

Background: G protein-coupled receptors mainly signal through heterotrimeric G-proteins.
Results: We have demonstrated that mutations in the extended Linker 2 impaired the activation of Gαs by β-adrenergic receptors.
Conclusion: We have proposed a potential novel conduit from β-adrenergic receptors to the helical domain of Gαs subunit via the extended Linker 2.
Significance: Our systematic mutagenesis studies provide insights into the activation mechanism of G-proteins by receptors.
Keywords: Cell Biology, Cell Signaling, Second Messenger, Signal Transduction, Signaling
Abstract
G protein-coupled receptors (GPCRs) relay extracellular signals mainly to heterotrimeric G-proteins (Gαβγ) and they are the most successful drug targets. The mechanisms of G-protein activation by GPCRs are not well understood. Previous studies have revealed a signal relay route from a GPCR via the C-terminal α5-helix of Gα to the guanine nucleotide-binding pocket. Recent structural and biophysical studies uncover a role for the opening or rotating of the α-helical domain of Gα during the activation of Gα by a GPCR. Here we show that β-adrenergic receptors activate eight Gαs mutant proteins (from a screen of 66 Gαs mutants) that are unable to bind Gβγ subunits in cells. Five of these eight mutants are in the αF/Linker 2/β2 hinge region (extended Linker 2) that connects the Ras-like GTPase domain and the α-helical domain of Gαs. This extended Linker 2 is the target site of a natural product inhibitor of Gq. Our data show that the extended Linker 2 is critical for Gα activation by GPCRs. We propose that a GPCR via its intracellular loop 2 directly interacts with the β2/β3 loop of Gα to communicate to Linker 2, resulting in the opening and closing of the α-helical domain and the release of GDP during G-protein activation.
Introduction
A structurally diverse repertoire of ligands, from photons to many hormones and neurotransmitters, activate G protein-coupled receptors (GPCRs)4 to elicit their physiological functions (1, 2). GPCRs comprise a large and diverse superfamily, and family members have been identified in organisms as evolutionarily distant as yeast and human. Heterotrimeric G-proteins (Gαβγ) directly relay the signals from GPCRs (3–5). These G-proteins are composed of α, β, and γ subunits. The β and γ subunits are tightly associated and can be regarded as one functional unit. G-proteins function as molecular binary switches with their biological activity determined by the bound nucleotide (3–5). Upon ligand-binding, GPCRs increase the exchange of GDP bound on the Gα subunit with GTP, in the presence of Gβγ subunits. This leads to the dissociation of the Gα subunit from the Gβγ dimer resulting in two functional subunits (Gα and Gβγ). Both Gα and Gβγ subunits signal to various cellular pathways.
Over the past 30 years, great progress has been made in understanding the mechanisms by which heterotrimeric G-proteins regulate their downstream targets (6, 7). Recently a series of crystal structures of GPCRs in the inactive and active states, bound with antagonists, inverse agonists, or agonists, have elucidated the structural basis for the modulation and activation of GPCRs by ligands (1, 8, 9). A crystal structure of the complex of β2-adrenergic receptor and Gs has revealed the structural changes in β2-adrenergic receptor and Gs, the interacting regions, and residues between a GPCR and a G-protein (10). However, the molecular mechanisms by which GPCRs activate G-proteins are still not completely understood (11, 12).
The structure of Gα subunits consists of two domains: a Ras-like GTPase domain and an α-helical domain (6) (Fig. 1, A and B). These two domains are linked by Linker 1 and Linker 2 (Fig. 1B). Between these two domains lies a deep cleft within which GDP or GTP is tightly bound (Fig. 1, A and B). The nucleotide is essentially occluded from the bulk solvent, leading to a proposal that the helical domain is the inhibitory barrier and provides the regulatory entry point by GPCRs or Gβγ subunits (13–16).
FIGURE 1.

Summary of mutated amino acid residues of Gαs. A, crystal structure of a Gα subunit shows the Ras-like GTPase domain and the α-helical domain. The αF, Linker 2, and β2 are indicated. The bound GDP is colored in magenta. B, diagram of a Gα subunit shows the relative locations of elements discussed in the paper. C, all residues on Gα contacting Gβγ are labeled (in red) based on crystal structures. The two images are the same rotated 180°. D, summary of mutated residues of Gαs (magenta and green letters). The secondary structure is assigned based on the crystal structure of Gαs·GTPγS (PDB code 1AZT). The three switch regions are indicated by black boxes. The two linker regions are indicated by arrows. Green amino acid letters indicate the residues mutated in the 8 Gαs mutants that are activated by β-ARs without Gβγ binding.
One of the major remaining problems in the biology of GPCR/G-protein signaling is to experimentally demonstrate whether GPCRs or Gβγ subunits play the catalytic exchange role in Gα protein activation (17). In theory, the question could be straightforwardly answered with purified proteins of GPCRs, Gα and Gβγ subunits. However, purified GPCRs had no guanine-nucleotide exchange effect on Gα in the absence of Gβγ subunits (18, 19). A GPCR could only activate Gα in the presence of Gβγ (18, 19), leading some to believe that GPCRs were not the real catalysts, although they were required to initiate the activation event from agonist binding. Yet, purified Gβγ subunits alone also could not accelerate the guanine nucleotide exchange on Gα subunits (3, 20). In fact, Gβγ subunits inhibit the basal nucleotide exchange activity of Gα subunits and behave as a guanine-nucleotide dissociation inhibitor (20). When the structure of Gβ was revealed to be similar to RCC1, a guanine-nucleotide exchange factor for the small GTPase Ran, Gβγ was proposed to be the real guanine-nucleotide exchange factor for Gα, whereas GPCRs served as a trigger (15, 21). Therefore, it remains a fundamental unsolved question: which one, a GPCR and/or Gβγ subunit, has the ability to catalyze the nucleotide exchange on Gα.
We reasoned that if GPCRs or Gβγ subunits are the nucleotide exchangers, we should be able to find mutants of GPCRs or Gβγ subunits that would accelerate the nucleotide exchange on Gα subunits in the absence of Gβγ or GPCRs, respectively. Alternatively, we might be able to find mutant Gα subunits that could be activated by GPCRs alone or by Gβγ alone. Here we describe our finding of a direct activation of some Gαs mutant proteins by β-adrenergic receptors without Gβγ subunits. Although our data do not exclude a possible additional catalytic role for Gβγ, it clearly demonstrates that GPCRs by themselves have the catalytic ability to activate Gα subunits. Furthermore, from this systematic study, a pivotal role for the αF/Linker 2/β2 region (extended Linker 2) in Gα activation has been uncovered. Therefore, we propose that the opening and closing of the two domains (the Ras-like GTPase domain and the helical domain) with Linker 2 as a pivot provide one of the mechanisms for Gα activation by GPCRs.
EXPERIMENTAL PROCEDURES
G-protein Purification
Gαs wild-type and mutant proteins were purified from Escherichia coli. The pGEX-6P-Gαs plasmid was transformed into bacteria strain BL21(DE3). One liter of bacterial culture was grown at room temperature until the absorbance at 600 nm was ∼1. G-protein expression was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside for 18 h at room temperature. The bacterial pellet was resuspended in lysis buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1% Triton X-100, 0.1 mg/ml of lysozyme, and 0.2 mm PMSF) and incubated on ice for 30 min. After sonication, the lysate was spun down at 10,000 × g for 120 min at 4 °C. Glutathione-agarose resin (0.5 ml, from Sigma) was added to the supernatant after pre-equilibration of the resin with lysis buffer. The mixture was gently agitated at 4 °C for 3 h. After washing 3 times with 10 ml of washing buffer (50 mm Tris, pH 8.0, 100 mm NaCl, and 0.2 mm PMSF), GST-tagged Gαs proteins were eluted with 0.5 ml of elution buffer (50 mm Tris, pH 8.0, 100 mm NaCl, 1 mm EDTA, 1 mm DTT, and 10% glycerol). preScission protease (Amersham Biosciences) was used to cleave GST off at 4 °C overnight.
