

Background: MEF2C is an important regulator of many developmental programs.
Results: Alternative splicing of the α exon of MEF2C regulates myogenesis. Loss of SRPK3 in rhabdomyosarcoma cells inhibits this splicing and blocks differentiation.
Conclusion: MEF2Cα2 promotes myogenesis, and restoration of MEF2Cα2 in rhabdomyosarcoma cells inhibits growth.
Significance: Defining the function and deregulation of MEF2Cα2 enhances the understanding of normal myogenesis and RMS tumorigenesis.
Keywords: Alternative Splicing, Histone Deacetylase (HDAC), Myogenesis, Rhabdomyosarcoma (RMS), Tumor Cell Biology, Tumor Immunology, MEF2C, SRPK3
Abstract
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children. Many cellular disruptions contribute to the progression of this pediatric cancer, including aberrant alternative splicing. The MEF2 family of transcription factors regulates many developmental programs, including myogenesis. MEF2 gene transcripts are subject to alternate splicing to generate protein isoforms with divergent functions. We found that MEF2Cα1 was the ubiquitously expressed isoform that exhibited no myogenic activity and that MEF2Cα2, the muscle-specific MEF2C isoform, was required for efficient differentiation. We showed that exon α in MEF2C was aberrantly alternatively spliced in RMS cells, with the ratio of α2/α1 highly down-regulated in RMS cells compared with normal myoblasts. Compared with MEF2Cα2, MEF2Cα1 interacted more strongly with and recruited HDAC5 to myogenic gene promoters to repress muscle-specific genes. Overexpression of the MEF2Cα2 isoform in RMS cells increased myogenic activity and promoted differentiation in RMS cells. We also identified a serine protein kinase, SRPK3, that was down-regulated in RMS cells and found that expression of SRPK3 promoted the splicing of the MEF2Cα2 isoform and induced differentiation. Restoration of either MEF2Cα2 or SPRK3 inhibited both proliferation and anchorage-independent growth of RMS cells. Together, our findings indicate that the alternative splicing of MEF2C plays an important role in normal myogenesis and RMS development. An improved understanding of alternative splicing events in RMS cells will potentially reveal novel therapeutic targets for RMS treatment.
Introduction
The myocyte enhancer factor 2 (MEF2) is a regulator of many developmental programs, including myogenesis (1). MEF2 is encoded by four vertebrate genes that encode MEF2A, MEF2B, MEF2C, and MEF2D. The MEF2 family is expressed in distinct but overlapping temporal and spatial expression patterns in the embryo and adult (2). Both MEF2C and MEF2D are implicated in myogenesis (3, 4), which is controlled by the concerted activity of the myogenic regulatory factors (MRFs),2 a group of four highly related basic helix loop helix transcription factors composed of Myf5, MyoD, Myf6, and myogenin (5). MEF2 factors alone do not possess myogenic activity but work in combination with the MRFs to drive the myogenic differentiation program (6).
MEF2 proteins control differentiation, proliferation, survival, and apoptosis in a wide range of cell types. The N terminus of the MEF2 proteins contains a highly conserved MADS box and an immediately adjacent motif termed the MEF2 domain. Together, these motifs mediate dimerization, DNA binding, and cofactor interactions (7). The C terminus of the MEF2 proteins is highly divergent among the family members and functions as the transcriptional activation domain. MEF2 proteins function as end points for multiple signaling pathways and confer a signal responsiveness to downstream target genes. MAP kinase pathways are known to converge on MEF2 (8, 9), resulting in a phosphorylation of the transcriptional activation domain of MEF2, which augments its transcriptional activity. Calcium signaling pathways also modulate MEF2 activity through multiple mechanisms (10–13). The activity of MEF2 is tightly controlled by class II HDACs, which bind to the MADS domain and promote the formation of multiprotein repressive complexes on MEF2-dependent genes (14). The phosphorylation of class II HDACs is mediated by calcium-regulated protein kinases, which promote the nuclear-cytoplasmic shuttling of the HDACs and subsequent activation of MEF2C (14, 15).
Each of the MEF2 genes are subject to extensive alternative splicing. MEF2C contains three alternative exons: the mutually exclusive exons α1/α2, the skipping/inclusion exon β, and the 3′ splice site region γ. The α1 domain is expressed ubiquitously, whereas the α2 domain is strongly expressed in skeletal muscle (16). The function of the α domain is unknown, although it has been shown that isoforms entirely lacking the α domain have enhanced activity (17). Inclusion of the β exon has been described in neural cells (16, 18), and the presence of the β exon in MEF2C has been found to strongly activate MEF2C-responsive reporters (19). The γ domain, generated by alternative splice site acceptors, has an inhibitory effect on the activity of MEF2C, and isoforms lacking this domain better synergize with MyoD (20). The use of alternative isoforms in skeletal muscle differentiation has been shown recently for MEF2D, which promotes late muscle differentiation through the use of alternative isoforms and generates a muscle-specific MEF2Dα2 isoform (21) that binds to the coactivator ASH2L and is resistant to phosphorylation by PKA and association with HDACs (22).
Rhabdomyosarcoma (RMS) is a highly malignant tumor that is the most common form of soft tissue tumors in children. It is thought to arise as a consequence of myogenic precursors failing to differentiate into normal muscle (23). There are two major histological categories of RMS, the embryonal RMS and alveolar (ARMS) subtypes. The more common form of the disease is the embryonal RMS subtype. ARMS, the more aggressive form of RMS, is characterized by chimeric transcripts that fuse the 5′ DNA binding domain of PAX3 or PAX7, respectively, to the transactivation domain of a forkhead transcription factor, creating novel PAX3/7-FOXO1 fusion proteins (24, 25).
Rhabdomyosarcoma tumors express the myogenic regulatory factors, but the MRFs are unable to promote differentiation (26). Indeed, MyoD and myogenin are used as diagnostic markers for RMS because they are expressed in almost every RMS tumor, including both major histological subtypes, embryonal RMS and ARMS (27). Many blocks to differentiation have been described and were the subject of a recent review (26). Exogenous expression of MEF2C (28) or MEF2D (29) can promote differentiation in RMS cells.
We showed that the muscle-specific MEF2C isoform (MEF2Cα2) was required for efficient differentiation of skeletal muscle cells and that this isoform was highly down-regulated in RMS cells. MEF2C isoforms containing the α2 exon have potent myogenic activity, as assayed by muscle-specific gene reporters, muscle-specific gene expression, and myotube formation, whereas isoforms containing α1 or lacking the α domain did not. Despite the robust expression of MEF2Cα1 in RMS cells, restoration of the MEF2Cα2 isoform promoted RMS differentiation and myotube formation. The MEF2Cα1 isoform had an enhanced association with HDAC5, which resulted in enhanced recruitment of class II HDACs to target promoters in the presence of MEF2Cα1. We found that the alterative splicing of the α1/α2 exon of MEF2C was controlled by the protein kinase SRPK3, which is specific for the SR (serine/arginine-rich domain) family of transcription factors, including the splicing factor ASF. We showed that SRPK3 was down-regulated in RMS cells. Exogenous expression of SRPK3 in RMS promoted the splicing of the MEF2Cα2 isoform, induced expression of muscle-specific genes, and drove the formation of myotubes. Exogenous expression of MEF2Cα2 or SRPK3 inhibited the proliferation and anchorage-independent growth of RMS cells.
