

Background: Little is known regarding the contribution of MAPKs to the development of ectodermal appendages.
Results: Mice with a deletion of p38α in ectoderm display defective secretion of dental enamel and the absence of dental cusps.
Conclusion: p38α mediates critical steps in tooth morphogenesis and enamel secretion.
Significance: This is the first in vivo study demonstrating that the p38 MAPK pathway is critical for tooth morphogenesis and enamel secretion.
Keywords: Bone Morphogenetic Protein (BMP), Microtubule-associated Protein (MAP), p38 MAPK, SMAD Transcription Factor, Tooth Development, Ameloblast, Ectodermal Dysplasia
Abstract
An improved understanding of the molecular pathways that drive tooth morphogenesis and enamel secretion is needed to generate teeth from organ cultures for therapeutic implantation or to determine the pathogenesis of primary disorders of dentition (Abdollah, S., Macias-Silva, M., Tsukazaki, T., Hayashi, H., Attisano, L., and Wrana, J. L. (1997) J. Biol. Chem. 272, 27678–27685). Here we present a novel ectodermal dysplasia phenotype associated with conditional deletion of p38α MAPK in ectodermal appendages using K14-cre mice (p38αK14 mice). These mice display impaired patterning of dental cusps and a profound defect in the production and biomechanical strength of dental enamel because of defects in ameloblast differentiation and activity. In the absence of p38α, expression of amelogenin and β4-integrin in ameloblasts and p21 in the enamel knot was significantly reduced. Mice lacking the MAP2K MKK6, but not mice lacking MAP2K MKK3, also show the enamel defects, implying that MKK6 functions as an upstream kinase of p38α in ectodermal appendages. Lastly, stimulation with BMP2/7 in both explant culture and an ameloblast cell line confirm that p38α functions downstream of BMPs in this context. Thus, BMP-induced activation of the p38α MAPK pathway is critical for the morphogenesis of tooth cusps and the secretion of dental enamel.
Introduction
Defining the molecular pathways that govern the development of teeth or the secretion of dental matrix proteins is fundamental to understand the basis of genetic disorders of dentition and ectodermal dysplasia syndromes (1–3). Additionally, this understanding forms the basis of strategies to generate teeth either in vitro for transplantation or in situ for regenerative applications. To date, a major focus of investigation in this area has been to define how the multiple secreted ligands derived from either the epithelium itself or from the underlying mesenchyme act at the molecular level (2).
In particular, multiple lines of evidence demonstrate that bone morphogenic proteins (BMPs)4 play a critical role in this process. BMPs are secreted with spatial and temporal coordination relative to the formation of the enamel knot, a signaling center in the invaginated oral epithelium critical for the morphogenesis of the dental cusps. In this context, BMP4 is expressed by the underlying mesenchyme, and BMP2 and BMP7 are expressed predominantly in the oral epithelium (4). Consistent with a role of BMPs in promoting formation of dental cusps, mice deficient for the secreted inhibitor of BMP signaling, ectodin, display expanded dental cusps (4, 5). Likewise, suppression of BMP signaling by transgenic expression of follistatin, a secreted inhibitor of BMP/TGFβ superfamily signaling, driven by the K14 promoter blocks the differentiation and enamel matrix synthetic capacity of ameloblasts, the cell type responsible for enamel secretion and the absence or near absence of dental cusp formation (6). In particular, BMP stimulation appears to up-regulate expression of the cell cycle inhibitor p21 in the oral epithelium near the enamel knot, because BMP4-soaked beads induce p21 expression. p21 is believed to contribute to the formation of dental cusps by slowing cellular proliferation in the ectodermal layer relative to that of the mesenchymal layer. p21 expression is itself inversely correlated with the expression of ectodin (7). Although p21-deficient mice have not been reported to have any defects in tooth cusp patterning, this may reflect redundancy with similar cell cycle inhibitors (8). Additionally, BMPs continue to play a role in the teeth of adult mice, because suppression of BMP signaling with transgenic expression of follistatin by the K14 promoter results in suppression of the differentiation and enamel matrix synthetic capacity of ameloblasts (6).
Generally BMPs signal through two main pathways: the SMAD transcription factors and MAPK pathways. In the context of osteoblast and chondrocyte differentiation, both the SMAD and the p38 MAPK pathways contribute to BMP function (9–11), whereas a prior study concluded that the p38 pathway and SMAD4 are functionally redundant in the oral epithelium, because inactivation of the p38 pathway produced no effect unless the SMAD pathway was also targeted with genetic ablation of SMAD4 (12). However, this conclusion was drawn on the basis of treatment of mandibular organ cultures with the p38 inhibitor SB203580, and the effect of genetic ablation of p38 signaling had not been studied. Moreover, in addition to a lack of definitive data regarding the role of MAPKs in mediating the effects of BMPs, nearly every other secreted ligand implicated in morphogenesis of ectodermal structures, including FGFs and WNTs, has the capacity to activate MAPKs (13). Thus, the relative contribution of each of these pathways to MAPK activation in this context remains to be fully elucidated.
Here, we have sought to determine the contribution of the p38 MAPK pathway to dental development and enamel production. In particular, we find that epithelial-specific deletion of p38α in p38αK14 mice results in defects in the morphogenesis of dental cusps and enamel secretion, because of defective signal transduction by BMPs in the absence of p38α.
EXPERIMENTAL PROCEDURES
Mice
Both the p38α floxed allele and p38αK14 mice were previously described (14, 15). Mice bearing Mkk3 and Mkk6 null alleles were also previously described (16, 17). Mkk6−/−, Mkk3−/−, and Tak1K14 mice were all maintained on the C57BL/6 background and housed in accordance with the policies of the institutional animal care and use committee. For all mice analyzed, age- and gender-matched WT C57BL/6 mice housed in the same facility or littermate control mice were used.
In Situ Hybridization and Immunohistochemistry
In situ hybridization was performed as previously described (9). Briefly, digoxigenin-labeled antisense probes were generated to the indicated targets, hybridized with paraffin sections, and visualized using an anti-digoxigenin HRP conjugate system. Immunohistochemistry was also performed as previously described (11).
Vickers Microhardness Testing
Maxillary and mandibular incisors were resin embedded, polished, and tested for enamel microhardness on a LECO M 400 HI testing machine (LECO, St. Joseph, MI), using a load of 25 g for 5 s with a Vickers tip. Thirty indentations per enamel sample were scored.
Scanning Electron Microscopy
Scanning electron microscopy was performed as previously described (18). Briefly, incisors were dried in air and acetone, fastened to stubs, sputter-coated, and visualized on a JEOL 6400 scanning electron microscope.
μCT Analysis of Dental Enamel
Skulls of the indicated mice were stripped of skin, immersed in 70% EtOH, and scanned in a μCT-35 (Scanco Medical Co.) at 70 kV, 114 μA, and 0.012 isotropic voxels. For quantitative analysis, individual mandibular incisors were contoured and analyzed using the Scanco software platform.