Gβγ Proteins Were Purified from Insect Hi5 Cells
One liter of Hi5 cell pellet was resuspended into 50 ml of lysis buffer (50 mm Tris, pH 8.0, 1 mm EDTA, and protease inhibitors: 10 μg/ml of leupeptin, 1 μg/ml of pepstatin A, 1 mm benzamidine, and 0.2 mm PMSF). After sonication, the lysate was spun down at 150,000 × g for 90 min at 4 °C. The membrane pellet was resuspended in 50 ml of lysis buffer. After homogenization, the lysate was centrifuged again. The final pellet was resuspended in 50 ml of extraction buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 2% dodecyl-β-d-maltoside (DM) and protease inhibitors). After centrifugation at 10,000 × g for 120 min at 4 °C, 1 ml of nickel-nitrilotriacetic acid-agarose beads pre-equilibrated with extraction buffer was added to the supernatant. The mixture was gently agitated overnight at 4 °C. After washing 3 times with 10 ml of washing buffer (50 mm Tris, pH 8.0, 100 mm NaCl, 5 mm imidazole, and 0.2 mm PMSF), Gβγ was eluted with 10 ml of elution buffer (50 mm Tris, pH 8.0, 100 mm NaCl, 10 mm EDTA, and 200 mm imidazole).
Gβγ Binding Assay (with Purified Gβ1γ2 Proteins)
Ten μg of Gαs protein (with an N-terminal GST tag, attached to glutathione beads) were incubated in 100 μl of binding buffer (20 mm Hepes, pH 8.0, 2 mm GDP, 1 mm DTT, 150 mm NaCl, and 0.02% DM) with 100 nm Gβ1γ2 proteins overnight at 4 °C. After centrifugation, beads were washed three times with binding buffer and then eluted with binding buffer containing 10 mm reduced glutathione. After SDS-PAGE, Western blots were performed with anti-Gαs and anti-Gβ antibodies (Santa Cruz Biotechnology, Inc.).
Gβγ Binding Assay (with Membrane Preparations)
Membrane preparations were made from 293T cells. After solubilization, the supernatant (in 20 mm Hepes, pH 8.0, 2 mm GDP, 1 mm DTT, 150 mm NaCl, 1% DM, and protease inhibitors) was pre-cleared with glutathione beads. 500 μl of supernatant was mixed with 10 μg of Gαs protein (with an N-terminal GST tag, attached to glutathione beads). The mixtures were rolled at 4 °C overnight. After centrifugation, beads were washed three times with binding buffer and then eluted with binding buffer containing 10 mm reduced glutathione. After SDS-PAGE, Western blots were performed with anti-Gαs and anti-Gβ antibodies.
Size Exclusion Chromatography
Size exclusion chromatography was used to examine the binding of Gαs and its mutants to Gβ1γ2 in solution. Gαs (1 μm) and Gβ1(C68S)γ2 (2 μm) were mixed and incubated on ice for 30 min. Samples were loaded on a Superdex 200 column (GE Healthcare Life Sciences) equilibrated with 20 mm Hepes, pH 8.0, 50 mm NaCl, and 1 mm EDTA at a flow rate of 0.5 ml/min. The elution was monitored at 280 nm, and 0.8-ml fractions were collected for subsequent SDS-PAGE analysis.
In Vivo cAMP Assay
Cells were plated on 6-well plates and treated with 1 mm isobutylmethylxanthine for 30 min at 37 °C. After washing twice with HEM buffer (20 mm Hepes, pH 7.4, 135 mm NaCl, 4.7 mm KCl, 1.2 mm MgSO4, 2.5 mm NaHCO3, 0.1 mm Ro-20–1724, 0.5 unit/ml of adenosine deaminase, and 1 mm isobutylmethylxanthine), cells were treated with 0, 1 nm, 10 nm, 100 nm, 1 μm, 10 μm, and 100 μm isoproterenol in HEM buffer for 5 min at 37 °C. After two additional washes with HEM buffer, cells were harvested and lysed with 0.5% Triton X-100 containing 1 mm isobutylmethylxanthine. The amount of cAMP was measured with a Direct Cyclic AMP Enzyme Immunoassay kit (Assay Designs, Inc.).
In Vitro Adenylyl Cyclase Activation Assay
Adenylyl cyclase activation assay of Sf9 membrane preparations was performed as previously described (22). Briefly, adenylyl cyclase isoform V (dog) was recombinantly expressed in Sf9 cells, and the adenylyl cyclase-containing membranes were prepared. Gαs protein was activated by adding a buffer consisting of 10 mm NaF, 10 mm MgCl2, 30 μm AlCl3 and incubated at 30 °C for 1 h. Activated Gαs proteins with membrane preparations of adenylyl cyclase V were incubated in buffer of 50 mm Tris, pH 7.6, 2 mm isobutylmethylxanthine, 1 mm ATP, 10 mm MgCl2, 20 mm creative phosphate, 100 units/ml of creative phosphokinase at 30 °C for 10 min. The samples were boiled for 3 min to stop the reaction. After spinning for 3 min at 16,000 × g, the supernatant was used for cAMP measurement with Direct Cyclic AMP Enzyme Immunoassay kit.
Purification of Turkey β1-Adrenergic Receptor Proteins
Turkey β1-adrenergic receptor protein was purified as described previously (23). Turkey β1-adrenergic receptor (residues 34–424 with a mutation C116L) cDNA was subcloned into the pVL1393 vector. Hi5 cells were infected with the recombinant baculovirus carrying the turkey β1-adrenergic receptor at a density of 2 × 106/ml. Sixty-hours later, cells were harvested and resuspended in lysis buffer (50 mm Tris-HCl, pH 8, 1 mm EDTA, 10 μg/ml of leupeptin, 9 mm benzamidine, 5 μg/ml of pepstatin A, and 2 mm PMSF). After homogenization, the cell lysate was centrifuged at 2,000 × g for 10 min and the supernatant was centrifuged again at 150,000 × g at 4 °C for 1.5 h. The membrane pellet was resuspended in lysis buffer and homogenized again. After spinning down at 150,000 × g at 4 °C for 1.5 h, the final pellet was then resuspended in membrane extraction buffer (50 mm Tris-HCl, pH 8, 350 mm NaCl, 3 mm imidazole, 2% DM, 10 μg/ml of leupeptin, 9 mm benzamidine, 5 μg/ml of pepstatin A, and 1 mm PMSF) and rolled at 4 °C for 3 h. After centrifugation at 150,000 × g at 4 °C for 1 h, solubilized membrane proteins were incubated with nickel-nitrilotriacetic acid-agarose (Qiagen) overnight. After washing with buffer containing 50 mm Tris-HCl, pH 8, 350 mm NaCl, 3 mm imidazole, 0.1% DM, 10 μg/ml of leupeptin, 9 mm benzamidine, 5 μg/ml of pepstatin A, and 1 mm PMSF, the receptor protein was eluted down by an imidazole gradient.
GTPγS Loading Assay
Agonist-stimulated GTPγS loading to G-proteins was performed as previously described (19). Gαs (with or without 1 μm Gβγ) and turkey β1-adrenergic receptor (20 nm) together with 10 μm alprenolol or isoproterenol in 200 μl of loading buffer (50 mm Tris, pH 8.0, 100 mm NaCl, 10 mm MgCl2, 1 mm EDTA, 0.02% DM, and 5 μm GDP) were incubated on ice for 20 min. After incubation at 30 °C (for wild-type and Gαs219) or at room temperature (for Gαs263 and Gαs195) for 5 min, 100 nm GTPγS was added. At various times, 40-μl aliquots were removed and added to 1 ml of termination buffer (20 mm Tris, pH 8.0, 100 mm NaCl, and 25 mm MgCl2, ice cold) and loaded onto nitrocellulose membrane (Schleicher & Schuell BioScience). After 3 washes with 1 ml of termination buffer, 4 ml of scintillation liquid were added to the membrane and 35S was counted to measure GTPγS loading. 5 mm GDP was used to determine nonspecific binding.
