoc test. *, p < 0.05 by Student's t test. Welch's t test was used to determine p = 0.003 and p = 0.39.
CN-POBS at 2.5 μm selectively blocks 65% of superoxide production from site IQ with no effects on oxidative phosphorylation, although at higher concentrations, it is less selective (48). CN-POBS was added 1 min prior to the substrate mix and significantly decreased the rate of superoxide/H2O2 production at “rest.” The rate attributed to site IQ after correction for the 65% effect of CN-POBS was 66 ± 29 pmol H2O2·min−1·mg of protein−1 (mean ± S.E.; n = 6) (Fig. 7A), indistinguishable from the estimate made using rotenone.
From the data obtained with CN-POBS and corroborated using rotenone, site IQ accounted for 23 ± 11% of the total rate of H2O2 production measured during “rest” (Fig. 7B and Table 3). This is the first evidence in any system that site IQ may be an important contributor to mitochondrial superoxide/H2O2 production ex vivo and in vivo. The sum of the rates of superoxide/H2O2 production from sites IF, IIIQo, and IQ was significantly different from the total rate of H2O2 production measured during “rest” (Fig. 7B), suggesting that yet other sites also produced superoxide/H2O2 at rest.
Flavin of Complex II, Site IIF, during “Rest”
The flavin site of complex II, site IIF, has a high capacity for superoxide/H2O2 production (20) (Fig. 3). When downstream electron transport is prevented, there is a peak of superoxide/H2O2 production from this site at succinate concentrations close to the physiological range (20), indicating that site IIF might also contribute to H2O2 generation at rest. The contribution of site IIF during “rest” ex vivo was assessed by adding malonate to inhibit electron transport at this site, followed by correction for consequent changes in the rate of superoxide production from sites IF, IIIQo, and IQ. The addition of malonate significantly decreased the rates of H2O2 production at “rest” (Table 3). The change in rate attributed to site IIF after correction was 80 ± 35 pmol H2O2·min−1·mg of protein−1 (mean ± S.E.; n = 7) (Fig. 7C and Table 3). Site IIF accounted for 24 ± 10% of the total rate of H2O2 production measured during “rest” (Fig. 7D and Table 3). This is the first evidence in any system that site IIF may be an important contributor to mitochondrial superoxide/H2O2 production ex vivo and in vivo.
The sum of the rates of superoxide/H2O2 production from sites IF, IIIQo, IQ, and IIF was not significantly different from the total rate of H2O2 production measured during “rest” (Fig. 7D). However, we still assessed the potential contributions of the remaining sites.
Electron-transferring Flavoprotein (ETF) and ETF:Q Oxidoreductase (ETF:QOR), Site EF, during “Rest”
β-Oxidation of fatty acids, primarily palmitoylcarnitine, is an important source of ATP for resting skeletal muscle. During oxidation of palmitoylcarnitine, electrons are transferred partly to NAD+ and partly to ETF and then through ETF:QOR to the Q-pool. Superoxide/H2O2 production in the ETF/ETF:QOR system (site EF) probably occurs at the flavin site of ETF (22). Site EF produces superoxide/H2O2 at a maximum rate of 210 pmol of H2O2·min−1·mg of protein−1 (22). However, during oxidation of palmitoylcarnitine plus carnitine as the sole added substrate under native conditions without inhibitors, the observed H2O2 production was mainly from sites IF, IIIQo, and IIF, leaving a small (but not statistically significant) shortfall that might have been from other sites, such as ETF (22). The contribution of site EF during “rest” ex vivo was assessed by omitting palmitoylcarnitine from the substrate mix, followed by correction for consequent changes in the rate of superoxide production from sites IF and IIIQo. Palmitoylcarnitine omission significantly decreased the rate of H2O2 production (Fig. 8A). There was no consequent change in the rates from sites IF and IIIQo (Table 3). The possibility that palmitoylcarnitine withdrawal could alter the rate of superoxide/H2O2 production from site IIF was tested by adding malonate in a mix without palmitoylcarnitine, which decreased H2O2 production to the same extent as in the presence of palmitoylcarnitine, showing that palmitoylcarnitine omission did not affect the rate from site IIF (data not shown). The rate attributed to site EF was 46 ± 21 pmol of H2O2·min−1·mg of protein−1 (mean ± S.E.; n = 8) (Fig. 8A and Table 3). Site EF accounted for 13 ± 6% of the total rate of H2O2 production measured during “rest” (Fig. 8B and Table 3).
FIGURE 8.

Contributions of sites EF, PF, OF, and GQ at “rest.” Mitochondria were incubated in medium mimicking rest (Table 1). A, total rate of H2O2 production and the rate in the absence of palmitoylcarnitine after correction for changes in the rates of sites IF and IIIQo (Table 3). The olive hatched area represents the contribution of site EF. B, total rate of H2O2 production at “rest” (from Fig. 2D) and the contributions of sites IF (from Fig. 5D), IIIQo (from Fig. 6D), IQ (from Fig. 7A), IIF (from Fig. 7C), and EF (from A). Inverted error bars indicate the propagated errors for each site, and the conventional error bar indicates the propagated sum of these errors. C, total rate of H2O2 production and the rates in the absence of pyruvate, 2-oxoglutarate, or glycerol 3-phosphate plus dihydroxyacetone phosphate (DHAP) to assess the contributions of sites PF, OF, and GQ. D, total rate of H2O2 production at “rest” (from Fig. 2D) and the contributions of sites IF (from Fig. 5D); IIIQo (from Fig. 6D); IQ (from Fig. 7A); IIF (from Fig. 7C); EF (from A); and GQ, PF, and OF (from C). Inverted error bars indicate the propagated errors for each site. Because some of the assessed contributions were negative, the sum of the contributions by each site was also plotted (error bar indicates the propagated sum of the errors for each assessed site) and used for the statistical test. Values are means ± S.E. (error bars) (n = 3–20). *, p < 0.05 by Student's t test. NS, not significantly different using one-way ANOVA followed by Tukey's post hoc test. Welch's t test was used to determine p = 0.42 and p = 0.15.
Other Sites, PF, OF, GQ, BF, and DQ, during “Rest”
The pyruvate dehydrogenase complex, site PF, and the 2-oxoglutarate dehydrogenase complex, site OF, have the greatest capacities for superoxide/H2O2 production in the NADH/NAD+ isopotential group (16) (Figs. 1 and 3). Omission of pyruvate from the substrate mixture slightly slowed the rate of H2O2 production during “rest,” although this effect was not statistically significant (p = 0.5). Fig. 8C shows that after correcting for consequent changes at sites IF, IIIQo, and IQ (Table 3), site PF was not significantly active at rest. Arithmetically, omission of pyruvate gave small negative rates for site PF after correction (Fig. 8D and Table 3), presumably reflecting noise in the assays and small errors in the assumptions.
Similarly, omission of 2-oxoglutarate or the glycerol 3-phosphate/dihydroxyacetone phosphate couple from the substrate mixture resulted in non-significant small increases in the total rates of H2O2 production after correction for consequent changes in other sites (Fig. 8C and Table 3). Therefore, these sites were also unlikely to contribute to the measured rates of H2O2 production. Again, these assessments gave small negative rates for sites OF and GQ after correction(Fig. 8D and Table 3).
Site DQ in dihydroorotate dehydrogenase (23) was considered to make zero contribution to total H2O2 production in our analysis because this site has low maximum capacity, the enzyme has low activity in skeletal muscle (49), and its substrate, dihydroorotate, is present at very low concentrations in rat skeletal muscle and was therefore not included in the substrate mix (Table 1). Similarly, site BF of the branched-chain 2-oxoacid dehydrogenase (16) was considered to make zero contribution because (i) branched-chain 2-oxoacids are present at very low concentrations in rat skeletal muscle mitochondria at rest and were therefore not included in the substrate mix (Table 1) and (ii) the addition of the transaminase inhibitor aminooxyacetate at 1 mm did not alter the measured rate of H2O2 production (data not shown), indicating that transamination of the added branched-chain amino acids to generate branched-chain 2-oxoacids and produce superoxide/H2O2 at site BF was not a major pathway.
Fig. 8D shows the final sum of the assessed rates of superoxide/H2O2 production from all of the sites, plotted both as a stack of positive and notionally negative contributions and as the sum of these values. This final value was indistinguishable from the total measured rate of H2O2 production (Fig. 8D and Table 3), so the observed rate of H2O2 production was entirely accounted for, within experimental error, by the sum of the assessed rates from each site.
