

Background: The α1 subunit harboring the A322D mutation is subject to excessive ERAD.
Results: VCP inhibition using Eeyarestatin I reduces the ERAD of α1(A322D) subunits, and coapplication of SAHA additively enhances their proteostasis.
Conclusion: Combining ERAD inhibition and folding enhancement yields significant functional rescue.
Significance: This combination represents a new, promising strategy to treat epilepsy resulting from GABAA receptor misfolding.
Keywords: Epilepsy, ER Quality Control, ER-associated Degradation, GABA Receptor, Protein Misfolding, Proteostasis, SAHA, VCP
Abstract
GABAA receptors are the primary inhibitory ion channels in the mammalian central nervous system. The A322D mutation in the α1 subunit results in its excessive endoplasmic reticulum-associated degradation at the expense of plasma membrane trafficking, leading to autosomal dominant juvenile myoclonic epilepsy. Presumably, valosin-containing protein (VCP)/p97 extracts misfolded subunits from the endoplasmic reticulum membrane to the cytosolic proteasome for degradation. Here we showed that inhibiting VCP using Eeyarestatin I reduces the endoplasmic reticulum-associated degradation of the α1(A322D) subunit without an apparent effect on its dynamin-1 dependent endocytosis and that this treatment enhances its trafficking. Furthermore, coapplication of Eeyarestatin I and suberanilohydroxamic acid, a known small molecule that promotes chaperone-assisted folding, yields an additive restoration of surface expression of α1(A322D) subunits in HEK293 cells and neuronal SH-SY5Y cells. Consequently, this combination significantly increases GABA-induced chloride currents in whole-cell patch clamping experiments than either chemical compound alone in HEK293 cells. Our findings suggest that VCP inhibition without stress induction, together with folding enhancement, represents a new strategy to restore proteostasis of misfolding-prone GABAA receptors and, therefore, a potential remedy for idiopathic epilepsy.
Introduction
Proteostasis, an optimal state of the cellular proteome (1, 2), is constantly challenged by intrinsic stress such as inherited misfolding-prone proteins (3), the environment (4), and aging (5). Proteostasis deficiency in ion channels leads to a variety of ion channel diseases called channelopathies, which are often caused by excessive endoplasmic reticulum-associated degradation (ERAD)2 and inefficient membrane trafficking of corresponding ion channel proteins harboring misfolding-prone mutations (3).
Valosin-containing protein (VCP), also called p97 in mammals, Cdc48p in yeast, CDC-48 in Caenorhabditis elegans, or TER94 in the fly, is a highly conserved, abundant ATPase of the ATPases associated with diverse cellular activities family (6). VCP is a homohexamer, forming a ring around a central pore (7). VCP has diverse physiological functions in ubiquitin-mediated degradation, autophagy, chromatin remodeling, cell cycle control, and more (8, 9). Probably the most prominent and studied role of VCP is in the ERAD regulation of a variety of substrates (10). The ERAD machinery is responsible for the degradation of terminally misfolded proteins in the endoplasmic reticulum (ER) (11–14). ERAD contains distinct but coupled steps, including substrate recognition, retrotranslocation, polyubiquitination, and proteasome targeting and degradation (11, 15). VCP plays an essential role in driving the retrotranslocation process, extracts misfolded proteins from the ER to the cytosol using energy, and targets them to the proteasome for degradation (10, 15).
VCP has been reported to differentially influence the maturation of various established ERAD substrates. RNA interference of VCP led to the accumulation of both mature and immature forms of the ΔF508 cystic fibrosis transmembrane conductance regulator, resulting in partial rescue of the mature ΔF508 cystic fibrosis transmembrane conductance regulator in IB3-1 cells (16). VCP inhibition rescued trafficking of lysosomal glucocerebrosidase harboring the L444P mutation in Gaucher disease patient-derived fibroblasts (17). In contrast, RNA interference of VCP did not increase the steady-state level of α1-antitrypsin Hong Kong variant or T-cell receptor T3 Δ chain (gene name CD3D) in HeLa cells (18). Therefore, the exact role of VCP in regulating protein maturation might depend on a specific ERAD substrate. Of particular interest is that the role of VCP in regulating GABAA receptor proteostasis has not yet been established.
GABAA receptors are the primary inhibitory neurotransmitter-gated ion channels in the mammalian central nervous system (19). They belong to and share common structural characteristics within the Cys loop receptor superfamily (20–22). The most common GABAA receptors in the human brain are a heteropentamer, consisting of two α1, two β2, and one γ2 subunits. Each subunit has four transmembrane (TM) helices (TM1-TM4, with the TM2 domain lining the interior of the pore), a large extracellular (or the ER luminal) N terminus, and a short extracellular (or the ER luminal) C terminus (Fig. 1A). The large cytosolic loop between TM3 and TM4 serves as a prominent candidate to interact with cytosolic molecular chaperones and degradation factors during the cellular protein folding and degradation steps.
FIGURE 1.