Tissue Culture and in Situ Hybridization
Oral epithelium from E14.5 embryos was dissected and cultured as previously described (4, 6). Tissue explants were treated with BSA (1 ng/ml; Sigma) or BMP2/7 (100 ng/ml; R&D Systems) in the presence or absence of SB203580, a p38α and β MAPK inhibitor (10 μm; EMD Millipore) for 16 h at 37 °C. After culture, explants were treated with cold MeOH for 2 min, fixed in 4% paraformaldehyde overnight at 4 °C and processed for whole mount in situ hybridization as previously described (6). The p21 probe was kindly provided by Dr. Irma Thesleff (7).
Cell Culture and Antibodies
The ameloblast-like cell (ALC) line was kindly provided by Dr. Toshihiro Sugiyama (19) and maintained in DMEM containing 10% dialyzed FBS, 2 mm l-glutamine, 1% HEPES, 1% nonessential amino acids, and 1% penicillin/streptomycin. The following antibodies were used: anti-GAPDH (Affinity Bioreagents); anti-phospho-MKK3/6, MKK3, phospho-p38, p38α, phospho-Smad1/5, Smad4, and RhoA (Cell Signaling Technology); and anti-β4 integrin and proliferating cell nuclear antigen (Santa Cruz Biotechnology).
p21 Luciferase Reporter Assay
For reporter gene assay, the ALC line was plated at 2 × 105 cells/well in 12-well plates. After 24 h post-transfection with the p21-luc reporter gene (a generous gift from Dr. Arthur Skoultchi) and Renilla along with vector or MKK6-CA mutant using Effectene transfection reagent (Qiagen), cells were treated with vehicle or BMP2/7 in the absence or presence of SB203580. Alternatively, cell were infected with lentivirus expressing control, murine Smad4 or RhoA shRNA and then transfected with the p21-luc reporter gene and Renilla. The luciferase activity of cell lysates was measured using a luciferase reporter assay system (Promega). Reporter activity was normalized to the protein content of the cell lysates.
Statistics
All statistical analysis was performed in the GraphPad prism software package. All values graphed are means ± standard deviation.
RESULTS
Expression and Activation of p38 Signaling Components in Ameloblasts
To examine activation of the p38 MAPK pathway in ameloblasts in vivo, immunohistochemistry was performed for the MAP2Ks upstream of p38, MKK3, and MMK6 and their phosphorylation levels in mandibular incisors (Fig. 1A). MKK3 and MKK6 are highly expressed and phosphorylated in both ameloblasts and odontoblasts. Likewise, phosphorylation of p38 MAPKs is observed in both presecretory and secretory ameloblasts and was nearly ablated in p38αK14 mice. (Fig. 1B) Thus, the p38 MAPK pathway is present and activated in ameloblasts in vivo.
FIGURE 1.
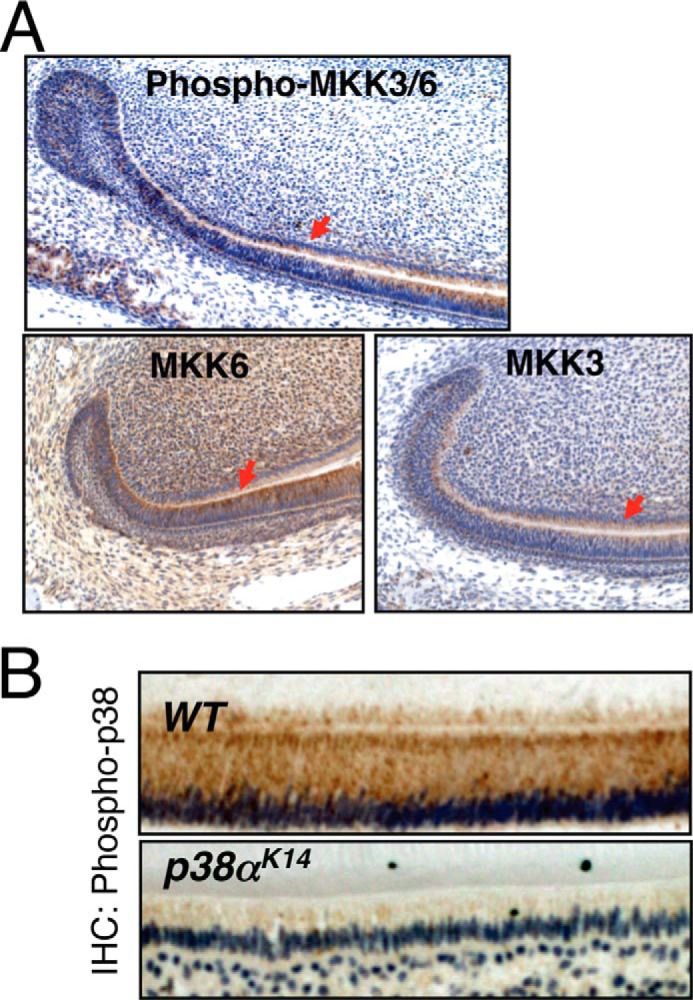
Expression and activation of p38 signaling components in ameloblasts. A, immunohistochemical staining for phospho-MKK3/6, MKK3, and MKK6 on mandibular incisors from 4-day-old WT mice. The expressions in ameloblasts are indicated with red arrows. B, immunohistochemical (IHC) staining for phospho-p38 on secretory ameloblasts from the mandibular incisors of 6-week-old p38αK14 and WT mice.
Mice Lacking p38α in Ectoderm Display Defects in Dental Enamel Production and Impaired Dental Cusp Morphogenesis
To determine the contribution of the p38 MAPK pathway to tooth development, mice with a skin-specific deletion of the MAPK p38α driven by the keratin 14 promoter (p38αK14 mice) were examined using μCT (Fig. 2A). Tooth enamel was visualized by windowing the scans to only display the most dense tissue, revealing a substantial reduction in total dental enamel and a complete absence of dental cusps in p38αK14 mice. Additionally, the reduced amount of enamel present displayed impaired biomechanical properties, because mice analyzed at 6 weeks of age displayed spontaneous cracking and fracture of dental enamel (Fig. 2, B and C). To confirm these qualitative impressions regarding the enamel defects in p38αK14 mice, individual mandibular incisors isolated from 3- or 6-week-old WT and p38αK14 mice were contoured, and the average enamel thickness and enamel volume relative to total tooth volume were measured using μCT (Fig. 2D). As expected, the average enamel thickness and enamel volume relative to total tooth volume were significantly reduced in p38αK14 mice compared with WT littermate controls. Displaying a histogram of the relative volume in voxels versus density reveals three peaks corresponding to soft tissue in the pulp, dentin, and tooth enamel. This result shows that the decrease of the total enamel volume in both qualitative (Fig. 2A) and quantitative (Fig. 2E) analyses does not result from shifting the enamel peak so that it falls below an arbitrary threshold, but the enamel peak is greatly reduced in size, without a shift in the mode toward lower densities. Consistent with this μCT analysis, histologic analysis of p38αK14 incisors shows occasional disruption of the ameloblast layer, implying that adhesion between ameloblasts and enamel is defective in the absence of p38α (Fig. 2F).