In summary, when rat skeletal muscle mitochondria were incubated in a complex medium mimicking the cytosol of rat skeletal muscle at rest, containing physiological concentrations of all substrates and effectors thought to be relevant, the total rate of H2O2 production was 337 ± 21 pmol of H2O2·min−1·mg of protein−1 (mean ± S.E.; n = 11). Ranked by magnitude, the sites that contributed to this H2O2 production were sites IIF (24 ± 10%), IQ (23 ± 11%), IF (20 ± 3%), IIIQo (15 ± 2%), and EF (13 ± 6%).
“Mild Aerobic Exercise”
During mild aerobic exercise, substrate concentrations are different, and the free Ca2+ concentration is 20-fold higher compared with rest (Table 1). When mitochondria were incubated in the medium mimicking mild aerobic exercise, ATP turnover was driven by extramitochondrial ATPases, respiration ran at 22% of the maximum rate (Fig. 2, A and B), and NAD(P)H and cytochrome b566 reduction levels were significantly lower than at “rest” (Fig. 4 and Table 3). Fig. 2, C and D, shows that the measured rate of H2O2 production during “mild aerobic exercise” was only a quarter of the rate at “rest.”
The contribution of each site to superoxide/H2O2 production during “mild aerobic exercise” was assessed exactly as described above for “rest” (Table 3). The contributions of sites IF and IIIQo were assessed using the calibration curves in Figs. 5C and 6C. Fig. 9 shows the final sum of the assessed rates of superoxide/H2O2 production from all sites, plotted both as a stack of positive and notionally negative contributions and as the sum of these values. This final value was indistinguishable from the total measured rate of H2O2 production (Fig. 9 and Table 3), so the observed rate of H2O2 production was entirely accounted for, within experimental error, by the sum of the assessed rates from each site. Compared with “rest,” the absolute contribution of each site decreased. The decrease was most marked for the sites in or connected to the QH2/Q isopotential group (Fig. 1), reflecting their sensitivity to the reduction level of the Q pool, which dropped by 50% (Fig. 4). Site IQ is also very sensitive to the protonmotive force (19), which will have decreased when ATP turnover increased. The absolute contribution of the only site in the NADH/NAD+ isopotential group that made a large contribution to H2O2 production at rest, site IF, decreased much less, because the reduction level of NAD(P)H dropped by only 20% (Fig. 4). As a result, the relative contribution of site IF to the rate of superoxide/H2O2 production increased significantly from 20% during “rest” to 44% during “mild aerobic exercise” (Table 3).
FIGURE 9.
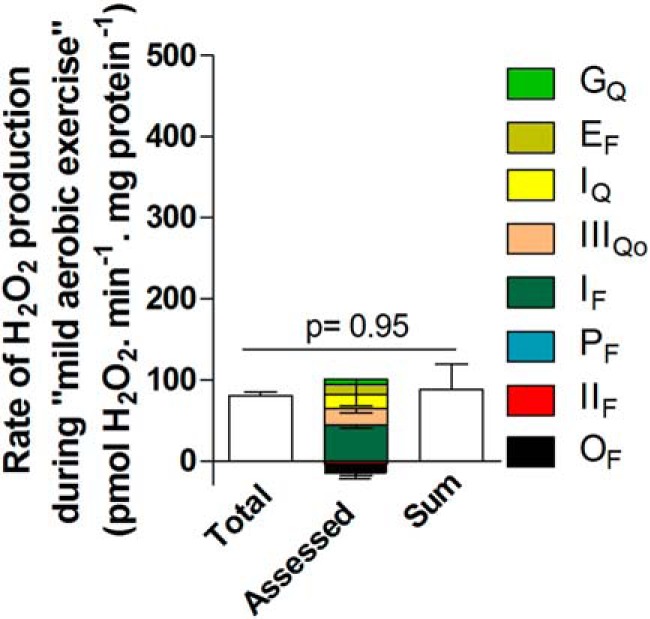
Contributions of different sites during “mild aerobic exercise.” Mitochondria were incubated in medium mimicking mild aerobic exercise (Table 1). The total rate of H2O2 production (from Fig. 2D) and the contributions of sites IF, IIIQo, IQ, EF, GQ, PF, IIF, and OF were assessed in the same way as for “rest” in Fig. 8 using the calibration curves in Figs. 5C and 6C. Inverted error bars indicate the propagated errors for each site. Values are means ± S.E. (error bars) (n = 3–9). Because some of the assessed contributions were negative, the sum of the contributions by each site was also plotted (error bar indicates the propagated sum of the errors for each assessed site) and used for the statistical test. Welch's t test was used to determine p = 0.95.
In summary, when rat skeletal muscle mitochondria were incubated in a complex medium mimicking the cytosol of rat skeletal muscle during mild aerobic exercise, containing physiological concentrations of all substrates and effectors thought to be relevant, the total rate of H2O2 production decreased to about 25% of the rate in the medium mimicking rest. The sites that contributed to this H2O2 production were, ranked by magnitude, sites IF (44 ± 4%), IQ (18 ± 15%), IIIQo (15 ± 4%), and maybe EF (12 ± 15%) and GQ (5 ± 7%).
“Intense Aerobic Exercise”
During intense aerobic exercise, free Ca2+ concentration is high; ATP concentration is maintained almost constant (∼6 mm); ADP concentration, which is more than 1 order of magnitude lower than ATP concentration, can increase 5-fold; and creatine phosphate hydrolysis generates an increased concentration of Pi (40). CO2 and lactate production increase, lowering intracellular pH (40, 50, 51), and substrate and effector concentrations also change (Table 1). When mitochondria were incubated in the medium mimicking intense aerobic exercise, ATP turnover was driven by sufficient extramitochondrial hexokinase to cause respiration to run at 90% of the maximum rate (Fig. 2, A and B), and NAD(P)H and cytochrome b566 reduction levels were significantly lower than at “rest” (Fig. 4 and Table 3). NAD(P)H reduction level was also significantly lower than during “mild aerobic exercise” (Fig. 4 and Table 3). Fig. 2, C and D, shows that the measured rate of H2O2 production during “intense aerobic exercise” was only one-sixth of the rate at “rest,” but there was no significant difference in rates between “mild aerobic exercise” and “intense aerobic exercise.”
The contribution of each site to superoxide/H2O2 production during “intense aerobic exercise” was assessed as described above for “rest” and “mild aerobic exercise” (Table 3). The contributions of sites IF and IIIQo were assessed using the new calibration curves in Fig. 10, C and F, because we found that the calibrations were different in this medium compared with the other two media (Figs. 5C and 6C), probably because of the lower pH.
Fig. 10, A and B, show the dependences of the observed rate of H2O2 production from site IF and the NAD(P)H redox state on the concentration of malate in the medium mimicking intense aerobic exercise. Fig. 10C replots the data from Fig. 10, A and B, to show the dependence of the measured rate of H2O2 production arising from superoxide production at site IF on NAD(P)H reduction state in this medium. Similarly, Fig. 10D shows the observed rate of H2O2 production from site IIIQo as a function of the ratio of added succinate and malonate in the presence of rotenone and myxothiazol. At each point, the rate of H2O2 production from site IF was calculated from the observed NAD(P)H reduction level using Fig. 10C and subtracted from the total observed rate to give the rate specifically from site IIIQo. Fig. 10E shows the cytochrome b566 redox state under the same conditions. Fig. 10F replots the data from Fig. 10, D and E, to show the dependence of the rate of H2O2 production arising from superoxide production at site IIIQo on the cytochrome b566 reduction state in this medium.
Fig. 11 shows the final sum of the assessed rates of superoxide/H2O2 production from all sites, plotted both as a stack of positive and notionally negative contributions and as the sum of these values. This final value was indistinguishable from the total measured rate of H2O2 production (p = 0.46) (Fig. 11 and Table 3), so the observed rate of H2O2 production was entirely accounted for, within experimental error, by the sum of the assessed rates from each site. However, because of the low rate of H2O2 production during “intense aerobic exercise,” the experimental errors were relatively high, particularly for the sites making smaller contributions (Table 3). Under this condition, site IF was the major contributor (99 ± 26%) and possibly the only one (Fig. 11). However, we could not discard the possibility that other sites were also active. Note that the total measured rates of H2O2 production were indistinguishable between “mild aerobic exercise” and “intense aerobic exercise” (Fig. 2D), but the sites contributing under each condition appeared to differ (Table 3).