Gαs and Receptor Binding Assay
Different concentrations of Gαs proteins (with an N-terminal GST tag, attached to glutathione beads) were incubated in 100 μl of binding buffer with 80 nm β-AR proteins (with a FLAG tag) at 4 °C for 1 h. GST (600 nm) was used as control. After centrifugation, beads were washed three times with washing buffer. After SDS-PAGE, Western blots were performed with anti-Gαs and anti-FLAG M2 antibodies.
RESULTS AND DISCUSSION
Identification of Gαs Mutants Defective in Gβγ Binding
To identify Gα subunits that could be activated by GPCRs in the absence of Gβγ subunits, we first characterized Gαs mutants that were defective in Gβγ binding. The crystal structures of Gα and Gαβγ have been solved (24, 25). The conformational differences between the free and Gβγ-bound forms of Gα-GDP mainly involve residues that directly interact with Gβ (24, 25). Based on the crystal structures of GαtGβ1γ1 and Gαi1Gβ1γ2 complexes, residues in the N-terminal, Switch I (Arg-185 to Ile-193, encompassing Linker 2 region), and Switch II region of Gα subunits are involved in contacting or binding of Gβγ (24, 25) (Fig. 1C, red residues). Furthermore, from the crystal structures of the Ras superfamily GTPases and their guanine nucleotide-exchange factors, such as Ras and Sos1, Arf1 and Sec7, Rac1 and Tiam1, EF-Tu and EF-Ts, as well as Ran and RCC1 (26–30), guanine nucleotide-exchange factors interact extensively with and remodel the Switch I and II regions of GTPases. Therefore, we have performed alanine scanning mutagenesis of every residue in these regions as well as some residues in Switch III and its adjacent regions (Table 1 and Fig. 1D).
TABLE 1.
Summary of the characterization of 66 GαS mutants
GTPγS binding and in vitro activation of adenylyl cyclase (AC) were shown as percentage of the values obtained with wild-type GαS. The increase of cAMP in cells is shown as the fold increase from each individual experiment. ΔN31 and ΔN38 are deletion mutants of the N-terminal 31 or 38 amino acids, respectively.
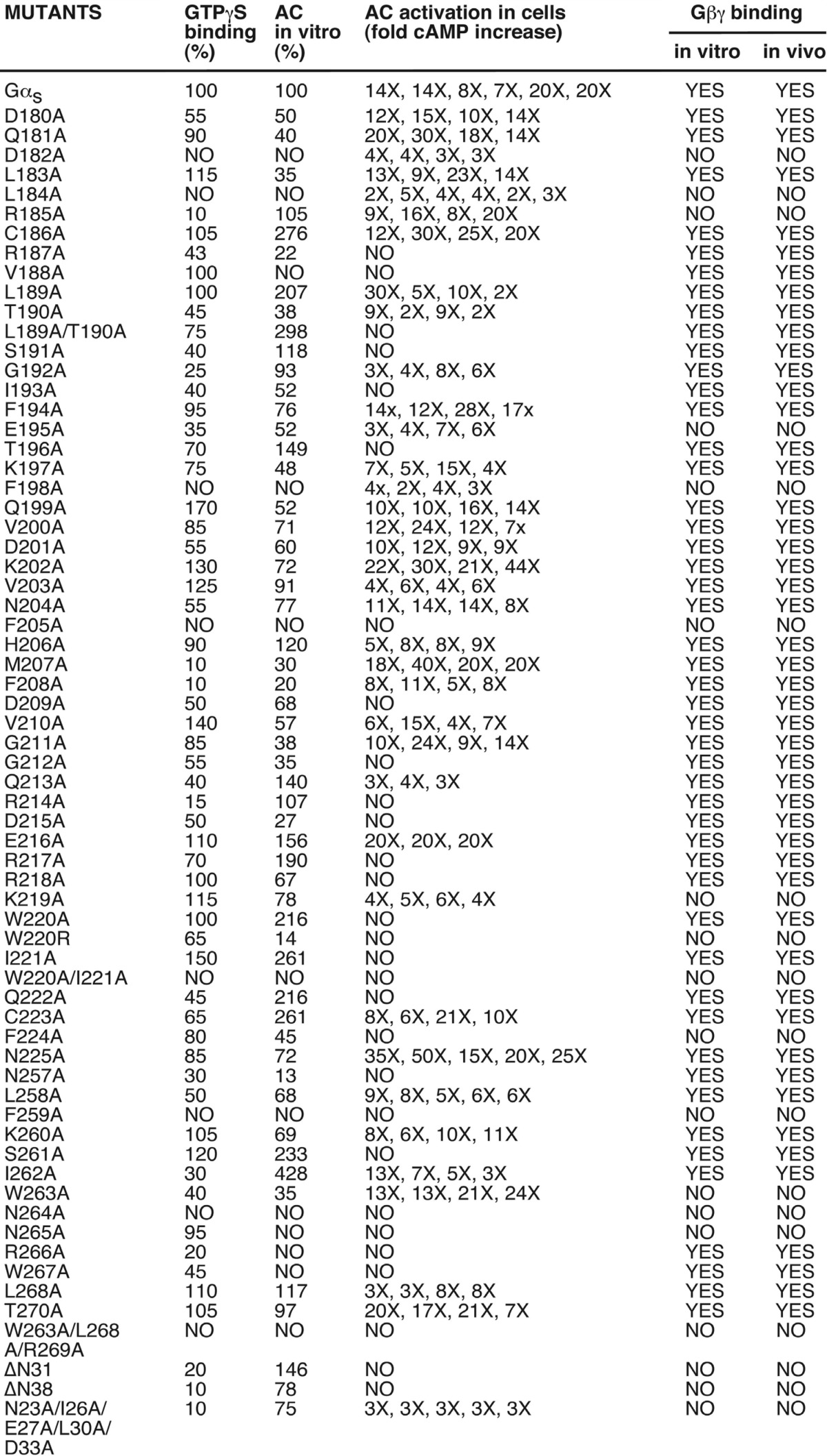
First we identified Gαs mutants that could not bind Gβγ. We purified recombinant proteins of wild-type Gαs and 66 mutant Gαs proteins from E. coli (Table 1 and some examples shown in Fig. 2A). (Here we used the short spliced variant of bovine Gαs. The difference between the short and long splice forms of Gαs is an insertion of 14 amino acids after residue Gly-70.) We also purified recombinant Gβ1γ2 proteins from insect Hi5 cells (Fig. 2A). To test the functionality of these purified Gαs mutant proteins, we performed in vitro GTPγS binding assays and adenylyl cyclase activation assays for all Gαs mutants (Table 1). Other than eight mutants (Table 1), all Gαs mutant proteins were able to bind to GTPγS, implying these purified proteins were stable and functional. Furthermore, using membrane preparations from Sf9 insect cells infected with recombinant baculoviruses carrying adenylyl cyclase type V, we performed in vitro adenylyl cyclase activation assays (Table 1, and some examples shown in Fig. 2, B–E). In addition to the 8 mutants that could not bind GTPγS in vitro, 4 more mutants could not activate adenylyl cyclase in vitro (Table 1). Three (Asn-265, Arg-266, and Trp-267) of these 4 residues are in the α3/β5 loop, which directly contacts the catalytic domain of adenylyl cyclase as revealed by the crystal structure of the complex of Gαs and the catalytic domains of adenylyl cyclase (31). Thus, most of the purified mutant Gαs proteins were stable and functional.
FIGURE 2.