FIGURE 2.

Dental phenotype of p38αK14 mice. A, skulls of 6-week-old p38αK14 and WT mice were scanned by μCT. Displayed are the lingual surfaces of the molars (top panels) and three-dimensional reconstructions windowed so that only enamel densities are visible (bottom panels). B, pictures of the molars from 3-week-old p38αK14 (middle panel) and WT mice (top panel) showing complete absence of molar cusps in p38αK14 mice. A view of the mandibular incisors shows spontaneous fracture of the enamel in p38αK14 mice (red arrow, bottom panel). C, the relative enamel volume and thickness of the indicated 6-week-old mice was determined by μCT analysis (top panels). Views of the two-dimensional slices from μCT analysis of p38αK14 and control mandibular incisors, showing a decrease in enamel thickness (bottom panels). D, quantitation of enamel thickness (mm) and enamel volume relative to total tooth volume in the mandibular incisors of 6- or 3-week-old p38αK14 and WT mice. ** indicates p < 0.01 by a two-tailed unpaired Student's t test. *, indicates p < 0.05. E, histogram plotting density on the x-axis versus the volume in voxels corresponding to density in mandibular incisors from 6-week-old p38αK14 and WT mice. The three peaks present correspond to soft tissue, dentin, and enamel densities from left to right. F, hematoxylin and eosin staining of 3-day-old mandibular incisors showing dissociation of soft tissue from dentin.
Next, the Vickers hardness test was performed to assess the biomechanical properties of the remaining enamel. As shown in Fig. 3A, the hardness of the enamel covering both the maxillary and mandibular incisors was significantly reduced in p38αK14 mice. Likewise, scanning electron microscopy confirmed the reduction in enamel thickness and demonstrated that, although the remaining enamel displays abnormal biomechanical properties, it shows a relatively normal ultrastructure (Fig. 3B). Additionally, we examined expression of ameloblast marker genes in mandibular incisors of p38αK14 mice using immunohistochemistry and in situ hybridization analyses. As expected, β4-integrin, amelogenin, and dentin sialophosphoprotein (DSPP) are expressed in WT ameloblasts. However, their expression levels were substantially reduced in p38αK14 ameloblasts (Fig. 3, C and D) (20).
FIGURE 3.

Defects in dental enamel production and ameloblast activity in p38αK14 mice. A, nanoindentation studies of incisors from 6-week-old p38αK14 and WT mice showing decreased enamel hardness, both by quantitative measure (left panels) and by visualization of the probe indentation site (right panels). ** indicates p < 0.01 by a two-tailed unpaired Student's t test; * indicates p < 0.05. VHN, Vickers hardness number. B, scanning electron microscopy analysis of mandibular incisors from 6-week-old p38αK14 and WT mice showing decreased thickness of the enamel layer (red arrows). Bar, 100 μm. C, immunohistochemical (IHC) staining for β4-integrin on mandibular incisors from 4-day-old WT and p38αK14 mice. D, in situ hybridization (ISH) for amelogenin and DSPP on mandibular incisors from 4-day-old WT and p38αK14 mice. The region of DSPP expression in ameloblasts is indicated with a red arrow. E, histogram plotting density on the x-axis versus the volume in voxels corresponding to that density in mandibular incisors from 4-week-old p38β−/− and WT mice. F, quantitation of enamel thickness and enamel volume relative to total tooth volume in the mandibular incisors of 4-week-old p38β−/− and WT mice.
Previously, both p38α and p38β were demonstrated to contribute to skeletal mineralization and osteoblast differentiation (11). Surprisingly, both tooth morphogenesis and quantitative measures of enamel production are normal in p38β−/− mice, suggesting that the contribution of p38α to tooth biology is highly selective (Fig. 3, E and F). Therefore, p38α activity is necessary for both the morphogenesis of dental cusps and effective enamel secretion.
MAP2K MKK6 but Not MAP2K MKK3 Contributes to the Formation of Dental Enamel
To determine the roles of the MAP2Ks upstream of p38, MKK3, and MKK6 in tooth formation, Mkk3−/−, Mkk6−/−, and Mkk3−/−Mkk6+/− mice were examined. Because Mkk3−/−Mkk6−/− mice are embryonic lethal, tooth phenotypes were analyzed in Mkk3−/−Mkk6+/− mice. In μCT analysis windowed to display only tooth enamel, Mkk6−/− mice showed relatively sparse and uneven enamel deposition most evident along the maxillary and mandibular incisors (Fig. 4A). This was confirmed by histogram analysis showing a reduced peak in the enamel density range (Fig. 4B). Accordingly, contouring of individual mandibular incisors demonstrated a decrease in both total enamel volume and enamel thickness (Fig. 4C). Similar to the phenotype of p38αK14 mice, scanning electron microscopy of mandibular incisors showed a decrease in the thickness of the enamel layer and an apparently normal ultrastructural enamel lattice pattern in Mkk6−/− mice (Fig. 4D). In contrast to these findings, both Mkk3−/− and Mkk3−/−Mkk6+/− mice showed no defects in dental enamel, suggesting that contribution of MKK6 to tooth development is highly selective (Fig. 4, E and F). Thus, MKK6-deficient mice display phenotypic defects consistent with a contribution to p38α activation in ameloblasts; however, the mild phenotype argues that other mechanisms of p38α activation are likely also involved.
FIGURE 4.

MKK6, but not MKK3, contributes to the formation of dental enamel. A, skulls of 3-week-old Mkk6−/− and WT mice were scanned by μCT. Displayed are three-dimensional reconstructions windowed so that only enamel densities are visible. B, histogram plotting density on the x-axis versus the volume in voxels corresponding to density in mandibular incisors from 3-week-old Mkk6−/− and WT mice. C, quantitation of enamel thickness (mm) and enamel volume relative to total tooth volume in the mandibular incisors of 3-week-old Mkk6−/− and WT mice. D, scanning electron microscopy analysis of Mkk6−/− and WT mandibular incisors, showing decreased enamel thickness in Mkk6−/− mice (red arrows). Bar, 100 μm. E, three-dimensional reconstruction of μCT scans of 5-week-old Mkk3−/− and WT mice, windowed to display only dental enamel. F, quantitation of enamel thickness (Th., mm) and enamel volume relative to total tooth volume in the mandibular incisors of 5-week-old Mkk3−/−, Mkk3−/−Mkk6+/−, and WT mice.