EXPERIMENTAL PROCEDURES
Cell Culture
RD cells (ATCC), SJRH30 (RH30) cells (ATCC), C2C12 myoblasts (ATCC), 10T1/2 cells (ATCC), and HEK293 cells (ATCC) were grown in DMEM supplemented with 10% fetal bovine serum (Hyclone) according to standard protocols. To induce differentiation of C2C12 myoblasts into myotubes, cells were grown to 70% confluence and the medium switched to DMEM supplemented with 2% horse serum (Hyclone). C2C12 cells were grown in differentiation medium for the number of days indicated in each experiment.
Cloning
Murine Mef2Cα2 (mMef2Cα2) and Srpk3 were PCR-amplified from cDNA reverse-transcribed from RNA isolated from C2C12 cells differentiated for 4 days. Human MEF2C isoforms (hMEF2C) were PCR-amplified from cDNA generated from RNA isolated from human myoblasts (a gift from Denis Guttridge, Ohio State University), RH30 cells, or HEK293 cells. A common primer set, MEF2C TOPO forward (5′ ATGGGGAGAAAAAAGATTCAGA 3′) and MEF2C TOPO reverse (5′ TCATGTTGCCCATCCTTCA 3′), was used to amplify both mMef2C and hMEF2C. Each of the PCR-amplified fragments was cloned into the pEF6/V5 His TOPO TA expression vector, and the clones were confirmed by sequencing.
Western Blot Analysis
Cell extracts were made by lysing PBS-washed cell pellets in radioimmune precipitation assay buffer supplemented with protease inhibitors (Complete protease inhibitor, Roche Diagnostics). Following incubation on ice, clear lysates were obtained by centrifugation. Protein concentrations were determined by Bradford assay (Bio-Rad). For each sample, 30 μg of protein was loaded on each gel. Proteins were transferred onto a PVDF membrane using a tank blotter (Bio-Rad). The membranes were then blocked with 5% milk in 1× Tris-buffered saline plus Tween 20 (TBST) and incubated with primary antibody overnight at 4 °C. Membranes were then washed with 1× TBST before incubation with the corresponding secondary antibody. Membranes were washed again with 1× TBST, incubated with chemiluminescent substrate according to the protocol of the manufacturer (SuperSignal, Pierce), and visualized by autoradiography. The antibodies used included anti-MEF2C (D80C1, Cell Signaling Technology), anti-HDAC5 (Cell Signaling Technology), anti-V5 (Rockland), anti-MHC (MF-20, Developmental Studies Hybridoma Bank), and anti-GAPDH (Millipore).
Gene Expression Analysis
RNA was isolated from cells by TRIzol extractions (Invitrogen). Following treatment with DNase (Promega), 2 μg of total RNA was reversed-transcribed with MultiScribeTM MuLV reverse transcriptase (Applied Biosystems). cDNA equivalent to 40 ng was used for quantitative PCR amplification (Applied Biosystems) with SYBR Green PCR master mix (Applied Biosystems). Samples to which no reverse transcriptase was added were included for each RNA sample. The relative levels of expression of genes were normalized according to those of hypoxanthine-guanine phosphoribosyltransferase. qPCR data were calculated using the comparative Ct method (Applied Biosystems). Standard deviations from the mean of the [Δ] Ct values were calculated from three independent RNA samples. Primers corresponding to the indicated genes were as described previously (30). Where possible, intron-spanning primers were used. All quantitative PCRs were performed in triplicate, and three independent RNA samples were assayed for each time point. For measurements of relative gene expression (-fold change), a -fold change was calculated for each sample pair by dividing the mRNA expression values of each sample pair. Each experimental -fold change was then normalized to the -fold change observed at hypoxanthine-guanine phosphoribosyltransferase.
Chromatin Immunoprecipitation
ChIP assays were performed and quantified as described previously (31) with the following modifications. 1 × 107 cells were used for each immunoprecipitation, and protein A-agarose beads (Invitrogen) were used to immunoprecipitate the antibody-antigen complexes. The following antibodies were used: HDAC5 (Cell Signaling Technology), HDAC4 (Cell Signaling Technology), and rabbit IgG (Santa Cruz Biotechnology) as a nonspecific control. Primers corresponding to the LMOD2 and CDKN1A promoters were as described previously (32). The real-time PCR was performed in triplicate. Values of [Δ][Δ] Ct were calculated using the following formula on the basis of the comparative Ct method: Ct, template (antibody) − Ct, template (IgG) = [Δ] Ct. Fold enrichments were determined using the formula: 2−[Δ]Ct. (experimental)/2 −[Δ] Ct (reference, CHR19). The standard error from the mean was calculated from replicate [Δ][Δ] Ct values obtained from at least three individual experiments.
Cell Transfections and Luciferase Assays
Cells were transfected with calcium phosphate according to standard protocols. The plasmids pEF6-mMef2Cα2, β−, γ+; pEF6-hMEF2Cα1, β−, γ+; pEF6-hMEF2Cα1, β−, γ−; pEF6-hMEF2Cα2, β−, γ−; pEF6-hMEF2Cα2, β−, γ+; and pEF6-hMEF2Cα−, β−, γ+ were used for expressing different isoforms of mMef2C and hMEF2C. pEF6-SRPK3 was used to express SRPK3. The plasmid pEMCIIs (provided by Andrew Lassar, Harvard Medical School) was used for expressing MyoD. Luciferase activity was determined using the Dual-Luciferase reporter assay system (Promega). RH30 or RD cells were seeded at a density of 5 × 103 cells/well in 96-well plates and transfected with 0.4 μg of DNA. Transfections were normalized to Renilla luciferase. Transfections were performed in triplicate, and all data sets were repeated at least twice.
Stable Cell Lines
Stable C2C12, RD, and RH30 cell lines overexpressing exogenous MEF2C or SRPK3 were constructed by transfecting cells with linearized pEF-V5 His vector (empty vector), linearized pEF-MEF2C, or linearized pEF-SRPK3 and selecting for blasticidin (10 μg/ml)-resistant colonies. Murine clones of Mef2Cα1 and Mef2Cα2 were used in murine cell lines, and human clones were used in human cell lines. Individual clones were isolated and propagated.
Immunohistochemistry
Cells were grown on coverslips, fixed with paraformaldehyde, incubated with goat serum supplemented with 1.0% Nonidet P-40 for 1 h, and washed with PBS. Primary antibodies against myosin heavy chain (1:100, MF20, Developmental Studies Hybridoma Bank) were incubated for 2 h at room temperature, washed with PBS, and detected by Alexa Fluor 488 goat anti-mouse antibody (1:500, Invitrogen). Cell nuclei were then stained by incubating with DAPI (1 μm, Invitrogen) for 5 min.
Proliferation
Cells were seeded in a 6-well plate at 6 × 104 cells/well and harvested every 2 days for cell counts with a hemocytometer. All counts were performed in triplicate, and individual experiments were repeated three times.
Soft Agar Assay
Soft agar assays were carried out in 60-mm dishes in which 2 ml of 0.7% Noble agar (USB) in 1× DMEM with 10% FBS was overlaid with 2 ml of 0.35% agar in 1× DMEM with 10% FBS containing 3 × 105 cells. RD and RH30 cells transected with pEF6 V5 His(vector), MEF2Cα2, and SRPK3 were grown to 70% confluence, trypsinized, and dispersed. Cells of each clone were plated in triplicate. 1 ml of culture medium was added to the top of each plate every 5 days, and cells were grown at 37 °C for 30 days. The plates were stained with 1 ml of 0.05% crystal violet (Fisher) for >1 h, and colonies were counted using a dissection microscope.
Statistics
qPCR data are presented as means ± S.D. Statistical comparisons were performed using unpaired two-tailed Student's t tests with a probability value of <0.05 taken to indicate significance.