Functional characterization of Gαs and its mutants. A, Coomassie Blue staining shows the purified Gαs proteins (some representatives), GST and Gβ1γ2. B–E, in vitro activation assays of adenylyyl cyclase by Gαs and its mutants (some representatives). F and G, in vitro binding of Gαs and representative mutants to Gβ1γ2. H, size exclusion chromatography of heterotrimer formation. Upper panels: elution profiles are shown for wild-type Gαs alone, GαsK219A alone, Gβ1γ2 alone, the mixture of wild-type Gαs/Gβ1γ2, and the mixture of GαsK219A/Gβ1γ2. Lower panels, SDS-PAGE analysis of the elution fractions. One representative experiment from three independent experiments is shown for each case. WB, Western blot.
We next tested the interaction of these Gαs mutants with Gβγ. For the in vitro binding experiments, we incubated wild-type Gαs and mutant Gαs proteins with purified Gβ1γ2 proteins. Glutathione beads were used to pulldown Gαs proteins. Co-precipitation of Gβ1γ2 was detected with anti-Gβ antibody. As shown in Table 1 (and some examples in Fig. 2F), wild-type Gαs and 48 mutant Gαs proteins pulled down Gβ1γ2, 18 other mutant Gαs proteins (including GαsE195A, GαsK219A, or GαsW263A) did not bind Gβ1γ2. To confirm these binding data and to show that the 18 mutant Gαs proteins could not interact with other Gβ subunits in addition to Gβ1, we used membrane preparations of 293T cells as the source of Gβγ subunits (293T cells at least express Gβ1, Gβ2, and Gβ4). The results were the same as the binding experiments with purified Gβ1γ2 (Table 1 and some examples shown in Fig. 2G). As a third approach, we used size exclusion chromatography to verify the inability of some Gαs mutants binding to Gβγ. As shown in Fig. 2H, wild-type Gαs, GαsK219A, and Gβ1γ2 eluted as single major peaks from the size exclusion column, demonstrating the homogeneity of the subunit preparations. When combined with excess Gβ1γ2, wild-type Gαs showed a two-peaks elution profile: one was the free Gβ1γ2 and the other was the complex of Gαs-Gβ1γ2 with a shorter retention time as compared with the isolated wild-type Gαs (Fig. 2H). On the other hand, GαsK219A showed a two-peak elution prolife with the same retention times as free Gβ1γ2 and free GαsK219A, indicating that Gβ1γ2 had little effect on the retention time of GαsK219A and that GαsK219A was unable to form a complex with Gβ1γ2 (Fig. 2H). Similar results were observed with GαsE195A and GαsW263A, which could not form trimers with Gβ1γ2. Together, these data demonstrate that 18 Gαs mutants (including GαsE195A, GαsK219A, and GαsW263A) are unable to interact with Gβγ subunits, and the locations of these 18 Gαs mutants are displaced in Fig. 3 (red and green residues).
FIGURE 3.
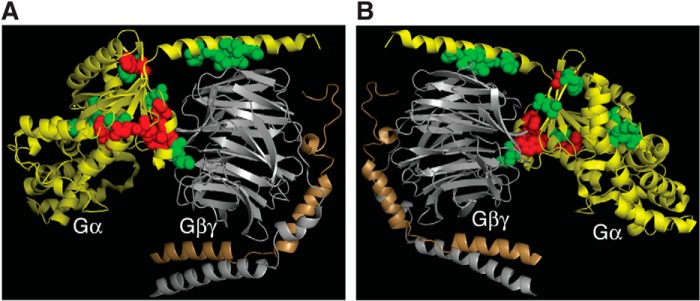
Spatial locations of 18 Gαs mutants that are defective in Gβγ binding. The structure displayed is Gαi1β1γ2 (PDB code 1GP2). Gα, yellow; Gβ, gray; Gγ, gold. Corresponding residues for the 18 Gαs mutants are highlighted (red and green). Green labeled residues are the 8 Gαs mutants that are defective in Gβγ binding but can still be activated by β-ARs. Red labeled residues are the 10 Gαs mutants that are defective in Gβγ binding and could not be activated by β-ARs. A and B are the same image rotated 180°.
Activation of Gαs Mutants by β-Adrenergic Receptors in Cells
Because our goal was to find Gαs mutants that could be potentially activated by GPCRs in the absence of Gβγ, we next investigated the activation of all 66 Gαs mutants by GPCRs in Gαs-deficient cells. Gαs-deficient mouse embryonic fibroblast (MEF) cells were derived from Gαs knock-out mouse embryos (32, 33). Because exon 2 of the Gαs gene was deleted, none of the two alternative spliced variants of Gαs were present in these Gαs−/− MEF cells (32, 33). We have made stable cell lines with these Gαs−/− MEF cells expressing Gαs and all 66 Gαs mutants (in pcDNA3.1/hygromycin vector). Stimulation of these cells with isoproterenol, which activates the endogenous Gs-coupled β-adrenergic receptors, increased cellular cAMP accumulation in cells expressing wild-type and 37 mutant Gαs proteins (Table 1, and some examples shown in Fig. 4). Among the 18 Gαs mutants that could not interact with Gβγ, 8 mutants (GαsD182A, GαsL184A, GαsR185A, GαsE195A, GαsF198A, GαsK219A, GαsW263A, and GαsN23A/I26A/E27A/L30A/D33A) could still mediate receptor activation of adenylyl cyclases leading to cAMP increase (Fig. 4, B-E, and green in Fig. 1D). As control, Gαs−/− MEF cells did not show any cAMP increase upon isoproterenol stimulation (Fig. 4A). The functionality of these Gαs mutant proteins was also verified by cholera toxin, a direct activator of Gαs (Fig. 4). These cellular studies demonstrate that 8 Gαs mutants are able to functionally couple to β-adrenergic receptors, are activated by the receptors, and stimulate the downstream effector adenylyl cyclases, leading to the cellular accumulation of cAMP, even though they could not bind to Gβγ.
FIGURE 4.
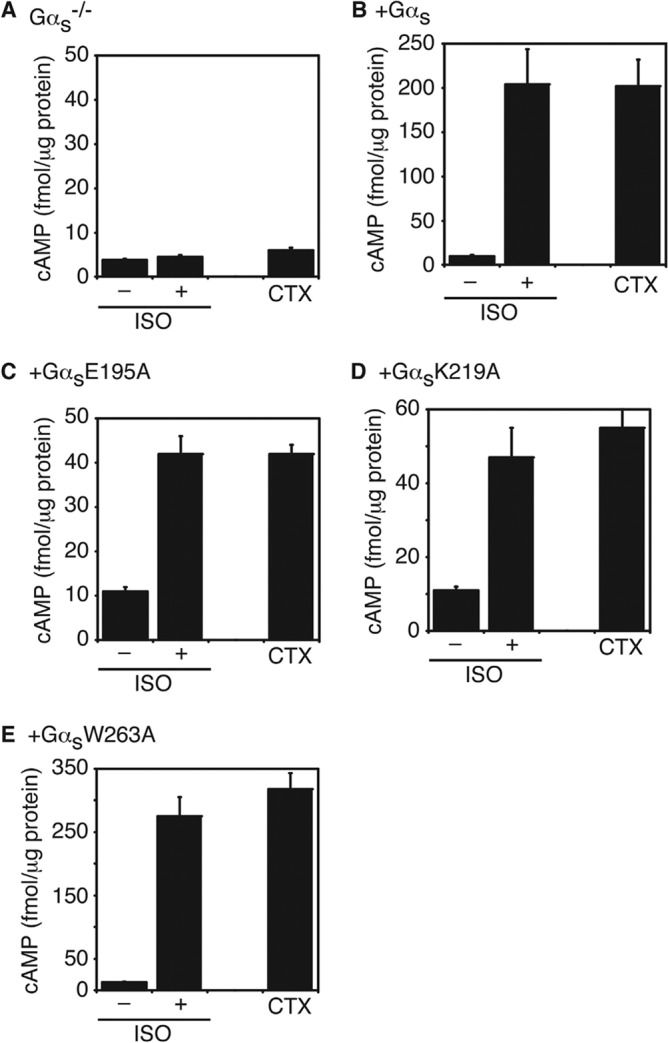
In vivo activation of adenylyl cyclase by Gαs and representative mutants. A, isoproterenol (ISO) or cholera toxin (CTX) failed to increase the cellular cAMP levels in Gαs−/− MEF cells. B–E, Both ISO and CTX increased the cellular cAMP levels in Gαs−/− cells stably re-expressing wild-type Gαs (B), GαsE195A (C), GαsK219A (D), or GαsW263A (E). Data shown are mean ± S.D. of three independent experiments.