FIGURE 11.
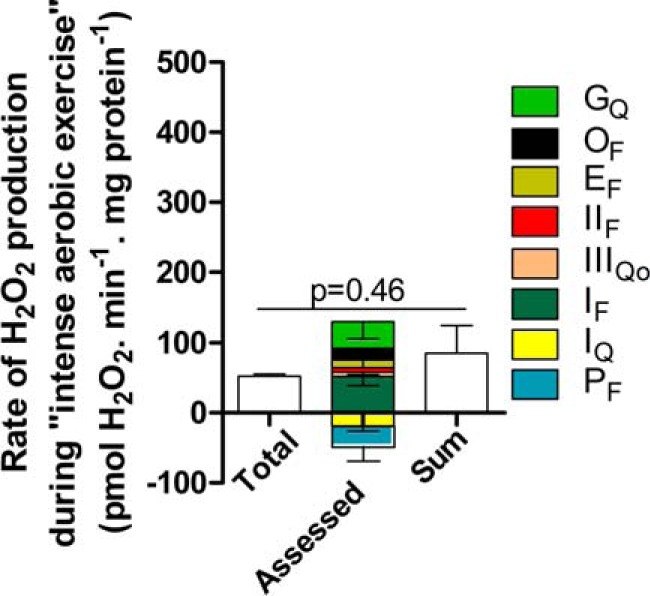
Contributions of different sites during “intense aerobic exercise.” Mitochondria were incubated in medium mimicking intense aerobic exercise (Table 1). The total rate of H2O2 production (from Fig. 2D) and the contributions of sites IF, IIIQo, IIF, EF, OF, GQ, IQ, and PF were assessed in the same way as for “rest” in Fig. 8 using the calibration curves in Fig. 10, C and F. Inverted error bars indicate the propagated errors for each site. Values are means ± S.E. (error bars) (n = 3). Because some of the assessed contributions were negative, the sum of the contributions by each site was also plotted (error bar indicates the propagated sum of the errors for each assessed site) and used for the statistical test. Welch's t test was used to determine p = 0.46.
In summary, when rat skeletal muscle mitochondria were incubated in a complex medium mimicking the cytosol of rat skeletal muscle during intense aerobic exercise, containing physiological concentrations of all substrates and effectors thought to be relevant, the total rate of H2O2 production decreased to about 15% of the rate in the medium mimicking rest. The main site that contributed to this H2O2 production was site IF (99 ± 26% of the total measured rate and 42 ± 10% of the sum of assessed rates).
Rates of H2O2 Production after Correction for Losses of H2O2 in the Mitochondrial Matrix
Peroxidases in the mitochondrial matrix degrade some H2O2 before it can escape and be registered by the extramitochondrial horseradish peroxidase/Amplex UltraRed assay. The losses can be greatly decreased by depleting mitochondrial glutathione. Previous work has established the relationship between observed rates of H2O2 production and total rates after correcting for H2O2 losses (24, 38), so we corrected the data presented above using Equation 1 to give a more realistic estimate of the true total rates and the rates from each site. Most of the sites produce superoxide/H2O2 exclusively to the matrix, so their relative contributions will not change substantially after correction. Importantly, however, ∼50% of the superoxide/H2O2 from sites IIIQo and GQ is produced toward the cytosol and avoids matrix peroxidase scavenging (17, 21). Therefore, the contribution of these sites becomes relatively smaller after this correction. Fig. 12 shows the corrected data and represents our best estimate of the total rate of mitochondrial superoxide/H2O2 production ex vivo and the contribution of each site during rest and mild and intense aerobic exercise. At “rest,” more than 50% of the total rate of superoxide/H2O2 production was shared between site IQ and site IIF, previously not considered to be physiologically relevant. During “exercise,” superoxide/H2O2 production decreased substantially, and the low capacity site IF accounted for half or more of the superoxide produced.
DISCUSSION
Mitochondria can produce superoxide or H2O2 from at least 10 different sites (16, 23). Which sites are the major producers in vivo is unknown. Currently, there are no methods available that can be used in intact cells or tissues or in vivo to identify unambiguously which sites are active or to measure the rates of production from each site. However, we recently developed methods to identify and quantify the rates from these sites in isolated mitochondria (24, 25). From these studies, we concluded that the overall rates and relative contributions of each site are strongly dependent on the substrate being oxidized. As a consequence, it is unreasonable to extrapolate from isolated mitochondria to tissues in vivo when the results in vitro are obtained using single conventional substrates at unphysiological concentrations.
The work described here represents a first step toward characterizing the mitochondrial sites of superoxide and H2O2 production in skeletal muscle in vivo. We analyzed the sites ex vivo in media designed to mimic the in vivo concentrations of all substrates and effectors we thought likely to be important in determining mitochondrial superoxide and H2O2 production in muscle at rest and during exercise. With this approach, extrapolations from ex vivo to in vivo are much more reasonable. Of course, the approach can still be criticized for assuming that other effects, such as fragmentation of mitochondria during isolation, do not substantially affect mitochondrial function and that other effectors that are not included in the media, such as components of unknown signaling pathways from the cytosol that do not work by altering the concentrations of the explicit metabolites, can be ignored. We used the approach specifically for rat skeletal muscle mitochondria, but it could be used with mitochondria from any cell type or any tissue from control or disease models as long as all metabolites and effectors are present at their physiological or pathological concentrations.
In the current paper, we identified and quantified for the first time all mitochondrial sites that are likely to contribute to mitochondrial superoxide and H2O2 production in skeletal muscle at rest and under mild aerobic and intense aerobic exercise. We found that the maximal capacities of the sites did not correlate with their native rates of superoxide and H2O2 production ex vivo. At “rest,” half of the total rate of H2O2 production was from sites IQ and IIF, two sites whose relevance in vivo was not previously appreciated. Also, the low capacity site IF produced H2O2 as fast as the highest capacity site, IIIQo.
In isolated mitochondria, site IQ can produce superoxide/H2O2 at high rates during both the forward and reverse reactions (43, 44). The contribution of site IQ in cells and in vivo is unknown. In some cells, the addition of rotenone decreases cellular production of reactive oxygen species (29, 52–54), which is consistent with a substantial contribution of site IQ. However, if the cells were oxidizing predominantly NAD-linked substrates, the addition of rotenone would block reduction of ubiquinone and decrease superoxide/H2O2 production from sites in the QH2/Q isopotential group, particularly sites IIIQo and IIF. If the cells were running reverse electron transport (29), the addition of rotenone would block reduction of NAD+ and decrease superoxide/H2O2 production from sites in the NADH/NAD+ isopotential group, particularly sites IF, PF, and OF. These alternative explanations greatly weaken any conclusion from rotenone inhibition experiments that site IQ is active in cells. Conversely, in many other cells, rotenone increases cellular production of reactive oxygen species (55–57), which appears to be inconsistent with a substantial role for site IQ. However, if the cells were oxidizing primarily NAD-linked substrates, the addition of rotenone would cause reduction of the NADH/NAD+ isopotential group and increase superoxide/H2O2 production from sites IF, PF, OF, etc., masking any decrease in the rate from site IQ. In general, whether rotenone addition causes an increase or decrease in observed production of reactive oxygen species may depend upon the balance of rates and capacities of sites in the NADH/NAD+ and QH2/Q isopotential groups and not only on the activity of site IQ itself. These considerations highlight the problems of valid interpretation when using electron transport chain inhibitors or genetic manipulations to define mitochondrial sites of superoxide/H2O2 production in cells if changes in the redox states of all other relevant sites are not properly accounted for as in Table 3.
Here, we used two different approaches to assess the contribution of site IQ. First, we used rotenone to inhibit the Q-site of complex I and corrected the observed overall changes in H2O2 production for the changes caused by alterations in the redox states of the NADH/NAD+ isopotential pool (site IF) and QH2/Q isopotential pool (site IIIQo), exposing the contribution of site IQ. Second, we used CN-POBS, which specifically suppresses superoxide production from site IQ without inhibiting electron transport (48) or altering the redox state of the NADH/NAD+ and QH2/Q isopotential pools (Table 3). Both approaches gave the same result (Fig. 7A). This is the first evidence that site IQ is active under a semiphysiological condition and therefore may also be significantly active in vivo.