p38α Regulates Cell Proliferation in the Enamel Knot
Patterning of dental cusps occurs in a specialized region of developing teeth termed the enamel knot. Because teeth form at the interface between invaginated oral epithelium and the underlying mesenchyme, local variations in cell proliferation create “ripples” in this interface, and these ripples result in dental cusps (21). First, to examine embryonic tooth development in p38αK14 mice, tooth development was examined in E13.5, E15.5, and E18.5 mouse embryos (Fig. 5A). Consistent with the major role of p38α in tooth morphogenesis being restricted to participating in cusp formation at the enamel knot, defects in morphology were not detected in the bud stage of E13.5 embryos prior to formation of the enamel knot, and time points thereafter displayed impairment in the folding of the mesenchymal/epithelial interface characteristic of cusp formation. To focus on the role of p38α in the formation of dental cusps, activation of the p38 MAPK pathway was characterized in the primary enamel knot of E14.5 WT and p38αK14 embryos using immunohistochemistry for phospho-MKK3/6. As shown in Fig. 5B, activation of the p38 pathway occurs throughout the oral epithelium in the tooth bud. Because altered proliferation is an important mechanism for dental cusp morphogenesis, we next assessed cell proliferation in the primary enamel knot using immunohistochemistry for proliferating cell nuclear antigen (Fig. 5C). WT mice show an absence of cell proliferation corresponding to the enamel knot (21), whereas a corresponding region was not present in p38αK14 mice (indicated with arrows). Induction of the cyclin-dependent kinase inhibitor p21 has been reported to contribute to the halt in proliferation at the enamel knot (7). Consistent with immunohistochemistry for proliferating cell nuclear antigen, induction of p21 is impaired in p38αK14 mice (indicated with an arrow), providing a mechanism for the dysregulated proliferation in enamel knots lacking p38α (Fig. 5D).
FIGURE 5.
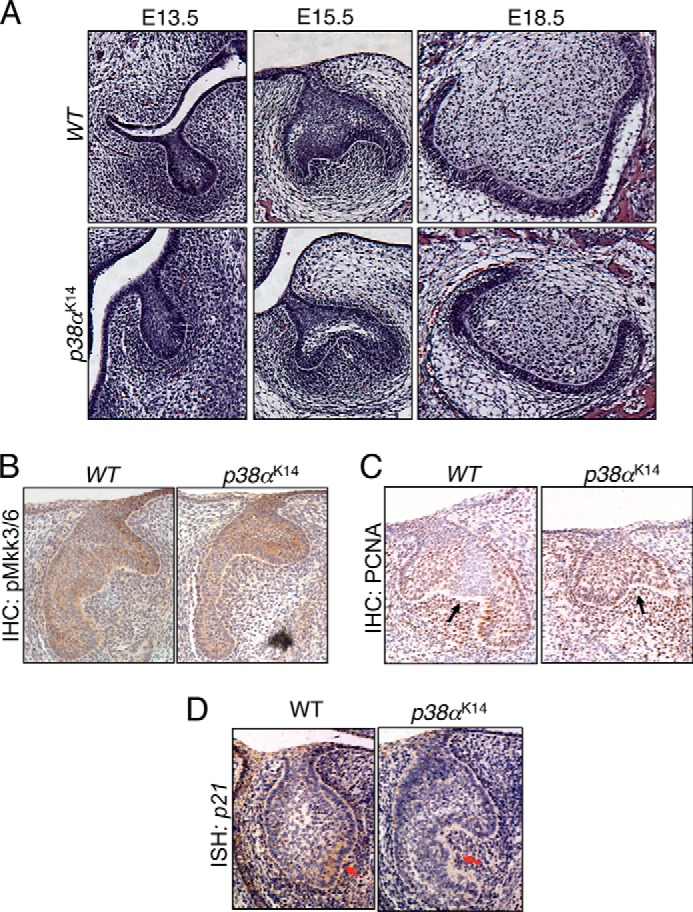
p38α acts in the enamel knot to influence dental cusp formation. A, coronal sections were cut from p38αK14 and WT embryos at the indicated ages. B–D, coronal sections were cut from E14.5 p38αK14 and WT embryos, and in situ hybridization (ISH) or immunohistochemistry (IHC) was performed as indicated. Arrows indicate cusp formation areas.
p38α Mediates BMP Signaling during Tooth Development
The defects in dental cusp formation and enamel secretion present in p38αK14 mice are very similar to those in mice with transgenic expression of the BMP inhibitor follistatin in dental epithelium (4, 6). Together with a previous report showing that p21 expression is directly regulated by BMPs in the enamel knot (7), this suggests that p38 may function primarily downstream of BMPs in the context of tooth development and enamel secretion. To directly examine BMP-induced activation of the p38 MAPK pathway in ameloblasts, the ameloblast-like cell line ALC (19) was stimulated with BMP2/7 at various time points, and the phosphorylation levels of p38 and MKK3/6 were examined by immunoblotting analysis (Fig. 6A). Phosphorylation levels of SMAD1/5, BMP receptor-regulated SMAD proteins, peaked at 15 min of stimulation and gradually decreased, whereas phosphorylation levels of MKK3/6 and p38 reached highest levels after 60 min of stimulation and were sustained thereafter. As expected, BMP2/7-induced phosphorylation of p38, but not MKK3/6, was blocked by the p38 inhibitor SB203580 (Fig. 6B).
FIGURE 6.

The p38 pathway transduces BMP signals in ameloblasts. A, ALC line was stimulated with BMP2/7 (100 ng/ml) for the indicated duration, and lysates were immunoblotted with the indicated antibodies. B, ALC line was pretreated with SB203580 (1 or 10 μm) for 1 h and then treated with BMP2/7 (100 ng/ml) for 30 min. Lysates were immunoblotted (IB) as indicated. C, after pretreatment with SB203580 (10 μm) for 1 h, ALC line was stimulated with BMP2/7 (100 ng/ml) for 3 h, and the relative levels of the indicated transcripts were determined by RT-PCR. Pretreatment with SB203580 resulted in a significant decrease for expression of amelogenin, ameloblastin, and p21 (p < 0.01, indicated with ** by a two-tailed Student's t test). D, ALC line was transfected with a luciferase reporter construct driven by the p21 promoter (p21-luc). After pretreatment with SB203580 for 30 min, cells were incubated with BMP2/7 for 48 h, and luciferase activity was measured. p21 luciferase activity was normalized to Renilla. Vehicle versus BMP, p < 0.01, indicted with **; p < 0.05, indicated with *, all by a two-tailed Student's t test. E and F, total RNAs were isolated from the ALC line expressing control vector or a constitutive active (CA) mutant of MKK6, and the effect on gene expression determined by RT-PCR (E). Alternatively, ALC line was transfected with p21-luc reporter gene and Renilla in the absence or presence of MKK6-CA and p21-luc activity was analyzed at 24 h after transfection (F). p21 luciferase activity was normalized to Renilla. ** indicates p < 0.01, and * indicates p < 0.05 by a two-tailed Student's t test. G, explants of embryonic oral epithelium isolated from E14.5 embryos were treated with vehicle, BMP2/7 alone, or BMP2/7 and SB203580 for 16 h, and whole mount in situ hybridization for p21 was performed. Shown are multiple representative images for each condition.