Activation of Purified Gαs Mutants by Purified β-Adrenergic Receptors in Vitro in the Absence of Gβγ
To directly demonstrate that a GPCR has the capability to accelerate the guanine-nucleotide exchange on Gα without Gβγ, we performed biochemical studies with purified recombinant proteins of a GPCR and Gαs mutants in vitro. We purified recombinant turkey β1-adrenergic receptors from insect Hi5 cells (23) (Fig. 5A). Gαs proteins were initially purified as GST fusion proteins and the GST tag was removed by protease cleavage (Fig. 5B). Among the 8 Gαs mutants identified above (able to be activated by β-AR in cells but unable to bind Gβγ), 5 (D182A, L184A, R185A, F198A, and N23A/I26A/E27A/L30A/D33A) were unstable after removal of the GST tag. Therefore, we examined the activation of the remaining 3 Gαs mutants (E195A, K219A, and W263A) by β-adrenergic receptor. The activation was monitored by the initial rate of GTPγS loading onto Gαs subunits. As reported previously, purified β-adrenergic receptors had no effect on the rate of GTPγS loading (∼90 fmol/min) onto wild-type Gαs proteins in the absence of Gβγ subunits (19, 34) (Fig. 5C). The rate of GTPγS loading on Gαs was the same in the presence of isoproterenol (an agonist for β-adrenergic receptor) or alprenolol (an antagonist). The initial rate of GTPγS loading to Gαs alone was similar to that of Gαs with β-adrenergic receptor in the presence of the antagonist alprenolol. In contrast, in the presence of purified Gβγ proteins, isoproterenol increased the initial rate of GTPγS loading onto Gαs (∼450 fmol/min) by 4-fold compared with that in the presence of alprenolol (∼115 fmol/min) (Fig. 5D). The fold-increase is similar to that reported in previous reconstituted systems, reflecting a relatively high basal nucleotide exchange rate of Gαs (19, 34).
FIGURE 5.

In vitro activation of Gαs by the β-adrenergic receptor. A, Coomassie Blue staining shows the purified turkey β-adrenergic receptor. B, Coomassie Blue staining shows some examples of purified G proteins (after GST cleavage). C, activation of Gαs by the β-adrenergic receptor in the presence of alprenolol (■) or isoproterenol (●). D, activation of Gαs + Gβ1γ2 by the β-adrenergic receptor in the presence of alprenolol (■) or isoproterenol (●). E, activation of GαsE195 by the β-adrenergic receptor in the presence of alprenolol (■) or isoproterenol (●). F, activation of GαsE195 + Gβ1γ2 by the β-adrenergic receptor in the presence of alprenolol (■) or isoproterenol (●). G, in vitro binding of Gαs and representative mutants to β-AR. Different concentrations (150, 300, and 500 nm) of Gαs and mutant proteins were used. H, left two panels, measurement of the initial rate of GTPγS loading onto wild-type Gαs (top panel) or GαsK219A mutant (bottom panel) in the presence of different concentrations of Gβ1γ2. Right two panels, measurement of the initial rate of GTPγS loading onto wild-type Gαs in the presence of Gβ1γ2 (top panel) or the GαsK219A mutant (bottom panel) in the absence of Gβ1γ2. One representative experiment from three independent experiments is shown for A-G. In H, data are shown as mean ± S.E.
With purified GαsE195A, we found that, without Gβγ subunits, isoproterenol increased the rate of GTPγS loading by about 2.4-fold compared to that with alprenolol (from ∼39 fmol/min with alprenolol to ∼95 fmol/min with isoproterenol) (Fig. 5E). Addition of Gβγ subunits did not significantly change the rates with either isoproterenol (∼105 fmol/min) or alprenolol (∼40 fmol/min) (Fig. 5F). Similar results were obtained with GαsK219A and GαsW263A mutants: without Gβγ subunits, isoproterenol increased the rate of GTPγS loading by 2∼3-fold on GαsK219A and GαsW263A mutants, compared to that with alprenolol. Furthermore, the mutations did not seem to alter the interaction between Gαs and β-AR (Fig. 5G). Moreover, increasing the concentrations of Gβγ enhanced the initial rate of GTPγS loading onto wild-type Gαs, but not GαsK219A (Fig. 5H). Additionally, increasing the concentrations of β-AR elevated the initial rate of GTPγS loading onto both wild-type Gαs (in the presence of Gβ1γ2) and GαsK219A (in the absence of Gβ1γ2) (Fig. 5H). These biochemical experiments clearly demonstrate that purified β1-adrenergic receptors can accelerate the guanine nucleotide exchange on Gαs in the absence of Gβγ subunits. We should note that previous studies with rhodopsin and transducin had indicated that high concentrations of rhodopsin might activate transducin in the absence of Gβγ, and that Gβ1 or Gγ1 knock-out mice still had some light response (35–40). Thus, GPCRs possess the ability to catalyze the nucleotide exchange on Gα subunits.
Role of Extended Linker 2 in Gα Activation
How do the 8 Gαs mutations (GαsD182A, GαsL184A, GαsR185A, GαsE195A, GαsF198A, GαsK219A, GαsW263A, and GαsN23A/I26A/E27A/L30A/D33A) alleviate the requirement of Gβγ for the activation of Gαs by β-adrenergic receptors? When mapped onto the crystal structure of the complex of β2-AR and Gs, 3 of these 8 residues (Asp-182, Leu-184, and Arg-185) are in αF helix (Fig. 6 A and B). Two of the 8 residues (E195 and F198) are in β2 sheet (Fig. 6B). αF and β2 flank Linker 2, which connects the Ras-like GTPases domain and the α-helical domain of Gαs (Figs. 6, A and B, and 1D). Also, within Linker 2, mutations of Arg-187, Leu-189/Thr-190, Ile-193, and Thr-196 all blocked β-AR induced cAMP increases in cells even though these mutants could bind to Gβγ, GTPγS, and activate adenylyl cyclase in vitro (Table 1). Hence, almost all residues in the extended Linker 2 are critical for Gα activation. Although some mutations (such as R185A and E195A) enable Gα activation by GPCR in the absence of Gβγ, other mutations (such as S191A and I193A) block Gα activation by GPCR in cells. When comparing the structures of inactive Gαi-GDP and active Gαi-GTPγS, or the structures of inactive Gαt-GDP and active Gαt-GTPγS, Linker 2 is moved toward the γ-phosphate, bringing the side chain oxygen of Thr-190 into the coordination sphere of the Mg2+ ion where it replaces one of the water molecules observed in the structure of the GDP form (24, 25). Furthermore, it has been shown that Linker 2 changes the conformation upon GPCR activation (41).
FIGURE 6.