The physiological contribution of superoxide/H2O2 production from site IIF has also been unappreciated, mostly because this site was not recognized as an important potential source until recently. However, the capacity of site IIF to produce superoxide/H2O2 in muscle mitochondria is great and is second only to site IIIQo (Fig. 3) (20). When succinate is used as a single substrate by isolated skeletal muscle mitochondria, the rate of production of superoxide/H2O2 by site IIF is highly dependent on the concentration of succinate, rising to a peak at about 400 μm (20), a concentration that approximates the physiological cytosolic level in skeletal muscle (Table 1), and then falling away at higher concentrations as succinate becomes inhibitory for this reaction. Our data indicate that at “rest,” ∼25% of the total H2O2 produced by muscle mitochondria originates from site IIF, indicating that this site is an important source of superoxide/H2O2 in vivo. Although the raw data are sparse, the concentration of succinate in skeletal muscle may rise from about 200 μm at rest to about 300 μm during exercise (Table 1). However, this increase ex vivo was not associated with a corresponding increase in the contribution of site IIF in “aerobic exercise” (Fig. 12), presumably because the activatory effect of higher succinate concentration was more than compensated for by the inhibitory effect of oxidation of the QH2/Q pool during “exercise” (Fig. 4). Nonetheless, under conditions in which succinate concentration rises, such as hypoxia-reperfusion (58), but the QH2/Q pool may not become oxidized, the contribution of site IIF in cardiac muscle or brain may be even greater.
The mechanisms underlying the decrease in total mitochondrial superoxide/H2O2 production during “aerobic exercise” are clear from our results. Despite increases in the concentrations of several substrates and effectors in the media mimicking exercise, including citrate, malate, pyruvate, succinate, glycerol 3-phosphate, acetylcarnitine, and free Ca2+ (Table 1), the redox centers in both the NADH/NAD+ and QH2/Q isopotential groups became more oxidized (Fig. 4 and Table 3). This shows that the dominant effect of the media mimicking exercise was increased supply of ADP caused by turnover of ATP, leading to an increased respiration rate and a more oxidized electron transport chain. Because of the steep dependence of superoxide/H2O2 production on the redox state of electron transport chain components (Figs. 5, 6, and 10), this led to the steep decline in total mitochondrial superoxide/H2O2 production seen in “exercise” in Fig. 2.
Similarly, the mechanisms underlying the decreased relative contributions of some sites are also clear. Because the oxidation of the NADH/NAD+ pool was less severe than the oxidation of the QH2/Q pool (Fig. 4 and Table 3), the decrease in the rate of superoxide production by site IF was less than the decrease at the other major sites, which are all linked to the QH2/Q pool, and the relative contribution of site IF increased. Although complex I has a high capacity for superoxide production at site IQ, the flavin site (site IF) has one of the lowest capacities in skeletal muscle mitochondria (Fig. 3). During “aerobic exercise” when the protonmotive force decreases and the NADH and QH2 pools become more oxidized, other sites contribute much less to overall H2O2 production, allowing the low capacity site IF to dominate (Fig. 12).
CONCLUSIONS
There is a lack of information about the specific mitochondrial sites of superoxide and hydrogen peroxide production that are physiologically or pathologically active in vivo or in intact cells. Our data provide the first realistic estimate of the sites active in skeletal muscle in vivo under three physiological conditions, rest, mild aerobic exercise, and intense aerobic exercise, and provide the first evidence that in addition to the sites currently recognized (sites IIIQo and IF), sites IQ and IIF may also be very important. These results illuminate the specific sites that may need to be normalized to prevent excessive mitochondrial superoxide and H2O2 production both physiologically and in many disease states.
This work was supported, in whole or in part, by National Institutes of Health Grant TL1 AG032116 (to C. L. Q.). This work was also supported by the Brazilian Government through the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) e ao Conselho de Nacional de Desenvolvimento Científico e Tecnológico programa Ciências Sem Fronteiras (CNPq-CSF) (to R. L. S. G.), the Glenn Foundation (to R. L. S. G. and I. V. P.), and the Carlsberg Foundation (to M. H.-M.).
- site IIIQo
- outer quinol-oxidizing site of respiratory complex III
- site IF
- flavin in the NADH-oxidizing site of respiratory complex I
- site IQ
- ubiquinone-reducing site of respiratory complex I
- site IIF
- flavin site of respiratory complex II
- site OF
- flavin in the 2-oxoglutarate dehydrogenase complex
- site PF
- flavin in the pyruvate dehydrogenase complex
- site BF
- flavin in the branched-chain 2-oxoacid (or α-ketoacid) dehydrogenase complex
- site GQ
- quinone reducing site in mitochondrial glycerol 3-phosphate dehydrogenase
- ETF
- electron-transferring flavoprotein
- ETF:QOR
- ETF:ubiquinone oxidoreductase
- site EF
- site in ETF:QOR, probably the flavin of ETF
- site DQ
- quinone reducing site in dihydroorotate dehydrogenase
- CDNB
- 1-chloro-2,4-dinitrobenzene
- VO2max
- whole body maximal O2 consumption rate
- CN-POBS
- N-cyclohexyl-4-(4-nitrophenoxy)benzenesulfonamide
- Q
- ubiquinone
- QH2
- ubiquinol
- Eh
- operating redox potential
- ANOVA
- analysis of variance.
REFERENCES
- 1. Loschen G., Flohé L., Chance B. (1971) Respiratory chain linked H2O2 production in pigeon heart mitochondria. FEBS Lett. 18, 261–264 [DOI] [PubMed] [Google Scholar]
- 2. Boveris A., Chance B. (1973) The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 134, 707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cadenas E., Davies K. J. (2000) Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 29, 222–230 [DOI] [PubMed] [Google Scholar]
- 4. Turrens J. F. (2003) Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brand M. D., Buckingham J. A., Esteves T. C., Green K., Lambert A. J., Miwa S., Murphy M. P., Pakay J. L., Talbot D. A., Echtay K. S. (2004) Mitochondrial superoxide and aging: uncoupling-protein activity and superoxide production. Biochem. Soc. Symp. 71, 203–213 [DOI] [PubMed] [Google Scholar]
- 6. Balaban R. S., Nemoto S., Finkel T. (2005) Mitochondria, oxidants, and aging. Cell 120, 483–495 [DOI] [PubMed] [Google Scholar]
- 7. Andreyev A. Y., Kushnareva Y. E., Starkov A. A. (2005) Mitochondrial metabolism of reactive oxygen species. Biochemistry 70, 200–214 [DOI] [PubMed] [Google Scholar]
- 8. Brookes P. S. (2005) Mitochondrial H+ leak and ROS generation: an odd couple. Free Radic. Biol. Med. 38, 12–23 [DOI] [PubMed] [Google Scholar]
- 9. Starkov A. A. (2008) The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N.Y. Acad. Sci. 1147, 37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kowaltowski A. J., de Souza-Pinto N. C., Castilho R. F., Vercesi A. E. (2009) Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 47, 333–343 [DOI] [PubMed] [Google Scholar]
- 11. Lambert A. J., Brand M. D. (2009) Reactive oxygen species production by mitochondria. Methods Mol. Biol. 554, 165–181 [DOI] [PubMed] [Google Scholar]
- 12. Murphy M. P. (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brand M. D. (2010) The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 45, 466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dröse S., Brandt U. (2012) Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 748, 145–169 [DOI] [PubMed] [Google Scholar]