RESULTS
The Muscle-specific α2 Exon of MEF2C Is Not Expressed in RMS Cells
To understand the blocks to differentiation in RMS cells, we undertook an analysis of the MEF2 family in RMS. During the course of this work, we found that both RD and RH30 cells highly expressed MEF2C (29), although MEF2C has also been reported to be down-regulated in RD cells (28). MEF2C has been shown to play an important role in myogenesis, and MEF2C is subject to alternative splicing by exclusion/inclusion of exon α1/2, exon β, and exon γ (Fig. 1A). Exon β has been reported to enhance MEF2C activity, whereas exon γ plays an inhibitory role. However, the function of the mutually exclusive exons α1 and α2 has not yet been characterized. To characterize the function of the MEF2C isoforms, we cloned MEF2C from RH30 cells, human myoblasts, C2C12 cells, and HEK293 cells. The isoforms recovered from each cell type are shown in Fig. 1B. As observed previously (16), the muscle-specific α2 exon was only found in mRNA from C2C12 cells and human myoblasts. The transcripts from C2C12 cells each contained the inhibitory γ domain, whereas human myoblast RNA produced transcripts with or without the γ domain. Both RD and RH30 cells contained the α1 exon with or without the γ domain. HEK293 cells expressed isoforms either with the α1 domain or lacking the α domain entirely. The transcripts containing the α1 domain lacked the γ domain, and the transcripts without the α domain contained the γ domain. Consistent with prior analyses that identified the β exon exclusively in neuronal tissue (16, 18), we identified no transcripts that contained the β domain from any of the cell types in our study.
FIGURE 1.

MEF2C isoforms in muscle and RMS. A, schematic of the MEF2C isoforms. The sequences of the exons are indicated below. m, murine sequence; h, human sequence. Amino acids that differ among the species are shown in red. B, MEF2C isoforms identified in the indicated cell lines. The number beside each isoform indicates the number of individual isoform clones identified/total number of clones recovered.
We sought to verify our results using RT-PCR to detect the expression of exons α and β by exon-specific primers in normal muscle and RMS cells. The location of the primers is shown in Fig. 2A. Consistent with the results shown in Fig. 1, we found that the MEF2Cα1 exon was ubiquitously expressed in both proliferating and differentiated C2C12 cells, human myoblasts, and the RMS cell lines (Fig. 2B). The MEF2Cα2 exon was only expressed in differentiated C2C12 cells and human myoblasts (Fig. 2B). Expression of the β exon could not be detected in any of the samples tested here (Fig. 2C). To verify detection of the β exon, we also assayed samples from the brain, induced pluripotent stem (iPS) cells, and neural progenitor cells derived from iPS cells (33). As anticipated, we found that brain and neural progenitor cells expressed the β exon, whereas iPS cells did not (Fig. 2C).
FIGURE 2.

Expression of the α and β exons of MEF2C in normal muscle and RMS. A, schematic of the exon structure of MEF2C with the location of the primers used to detect the indicated exons. B, the α1 exon of MEF2C is expressed ubiquitously in skeletal muscle, but the α2 exon is strongly up-regulated during differentiation. Exon expression was detected by RT-PCR on the indicated samples. U.D., undifferentiated myoblasts; D, days of differentiation; h.m., human myoblasts. C, the β exon is not expressed in muscle or RMS cells. Exon expression was detected by RT-PCR on the indicated samples as in B and from iPS cells, neural progenitor cells (NPC), and brain. D, expression of the α1 exon does not change during myoblast differentiation. Gene expression was assayed by qRT-PCR. Error bars show mean ± S.D. E, the α1 exon is highly expressed in RMS cells, as assayed as in D. ***, p < 0.001. F, the α2 exon is up-regulated during differentiation, as assayed as in D. Data are shown as the ratio of α2 expression relative to the expression of α1. ***, p < 0.001. G, the α2 exon is highly down-regulated in RMS and not induced by differentiation, as assayed as in F. ***, p < 0.001.
To further clarify our results, we used quantitative RT-PCR (qRT-PCR) to quantitate the expression pattern of MEF2Cα1/α2 isoforms during normal myogenesis and in RMS cells. Using primers specific to the α1 or α2 domain of MEF2C, we examined expression in C2C12 cells throughout a time course of differentiation. We found that the transcript for MEF2Cα1 was expressed in proliferating C2C12 cells (undifferentiated), and expression did not change significantly when cells were differentiated (Fig. 2D). In RMS cells, expression of MEF2Cα1 was compared with the expression levels found in human myoblast RNA. We found that both RD and RH30 cells expressed very high transcript levels of MEF2Cα1 (Fig. 2E). When the expression of MEFCα2 was examined, the expression was very low in proliferating C2C12 cells, but the ratio of α2 expression versus α1 expression increased sharply upon differentiation (Fig. 2F). For RMS cells, very low expression of MEF2Cα2 was observed compared with the expression observed in human myoblast RNA, and the ratio of α2/α1 expression did not increase significantly upon differentiation (Fig. 2G).
MEF2Cα2 Has Myogenic Activity whereas MEF2Cα1 Does Not
We next compared the myogenic activity of the MEF2C isoforms on a muscle-specific reporter. We chose a muscle-specific reporter that contains the Leiomodin2 (Lmod2) promoter fused to luciferase, Lmod2-luc, which we have used previously to characterize the activity of the MRFs and MEF2D (29, 34). The Lmod2-luc reporter shows very low activity in proliferating cells and is strongly up-regulated upon differentiation. Transfection of MyoD or myogenin activate the reporter. Therefore, we assayed for the activity of the Lmod2-luc reporter with MyoD alone and in combination with each of the MEF2 isoforms in 10T1/2 cells, a fibroblast cell line considered poised for activation of muscle-specific genes. We found that MEF2Cα1 did not enhance the activity of the reporter and, in fact, had a mild inhibitory effect (Fig. 3A). The addition of the γ domain was also modestly inhibitory, as seen previously. The isoform lacking the α domain did not significantly inhibit or activate the Lmod2-luc reporter. The MEF2Cα2 isoform strongly activated the Lmod2-luc reporter. Addition of the γ domain again lead to a modest inhibition, but the γ domain-containing isoform still robustly activated the Lmod2-luc reporter. The work indicates that transcripts including α2 are required for MEF2C myogenic enhancing activity, among which the isoform without the γ domain is modestly stronger than that with the γ domain. Transcripts with the α1 exon appear to inhibit the myogenic activity of MyoD.
FIGURE 3.

MEF2Cα2 robustly enhances MRF activity, whereas MEF2Cα1 does not. A, MEF2Cα2 stimulates the activity of MyoD on a muscle-specific luciferase reporter construct. 10T1/2 cells were transfected with the indicated constructs. Values are represented with respect to a luciferase vector with no promoter (pGL3 basic). pGL3 (+), luciferase vector with the constitutive CMV promoter; Lmod2-luc, luciferase vector with a ∼300-bp Leiomodin 2 (Lmod2) promoter. Error bars show mean ± S.D. **, p < 0.01; ***, p < 0.001. B, confirmation of the expression of MEF2Cα1 and MEF2Cα2. 10T1/2 cells were transfected with expression constructs for MyoD, MEF2Cα1, and MEF2Cα2 as indicated, and gene expression was determined by qRT-PCR for the indicated genes. Vector, vector-only transfection where the expression level was set to 1. Error bars show mean ± S.D. ***, p < 0.001. C, MEF2Cα2 activates endogenous MRF target gene expression. 10T1/2 cells were transfected with expression constructs for MyoD, MEF2Cα1, and MEF2Cα2 and analyzed for the indicated genes as in B.