Diagrams of Gα activation by GPCRs. A, ribbon presentation of 8 Gαs mutants that are defective in Gβγ binding, but are still activated by β-adrenergic receptors in cells. The structure displayed is the complex of β2-AR·Gs (PDB code 3SN6). β2-AR, dark blue; Gαs, yellow; Gβ, gray; Gγ, gold. Residues in the 8 Gαs mutants (that are defective in Gβγ binding, but are still activated by β-adrenergic receptors in cells) are in green. B, detailed view of the 5 residues in the αF/Linker 2/β2 region. C, detailed view of the residues from the other 3 Gαs mutants. These figures were drawn with MolScript and Raster three-dimensional programs. D, diagram of the clam-shell model of Gα activation. The helical domain opens away from the Ras-like GTPase domain, allowing GDP released. E, diagram of the rolling-top model of Gα activation. The helical domain (the top) slides and rotates away from the Ras-like GTPase domain, leading to GDP release. Dashed lines are unmodeled residues in the β2-AR/Gs structure. F, zoomed view of the link from ICL2 of β2-AR to the αF/Linker 2/β2 region of Gαs from the structure of the β2-AR·Gs complex. Some parts of Gαs were removed for clarity. ICL2, intracellular loop 2 of β2-AR. G, diagram of Gαq with its inhibitor of YM-254890.
A critical role for the extended Linker 2 in Gα activation is consistent with the observation from the crystal structure of the complex of β2-AR and Gs, which shows a large rotation (∼127°) of the α-helical domain relative to the Ras-like GTPase domain upon G-protein activation (Fig. 6, A and E) (10). αF/Linker 2, through the β2-strand, is connected to the β2/β3 loop, which interacts with intracellular loop 2 of β2-AR (Fig. 6F). Furthermore, experiments using various biophysical measurements suggest a “clam-shell” like opening model for Gα activation by GPCRs, in which the helical domain opens away from the Ras-like GTPase domain (42–45) (Fig. 6D). Here the clustering of several Gαs mutants critical for GPCR activation of Gαs around the extended Linker 2 suggests a possibility that Linker 2 may serve as the “hinge” in this clam-shell model (Fig. 6D). Similarly, in the “rolling-top” model (in which the α-helical domain slides and rotates away along the sideways from the Ras-like GTPase domain) observed in the β2-AR·Gs complex structure, this hinge is stretched (some residues in the linkers were disordered and thus were unmodeled in the structure) (10)(Fig. 6E). Mutations of residues in this hinge could make it easy to open the interface between the two domains of Gα or to enhance the interdomain motions. Preventing this interdomain opening by cross-linking inhibits GPCR-catalyzed G-protein activation (42). We should note that, in addition to Linker 2, Linker 1 also connects the Ras-like GTPase domain and the α-helical domain. Indeed a point mutation (G56P) in Linker 1 of transducin increased the basal exchange rate and exhibited some degrees of activation at high levels of rhodopsin in the absence of β1γ1 (35). The role of Linker 1 in Gα-protein activation by GPCRs requires future systematic investigation. Here we propose that a GPCR via its intracellular loop 2 directly interacts with the β2/β3 loop of Gα to communicate to Linker 2 and αF, resulting in the rotation of the helical domain and the release of GDP (Fig. 6, E and F).
Linker 2 as the Target Site of a Natural Product Inhibitor for Gq
The crystal structure of Gq and a small molecule natural product inhibitor shows the inhibitor binds to a hydrophobic cleft and directly contacts Linker 2 (Fig. 6G) (46). This inhibitor, YM-254890, inhibits the guanine-nucleotide exchange reaction by preventing the GDP release (46). Also, this inhibitor blocks the AlF4−-induced conformational change in Gαq (46). It is proposed that this inhibitor stabilizes an inactive GDP-bound form (46). These structural data suggest that Linker 2 might be critical for G-protein activation, and that the Linker 2 region is a potential therapeutic targeting site.
Role of αN, α3, and Switch Regions in Gα Activation in Cells
In addition to the extended Linker 2, our data have uncovered some other residues that are essential for Gαs activation in cells by β-ARs. The N-terminal residues (Asn-23, Ile-26, Glu-27, Leu-30, and Asp-33) and Trp-263 are located at or close to the receptor-interacting surface (10, 15, 47, 48)(Fig. 6, A and C). Trp-263 is close to the α3/β5 loop. This region is analogous to GEF contact sites in other GTPases such as Ras and EF-Tu (26, 27). The crystal structure of β2-AR/Gs has revealed that β2-AR interacts with the αN/β1 hinge; this may explain the role of the 5 N-terminal residues (Asn-23, Ile-26, Glu-27, Leu-30, and Asp-33) in the activation of Gα by GPCR.
Lys-219 is located within Switch II (Figs. 1D and 6C). Lys-206 of Gαt (corresponding to Lys-219 in Gαs) had been identified to be important for Gαt activation by rhodopsin (49). Several residues near Lys-219 of Gαs such as Gly-212 and Gln-213 are critical for triggering conformational changes in Switch II through an interaction with the γ-phosphate in GTP and for stabilizing the transition state for GTP hydrolysis, respectively (13, 14, 50, 51). Indeed, most Gαs mutations in the Switch I and II regions (such as Ile-193, Thr-196, Asp-209, Gly-212, Arg-214, Asp-215, Arg-217, Arg-218, Trp-220, Ile-221, and Gln-222) could not be activated by β-adrenergic receptors in cells, despite their ability to bind to Gβγ subunits, bind to GTPγS, and activate adenylyl cyclases in vitro (Table 1). Arg-187 and Thr-190 (in Switch I) as well as Gly-212 (in Switch II) contact the γ-phosphate of GTP (6). Together, these data demonstrate that αN, α3, Switch I and Switch II are critical for Gα activation by GPCRs.
The critical role of Switch I and II regions in Gα activation is similar to the activation of Ras-superfamily GTPases by their GEFs. GEFs for small GTPases have different structures (26–30). They contact GTPases at the same as well as different amino acid residues and induce different conformational changes on GTPases to drive out the GDP. However, they all utilize a two-sided attack to release positive charges (the Mg2+ ion and the invariant lysine residue in the P loop (phosphate-binding loop)) from their interaction with the phosphates of the nucleotide (52). These GEFs interact extensively with and remodel Switch I and II regions of GTPases, which form part of the binding pocket for Mg2+ and the γ-phosphate of GTP. Thus, although GPCRs are unusual GEFs because they do not contact these switch regions directly, they still require/remodel these switch regions for the activation of Gα.
Possible Role of Gβγ in Gα Activation
Our mutagenesis studies unexpectedly reveal a role for residues Trp-263 (in α3) and Asn-265 (in the α3-β5 loop) in Gβγ binding (Table 1). From the structure of the Gαi·GDP/Gβγ complex, this region is not directly involved in contacting with Gβγ. However, this region is right next to Switch III and forms an elaborate inter-dependent network of polar interactions with the Switch II region, which is critical for interacting with Gβγ (24). Although we did not address the role of Gβγ in the guanine-nucleotide exchange on Gα, we should mention that there were two models that proposed a catalytic role for Gβγ (15, 16, 21). Gβγ subunits contact the switch regions of Gα subunits (24, 25), and have a structure similar to RCC1 (30). Hence, Gβγ subunits would be more like GEFs if only Gβγ subunits could be proven to possess GEF activity without GPCRs. The role of Gβγ in Gα activation could be complex. Gβγ stabilizes GDP binding on Gα, thus serving as a guanine-nucleotide dissociation inhibitor. GPCR activation could release the inhibition of Gβγ. However, the nucleotide exchange rate of Gα, in the absence of Gβγ, is still slower than that in the presence of GPCRs. This would indicate that, in addition to releasing Gβγ, GPCRs act on the nucleotide exchange on Gα. Gβγ subunits could serve an additional catalytic role to augment the complex formation between GPCRs and Gα subunits, similar to the role of ELMO in facilitating nucleotide exchange on Rac by Dock180 (53). Indeed, our data (Table 1, Fig. 5, E and F) show that, whereas GαsE195A could be activated by β-ARs in the absence of Gβγ, the extent of activation was suboptimal relative to wild-type Gαs in the presence of Gβγ. This implies that Gβγ somehow contributes to the activation process.