- 15. Sies H. (2014) Role of metabolic H2O2 generation: redox signaling and oxidative stress. J. Biol. Chem. 289, 8735–8741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Quinlan C. L., Goncalves R. L., Hey-Mogensen M., Yadava N., Bunik V. I., Brand M. D. (2014) The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 289, 8312–8325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Quinlan C. L., Gerencser A. A., Treberg J. R., Brand M. D. (2011) The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J. Biol. Chem. 286, 31361–31372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Treberg J. R., Quinlan C. L., Brand M. D. (2011) Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J. Biol. Chem. 286, 27103–27110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lambert A. J., Brand M. D. (2004) Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem. J. 382, 511–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Quinlan C. L., Orr A. L., Perevoshchikova I. V., Treberg J. R., Ackrell B. A., Brand M. D. (2012) Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 287, 27255–27264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Orr A. L., Quinlan C. L., Perevoshchikova I. V., Brand M. D. (2012) A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J. Biol. Chem. 287, 42921–42935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perevoshchikova I. V., Quinlan C. L., Orr A. L., Gerencser A. A., Brand M. D. (2013) Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 61, 298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hey-Mogensen M., Goncalves R. L., Orr A. L., Brand M. D. (2014) Production of superoxide/H2O2 by dihydroorotate dehydrogenase in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 72, 149–155 [DOI] [PubMed] [Google Scholar]
- 24. Quinlan C. L., Treberg J. R., Perevoshchikova I. V., Orr A. L., Brand M. D. (2012) Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radic. Biol. Med. 53, 1807–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Quinlan C. L., Perevoshchikova I. V., Hey-Mogensen M., Orr A. L., Brand M. D. (2013) Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 1, 304–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sakellariou G. K., Vasilaki A., Palomero J., Kayani A., Zibrik L., McArdle A., Jackson M. J. (2013) Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid. Redox Signal. 18, 603–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vanden Hoek T. L., Becker L. B., Shao Z., Li C., Schumacker P. T. (1998) Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J. Biol. Chem. 273, 18092–18098 [DOI] [PubMed] [Google Scholar]
- 28. Powers S. K., Duarte J., Kavazis A. N., Talbert E. E. (2010) Reactive oxygen species are signalling molecules for skeletal muscle adaptation. Exp. Physiol. 95, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee S., Tak E., Lee J., Rashid M. A., Murphy M. P., Ha J., Kim S. S. (2011) Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res. 21, 817–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Powers S. K., Jackson M. J. (2008) Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol. Rev. 88, 1243–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Callahan L. A., She Z. W., Nosek T. M. (2001) Superoxide, hydroxyl radical, and hydrogen peroxide effects on single-diaphragm fiber contractile apparatus. J. Appl. Physiol. 90, 45–54 [DOI] [PubMed] [Google Scholar]
- 32. Sakellariou G. K., Jackson M. J., Vasilaki A. (2014) Redefining the major contributors to superoxide production in contracting skeletal muscle: the role of NAD(P)H oxidases. Free Radic. Res. 48, 12–29 [DOI] [PubMed] [Google Scholar]
- 33. Wardman P. (2007) Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic. Biol. Med. 43, 995–1022 [DOI] [PubMed] [Google Scholar]
- 34. Halliwell B., Whiteman M. (2004) Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br. J. Pharmacol. 142, 231–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Affourtit C., Quinlan C. L., Brand M. D. (2012) Measurement of proton leak and electron leak in isolated mitochondria. Methods Mol. Biol. 810, 165–182 [DOI] [PubMed] [Google Scholar]
- 36. Quinlan C. L., Perevoschikova I. V., Goncalves R. L., Hey-Mogensen M., Brand M. D. (2013) The determination and analysis of site-specific rates of mitochondrial reactive oxygen species production. Methods Enzymol. 526, 189–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Crofts A. R., Meinhardt S. W., Jones K. R., Snozzi M. (1983) The role of the quinone pool in the cyclic electron-transfer chain of Rhodopseudomonas sphaeroides: a modified Q-cycle mechanism. Biochim. Biophys. Acta 723, 202–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Treberg J. R., Quinlan C. L., Brand M. D. (2010) Hydrogen peroxide efflux from muscle mitochondria underestimates matrix superoxide production: a correction using glutathione depletion. FEBS J. 277, 2766–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brooks G. A., Mercier J. (1994) Balance of carbohydrate and lipid utilization during exercise: the “crossover” concept. J. Appl. Physiol. 76, 2253–2261 [DOI] [PubMed] [Google Scholar]
- 40. Cieslar J. H., Dobson G. P. (2000) Free [ADP] and aerobic muscle work follow at least second order kinetics in rat gastrocnemius in vivo. J. Biol. Chem. 275, 6129–6134 [DOI] [PubMed] [Google Scholar]
- 41. Chance B., Sies H., Boveris A. (1979) Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 59, 527–605 [DOI] [PubMed] [Google Scholar]
- 42. St-Pierre J., Buckingham J. A., Roebuck S. J., Brand M. D. (2002) Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 277, 44784–44790 [DOI] [PubMed] [Google Scholar]
- 43. Hansford R. G., Hogue B. A., Mildaziene V. (1997) Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J. Bioenerg. Biomembr. 29, 89–95 [DOI] [PubMed] [Google Scholar]
- 44. Lambert A. J., Brand M. D. (2004) Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I). J. Biol. Chem. 279, 39414–39420 [DOI] [PubMed] [Google Scholar]
- 45. Hinkle P. C., Butow R. A., Racker E., Chance B. (1967) Partial resolution of the enzymes catalyzing oxidative phosphorylation. XV. Reverse electron transfer in the flavin-cytochrome beta region of the respiratory chain of beef heart submitochondrial particles. J. Biol. Chem. 242, 5169–5173 [PubMed] [Google Scholar]
- 46. Starkov A. A., Fiskum G. (2003) Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 86, 1101–1107 [DOI] [PubMed] [Google Scholar]
- 47. Muller F. L., Liu Y., Abdul-Ghani M. A., Lustgarten M. S., Bhattacharya A., Jang Y. C., Van Remmen H. (2008) High rates of superoxide production in skeletal-muscle mitochondria respiring on both complex I- and complex II-linked substrates. Biochem. J. 409, 491–499 [DOI] [PubMed] [Google Scholar]
- 48. Orr A. L., Ashok D., Sarantos M. R., Shi T., Hughes R. E., Brand M. D. (2013) Inhibitors of ROS production by the ubiquinone-binding site of mitochondrial complex I identified by chemical screening. Free Radic. Biol. Med. 65, 1047–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Löffler M., Becker C., Wegerle E., Schuster G. (1996) Catalytic enzyme histochemistry and biochemical analysis of dihydroorotate dehydrogenase/oxidase and succinate dehydrogenase in mammalian tissues, cells and mitochondria. Histochem. Cell Biol. 105, 119–128 [DOI] [PubMed] [Google Scholar]
- 50. Hultman E., Spriet L. L. (1986) Skeletal muscle metabolism, contraction force and glycogen utilization during prolonged electrical stimulation in humans. J. Physiol. 374, 493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Horská A., Brant L. J., Ingram D. K., Hansford R. G., Roth G. S., Spencer R. G. (1999) Effect of long-term caloric restriction and exercise on muscle bioenergetics and force development in rats. Am. J. Physiol. 276, E766–E773 [DOI] [PubMed] [Google Scholar]