To confirm these results, we next assayed for the activity of the MEF2C isoforms on endogenous gene expression. 10T1/2 cells were transfected with constructs expressing MyoD in combination with constructs expressing either MEF2Cα1 or MEF2Cα2. Gene expression analysis confirmed the expression of each MEF2C isoform (Fig. 3B). We found that transfection of MEF2Cα2 with MyoD strongly induced muscle-specific gene expression, including myosin light chain, phosphorylatable, fast (Mylpf); creatine kinase, muscle (Ckm); and troponin T, type 1 (Tnnt1), whereas MEF2Cα1 had no activity (Fig. 3C).
Our data are consistent with previous findings showing that muscle expresses both MEF2Cα2 and MEF2Cα1 (16, 18). To determine the effect of each isoform in muscle, MEF2Cα1 and MEF2Cα2 were individually ectopically expressed in C2C12 cells. Proliferating C2C12 cells were transfected with plasmids expressing MEF2Cα1 or MEF2Cα2 and then induced to differentiate. Expression of the individual isoforms was confirmed by qRT-PCR (Fig. 4A). Expression of the exogenous epitope-tagged MEF2C isoforms was also confirmed by Western blot analysis (Fig. 4B). When differentiation-specific gene expression was examined, we found that exogenous expression of MEF2Cα2 stimulated the expression of actin (Acta1), troponin 1 type 2 (Tnni2), and leiomodin 2 (Lmod2), whereas the MEF2Cα1 isoform had a modest inhibitory effect (Fig. 4C). The effect on differentiation was also assayed by immunostaining for expression of myosin heavy chain (MHC), which is commonly used as a marker for myogenesis. We found that ectopic expression of MEF2Cα2 significantly stimulated the formation of myosin heavy chain-positive myotubes, whereas the expression of MEF2Cα1 was inhibitory for myotube formation (Fig. 4D).
FIGURE 4.

MEF2Cα2 promotes myogenesis in C2C12 cells. A, C2C12 cells were transfected with constructs expressing vector, MEF2Cα1, or MEF2Cα2. Expression of the isoforms was confirmed by qRT-PCR. Error bars show mean ± S.D. ***, p < 0.001. B, protein expression of the epitope-tagged MEF2C isoforms was confirmed by Western blot analysis. C, differentiation-specific gene expression is induced by overexpression of MEF2Cα2. Gene expression was assayed for the indicated genes by qRT-PCR. Error bars show mean ± S.D. ***, p < 0.001. D, overexpression of MEF2Cα2 promotes myotube formation, whereas MEF2Cα1 is inhibitory. The cell lines in A were differentiated for 3 days, immunostained with antibodies against MHC, and counterstained with DAPI. Images were taken at ×200 magnification. Scale bars = 5 μm. The data are quantitated in the bottom panels.
To determine whether MEF2Cα2 could rescue the MRF-dependent activation of muscle-specific genes in RMS, we first asked whether MEF2Cα2 could activate the Lmod2 reporter in RD cells. We found that MEF2Cα2 robustly induced the Lmod2 reporter, whereas the MEF2Cα1 isoform was not able to activate the reporter (Fig. 5A). Consistent with our results in Fig. 3, we found that MEF2Cα2 robustly stimulated the Lmod-luc reporter in RD cells. The isoform lacking an α domain did modestly activate the reporter (∼2-fold) but not nearly to the degree as the MEF2Cα2 isoform. Next we examined the effect on the expression of differentiation-specific genes in RD cells and found that MEF2Cα2 stimulated the expression of LMOD2, TNNI2, and CDKN1A (p21) (Fig. 5B). The cell cycle regulator p21 is required for terminal differentiation (35) and is regulated in part by MyoD in muscle (36). To determine whether the MEF2Cα2 isoform could promote differentiation in RMS cells, exogenous MEF2Cα1 and MEF2Cα2 were expressed in RD cells, and myotube formation was assayed by MHC immunohistochemistry. We found that MEF2Cα2 expression markedly induced differentiation in RD cells (Fig. 5C).
FIGURE 5.

MEF2Cα2 promotes differentiation in RMS cells. A, MEF2Cα2 activates a muscle-specific reporter in RMS cells. RD cells were transfected with the indicated constructs. Values are represented with respect to a luciferase vector with no promoter (pGL3 basic). pGL3 (+), luciferase vector with the constitutive CMV promoter; Lmod2-luc, luciferase vector with a ∼300-bp Leiomodin 2 (Lmod2) promoter. Error bars show mean ± S.D. ***, p < 0.001. B, MEF2Cα2 promotes differentiation-specific gene expression in RD cells. RD cells were transfected with constructs expressing vector, MEF2Cα1, and MEF2Cα2. Gene expression was assayed for the indicated genes by qRT-PCR. Error bars show mean ± S.D. ***, p < 0.001. C, MEF2Cα2 promotes the expression of MHC in RD cells. RD cells expressing vector, MEF2Cα1, and MEF2Cα2 were immunostained for MHC and counterstained with DAPI. Images were taken at ×100 magnification. Scale bars = 10 μm.
MEF2Cα1 Associates Preferentially with HDAC5
To begin to address how the α2 exon of MEF2C promotes myogenesis whereas the α1 exon does not, we asked whether the association with HDACs with each isoform was distinct. MEF2 is well known to interact with histone deacetylases (1), and the differential phosphorylation of the α1/α2 exon of MEF2D alters the association with HDACs (22). Therefore, we asked whether a difference in HDAC association could be observed for MEF2Cα1 versus MEF2Cα2. HEK293 cells, which express endogenous MEF2Cα1, were transfected with constructs expressing MEF2Cα1 or MEF2Cα2. MEF2C proteins were immunoprecipitated with antibodies against MEF2C, and the immunoprecipitate was probed for HDAC5. We found that the cells transfected with a plasmid expressing MEF2Cα2 immunoprecipitated HDAC5 less robustly than cells transfected with a plasmid expressing MEF2Cα1 (Fig. 6A). The antibody used for the immunoprecipitation could immunoprecipitate both MEF2Cα1 and MEF2Cα2, so it is possible that the differential association of HDAC5 might be more significant than that indicated by our experiment because HEK293 cells have endogenous levels of MEF2Cα1. Selective immunoprecipitation of the isoforms using epitope tags on the constructs was attempted, but nonspecific bands in the immunoprecipitate precluded analysis of HDAC association.
FIGURE 6.

MEF2Cα1 recruits HDACs to target promoters. A, MEF2Cα1 interacts with HDAC5 more robustly than MEF2Cα2. HEK293 cells were transfected with expression constructs for MEF2Cα1 and MEF2Cα2, immunoprecipitated (IP) with an antibody against MEF2C, and then the blot was probed with antibodies against HDAC5. EXT, extract; exp., exposure. B, MEF2Cα2 inhibits recruitment of HDAC5 to target promoters. ChIP assays were performed on RD cells transfected with vector control or an expression construct for MEF2Cα2 with antibodies against HDAC5, and immunoprecipitated DNA was probed with primers corresponding to the indicated promoters. Error bars show mean ± S.D. ***, p < 0.001. C, HDAC4 recruitment to target promoters is also inhibited by MEF2Cα2. ChIP assays were performed as in B but with antibodies against HDAC4.