Previously two models were proposed for a catalytic role for Gβγ subunits in activating Gα subunits. In the “lever” model (15, 21), GPCRs cause a tilt (or a rotation) of Gβγ relative to Gα. The membrane proximal part (interacting with the Gα N-terminal helix) of Gβγ moves closer to Gα. The opposite end of Gβγ moves away from Gα and, at the same time, pulls along the interacting parts of Gα. These interacting residues from Switch I and II of Gα are part of the lip of the nucleotide binding pocket. Therefore, GDP can exit permitting exchange by GTP. In the second “gear shift” model (16), GPCRs also cause a tilt of Gβγ. However, the rotating direction is opposite of that in the lever model. Gβγ moves closer to Gα. The N terminus of Gγ (the membrane distal end of Gβγ) moves to contact and displace the helical domain of Gα, creating an exit route for GDP. The exact role of Gβγ in the exchange reaction needs further investigation.
Conclusions
Upon ligand-binding, GPCRs initiate the exchange of GDP bound on Gα subunits of G-proteins with GTP. However, the detailed molecular mechanisms by which GPCRs activate G-proteins are not well understood. Here we reveal some new insights on this process. We demonstrate that a GPCR could activate some Gα mutants in the absence of Gβγ, and that the extended Linker 2 in Gα is critical for Gα activation by GPCRs. These data will advance our understanding of this critical cellular signaling process.
The closed and open states of the cleft (the GDP/GTP binding site) are determined by relative pivoting of the Ras-GTPase domain and the α-helical domain at Linker 2. The closed state of the cleft is represented in all crystal structures of Gα subunits. One of the open states of the cleft is captured in the β2-AR/Gs crystal structure. This model of activation of Gα subunits suggests that factors binding at the interface of the Ras-GTPase domain and the helical domain might facilitate cleft closure, thus serving as an inhibitor of Gα activation (Fig. 6G). Given this pivot model, the two domains of Gα could be either clam-shell opening or rolling-top expansion including a relative rotation of the GTPase domain and the helical domain around an axis through the linkers (Fig. 6, D and E). Results from several biophysical studies are consistent with the clam-shell opening, and that the crystal structure of the β2-AR·Gs complex is in line with the rolling-top movement. Therefore, in addition to the established role for α5-helix of Gα connecting GPCRs to the guanine-nucleotide binding pocket, we show here that the extended Linker 2 connects GPCRs to the nucleotide binding pocket as well as the α-helical domain of Gα. In conclusion, our data demonstrate that GPCRs possess the capability to catalyze the nucleotide exchange on Gα subunits, and that the extended Linker 2 is critical for Gα activation by GPCRs.
Acknowledgments
We thank Drs. Lonny Levin and Tom Sakmar and members of our laboratory for helpful comments and discussion.
This work was supported, in whole or in part, by National Institutes of Health Grant HL 91525 (to X.-Y. H.).
- GPCR
- G protein-coupled receptor
- DM
- dodecyl-β-d-maltoside
- MEF
- mouse embryonic fibroblast
- β-AR
- β-adrenergic receptor
- GEF
- guanine-nucleotide exchange factor.
REFERENCES
- 1. Rosenbaum D. M., Rasmussen S. G., Kobilka B. K. (2009) The structure and function of G-protein-coupled receptors. Nature 459, 356–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pierce K. L., Premont R. T., Lefkowitz R. J. (2002) Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650 [DOI] [PubMed] [Google Scholar]
- 3. Gilman A. G. (1987) G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649 [DOI] [PubMed] [Google Scholar]
- 4. Bourne H. R., Sanders D. A., McCormick F. (1990) The GTPase superfamily: a conserved switch for diverse cell functions. Nature 348, 125–132 [DOI] [PubMed] [Google Scholar]
- 5. Simon M. I., Strathmann M. P., Gautam N. (1991) Diversity of G proteins in signal transduction. Science 252, 802–808 [DOI] [PubMed] [Google Scholar]
- 6. Sprang S. R., Chen Z., Du X. (2007) Structural basis of effector regulation and signal termination in heterotrimeric Gα proteins. Adv. Protein Chem. 74, 1–65 [DOI] [PubMed] [Google Scholar]
- 7. Tesmer J. J. (2010) The quest to understand heterotrimeric G protein signaling. Nat. Struct. Mol. Biol. 17, 650–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choe H. W., Park J. H., Kim Y. J., Ernst O. P. (2011) Transmembrane signaling by GPCRs: insight from rhodopsin and opsin structures. Neuropharmacology 60, 52–57 [DOI] [PubMed] [Google Scholar]
- 9. Katritch V., Cherezov V., Stevens R. C. (2013) Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ramachandran S., Cerione R. A. (2006) How GPCRs hit the switch. Nat. Struct. Mol. Biol. 13, 756–757 [DOI] [PubMed] [Google Scholar]
- 12. Schwartz T. W., Sakmar T. P. (2011) Structural biology: snapshot of a signalling complex. Nature 477, 540–541 [DOI] [PubMed] [Google Scholar]
- 13. Noel J. P., Hamm H. E., Sigler P. B. (1993) The 2.2 A crystal structure of transducin-α complexed with GTPγS. Nature 366, 654–663 [DOI] [PubMed] [Google Scholar]
- 14. Coleman D. E., Berghuis A. M., Lee E., Linder M. E., Gilman A. G., Sprang S. R. (1994) Structures of active conformations of Gαi1 and the mechanism of GTP hydrolysis. Science 265, 1405–1412 [DOI] [PubMed] [Google Scholar]
- 15. Iiri T., Farfel Z., Bourne H. R. (1998) G-protein diseases furnish a model for the turn-on switch. Nature 394, 35–38 [DOI] [PubMed] [Google Scholar]
- 16. Cherfils J., Chabre M. (2003) Activation of G-protein Galpha subunits by receptors through Gα-Gβ and Gα-Gγ interactions. Trends Biochem. Sci 28, 13–17 [DOI] [PubMed] [Google Scholar]
- 17. Bourne H. R. (1997) How receptors talk to trimeric G proteins. Curr. Opin. Cell Biol. 9, 134–142 [DOI] [PubMed] [Google Scholar]
- 18. Fung B. K. (1983) Characterization of transducin from bovine retinal rod outer segments: I. separation and reconstitution of the subunits. J. Biol. Chem. 258, 10495–10502 [PubMed] [Google Scholar]
- 19. Florio V. A., Sternweis P. C. (1989) Mechanisms of muscarinic receptor action on Go in reconstituted phospholipid vesicles. J. Biol. Chem. 264, 3909–3915 [PubMed] [Google Scholar]
- 20. Higashijima T., Ferguson K. M., Sternweis P. C., Smigel M. D., Gilman A. G. (1987) Effects of Mg2+ and the βγ-subunit complex on the interactions of guanine nucleotides with G proteins. J. Biol. Chem. 262, 762–766 [PubMed] [Google Scholar]
- 21. Rondard P., Iiri T., Srinivasan S., Meng E., Fujita T., Bourne H. R. (2001) Mutant G protein α subunit activated by Gβγ: a model for receptor activation? Proc. Natl. Acad. Sci. U.S.A. 98, 6150–6155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Steegborn C., Litvin T. N., Hess K. C., Capper A. B., Taussig R., Buck J., Levin L. R., Wu H. (2005) A novel mechanism for adenylyl cyclase inhibition from the crystal structure of its complex with catechol estrogen. J. Biol. Chem. 280, 31754–31759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang J., Chen S., Zhang J. J., Huang X. Y. (2013) Crystal structure of oligomeric β1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat Struct Mol Biol 20, 419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wall M. A., Coleman D. E., Lee E., Iñiguez-Lluhi J. A., Posner B. A., Gilman A. G., Sprang S. R. (1995) The structure of the G protein heterotrimer Gi α1β1γ2. Cell 83, 1047–1058 [DOI] [PubMed] [Google Scholar]