- 52. Zamzami N., Marchetti P., Castedo M., Decaudin D., Macho A., Hirsch T., Susin S. A., Petit P. X., Mignotte B., Kroemer G. (1995) Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 182, 367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chandel N. S., Maltepe E., Goldwasser E., Mathieu C. E., Simon M. C., Schumacker P. T. (1998) Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. U.S.A. 95, 11715–11720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Aon M. A., Cortassa S., Marbán E., O'Rourke B. (2003) Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 278, 44735–44744 [DOI] [PubMed] [Google Scholar]
- 55. Barrientos A., Moraes C. T. (1999) Titrating the effects of mitochondrial complex I impairment in the cell physiology. J. Biol. Chem. 274, 16188–16197 [DOI] [PubMed] [Google Scholar]
- 56. Nakamura K., Bindokas V. P., Kowlessur D., Elas M., Milstien S., Marks J. D., Halpern H. J., Kang U. J. (2001) Tetrahydrobiopterin scavenges superoxide in dopaminergic neurons. J. Biol. Chem. 276, 34402–34407 [DOI] [PubMed] [Google Scholar]
- 57. Li N., Ragheb K., Lawler G., Sturgis J., Rajwa B., Melendez J. A., Robinson J. P. (2003) Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 278, 8516–8525 [DOI] [PubMed] [Google Scholar]
- 58. Ariza A. C., Deen P. M., Robben J. H. (2012) The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front. Endocrinol. (Lausanne) 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nicholls D. G., Ferguson S. J. (2002) Bioenergetics 3, pp. 105–106, Academic Press, London [Google Scholar]
- 60. Owen O. E., Markus H., Sarshik S., Mozzoli M. (1973) Relationship between plasma and muscle concentrations of ketone bodies and free fatty acids in fed, starved and alloxan-diabetic states. Biochem. J. 134, 499–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cieslar J., Huang M. T., Dobson G. P. (1998) Tissue spaces in rat heart, liver, and skeletal muscle in vivo. Am. J. Physiol. 275, R1530–R1536 [DOI] [PubMed] [Google Scholar]
- 62. Ardawi M. S., Jamal Y. S. (1990) Glutamine metabolism in skeletal muscle of glucocorticoid-treated rats. Clin. Sci. 79, 139–147 [DOI] [PubMed] [Google Scholar]
- 63. Adibi S. A. (1971) Interrelationships between level of amino acids in plasma and tissues during starvation. Am. J. Physiol. 221, 829–838 [DOI] [PubMed] [Google Scholar]
- 64. Børsheim E., Kobayashi H., Traber D. L., Wolfe R. R. (2006) Compartmental distribution of amino acids during hemodialysis-induced hypoaminoacidemia. Am. J. Physiol. Endocrinol. Metab. 290, E643–E652 [DOI] [PubMed] [Google Scholar]
- 65. Williams J. A., Withrow C. D., Woodbury D. M. (1971) Effects of ouabain and diphenylhydantoin on transmembrane potentials, intracellular electrolytes, and cell pH of rat muscle and liver in vivo. J. Physiol. 212, 101–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Heymsfield S. B., Stevens V., Noel R., McManus C., Smith J., Nixon D. (1982) Biochemical composition of muscle in normal and semistarved human subjects: relevance to anthropometric measurements. Am. J. Clin. Nutr. 36, 131–142 [DOI] [PubMed] [Google Scholar]
- 67. Vinnakota K. C., Bassingthwaighte J. B. (2004) Myocardial density and composition: a basis for calculating intracellular metabolite concentrations. Am. J. Physiol. Heart. Circ. Physiol. 286, H1742–H1749 [DOI] [PubMed] [Google Scholar]
- 68. Brocks D. G., Siess E. A., Wieland O. H. (1980) Validity of the digitonin method for metabolite compartmentation in isolated hepatocytes. Biochem. J. 188, 207–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wiesner R. J., Kreutzer U., Rösen P., Grieshaber M. K. (1988) Subcellular distribution of malate-aspartate cycle intermediates during normoxia and anoxia in the heart. Biochim. Biophys. Acta 936, 114–123 [DOI] [PubMed] [Google Scholar]
- 70. Siess E. A., Brocks D. G., Lattke H. K., Wieland O. H. (1977) Effect of glucagon on metabolite compartmentation in isolated rat liver cells during gluconeogenesis from lactate. Biochem. J. 166, 225–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kauppinen R. A., Hiltunen J. K., Hassinen I. E. (1982) Compartmentation of citrate in relation to the regulation of glycolysis and the mitochondrial transmembrane proton electrochemical potential gradient in isolated perfused rat heart. Biochim. Biophys. Acta 681, 286–291 [DOI] [PubMed] [Google Scholar]
- 72. Soboll S., Horst C., Hummerich H., Schumacher J. P., Seitz H. J. (1992) Mitochondrial metabolism in different thyroid states. Biochem. J. 281, 171–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sundqvist K. E., Heikkilä J., Hassinen I. E., Hiltunen J. K. (1987) Role of NADP+ (corrected)-linked malic enzymes as regulators of the pool size of tricarboxylic acid-cycle intermediates in the perfused rat heart. Biochem. J. 243, 853–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hensgens H. E., Meijer A. J., Williamson J. R., Gimpel J. A., Tager J. M. (1978) Proline metabolism in isolated rat liver cells. Biochem. J. 170, 699–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Idell-Wenger J. A., Grotyohann L. W., Neely J. R. (1978) Coenzyme A and carnitine distribution in normal and ischemic hearts. J. Biol. Chem. 253, 4310–4318 [PubMed] [Google Scholar]
- 76. Akerboom T. P., Bookelman H., Zuurendonk P. F., van der Meer R., Tager J. M. (1978) Intramitochondrial and extramitochondrial concentrations of adenine nucleotides and inorganic phosphate in isolated hepatocytes from fasted rats. Eur. J. Biochem. 84, 413–420 [DOI] [PubMed] [Google Scholar]
- 77. Schiaffino S., Hanzlíková V., Pierobon S. (1970) Relations between structure and function in rat skeletal muscle fibers. J. Cell Biol. 47, 107–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mathieu-Costello O., Ju Y., Trejo-Morales M., Cui L. (2005) Greater capillary-fiber interface per fiber mitochondrial volume in skeletal muscles of old rats. J. Appl. Physiol. 99, 281–289 [DOI] [PubMed] [Google Scholar]
- 79. Desplanches D., Kayar S. R., Sempore B., Flandrois R., Hoppeler H. (1990) Rat soleus muscle ultrastructure after hindlimb suspension. J. Appl. Physiol. 69, 504–508 [DOI] [PubMed] [Google Scholar]
- 80. Kirkwood S. P., Munn E. A., Brooks G. A. (1986) Mitochondrial reticulum in limb skeletal muscle. Am. J. Physiol. 251, C395–C402 [DOI] [PubMed] [Google Scholar]
- 81. van Ekeren G. J., Sengers R. C., Stadhouders A. M. (1992) Changes in volume densities and distribution of mitochondria in rat skeletal muscle after chronic hypoxia. Int. J. Exp. Pathol. 73, 51–60 [PMC free article] [PubMed] [Google Scholar]
- 82. Else P. L., Hulbert A. J. (1985) Mammals: an allometric study of metabolism at tissue and mitochondrial level. Am. J. Physiol. 248, R415–R421 [DOI] [PubMed] [Google Scholar]
- 83. Westerblad H., Allen D. G. (1993) The contribution of [Ca2+]i to the slowing of relaxation in fatigued single fibres from mouse skeletal muscle. J. Physiol. 468, 729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ingalls C. P., Warren G. L., Armstrong R. B. (1999) Intracellular Ca2+ transients in mouse soleus muscle after hindlimb unloading and reloading. J. Appl. Physiol. 87, 386–390 [DOI] [PubMed] [Google Scholar]
- 85. Bruton J., Tavi P., Aydin J., Westerblad H., Lännergren J. (2003) Mitochondrial and myoplasmic [Ca2+] in single fibres from mouse limb muscles during repeated tetanic contractions. J. Physiol. 551, 179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Berchtold M. W., Brinkmeier H., Müntener M. (2000) Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol. Rev. 80, 1215–1265 [DOI] [PubMed] [Google Scholar]
- 87. Aragón J. J., Lowenstein J. M. (1980) The purine-nucleotide cycle. Comparison of the levels of citric acid cycle intermediates with the operation of the purine nucleotide cycle in rat skeletal muscle during exercise and recovery from exercise. Eur. J. Biochem. 110, 371–377 [DOI] [PubMed] [Google Scholar]