To examine the importance of the p38 MAPK pathway for BMP-induced gene expression in ameloblasts, the ALC line was incubated with BMP2/7 for 3 h in the presence or absence of SB203580, and gene induction was measured by RT-PCR analysis. BMP2/7 stimulation modestly induced expression of the enamel matrix components amelogenin and ameloblastin, and this was blocked by addition of SB203580 (Fig. 6C). This indicates that p38 activation is necessary for BMP-induced expression of amelogenin and ameloblastin. Additionally, BMP2/7-induced expression of p21 was also blocked by SB203580, which is consistent with the defective p21 expression in the enamel knot of p38αK14 mice (Fig. 5D). Likewise, p21 transcriptional activity was modestly increased by BMP2/7 stimulation in the ALC line, and this was inhibited by SB203580, indicating that the p38 MAPK pathway is required for BMP-induced expression and activation of p21 in ameloblasts (Fig. 6, C and D). To directly examine the contribution of p38 activation to induce these genes, a constitutively active MKK6 mutant (MKK6-CA) that enforced activation of the p38 pathway was stably expressed in the ALC line via lentivirus-mediated delivery. As expected, exogenous expression of MKK6-CA was sufficient to induce expression of amelogenin and ameloblastin (Fig. 6E). A similar effect of constitutively active MKK6 to induce transcriptional activation of p21 was also observed (Fig. 6F). Thus, p38 activation is essential for gene induction by BMPs in ameloblasts.
To demonstrate the relevance of this observation in a physiologic system, we conducted ex vivo culture of explants of embryonic dental epithelium isolated from E14.5 WT embryos. The explants were stimulated with BMP2/7 in the presence or absence of SB203580, and whole mount in situ staining for p21 was performed to assess BMP-induced expression of p21 (Fig. 6G). BMP2/7 stimulation induced p21 expression in the oral epithelium, and this was blocked by addition of SB203580. Thus, both in vivo and in vitro evidence demonstrates that p38 acts in oral epithelium to induce p21 expression.
SMAD4-dependent Pathways Are Dispensable for Induction of Ameloblastin, Amelogenin, and p21 in Vitro
Because stimulation with BMPs activates both MAPK and SMAD pathways, we examined whether the SMAD pathway is required for the ability of BMP2/7 to induce expression of these targets. To this end, SMAD4, which is classically required for SMAD signaling, was deleted in the ALC line via lentivirus-mediated delivery of murine Smad4 shRNA, although recent work has proven the existence of SMAD4-independent SMAD signaling pathways (Fig. 7A) (10). SMAD4 deficiency had no effect on the ability of BMP2/7 to induce transcriptional activation of p21 or expression of amelogenin or ameloblastin (Fig. 7, B and C). Thus, BMP-induced SMAD activation is likely to be dispensable for gene induction and p21 activation in ameloblasts.
FIGURE 7.
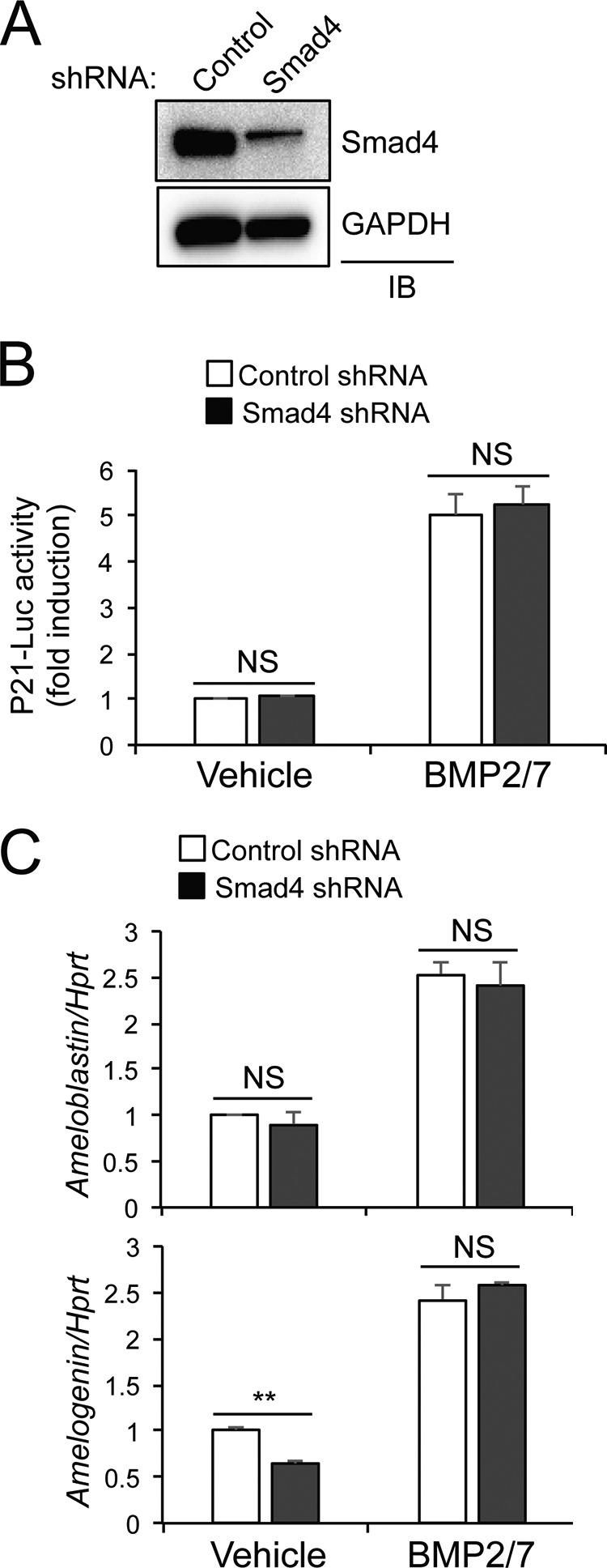
Contribution of SMAD4-dependent pathways to gene expression in ameloblasts. A, ALC line was infected with lentivirues expressing control or murine Smad4 shRNA. After puromycin selection, SMAD4 levels were determined by immunoblotting (IB) analysis. B and C, Smad4-sufficent or -deficient ALC line was transfected with p21-luc reporter gene and Renilla, and 24 h after transfection, cells were treated with BMP2/7 for 24 h. p21 luciferase activity was normalized to Renilla (B). Alternatively, total RNAs were isolated, and the effect on gene expression was determined by RT-PCR (C). ** indicates p < 0.01; * indicates p < 0.05 by a two-tailed Student's t test; NS indicates p > 0.05.