- 25. Lambright D. G., Sondek J., Bohm A., Skiba N. P., Hamm H. E., Sigler P. B. (1996) The 2.0-Å crystal structure of a heterotrimeric G protein. Nature 379, 311–319 [DOI] [PubMed] [Google Scholar]
- 26. Kawashima T., Berthet-Colominas C., Wulff M., Cusack S., Leberman R. (1996) The structure of the Escherichia coli EF-Tu·EF-Ts complex at 2.5-Å resolution. Nature 379, 511–518 [DOI] [PubMed] [Google Scholar]
- 27. Boriack-Sjodin P. A., Margarit S. M., Bar-Sagi D., Kuriyan J. (1998) The structural basis of the activation of Ras by Sos. Nature 394, 337–343 [DOI] [PubMed] [Google Scholar]
- 28. Goldberg J. (1998) Structural basis for activation of ARF GTPase: mechanisms of guanine nucleotide exchange and GTP-myristoyl switching. Cell 95, 237–248 [DOI] [PubMed] [Google Scholar]
- 29. Worthylake D. K., Rossman K. L., Sondek J. (2000) Crystal structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature 408, 682–688 [DOI] [PubMed] [Google Scholar]
- 30. Renault L., Kuhlmann J., Henkel A., Wittinghofer A. (2001) Structural basis for guanine nucleotide exchange on Ran by the regulator of chromosome condensation (RCC1). Cell 105, 245–255 [DOI] [PubMed] [Google Scholar]
- 31. Tesmer J. J., Sunahara R. K., Gilman A. G., Sprang S. R. (1997) Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsα:GTPγS (see comments). Science 278, 1907–1916 [DOI] [PubMed] [Google Scholar]
- 32. Bastepe M., Gunes Y., Perez-Villamil B., Hunzelman J., Weinstein L. S., Jüppner H. (2002) Receptor-mediated adenylyl cyclase activation through XLα(s), the extra-large variant of the stimulatory G protein α-subunit. Mol. Endocrinol. 16, 1912–1919 [DOI] [PubMed] [Google Scholar]
- 33. Sun Y., Huang J., Xiang Y., Bastepe M., Jüppner H., Kobilka B. K., Zhang J. J., Huang X. Y. (2007) Dosage-dependent switch from G protein-coupled to G protein-independent signaling by a GPCR. EMBO J. 26, 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pedersen S. E., Ross E. M. (1982) Functional reconstitution of β-adrenergic receptors and the stimulatory GTP-binding protein of adenylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 79, 7228–7232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Singh G., Ramachandran S., Cerione R. A. (2012) A constitutively active Gα subunit provides insights into the mechanism of G protein activation. Biochemistry 51, 3232–3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Phillips W. J., Wong S. C., Cerione R. A. (1992) Rhodopsin/transducin interactions: II. influence of the transducin-βγ subunit complex on the coupling of the transducin-α subunit to rhodopsin. J. Biol. Chem. 267, 17040–17046 [PubMed] [Google Scholar]
- 37. Herrmann R., Heck M., Henklein P., Hofmann K. P., Ernst O. P. (2006) Signal transfer from GPCRs to G proteins: role of the Gα N-terminal region in rhodopsin-transducin coupling. J. Biol. Chem. 281, 30234–30241 [DOI] [PubMed] [Google Scholar]
- 38. Lobanova E. S., Finkelstein S., Herrmann R., Chen Y. M., Kessler C., Michaud N. A., Trieu L. H., Strissel K. J., Burns M. E., Arshavsky V. Y. (2008) Transducin γ-subunit sets expression levels of α- and β-subunits and is crucial for rod viability. J. Neurosci. 28, 3510–3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kolesnikov A. V., Rikimaru L., Hennig A. K., Lukasiewicz P. D., Fliesler S. J., Govardovskii V. I., Kefalov V. J., Kisselev O. G. (2011) G-protein βγ-complex is crucial for efficient signal amplification in vision. J. Neurosci. 31, 8067–8077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nikonov S. S., Lyubarsky A., Fina M. E., Nikonova E. S., Sengupta A., Chinniah C., Ding X. Q., Smith R. G., Pugh E. N., Jr., Vardi N., Dhingra A. (2013) Cones respond to light in the absence of transducin β subunit. J. Neurosci. 33, 5182–5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oldham W. M., Van Eps N., Preininger A. M., Hubbell W. L., Hamm H. E. (2007) Mapping allosteric connections from the receptor to the nucleotide-binding pocket of heterotrimeric G proteins. Proc. Natl. Acad. Sci. U.S.A. 104, 7927–7932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van Eps N., Preininger A. M., Alexander N., Kaya A. I., Meier S., Meiler J., Hamm H. E., Hubbell W. L. (2011) Interaction of a G protein with an activated receptor opens the interdomain interface in the α subunit. Proc. Natl. Acad. Sci. U.S.A. 108, 9420–9424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chung K. Y., Rasmussen S. G., Liu T., Li S., DeVree B. T., Chae P. S., Calinski D., Kobilka B. K., Woods V. L., Jr., Sunahara R. K. (2011) Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature 477, 611–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Westfield G. H., Rasmussen S. G., Su M., Dutta S., DeVree B. T., Chung K. Y., Calinski D., Velez-Ruiz G., Oleskie A. N., Pardon E., Chae P. S., Liu T., Li S., Woods V. L., Jr., Steyaert J., Kobilka B. K., Sunahara R. K., Skiniotis G. (2011) Structural flexibility of the Gαs α-helical domain in the β2-adrenoceptor Gs complex. Proc. Natl. Acad. Sci. U.S.A. 108, 16086–16091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jones J. C., Jones A. M., Temple B. R., Dohlman H. G. (2012) Differences in intradomain and interdomain motion confer distinct activation properties to structurally similar Gα proteins. Proc. Natl. Acad. Sci. U.S.A. 109, 7275–7279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nishimura A., Kitano K., Takasaki J., Taniguchi M., Mizuno N., Tago K., Hakoshima T., Itoh H. (2010) Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proc. Natl. Acad. Sci. U.S.A. 107, 13666–13671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grishina G., Berlot C. H. (2000) A surface-exposed region of Gαs in which substitutions decrease receptor-mediated activation and increase receptor affinity. Mol. Pharmacol. 57, 1081–1092 [PubMed] [Google Scholar]
- 48. Zhang Z., Melia T. J., He F., Yuan C., McGough A., Schmid M. F., Wensel T. G. (2004) How a G protein binds a membrane. J. Biol. Chem. 279, 33937–33945 [DOI] [PubMed] [Google Scholar]
- 49. Onrust R., Herzmark P., Chi P., Garcia P. D., Lichtarge O., Kingsley C., Bourne H. R. (1997) Receptor and βγ binding sites in the α subunit of the retinal G protein transducin. Science 275, 381–384 [DOI] [PubMed] [Google Scholar]
- 50. Lambright D. G., Noel J. P., Hamm H. E., Sigler P. B. (1994) Structural determinants for activation of the α-subunit of a heterotrimeric G protein. Nature 369, 621–628 [DOI] [PubMed] [Google Scholar]
- 51. Sondek J., Lambright D. G., Noel J. P., Hamm H. E., Sigler P. B. (1994) GTPase mechanism of G proteins from the 1.7-Å crystal structure of transducin α-GDP-AIF-4. Nature 372, 276–279 [DOI] [PubMed] [Google Scholar]
- 52. Vetter I. R., Wittinghofer A. (2001) The guanine nucleotide-binding switch in three dimensions. Science 294, 1299–1304 [DOI] [PubMed] [Google Scholar]
- 53. Brugnera E., Haney L., Grimsley C., Lu M., Walk S. F., Tosello-Trampont A. C., Macara I. G., Madhani H., Fink G. R., Ravichandran K. S. (2002) Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat. Cell. Biol. 4, 574–582 [DOI] [PubMed] [Google Scholar]