- 88. Pichard C., Vaughan C., Struk R., Armstrong R. L., Jeejeebhoy K. N. (1988) Effect of dietary manipulations (fasting, hypocaloric feeding, and subsequent refeeding) on rat muscle energetics as assessed by nuclear magnetic resonance spectroscopy. J. Clin. Invest. 82, 895–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. MacDermott M. (1990) The intracellular concentration of free magnesium in extensor digitorum longus muscles of the rat. Exp. Physiol. 75, 763–769 [DOI] [PubMed] [Google Scholar]
- 90. Baraban S. C., Bellingham M. C., Berger A. J., Schwartzkroin P. A. (1997) Osmolarity modulates K+ channel function on rat hippocampal interneurons but not CA1 pyramidal neurons. J. Physiol. 498, 679–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gillin A. G., Sands J. M. (1992) Characteristics of osmolarity-stimulated urea transport in rat IMCD. Am. J. Physiol. 262, F1061–F1067 [DOI] [PubMed] [Google Scholar]
- 92. Makara J. K., Petheö G. L., Tóth A., Spät A. (2000) Effect of osmolarity on aldosterone production by rat adrenal glomerulosa cells. Endocrinology 141, 1705–1710 [DOI] [PubMed] [Google Scholar]
- 93. Parrilla R. (1978) The effect of starvation in the rat on metabolite concentrations in blood, liver and skeletal muscle. Pflugers Arch. 374, 9–14 [DOI] [PubMed] [Google Scholar]
- 94. Chen V., Ianuzzo C. D., Fong B. C., Spitzer J. J. (1984) The effects of acute and chronic diabetes on myocardial metabolism in rats. Diabetes 33, 1078–1084 [DOI] [PubMed] [Google Scholar]
- 95. Fellenius E., Björkroth U., Kiessling K. H. (1973) Metabolic changes induced by ethanol in muscle and liver tissue of the rat in vivo. Acta Chem. Scand. 27, 2361–2366 [DOI] [PubMed] [Google Scholar]
- 96. Paul H. S., Adibi S. A. (1978) Leucine oxidation in diabetes and starvation: effects of ketone bodies on branched-chain amino acid oxidation in vitro. Metabolism 27, 185–200 [DOI] [PubMed] [Google Scholar]
- 97. Konijn A. M., Carmel N., Kaufmann N. A. (1976) The redox state and the concentration of ketone bodies in tissues of rats fed carbohydrate free diets. J. Nutr. 106, 1507–1514 [DOI] [PubMed] [Google Scholar]
- 98. Zorzano A., Balon T. W., Brady L. J., Rivera P., Garetto L. P., Young J. C., Goodman M. N., Ruderman N. B. (1985) Effects of starvation and exercise on concentrations of citrate, hexose phosphates and glycogen in skeletal muscle and heart: evidence for selective operation of the glucose-fatty acid cycle. Biochem. J. 232, 585–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Brass E. P., Hoppel C. L. (1978) Carnitine metabolism in the fasting rat. J. Biol. Chem. 253, 2688–2693 [PubMed] [Google Scholar]
- 100. Scharff R., Wool I. G. (1966) Effect of diabetes on the concentration of amino acids in plasma and heart muscle of rats. Biochem. J. 99, 173–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Goodman M. N., Ruderman N. B., Aoki T. T. (1978) Glucose and amino acid metabolism in perfused skeletal muscle: effect of dichloroacetate. Diabetes 27, 1065–1074 [DOI] [PubMed] [Google Scholar]
- 102. Dohm G. L., Beecher G. R., Warren R. Q., Williams R. T. (1981) Influence of exercise on free amino acid concentrations in rat tissues. J. Appl. Physiol. 50, 41–44 [DOI] [PubMed] [Google Scholar]
- 103. Olde Damink S. W., de Blaauw I., Deutz N. E., Soeters P. B. (1999) Effects in vivo of decreased plasma and intracellular muscle glutamine concentration on whole-body and hindquarter protein kinetics in rats. Clin. Sci. 96, 639–646 [DOI] [PubMed] [Google Scholar]
- 104. Ruderman N. B., Schmahl F. W., Goodman M. N. (1977) Regulation of alanine formation and release in rat muscle in vivo: effect of starvation and diabetes. Am. J. Physiol. 233, E109–E114 [DOI] [PubMed] [Google Scholar]
- 105. Manchester K. L., Wool I. G. (1963) Insulin and incorporation of amino acids into protein of muscle. 1. Accumulation and incorporation studies with the perfused rat heart. Biochem. J. 89, 202–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Goldstein L., Perlman D. F., McLaughlin P. M., King P. A., Cha C. J. (1983) Muscle glutamine production in diabetic ketoacidotic rats. Biochem. J. 214, 757–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hutson S. M., Zapalowski C., Cree T. C., Harper A. E. (1980) Regulation of leucine and α-ketoisocaproic acid metabolism in skeletal muscle. Effects of starvation and insulin. J. Biol. Chem. 255, 2418–2426 [PubMed] [Google Scholar]
- 108. Garber A. J., Karl I. E., Kipnis D. M. (1976) Alanine and glutamine synthesis and release from skeletal muscle. I. Glycolysis and amino acid release. J. Biol. Chem. 251, 826–835 [PubMed] [Google Scholar]
- 109. Karl I. E., Garber A. J., Kipnis D. M. (1976) Alanine and glutamine synthesis and release from skeletal muscle. III. Dietary and hormonal regulation. J. Biol. Chem. 251, 844–850 [PubMed] [Google Scholar]
- 110. Millward D. J., Nnanyelugo D. O., James W. P., Garlick P. J. (1974) Protein metabolism in skeletal muscle: the effect of feeding and fasting on muscle RNA, free amino acids and plasma insulin concentrations. Br. J. Nutr. 32, 127–142 [DOI] [PubMed] [Google Scholar]
- 111. Turinsky J., Long C. L. (1990) Free amino acids in muscle: effect of muscle fiber population and denervation. Am. J. Physiol. 258, E485–E491 [DOI] [PubMed] [Google Scholar]
- 112. Pastoris O., Dossena M., Foppa P., Arnaboldi R., Gorini A., Villa R. F., Benzi G. (1995) Modifications by chronic intermittent hypoxia and drug treatment on skeletal muscle metabolism. Neurochem. Res. 20, 143–150 [DOI] [PubMed] [Google Scholar]
- 113. Wijekoon E. P., Skinner C., Brosnan M. E., Brosnan J. T. (2004) Amino acid metabolism in the Zucker diabetic fatty rat: effects of insulin resistance and of type 2 diabetes. Can. J. Physiol. Pharmacol. 82, 506–514 [DOI] [PubMed] [Google Scholar]
- 114. Bertocci L. A., Lujan B. F. (1999) Incorporation and utilization of [3-13C]lactate and [1,2-13C]acetate by rat skeletal muscle. J. Appl. Physiol. 86, 2077–2089 [DOI] [PubMed] [Google Scholar]
- 115. Spydevold S., Davis E. J., Bremer J. (1976) Replenishment and depletion of citric acid cycle intermediates in skeletal muscle. Indication of pyruvate carboxylation. Eur. J. Biochem. 71, 155–165 [DOI] [PubMed] [Google Scholar]
- 116. Lee S. H., Davis E. J. (1979) Carboxylation and decarboxylation reactions. Anaplerotic flux and removal of citrate cycle intermediates in skeletal muscle. J. Biol. Chem. 254, 420–430 [PubMed] [Google Scholar]
- 117. Garber A. J. (1978) Skeletal muscle protein and amino acid metabolism in experimental chronic uremia in the rat: accelerated alanine and glutamine formation and release. J. Clin. Invest. 62, 623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Garber A. J. (1978) The regulation of skeletal muscle alanine and glutamine formation and release in experimental chronic uremia in the rat: subsensitivity of adenylate cyclase and amino acid release to epinephrine and serotonin. J. Clin. Invest. 62, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Goodman M. N., Lowenstein J. M. (1977) The purine nucleotide cycle: studies of ammonia production by skeletal muscle in situ and in perfused preparations. J. Biol. Chem. 252, 5054–5060 [PubMed] [Google Scholar]
- 120. Walser M., Lund P., Ruderman N. B., Coulter A. W. (1973) Synthesis of essential amino acids from their α-[keto] analogues by perfused rat liver and muscle. J. Clin. Invest. 52, 2865–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Albe K. R., Butler M. H., Wright B. E. (1990) Cellular concentrations of enzymes and their substrates. J. Theor. Biol. 143, 163–195 [DOI] [PubMed] [Google Scholar]
- 122. Tischler M. E., Henriksen E. J., Cook P. H. (1988) Role of glucocorticoids in increased muscle glutamine production in starvation. Muscle Nerve 11, 752–756 [DOI] [PubMed] [Google Scholar]
- 123. Kasperek G. J. (1989) Regulation of branched-chain 2-oxo acid dehydrogenase activity during exercise. Am. J. Physiol. 256, E186–E190 [DOI] [PubMed] [Google Scholar]
- 124. Hutson S. M., Harper A. E. (1981) Blood and tissue branched-chain amino and α-[keto] acid concentrations: effect of diet, starvation, and disease. Am. J. Clin. Nutr. 34, 173–183 [DOI] [PubMed] [Google Scholar]