To understand whether the differential association of HDAC5 observed would influence HDAC recruitment to target genes, we performed ChIP assays for HDAC5 in RD cells expressing MEF2Cα1 transfected with a vector control or with a construct expressing exogenous MEF2Cα2. We found that HDAC5 could be detected on muscle-specific promoters in RD cells transfected with vector, but this association was decreased when MEF2Cα2 was expressed (Fig. 6B). The decrease in HDAC recruitment was also observed at the CDKN1A (p21) promoter. We also examined the recruitment of HDAC4, an additional class II HDAC, by ChIP assays and found that HDAC4 association was also disrupted by MEF2Cα2 expression (Fig. 6C). Our data indicate that MEF2Cα2 may promote muscle gene expression at least in part by reducing the recruitment of HDACs to target promoters and, therefore, promoting gene activation.
SRPK3 Is Down-regulated in RMS Cells
Because our data suggested that the lack of expression of MEF2Cα2 in RMS cells might contribute to the block to differentiation in these cells, we sought to understand why the MEF2Cα2 isoform was not expressed in RMS cells. To address this, we attempted to identify the splicing factors that controlled the α isoform selection. Two bioinformatic databases, Expasy (37) and Uniprot (38), were used to predict the splicing factors that might recognize the α exon splice sites in MEF2C. Both programs predicted the serine/arginine-rich splicing factor 1, SRSF1 (ASF), which is activated by phosphorylation. To initiate our analysis, we assayed for the expression of ASF in skeletal muscle and RMS cells. We found that the expression of ASF is modestly up-regulated during myogenesis (Fig. 7A), consistent with a role for promoting the MEF2Cα1-to-MEF2Cα2 switch. However, when RMS cells were analyzed for expression of ASF, we found that ASF was highly up-regulated compared with human myoblasts (Fig. 7B). This result is consistent with many other studies showing that ASF is often highly up-regulated in cancer (39).
FIGURE 7.
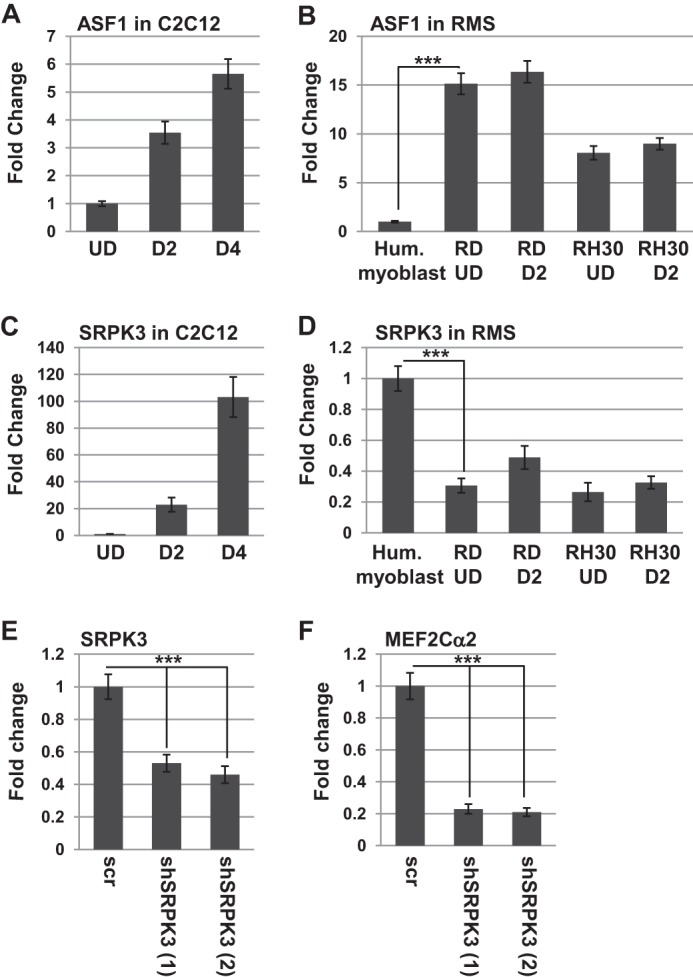
Expression of splicing factors in skeletal muscle and RMS. A, ASF is modestly up-regulated upon differentiation in C2C12 cells. Gene expression was assayed by qRT-PCR, and the days of differentiation (D) are indicated. Error bars show mean ± S.D. UD, undifferentiated. B, ASF is highly expressed in RMS cells. Expression was assayed as in A. Hum., human. C, SRPK3 is robustly up-regulated upon differentiation of C2C12 cells, as assayed as in A. D, SRPK3 is highly down-regulated in RMS cells, as assayed as in A. ***, p < 0.001. E, depletion of SRPK3. C2C12 cells were transfected individually with multiple shRNA constructs (shSRPK3), and stable transformants were selected. The two constructs shown are represented by (1) and (2). Gene expression was assayed after 2 days of differentiation by qRT-PCR. Error bars show mean ± S.D. scr, scrambled. ***, p < 0.001. F, SRPK3 is required for MEF2Cα2 splicing. Gene expression was assayed on the shSRPK3 depletions as in E.
We next looked for expression of upstream kinases required for activation of ASF. We choose SRPK3, a muscle-specific protein kinase that is regulated by MEF2C in skeletal muscle (40). As shown previously (40), SRPK3 was strongly up-regulated during normal myogenesis (Fig. 7C). We also found that SRPK3 was down-regulated in RMS cells (Fig. 7D). To determine whether SRPK3 was required for splicing of the MEF2Cα2 isoform, SRPK3 was depleted from C2C12 cells using shRNA constructs. Multiple shRNA constructs were used independently, and the results of two individual constructs are shown. We found that the constructs depleted SRPK3 (Fig. 7E) and inhibited splicing of the MEF2Cα2 isoform when assayed after 2 days of differentiation (Fig. 7F).
SRPK3 Activates the Splicing of MEF2Cα2 and Promotes Differentiation in RMS Cells
To determine whether the down-regulation of SRPK3 contributed to the isoform selection in MEF2C and the block to differentiation in RMS cells, we ectopically expressed SRPK3 in RD cells. The expression of SRPK3 was confirmed by qRT-PCR (Fig. 8A). The expression of the MEF2Cα1 and MEF2Cα2 isoforms was then analyzed, and we found that expression of exogenous SRPK3 did not significantly alter the expression of the MEF2Cα1 isoform, but it did strongly enhance expression of the MEF2Cα2 isoform (Fig. 8B). To determine whether the expression of SRPK3 could promote differentiation, we assayed these cells for differentiation-specific gene expression, including LMOD2, ACTA1, TNNT1, and CDKN1A. We found that each of these genes was up-regulated in cells expressing SRPK3 (Fig. 8C), strongly suggesting that SRPK3 promotes differentiation-specific splicing, which allows expression of the appropriate transcripts required for differentiation. Differentiation was also assayed by immunostaining for MHC in RD cells transfected with expression constructs for vector, MEF2Cα2, or SRPK3. We found that MEF2Cα2 or SRPK3 promoted a robust MHC signal and the appearance of myotubes (Fig. 8D). To determine whether SRPK3 and MEF2Cα2 could also promote differentiation in ARMS cells, the above experiment was repeated in RH30 cells, and, again, robust expression of MHC was observed (Fig. 8D).
FIGURE 8.

SRPK3 and MEF2Cα2 promote differentiation of RMS cells. A, SRPK3 overexpression in RD cells. RD cells were transfected with a vector control or an expression construct for SRPK3 and assayed for gene expression by qRT-PCR. B, SRPK3 induces the expression of MEF2Cα2. Gene expression was assayed by qRT-PCR. Error bars show mean ± S.D. ***, p < 0.001. C, SRPK3 induces differentiation-specific gene expression in RD cells. Cells as in A were assayed for gene expression by qRT-PCR for the indicated genes. Error bars show mean ± S.D. ***, p < 0.001. D, SRPK3 or MEF2Cα2 induce MHC expression in RMS cells. RD (left panel) and RH30 cells (right panel) were transfected with a vector control or expression constructs for SRPK3 or MEF2Cα2, immunostained for MHC, and counterstained with DAPI. Images were taken at ×100 magnification. Scale bars = 10 μm.