RhoA Mediates Activation of the p38 Pathway Downstream of BMP Signaling
Next, we sought to identify upstream signal transducers of the MKK6/p38α signaling axis in BMP signaling. Given that MAP3K TAK1 functions as a critical upstream activator of the MKK3/6-p38α/β MAPK signaling axis in long bones (11), we examined whether TAK1 plays a similar role upstream of p38 in ameloblasts in vivo by using Tak1K14 mice. Consistent with a previous study (22), these mice develop a lethal dermatitis within 2 weeks of age. Despite limiting analysis of their teeth to the first 2 weeks of life, Tak1K14 mice show no apparent defects in cusp morphogenesis (Fig. 8A). Thus, although TAK1 is able to activate the p38 MAPK pathway in long bones, it is dispensable for the ectodermal contribution to tooth development. A number of mice lacking other MAP3Ks, including Mekk2 (MAP3K2), Tpl2 (MAP3K8), and Taok3 (MAP3K18), were also obtained, and their tooth phenotypes were examined. Similar to Tak1K14 mice, no defects were found to display detectable by μCT in adult mice (data not shown), whereas Mlk3 (MAP3K11)-deficient mice display severe defects in dentin formation as we previously reported (23). Taken together, these findings demonstrate that the MAP3Ks mediating p38 activation in ameloblasts are distinct from those in other tissues, although further investigation will be needed to identify the exact genes responsible.
FIGURE 8.

RhoA mediates activation of the p38 pathway by BMP2/7 in ameloblasts. A, three-dimensional reconstruction of μCT scans from 2-week-old Tak1K14 and WT mice, showing comparable dental cusp morphology. B, ALC cells were infected with lentiviruses expressing control or murine RhoA shRNA. After puromycin selection, RhoA levels were measured by immunoblotting (IB) analysis. C, RhoA-sufficient or -deficient ALC cells were stimulated with BMP2/7 (100 ng/ml) for the indicated duration and immunoblotted with the indicated antibodies. D, RhoA-sufficient or -deficient ALC cells were stimulated with BMP2/7 (100 ng/ml) for 3 h, and RT-PCR was performed to measure gene expression. E, RhoA-sufficient or -deficient ALC cells were transfected with the p21-luc reporter gene and Renilla, and 24 h later cells were stimulated with BMP2/7 for 48 h. p21 luciferase activity was normalized to Renilla. ** indicates p < 0.01 by a two-tailed Student's t test.
Therefore, we examined whether a small G protein, which has been known to activate various MAP3Ks, might be required for activation of the p38 pathway in ameloblasts. In particular, we tested involvement of RhoA in ameloblast gene induction and p21 activity, given that it has been described to contribute to dental enamel formation (24, 25). Intriguingly, knockdown of RhoA results in a significant reduction in BMP-induced phosphorylation of both MKK3/6-p38 and SMAD1/5 in the ALC line (Fig. 8, B and C). Likewise, BMP2/7-induced expression of ameloblastin, amelogenin, and p21 was reduced in RhoA-deficient ALC line (Fig. 8D). BMP2/7-induced transcriptional activation of p21 was also ablated by RhoA knockdown in the ALC line (Fig. 8E).
DISCUSSION
Here we demonstrate that p38α MAPK is a crucial mediator of the effects of BMPs in tooth development and enamel secretion. In these contexts, p38α is required for expression of p21 in embryonic enamel knot and for expression of amelogenin and β4-integrin in ameloblasts. Biochemical analysis using ALC ameloblast-like cell line showed that the RhoA-MKK6-p38α signaling axis is crucial for induction of p21, amelogenin, and ameloblastin genes and p21 transcriptional activation by BMP2/7, and in this in vitro system, the p38 pathway plays a dominant role over SMAD4-dependent pathway in expression of these targets and p21 activation. However, this conclusion is subject to the limitations of the existence of SMAD4-independent SMAD signaling pathways and the in vitro knockdown system used.
The role of p38α in the formation of ectodermal appendages is not limited to teeth, because p38αK14 mice display defects in the development of hair, including loss of coat hair and a complete absence of awl hairs, one of the morphologic classes of pellage hairs in mice up to 3 weeks of age. The meibomian glands, footpad glands, and nails in p38αK14 mice were morphologically intact. Thus, p38α selectively mediates the development of a subset of ectodermal appendages (data not shown).
Consistent with the in vivo finding of defective tooth cusp morphogenesis in the absence of p38α, in vitro models demonstrate that the p38 MAPK pathway mediates expression of p21, amelogenin, and ameloblastin in response to BMP2/7. This finding is also consistent with reports demonstrating that p21 expression is regulated by the p38 MAPK pathway in other tissues (26). Interestingly, in the context of gamma radiation-induced arrest in the cell cycle, the effect of p38 on p21 mRNA levels is post-transcriptional, mediated via phosphorylation and accumulation of the mRNA stability factor HuR (27). This raises the possibility that a similar mechanism may apply to p38 regulation of p21 expression in the enamel knot.
In addition to mice lacking p38α in oral epithelium, the dentition of mice lacking p38β was also examined, and no defects were detected. Thus, whereas both p38α and p38β contribute to osteoblast differentiation and long bone mineralization, only p38α functions in ameloblasts. Consistent with the lack of a dental phenotype in p38β−/− mice, immunohistochemical staining with an antibody recognizing pan isoform phospho-p38 is nearly absent in p38αK14 mice, arguing that p38α is the predominant p38 isoform active in ameloblasts.
The finding that expression of β4-integrin, amelogenin and DSPP in ameloblasts are all defective in the absence of p38α indicates that the functional defects in adult p38αK14 ameloblasts are likely to be multifactorial, involving defective expression of both enamel matrix genes and adhesion molecules. These defects, along with the finding that the p38 MAPK pathway is activated in both neonatal and adult ameloblasts, suggest that the requirement for p38 activation in ameloblasts is continual and that the reduction in enamel secretion in p38αK14 mice is unlikely to be secondary to developmental effects. Notably, β4-intergrin deficiency in either humans or mice results in impaired epidermal basement membrane adherence, absence of hemidesmosomes, and the clinical syndrome of epidermolysis bullosa (28, 29). Deficient expression of molecules such as β4-integrin involved in basement membrane adhesion may explain the observation that the ameloblast cell layer is disrupted and displays intussusception relative to the enamel surface.