- 125. May R. C., Hara Y., Kelly R. A., Block K. P., Buse M. G., Mitch W. E. (1987) Branched-chain amino acid metabolism in rat muscle: abnormal regulation in acidosis. Am. J. Physiol. 252, E712–E718 [DOI] [PubMed] [Google Scholar]
- 126. Berger M., Hagg S. A., Goodman M. N., Ruderman N. B. (1976) Glucose metabolism in perfused skeletal muscle: effects of starvation, diabetes, fatty acids, acetoacetate, insulin and exercise on glucose uptake and disposition. Biochem. J. 158, 191–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Maizels E. Z., Ruderman N. B., Goodman M. N., Lau D. (1977) Effect of acetoacetate on glucose metabolism in the soleus and extensor digitorum longus muscles of the rat. Biochem. J. 162, 557–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Dawson K. D., Baker D. J., Greenhaff P. L., Gibala M. J. (2005) An acute decrease in TCA cycle intermediates does not affect aerobic energy delivery in contracting rat skeletal muscle. J. Physiol. 565, 637–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Goodman M. N., Berger M., Ruderman N. B. (1974) Glucose metabolism in rat skeletal muscle at rest. Effect of starvation, diabetes, ketone bodies and free fatty acids. Diabetes 23, 881–888 [DOI] [PubMed] [Google Scholar]
- 130. Saha A. K., Laybutt D. R., Dean D., Vavvas D., Sebokova E., Ellis B., Klimes I., Kraegen E. W., Shafrir E., Ruderman N. B. (1999) Cytosolic citrate and malonyl-CoA regulation in rat muscle in vivo. Am. J. Physiol. 276, E1030–E1037 [DOI] [PubMed] [Google Scholar]
- 131. Hintz C. S., Chi M. M., Fell R. D., Ivy J. L., Kaiser K. K., Lowry C. V., Lowry O. H. (1982) Metabolite changes in individual rat muscle fibers during stimulation. Am. J. Physiol. 242, C218–C228 [DOI] [PubMed] [Google Scholar]
- 132. Rennie M. J., Winder W. W., Holloszy J. O. (1976) A sparing effect of increased plasma fatty acids on muscle and liver glycogen content in the exercising rat. Biochem. J. 156, 647–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Hagg S. A., Taylor S. I., Ruberman N. B. (1976) Glucose metabolism in perfused skeletal muscle: pyruvate dehydrogenase activity in starvation, diabetes and exercise. Biochem. J. 158, 203–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Sacktor B., Wormser-Shavit E., White J. I. (1965) Diphosphopyridine nucleotide-linked cytoplasmic metabolites in rat leg muscle in situ during contraction and recovery. J. Biol. Chem. 240, 2678–2681 [PubMed] [Google Scholar]
- 135. Howarth R. E., Baldwin R. L. (1971) Concentrations of selected enzymes and metabolites in rat skeletal muscle: effects of food restriction. J. Nutr. 101, 485–494 [DOI] [PubMed] [Google Scholar]
- 136. Dale R. A. (1965) Effects of sampling procedures on the contents of some intermediate metabolities of glycolysis in rat tissues. J. Physiol. 181, 701–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Kondoh Y., Kawase M., Kawakami Y., Ohmori S. (1992) Concentrations of d-lactate and its related metabolic intermediates in liver, blood, and muscle of diabetic and starved rats. Res. Exp. Med. (Berl.) 192, 407–414 [DOI] [PubMed] [Google Scholar]
- 138. Koves T. R., Ussher J. R., Noland R. C., Slentz D., Mosedale M., Ilkayeva O., Bain J., Stevens R., Dyck J. R., Newgard C. B., Lopaschuk G. D., Muoio D. M. (2008) Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7, 45–56 [DOI] [PubMed] [Google Scholar]
- 139. Minatogawa Y., Hue L. (1984) Fructose 2,6-bisphosphate in rat skeletal muscle during contraction. Biochem. J. 223, 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Peterson R. D., Gaudin D., Bocek R. M., Beatty C. H. (1964) α-glycerophosphate metabolism in muscle under aerobic and hypoxic conditions. Am. J. Physiol. 206, 599–602 [DOI] [PubMed] [Google Scholar]
- 141. Klingenberg M., Buecher T. (1960) Biological oxidations. Annu. Rev. Biochem. 29, 669–708 [DOI] [PubMed] [Google Scholar]
- 142. Veech R. L., Raijman L., Dalziel K., Krebs H. A. (1969) Disequilibrium in the triose phosphate isomerase system in rat liver. Biochem. J. 115, 837–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Veech R. L., Lawson J. W., Cornell N. W., Krebs H. A. (1979) Cytosolic phosphorylation potential. J. Biol. Chem. 254, 6538–6547 [PubMed] [Google Scholar]
- 144. Arnold H., Pette D. (1970) Binding of aldolase and triosephosphate dehydrogenase to F-actin and modification of catalytic properties of aldolase. Eur. J. Biochem. 15, 360–366 [DOI] [PubMed] [Google Scholar]
- 145. Bhuiyan A. K., Bartlett K., Sherratt H. S., Agius L. (1988) Effects of ciprofibrate and 2-[5-(4-chlorophenyl)pentyl]oxirane-2-carboxylate (POCA) on the distribution of carnitine and CoA and their acyl-esters and on enzyme activities in rats: relation between hepatic carnitine concentration and carnitine acetyltransferase activity. Biochem. J. 253, 337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Noland R. C., Koves T. R., Seiler S. E., Lum H., Lust R. M., Ilkayeva O., Stevens R. D., Hegardt F. G., Muoio D. M. (2009) Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J. Biol. Chem. 284, 22840–22852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Marquis N. R., Fritz I. B. (1964) Enzymological determination of free carnitine concentrations in rat tissues. J. Lipid Res. 5, 184–187 [PubMed] [Google Scholar]
- 148. Pearson D. J., Tubbs P. K. (1967) Carnitine and derivatives in rat tissues. Biochem. J. 105, 953–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Howarth K. R., LeBlanc P. J., Heigenhauser G. J., Gibala M. J. (2004) Effect of endurance training on muscle TCA cycle metabolism during exercise in humans. J. Appl. Physiol. 97, 579–584 [DOI] [PubMed] [Google Scholar]
- 150. Thyfault J. P., Cree M. G., Tapscott E. B., Bell J. A., Koves T. R., Ilkayeva O., Wolfe R. R., Dohm G. L., Muoio D. M. (2010) Metabolic profiling of muscle contraction in lean compared with obese rodents. Am. J. Physiol. 299, R926–R934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Challiss R. A., Vranic M., Radda G. K. (1989) Bioenergetic changes during contraction and recovery in diabetic rat skeletal muscle. Am. J. Physiol. 256, E129–E137 [DOI] [PubMed] [Google Scholar]
- 152. Essén-Gustavsson B., Blomstrand E. (2002) Effect of exercise on concentrations of free amino acids in pools of type I and type II fibres in human muscle with reduced glycogen stores. Acta Physiol. Scand. 174, 275–281 [DOI] [PubMed] [Google Scholar]
- 153. Awapara J. (1956) The taurine concentration of organs from fed and fasted rats. J. Biol. Chem. 218, 571–576 [PubMed] [Google Scholar]
- 154. Hitchins S., Cieslar J. M., Dobson G. P. (2001) 31P NMR quantitation of phosphorus metabolites in rat heart and skeletal muscle in vivo. Am. J. Physiol. Heart Circ. Physiol. 281, H882–H887 [DOI] [PubMed] [Google Scholar]
- 155. Kushmerick M. J., Meyer R. A. (1985) Chemical changes in rat leg muscle by phosphorus nuclear magnetic resonance. Am. J. Physiol. 248, C542–C549 [DOI] [PubMed] [Google Scholar]
- 156. Dumas J. F., Bielicki G., Renou J. P., Roussel D., Ducluzeau P. H., Malthièry Y., Simard G., Ritz P. (2005) Dexamethasone impairs muscle energetics, studied by 31P NMR, in rats. Diabetologia 48, 328–335 [DOI] [PubMed] [Google Scholar]
- 157. Geers C., Gros G. (1990) Effects of carbonic anhydrase inhibitors on contraction, intracellular pH and energy-rich phosphates of rat skeletal muscle. J. Physiol. 423, 279–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Munkvik M., Lunde P. K., Sejersted O. M. (2009) Causes of fatigue in slow-twitch rat skeletal muscle during dynamic activity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R900–R910 [DOI] [PubMed] [Google Scholar]
- 159. Chung Y., Sharman R., Carlsen R., Unger S. W., Larson D., Jue T. (1998) Metabolic fluctuation during a muscle contraction cycle. Am. J. Physiol. 274, C846–C852 [DOI] [PubMed] [Google Scholar]