Finally, we sought to understand whether SRPK3 or MEF2Cα2 could inhibit the proliferation and tumorigenic growth of RMS cells. RD cells expressing exogenous SRPK3 or MEF2Cα2 were assayed for proliferation, and we found that these cells had reduced proliferation rates when compared with the vector only controls (Fig. 9A). To extend this result to the ARMS subtype of RMS, the proliferation assay was repeated in RH30 cells. We found that exogenous expression of SRPK3 or MEF2Cα2 also inhibited the proliferation of RH30 cells (Fig. 9B). To determine whether SRPK3 or MEF2Cα2 could inhibit anchorage-independent growth of these cells, growth in soft agar medium was assayed. We found that RD cells expressing exogenous MEF2Cα2 or SRPK3 formed fewer colonies in soft agar media (Fig. 9C) and that the colonies that did form were smaller in size than those observed for the vector only controls (Fig. 9D). The data suggest that restoration of differentiation-specific splicing may inhibit RMS tumor growth.
FIGURE 9.

SRPK3 and MEF2Cα2 inhibit growth of RMS cells. A, SRPK3 or MEF2Cα2 inhibit the proliferation of RD cells. RD cells expressing the indicated constructs were seeded at equivalent densities and harvested for cell counts every 2 days. Error bars show mean ± S.D. B, SRPK3 or MEF2Cα2 inhibit the proliferation of RH30 cells. Proliferation was assayed as in A. C, SRPK3 or MEF2Cα2 inhibit the number of anchorage-independent colonies formed. Error bars show mean ± S.D. ***, p < 0.001. D, SRPK3 or MEF2Cα2 inhibit the size of anchorage-independent colonies formed. The largest colonies observed for each cell line are shown.
DISCUSSION
We show here that the α2 exon of MEF2C confers myogenic activity on MEF2C and results in differential HDAC recruitment to target promoters. The expression of the MEF2Cα1 exon in RMS cells contributes to the lack of differentiation observed in those cells. The splicing of the α2 exon is promoted by SRPK3, and restoration of SRPK3 or MEF2Cα2 in RMS cells enhances differentiation and inhibits proliferation and tumorigenic growth. MEF2C has been shown previously to induce differentiation in RMS cells (28), and our results reveal that the deficiency in MEF2C activity is due to the lack of appropriate muscle-specific splicing.
Defects in alternative splicing have been observed previously in RMS cells. The oncogenes Murine Double Minute 2 (MDM2) and MDM4 exhibit genotoxic stress-inducible splice forms in high-risk metastatic disease represented by both embryonal RMS and ARMS. Expression of these alterative isoforms promotes metastatic behavior of tumor cells (41). Multiple splicing isoforms of PAX3, PAX7, and the PAX-FOXO1 fusions have also been observed in RMS, and differences in the PAX7 splicing pattern between murine skeletal muscle and RMS tumors has been observed (42). To our knowledge, our work is the first to implicate the deregulation of a splicing factor in RMS. We show here that SRPK3 is required for the isoform switch between MEF2Cα1 and MEF2Cα2 but likely controls the splicing of many other genes required for normal muscle differentiation.
A recent study has shown that the expression and alternative splicing of the MEF2 genes are deregulated in muscle from neuromuscular disorder patients, including myotonic dystrophy type 1 (DM1) and myotonic dystrophy type 2 (DM2) (43). In DM, expression of a 224-bp isoform encompassing exons 4A and 4B (corresponding to MEF2Cα1) has been found to be expressed in muscle, whereas normal muscle contained a 217-bp isoform encompassing exon 5a (corresponding to MEF2Cα2) (43). Our work suggests that expression of MEF2Cα1 in diseased muscle would prevent appropriate differentiation-specific gene expression and contribute to the muscle dysfunction observed in the patients.
In a related study, MEF2C has been found to be deregulated in cardiac tissue of DM1 patients (44). A screen of microRNAs revealed that several miRNAs were differentially expressed in a mouse model of DM1 and that many of these miRNAs were direct MEF2 transcriptional targets. A down-regulation of MEF2C and MEF2A was observed in both the mouse models and in human DM1 cardiac tissues, and restoration of MEF2C promoted expression of miRNA and mRNA targets in DM1. Cardiac tissue is thought to express the α1 isoform of MEF2C, and it will be interesting to understand how alternative splicing of MEF2C contributes to the dysfunction of MEF2C observed in both cardiac and skeletal muscle tissue in DM1 patients.
It is intriguing that the MEF2Dα2 isoform has been shown recently to activate differentiation-specific transcription (22), whereas the ubiquitously expressed MEF2Dα1 form does not, similar to what we observed with MEF2C. In the case of MEF2D, the activity has been shown to be due to differential phosphorylation of the α1 versus α2 exon mediated by PKA (22, 45). Phosphorylation of the MEF2Dα1 isoform induces association with histone deacetylases (22). We also see that the α1 exon of MEF2C interacts preferentially with HDAC5 and induces the recruitment of HDAC5 and HDAC4 to target promoters. The basis of the differentiation interaction with HDAC5 is currently unclear for MEF2C, but it may also involve differential phosphorylation. The phosphorylation of MEF2C is unlikely to be mediated by PKA because MEF2C has been reported to be a poor substrate for PKA (45), and the α1 exon of MEF2C does not contain consensus sites for PKA phosphorylation.
Besides the modulation of MEF2C by HDACs, we cannot rule out the potential regulation of α1/α2 through differential interactions with other transcription factors and cofactors. Many factors have been shown to modulate the activity of MEF2C during myogenesis, including the myogenic regulatory factors MyoD and myogenin (46), the histone acetyltransferase P300 (47), the steroid nuclear receptor coactivator NCOA2/GRIP-1 (48), and the mastermind-like transcriptional coactivator (49). The calcineurin inhibitor Cabin1 sequesters MEF2C in a transcriptionally inactive state that is released by an increase in intracellular calcium concentration (50). The differential interaction of MEF2Cα1 and MEF2Cα2 with any of these factors may contribute to the differences in myogenic activity we observe here. Intriguingly, the MEF2Cα1 domain has been shown previously to be the target of the inhibitory effect of the Notch signaling pathway, which represses myogenesis (51). The SVGHSPESEDKY region, which is uniquely present in MEF2Cα1 and not in MEF2Cα2, MEF2A, MEF2B, or MEF2D, has been shown to be required for Notch-mediated repression. Activated Notch signaling is common in many cancers, and activated Notch has also been observed in RMS cells (52). Therefore, differential interactions of the MEF2C α1/α2 isoforms with the Notch signaling pathway may also contribute to the differential activity of the isoforms. The data shown here confirm that MEF2C constructs entirely lacking the α domain have higher activity than the MEF2Cα1 isoform found in RMS cells. Further understanding of how elevated Notch signaling and MEF2Cα1 expression in RMS cells may contribute to the pathology of RMS is an important future direction for these studies.
MEF2C is a direct transcriptional activator of many important developmental genes, including c-jun (53) and matrix metalloproteinase 10 (MMP10) (54). MEF2C is also a direct transcriptional activator of several miRNAs, including miR-1, miR-21, miR-29, miR-30, and miR-133 (44). It will be important to understand which isoform of MEF2C directs transcription of these important targets in each system and how the differential expression and regulation of the isoforms contributes to the appropriate expression of MEF2C target genes.