Additionally, the contribution of MKK3 and MKK6 MAP2Ks upstream of p38α to tooth biology was examined. Because both MKK3 and MKK6 can mediate the two activation loop phosphorylation events required for p38α activation, they are believed to be partially redundant at the biochemical level (30). However, in the setting of skeletal mineralization, both MKK3 and MKK6 contributed to long bone mineralization, whereas only MKK3 contributed significantly to calvarial mineralization. This implies that two MAP2Ks can have partially overlapping biochemical roles but distinct functions in vivo (11). In our analysis of dentition, Mkk6−/− but not Mkk3−/− mice displayed a mild defect in enamel production, and both Mkk3−/− and Mkk6−/− mice had normal dental cusp formation and coat hair development. Because only MKK6 but not MKK3 has a role in dental enamel production, and the phenotype of Mkk6−/− is substantially less severe than that of p38αK14 mice, one may propose that another MAP2K plays a partially redundant role in enamel production. One obvious candidate for this is MKK4, which has been observed to contribute to residual p38 activation by UV light in Mkk3−/−Mkk6−/− embryonic fibroblasts (31). If MKK4 acts in ameloblasts to regulate enamel production, this would be the first observation of a physiologic role for MKK4 in p38 activation.
Although ectodysplasins are also associated with morphogenesis of enamel knot and development of certain types of pellage hair, several lines of evidence argue that p38α does mediate their effects in vivo (32). Defects in EDA signaling result in impaired morphogenesis of lacrimal and meibomian glands, but they are intact in p38αK14 mice (33). Similarly, the transcriptional hallmark of impaired EDA signaling in the enamel knot is dysregulated expression of Shh (sonic hedgehog), not p21 as we observe in p38αK14 mice (34). This apparent lack of a role for p38α in ectodysplasin signaling may correlate with in vitro observations that, contrary to many other TNF family receptors, overexpression of the ectodysplasin receptor EDAR was not associated with induction of JNK or p38 MAPK activity in vitro (35).
Lastly, whereas TAK1 is necessary for the activation of p38 by BMPs in osteoblast and chondrocyte differentiation (9, 11), TAK1K14 mice do not recapitulate the phenotype of p38αK14 mice. A similar lack of effect was observed in mice lacking the MAP3Ks Tpl2, Mekk2, or Taok3. Mice lacking the MAP3K Mlk3 have a profound defect in dentin production and corresponding spontaneous tooth fracture, which complicates assessment of ameloblast function (23). However, Mlk3-deficient mice clearly do not display the defects in tooth cusp formation seen in p38αK14 mice. This argues that substantial differences exist between the means of proximal activation of the p38 MAPK pathway in the oral epithelium versus other tissues. This discordance between the BMP signaling pathways in ameloblasts and those observed in other contexts suggests that an alternative pathway to the well studied SMAD or TAK1-dependent MAPK pathways is involved. Supporting this conclusion, RhoA was found to contribute to both SMAD and p38 activation in ameloblasts. This raises the possibility that the RhoA/ROCK pathway is mediating activation of the p38 pathway in this context (36). Although mice expressing a dominant negative RhoA transgene have defects in dental enamel, it is unclear whether they recapitulate the same defects in morphogenesis as seen in p38αK14 mice (24, 25). Further in vivo studies with RhoA conditional mice and mice deficient in ROCK1 and 2 are necessary to further substantiate that RhoA acts upstream of the p38 pathway in the oral epithelium.
Acknowledgment
We thank Xiu-Ping Wang for sharing reagents and for insightful advice.
This work was supported, in whole or in part, by National Institutes of Health Grant AI074957 (to J. M. P.). L. H. G. is a member of the board of directors of and holds equity in Bristol-Myers Squibb.
- BMP
- bone morphogenic proteins
- En
- embryonic day n
- ALC
- ameloblast-like cell
- DSPP
- dentin sialophosphoprotein
- μCT
- micro-computed tomography.
REFERENCES
- 1. Abdollah S., Macías-Silva M., Tsukazaki T., Hayashi H., Attisano L., Wrana J. L. (1997) TbetaRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J. Biol. Chem. 272, 27678–27685 [DOI] [PubMed] [Google Scholar]
- 2. Jernvall J., Thesleff I. (2012) Tooth shape formation and tooth renewal: evolving with the same signals. Development 139, 3487–3497 [DOI] [PubMed] [Google Scholar]
- 3. Mikkola M. L. (2008) TNF superfamily in skin appendage development. Cytokine Growth Factor Rev. 19, 219–230 [DOI] [PubMed] [Google Scholar]
- 4. Wang X. P., Suomalainen M., Jorgez C. J., Matzuk M. M., Wankell M., Werner S., Thesleff I. (2004) Modulation of activin/bone morphogenetic protein signaling by follistatin is required for the morphogenesis of mouse molar teeth. Dev. Dyn. 231, 98–108 [DOI] [PubMed] [Google Scholar]
- 5. Laurikkala J., Kassai Y., Pakkasjärvi L., Thesleff I., Itoh N. (2003) Identification of a secreted BMP antagonist, ectodin, integrating BMP, FGF, and SHH signals from the tooth enamel knot. Dev. Biol. 264, 91–105 [DOI] [PubMed] [Google Scholar]
- 6. Wang X. P., Suomalainen M., Jorgez C. J., Matzuk M. M., Werner S., Thesleff I. (2004) Follistatin regulates enamel patterning in mouse incisors by asymmetrically inhibiting BMP signaling and ameloblast differentiation. Dev. Cell 7, 719–730 [DOI] [PubMed] [Google Scholar]
- 7. Jernvall J., Aberg T., Kettunen P., Keränen S., Thesleff I. (1998) The life history of an embryonic signaling center: BMP-4 induces p21 and is associated with apoptosis in the mouse tooth enamel knot. Development 125, 161–169 [DOI] [PubMed] [Google Scholar]
- 8. Deng C., Zhang P., Harper J. W., Elledge S. J., Leder P. (1995) Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82, 675–684 [DOI] [PubMed] [Google Scholar]