Although the MEF2Cα2 isoform has been known to be expressed in skeletal muscle, our results reveal the requirement for the α2 exon for myogenesis and show that the differentiation defect in RMS cells extends to the muscle-specific splicing patterns required for differentiation. It will be important to further understand the deregulation of splicing factors such as SRPK3 in RMS because targeting these changes may offer novel therapeutic approaches for treating RMS. Defining the molecular basis for the myogenic activity of the α2 exon of MEF2C and the differential recruitment of HDAC5 will also be important in understanding normal skeletal muscle differentiation. Understanding the function of the α exon of MEF2C and how the appropriate splicing is achieved also contributes to the understanding of muscle dysfunction in neuromuscular disease patients and may potentially offer new therapeutic approaches for this disease as well.
Acknowledgments
We thank Keith Gagnon (Southern Illinois University) for the iPS and neural progenitor cell cDNA and James Maclean II (Southern Illinois University) for the cDNA from brain.
This work was supported, in whole or in part, by NIAMS/National Institutes of Health Grant RAR 060017A. This work was also supported by Grant 159609 from the American Cancer Society, Illinois Division (to J. D.).
- MRF
- myogenic regulatory factor
- HDAC
- histone deacetylase
- RMS
- rhabdomyosarcoma
- ARMS
- alveolar rhabdomyosarcoma
- iPS cell
- induced pluripotent stem cell
- qRT-PCR
- quantitative RT-PCR
- MHC
- myosin heavy chain
- miRNA
- microRNA
- ASF
- alternative splicing factor.
REFERENCES
- 1. Potthoff M. J., Olson E. N. (2007) MEF2: a central regulator of diverse developmental programs. Development 134, 4131–4140 [DOI] [PubMed] [Google Scholar]
- 2. Edmondson D. G., Lyons G. E., Martin J. F., Olson E. N. (1994) Mef2 gene expression marks the cardiac and skeletal muscle lineages during mouse embryogenesis. Development 120, 1251–1263 [DOI] [PubMed] [Google Scholar]
- 3. Potthoff M. J., Arnold M. A., McAnally J., Richardson J. A., Bassel-Duby R., Olson E. N. (2007) Regulation of skeletal muscle sarcomere integrity and postnatal muscle function by Mef2c. Mol. Cell. Biol. 27, 8143–8151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Penn B. H., Bergstrom D. A., Dilworth F. J., Bengal E., Tapscott S. J. (2004) A MyoD-generated feed-forward circuit temporally patterns gene expression during skeletal muscle differentiation. Genes Dev. 18, 2348–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kablar B., Rudnicki M. A. (2000) Skeletal muscle development in the mouse embryo. Histol. Histopathol. 15, 649–656 [DOI] [PubMed] [Google Scholar]
- 6. Molkentin J. D., Black B. L., Martin J. F., Olson E. N. (1995) Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell 83, 1125–1136 [DOI] [PubMed] [Google Scholar]
- 7. Black B. L., Olson E. N. (1998) Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol. 14, 167–196 [DOI] [PubMed] [Google Scholar]
- 8. Han J., Jiang Y., Li Z., Kravchenko V. V., Ulevitch R. J. (1997) Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature 386, 296–299 [DOI] [PubMed] [Google Scholar]
- 9. Dodou E., Treisman R. (1997) The Saccharomyces cerevisiae MADS-box transcription factor Rlm1 is a target for the Mpk1 mitogen-activated protein kinase pathway. Mol. Cell. Biol. 17, 1848–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Youn H. D., Grozinger C. M., Liu J. O. (2000) Calcium regulates transcriptional repression of myocyte enhancer factor 2 by histone deacetylase 4. J. Biol. Chem. 275, 22563–22567 [DOI] [PubMed] [Google Scholar]
- 11. D'Andrea M., Pisaniello A., Serra C., Senni M. I., Castaldi L., Molinaro M., Bouché M. (2006) Protein kinase C θ co-operates with calcineurin in the activation of slow muscle genes in cultured myogenic cells. J. Cell. Physiol. 207, 379–388 [DOI] [PubMed] [Google Scholar]
- 12. Shalizi A., Gaudillière B., Yuan Z., Stegmüller J., Shirogane T., Ge Q., Tan Y., Schulman B., Harper J. W., Bonni A. (2006) A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science 311, 1012–1017 [DOI] [PubMed] [Google Scholar]
- 13. McKinsey T. A., Zhang C. L., Olson E. N. (2002) MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem. Sci. 27, 40–47 [DOI] [PubMed] [Google Scholar]
- 14. Zhang C. L., McKinsey T. A., Chang S., Antos C. L., Hill J. A., Olson E. N. (2002) Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110, 479–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu J., McKinsey T. A., Nicol R. L., Olson E. N. (2000) Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 97, 4070–4075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hakim N. H., Kounishi T., Alam A. H., Tsukahara T., Suzuki H. (2010) Alternative splicing of Mef2c promoted by Fox-1 during neural differentiation in P19 cells. Genes Cells 15, 255–267 [DOI] [PubMed] [Google Scholar]
- 17. Infantino V., Convertini P., Menga A., Iacobazzi V. (2013) MEF2C exon α: role in gene activation and differentiation. Gene 531, 355–362 [DOI] [PubMed] [Google Scholar]
- 18. McDermott J. C., Cardoso M. C., Yu Y. T., Andres V., Leifer D., Krainc D., Lipton S. A., Nadal-Ginard B. (1993) hMEF2C gene encodes skeletal muscle- and brain-specific transcription factors. Mol. Cell. Biol. 13, 2564–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhu B., Ramachandran B., Gulick T. (2005) Alternative pre-mRNA splicing governs expression of a conserved acidic transactivation domain in myocyte enhancer factor 2 factors of striated muscle and brain. J. Biol. Chem. 280, 28749–28760 [DOI] [PubMed] [Google Scholar]
- 20. Zhu B., Gulick T. (2004) Phosphorylation and alternative pre-mRNA splicing converge to regulate myocyte enhancer factor 2C activity. Mol. Cell. Biol. 24, 8264–8275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martin J. F., Miano J. M., Hustad C. M., Copeland N. G., Jenkins N. A., Olson E. N. (1994) A Mef2 gene that generates a muscle-specific isoform via alternative mRNA splicing. Mol. Cell. Biol. 14, 1647–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sebastian S., Faralli H., Yao Z., Rakopoulos P., Palii C., Cao Y., Singh K., Liu Q. C., Chu A., Aziz A., Brand M., Tapscott S. J., Dilworth F. J. (2013) Tissue-specific splicing of a ubiquitously expressed transcription factor is essential for muscle differentiation. Genes Dev. 27, 1247–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Merlino G., Helman L. J. (1999) Rhabdomyosarcoma: working out the pathways. Oncogene 18, 5340–5348 [DOI] [PubMed] [Google Scholar]
- 24. Barr F. G., Galili N., Holick J., Biegel J. A., Rovera G., Emanuel B. S. (1993) Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 3, 113–117 [DOI] [PubMed] [Google Scholar]
- 25. Galili N., Davis R. J., Fredericks W. J., Mukhopadhyay S., Rauscher F. J., 3rd, Emanuel B. S., Rovera G., Barr F. G. (1993) Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 5, 230–235 [DOI] [PubMed] [Google Scholar]
- 26. Keller C., Guttridge D. C. (2013) Mechanisms of impaired differentiation in rhabdomyosarcoma. FEBS J. 280, 4323–4334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sartori F., Alaggio R., Zanazzo G., Garaventa A., Di Cataldo A., Carli M., Rosolen A. (2006) Results of a prospective minimal disseminated disease study in human rhabdomyosarcoma using three different molecular markers. Cancer 106, 1766–1775 [DOI] [PubMed] [Google Scholar]
- 28. MacQuarrie K. L., Yao Z., Fong A. P., Diede S. J., Rudzinski E. R., Hawkins D. S., Tapscott S. J. (2013) Comparison of genome-wide binding of MyoD in normal human myogenic cells and rhabdomyosarcomas identifies regional and local suppression of promyogenic transcription factors. Mol. Cell. Biol. 33, 773–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang M., Truscott J., Davie J. (2013) Loss of MEF2D expression inhibits differentiation and contributes to oncogenesis in rhabdomyosarcoma cells. Mol. Cancer 12, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Londhe P., Davie J. K. (2011) γ Interferon modulates myogenesis through the major histocompatibility complex class II transactivator, CIITA. Mol. Cell. Biol. 31, 2854–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Londhe P., Davie J. K. (2011) Sequential association of myogenic regulatory factors and E proteins at muscle-specific genes. Skelet. Muscle 1, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu B., Zhang M., Byrum S. D., Tackett A. J., Davie J. K. (2014) TBX2 blocks myogenesis and promotes proliferation in rhabdomyosarcoma cells. Int. J. Cancer 135, 785–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kasukawa Y., Stabnov L., Miyakoshi N., Baylink D. J., Mohan S. (2002) Insulin-like growth factor I effect on the number of osteoblast progenitors is impaired in ovariectomized mice. J. Bone Miner. Res. 17, 1579–1587 [DOI] [PubMed] [Google Scholar]
- 34. Davie J. K., Cho J. H., Meadows E., Flynn J. M., Knapp J. R., Klein W. H. (2007) Target gene selectivity of the myogenic basic helix-loop-helix transcription factor myogenin in embryonic muscle. Dev. Biol. 311, 650–664 [DOI] [PubMed] [Google Scholar]
- 35. Parker S. B., Eichele G., Zhang P., Rawls A., Sands A. T., Bradley A., Olson E. N., Harper J. W., Elledge S. J. (1995) p53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science 267, 1024–1027 [DOI] [PubMed] [Google Scholar]
- 36. Halevy O., Novitch B. G., Spicer D. B., Skapek S. X., Rhee J., Hannon G. J., Beach D., Lassar A. B. (1995) Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science 267, 1018–1021 [DOI] [PubMed] [Google Scholar]
- 37. Artimo P., Jonnalagedda M., Arnold K., Baratin D., Csardi G., de Castro E., Duvaud S., Flegel V., Fortier A., Gasteiger E., Grosdidier A., Hernandez C., Ioannidis V., Kuznetsov D., Liechti R., Moretti S., Mostaguir K., Redaschi N., Rossier G., Xenarios I., Stockinger H. (2012) ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 40, W597–W603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. UniProt C. (2014) Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 42, D191–D198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Das S., Krainer A. R. (2014) Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol. Cancer Res. 12, 1195–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nakagawa O., Arnold M., Nakagawa M., Hamada H., Shelton J. M., Kusano H., Harris T. M., Childs G., Campbell K. P., Richardson J. A., Nishino I., Olson E. N. (2005) Centronuclear myopathy in mice lacking a novel muscle-specific protein kinase transcriptionally regulated by MEF2. Genes Dev. 19, 2066–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jacob A. G., O'Brien D., Singh R. K., Comiskey D. F., Jr., Littleton R. M., Mohammad F., Gladman J. T., Widmann M. C., Jeyaraj S. C., Bolinger C., Anderson J. R., Barkauskas D. A., Boris-Lawrie K., Chandler D. S. (2013) Stress-induced isoforms of MDM2 and MDM4 correlate with high-grade disease and an altered splicing network in pediatric rhabdomyosarcoma. Neoplasia 15, 1049–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Du S., Lawrence E. J., Strzelecki D., Rajput P., Xia S. J., Gottesman D. M., Barr F. G. (2005) Co-expression of alternatively spliced forms of PAX3, PAX7, PAX3-FKHR and PAX7-FKHR with distinct DNA binding and transactivation properties in rhabdomyosarcoma. Int. J. Cancer 115, 85–92 [DOI] [PubMed] [Google Scholar]
- 43. Bachinski L. L., Sirito M., Böhme M., Baggerly K. A., Udd B., Krahe R. (2010) Altered MEF2 isoforms in myotonic dystrophy and other neuromuscular disorders. Muscle Nerve 42, 856–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kalsotra A., Singh R. K., Gurha P., Ward A. J., Creighton C. J., Cooper T. A. (2014) The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell Rep. 6, 336–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Du M., Perry R. L., Nowacki N. B., Gordon J. W., Salma J., Zhao J., Aziz A., Chan J., Siu K. W., McDermott J. C. (2008) Protein kinase A represses skeletal myogenesis by targeting myocyte enhancer factor 2D. Mol. Cell. Biol. 28, 2952–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Molkentin J. D., Olson E. N. (1996) Combinatorial control of muscle development by basic helix-loop-helix and MADS-box transcription factors. Proc. Natl. Acad. Sci. U.S.A. 93, 9366–9373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sartorelli V., Huang J., Hamamori Y., Kedes L. (1997) Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol. Cell. Biol. 17, 1010–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen S. L., Dowhan D. H., Hosking B. M., Muscat G. E. (2000) The steroid receptor coactivator, GRIP-1, is necessary for MEF-2C-dependent gene expression and skeletal muscle differentiation. Genes Dev. 14, 1209–1228 [PMC free article] [PubMed] [Google Scholar]
- 49. McElhinny A. S., Li J. L., Wu L. (2008) Mastermind-like transcriptional co-activators: emerging roles in regulating cross talk among multiple signaling pathways. Oncogene 27, 5138–5147 [DOI] [PubMed] [Google Scholar]
- 50. Youn H. D., Liu J. O. (2000) Cabin1 represses MEF2-dependent Nur77 expression and T cell apoptosis by controlling association of histone deacetylases and acetylases with MEF2. Immunity 13, 85–94 [DOI] [PubMed] [Google Scholar]
- 51. Wilson-Rawls J., Molkentin J. D., Black B. L., Olson E. N. (1999) Activated notch inhibits myogenic activity of the MADS-Box transcription factor myocyte enhancer factor 2C. Mol. Cell. Biol. 19, 2853–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Raimondi L., Ciarapica R., De Salvo M., Verginelli F., Gueguen M., Martini C., De Sio L., Cortese G., Locatelli M., Dang T. P., Carlesso N., Miele L., Stifani S., Limon I., Locatelli F., Rota R. (2012) Inhibition of Notch3 signalling induces rhabdomyosarcoma cell differentiation promoting p38 phosphorylation and p21(Cip1) expression and hampers tumour cell growth in vitro and in vivo. Cell Death Differ. 19, 871–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nadruz W., Jr., Kobarg C. B., Constancio S. S., Corat P. D., Franchini K. G. (2003) Load-induced transcriptional activation of c-jun in rat myocardium: regulation by myocyte enhancer factor 2. Circ. Res. 92, 243–251 [DOI] [PubMed] [Google Scholar]
- 54. Chang S., Young B. D., Li S., Qi X., Richardson J. A., Olson E. N. (2006) Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 126, 321–334 [DOI] [PubMed] [Google Scholar]