- 9. Shim J. H., Greenblatt M. B., Xie M., Schneider M. D., Zou W., Zhai B., Gygi S., Glimcher L. H. (2009) TAK1 is an essential regulator of BMP signalling in cartilage. EMBO J. 28, 2028–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Retting K. N., Song B., Yoon B. S., Lyons K. M. (2009) BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 136, 1093–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Greenblatt M. B., Shim J. H., Zou W., Sitara D., Schweitzer M., Hu D., Lotinun S., Sano Y., Baron R., Park J. M., Arthur S., Xie M., Schneider M. D., Zhai B., Gygi S., Davis R., Glimcher L. H. (2010) The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J. Clin. Invest. 120, 2457–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu X., Han J., Ito Y., Bringas P., Jr., Deng C., Chai Y. (2008) Ectodermal Smad4 and p38 MAPK are functionally redundant in mediating TGF-beta/BMP signaling during tooth and palate development. Dev. Cell 15, 322–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shim J. H., Greenblatt M. B., Zou W., Huang Z., Wein M. N., Brady N., Hu D., Charron J., Brodkin H. R., Petsko G. A., Zaller D., Zhai B., Gygi S., Glimcher L. H., Jones D. C. (2013) Schnurri-3 regulates ERK downstream of WNT signaling in osteoblasts. J. Clin. Invest. 123, 4010–4022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim C., Sano Y., Todorova K., Carlson B. A., Arpa L., Celada A., Lawrence T., Otsu K., Brissette J. L., Arthur J. S., Park J. M. (2008) The kinase p38 alpha serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression. Nat. Immunol. 9, 1019–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nishida K., Yamaguchi O., Hirotani S., Hikoso S., Higuchi Y., Watanabe T., Takeda T., Osuka S., Morita T., Kondoh G., Uno Y., Kashiwase K., Taniike M., Nakai A., Matsumura Y., Miyazaki J., Sudo T., Hongo K., Kusakari Y., Kurihara S., Chien K. R., Takeda J., Hori M., Otsu K. (2004) p38alpha mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol. Cell. Biol. 24, 10611–10620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wysk M., Yang D. D., Lu H. T., Flavell R. A., Davis R. J. (1999) Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for tumor necrosis factor-induced cytokine expression. Proc. Natl. Acad. Sci. U.S.A. 96, 3763–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tanaka N., Kamanaka M., Enslen H., Dong C., Wysk M., Davis R. J., Flavell R. A. (2002) Differential involvement of p38 mitogen-activated protein kinase kinases MKK3 and MKK6 in T-cell apoptosis. EMBO Reports 3, 785–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bartlett J. D., Dobeck J. M., Tye C. E., Perez-Moreno M., Stokes N., Reynolds A. B., Fuchs E., Skobe Z. (2010) Targeted p120-catenin ablation disrupts dental enamel development. PLoS One 5, e12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakata A., Kameda T., Nagai H., Ikegami K., Duan Y., Terada K., Sugiyama T. (2003) Establishment and characterization of a spontaneously immortalized mouse ameloblast-lineage cell line. Biochem. Biophys. Res. Commun. 308, 834–839 [DOI] [PubMed] [Google Scholar]
- 20. D'Souza R. N., Cavender A., Sunavala G., Alvarez J., Ohshima T., Kulkarni A. B., MacDougall M. (1997) Gene expression patterns of murine dentin matrix protein 1 (Dmp1) and dentin sialophosphoprotein (DSPP) suggest distinct developmental functions in vivo. J. Bone Miner. Res. 12, 2040–2049 [DOI] [PubMed] [Google Scholar]
- 21. Obara N., Lesot H. (2007) Asymmetrical growth, differential cell proliferation, and dynamic cell rearrangement underlie epithelial morphogenesis in mouse molar development. Cell Tissue Res. 330, 461–473 [DOI] [PubMed] [Google Scholar]
- 22. Omori E., Matsumoto K., Sanjo H., Sato S., Akira S., Smart R. C., Ninomiya-Tsuji J. (2006) TAK1 is a master regulator of epidermal homeostasis involving skin inflammation and apoptosis. J. Biol. Chem. 281, 19610–19617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zou W., Greenblatt M. B., Shim J. H., Kant S., Zhai B., Lotinun S., Brady N., Hu D. Z., Gygi S. P., Baron R., Davis R. J., Jones D., Glimcher L. H. (2011) MLK3 regulates bone development downstream of the faciogenital dysplasia protein FGD1 in mice. J. Clin. Invest. 121, 4383–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xue H., Li Y., Everett E. T., Ryan K., Peng L., Porecha R., Yan Y., Lucchese A. M., Kuehl M. A., Pugach M. K., Bouchard J., Gibson C. W. (2013) Ameloblasts require active RhoA to generate normal dental enamel. Eur. J. Oral Sci. 121, 293–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Y., Pugach M. K., Kuehl M. A., Peng L., Bouchard J., Hwang S. Y., Gibson C. W. (2011) Dental enamel structure is altered by expression of dominant negative RhoA in ameloblasts. Cells Tissues Organs 194, 227–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lavelle D., DeSimone J., Hankewych M., Kousnetzova T., Chen Y. H. (2003) Decitabine induces cell cycle arrest at the G1 phase via p21(WAF1) and the G2/M phase via the p38 MAP kinase pathway. Leuk. Res. 27, 999–1007 [DOI] [PubMed] [Google Scholar]
- 27. Lafarga V., Cuadrado A., Lopez de Silanes I., Bengoechea R., Fernandez-Capetillo O., Nebreda A. R. (2009) p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G1/S checkpoint. Mol. Cell. Biol. 29, 4341–4351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van der Neut R., Krimpenfort P., Calafat J., Niessen C. M., Sonnenberg A. (1996) Epithelial detachment due to absence of hemidesmosomes in integrin beta 4 null mice. Nat. Genet. 13, 366–369 [DOI] [PubMed] [Google Scholar]
- 29. Vidal F., Aberdam D., Miquel C., Christiano A. M., Pulkkinen L., Uitto J., Ortonne J. P., Meneguzzi G. (1995) Integrin beta 4 mutations associated with junctional epidermolysis bullosa with pyloric atresia. Nat. Genet. 10, 229–234 [DOI] [PubMed] [Google Scholar]
- 30. Zarubin T., Han J. (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res. 15, 11–18 [DOI] [PubMed] [Google Scholar]
- 31. Brancho D., Tanaka N., Jaeschke A., Ventura J. J., Kelkar N., Tanaka Y., Kyuuma M., Takeshita T., Flavell R. A., Davis R. J. (2003) Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 17, 1969–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kere J., Srivastava A. K., Montonen O., Zonana J., Thomas N., Ferguson B., Munoz F., Morgan D., Clarke A., Baybayan P., Chen E. Y., Ezer S., Saarialho-Kere U., de la Chapelle A., Schlessinger D. (1996) X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat. Genet. 13, 409–416 [DOI] [PubMed] [Google Scholar]
- 33. Grüneberg H. (1971) The glandular aspects of the tabby syndrome in the mouse. J. Embryol. Exp. Morphol. 25, 1–19 [PubMed] [Google Scholar]
- 34. Tucker A. S., Headon D. J., Schneider P., Ferguson B. M., Overbeek P., Tschopp J., Sharpe P. T. (2000) Edar/Eda interactions regulate enamel knot formation in tooth morphogenesis. Development 127, 4691–4700 [DOI] [PubMed] [Google Scholar]
- 35. Koppinen P., Pispa J., Laurikkala J., Thesleff I., Mikkola M. L. (2001) Signaling and subcellular localization of the TNF receptor Edar. Exp. Cell Res. 269, 180–192 [DOI] [PubMed] [Google Scholar]
- 36. Wang Y. K., Yu X., Cohen D. M., Wozniak M. A., Yang M. T., Gao L., Eyckmans J., Chen C. S. (2012) Bone morphogenetic protein-2-induced signaling and osteogenesis is regulated by cell shape, RhoA/ROCK, and cytoskeletal tension. Stem Cells Dev. 21, 1176–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]