VCP interacts stronger with α1(A322D) subunits than with WT α1 subunits in HEK293 cells stably expressing WT α1β2γ2 or α1(A322D)β2γ2 GABAA receptors. A, topology of the α1 subunit. The A322D mutation is in the TM3 domain, labeled by an asterisk. B, effect of lactacystin on α1 subunit variant maturation. Treatment with lactacystin (5 μm, 24 h), a potent proteasome inhibitor, does not increase the endo H-resistant post-ER glycoform of the misfolding-prone α1(A322D) subunit in HEK293 cells (n = 2). Endo H-resistant α1 subunit bands represent properly folded, post-ER α1 subunit glycoforms that traffic at least to the Golgi compartment, whereas endo H-sensitive α1 subunit bands represent immature α1 subunit glycoforms that are retained in the ER. The peptide-N-glycosidase F enzyme cleaves between the innermost N-acetyl-d-glucosamine and asparagine residues from N-linked glycoproteins, serving as a control for unglycosylated α1 subunits (lane 3). After endo H digestion, subunits with a molecular weight equal to unglycosylated α1 subunits were considered endo H-sensitive (bottom arrow), whereas those with a higher molecular weight were considered endo H-resistant (top arrow) (lanes 2 and 5). The curly bracket indicates the top endo H-resistant bands in lanes 2, 5, 7, and 9 (no endo H-resistant bands were visible in lanes 7 and 9). β-actin served as a protein loading control. Ctl, no enzyme digestion control; Eh, endo H; Pf, peptide-N-glycosidase F; IB, immunoblot. C and D, the cellular interaction (direct or indirect) between the α1 subunit and VCP was verified by immunoprecipitation and Western blot analyses. HEK293 cells expressing WT α1β2γ2 or α1(A322D)β2γ2 GABAA receptors were lysed and immunoprecipitated with mouse anti-α1 subunit antibody or normal mouse IgG for negative, nonspecific binding control before being subjected to SDS-PAGE and Western blot analysis (C) (n = 3). The ratio of the VCP/α1 subunit post-immunoprecipitation, as a measure of the interaction between VCP and α1 subunit, was quantified, normalized to that of the WT, and is shown in D. EV, empty vector; IP, immunoprecipitation. Each data point in D is reported as mean ± S.E. **, p < 0.01.
GABAA receptors have a strong genetic association with idiopathic epilepsy (23–27). The missense A322D mutation in the TM3 domain of the α1 subunit of GABAA receptors leads to autosomal dominant juvenile myoclonic epilepsy, a common form of idiopathic generalized epilepsy representing 5–10% of all epilepsy cases (28). The A322D mutation results in the misfolding and, therefore, rapid degradation of the α1(A322D) subunit, mainly by ERAD (29). The consequence is that few α1(A322D) subunits are transported to the plasma membrane, reducing the level of functional pentameric GABAA receptors in the cell membrane. The A322D mutation leads to substantially reduced GABA-induced current in electrophysiological experiments. The few mutant receptors that reach the plasma membrane generate GABA-induced currents with different kinetics properties compared with WT receptors (30, 31). The cellular ERAD machinery regulating the rapid degradation of α1(A322D) subunits, however, is largely unexplored in the literature.
Presumably, the misfolded α1(A322D) subunit is recognized by the ER quality control machinery, polyubiquitinated, extracted from the ER membrane to the cytosol, and targeted to the proteasome for degradation. Here, we studied VCP as our first step to characterize the ERAD network for GABAA receptors because VCP plays an essential role in the substrate extraction step. Moreover, VCP appears in the interactome list for the ρ1 subunit of GABAA receptors (32), and human ρ1 and α1 subunits share high sequence homology (32.5% identity and 63.8% similarity).
We hypothesized that VCP extracts misfolded α1(A322D) subunits for their fast degradation, which outcompetes their folding and trafficking. Therefore, inhibiting VCP allows α1(A322D) subunits to have more time to fold in the ER for subsequent trafficking to the plasma membrane. We have demonstrated previously that SAHA, a potent histone deacetylase inhibitor, increases functional α1(A322D) subunit cell surface levels, partially by promoting BiP and calnexin-assisted folding (33). In this study, we investigated how VCP inhibition influences the degradation and trafficking of α1(A322D) subunits. Furthermore, we determined whether ERAD inhibition and folding enhancement by using SAHA have an additive effect to restore the function of epilepsy-associated GABAA receptors.
EXPERIMENTAL PROCEDURES
Reagents
Eeyarestatin I (EerI) and Dynole 34-2 were obtained from Tocris Bioscience. SAHA and lactacystin were from Cayman Chemical, and thapsigargin was from Enzo Life Science. The pCMV6 plasmids containing the human GABAA receptor α1, β2 (isoform 2), and γ2 (isoform 2) subunits and the pCMV6 entry vector plasmid (pCMV6-EV) were obtained from Origene. The human GABAA receptor α1 subunit missense mutation A322D was constructed using the QuikChange II site-directed mutagenesis kit (Agilent Genomics), and the cDNA sequences were confirmed by DNA sequencing.
The mouse monoclonal anti-α1 (clone BD24) and anti-β2/3 (clone 62-3G1) antibodies were obtained from Millipore, and the rabbit polyclonal anti-γ2 antibody was from R&D systems. The mouse monoclonal anti-β-actin antibody came from Sigma. The rabbit polyclonal anti-calnexin and anti-Hsp70, mouse monoclonal anti-Hsp90, and rat polyclonal anti-Grp94 antibodies were obtained from Enzo Life Sciences. The rabbit monoclonal anti-VCP and anti-BiP antibodies were obtained from Epitomics. The rabbit polyclonal anti-ubiquitin antibody was obtained from Cell Signaling Technology.
Cell Culture and Transfection
HEK293 cells and SH-SY5Y cells came from the ATCC and were maintained in DMEM (Hyclone) with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich) and 1% penicillin-streptomycin (Hyclone) at 37 °C in 5% CO2. Monolayers were passaged upon reaching confluency with trypsin 0.05% (Hyclone). Cells were grown in 6-well plates or 10-cm dishes and allowed to reach ∼70% confluency before transient transfection using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. Stable cell lines for α1β2γ2 and α1(A322D)β2γ2 receptors were generated using the G-418 selection method. Briefly, cells were transfected with α1:β2:γ2 (1:1:1) and α1(A322D):β2:γ2 (1:1:1) plasmids and then maintained in DMEM supplemented with 0.6 mg/ml G418 (Enzo Life Sciences) for 15 days. G-418 resistant cells were selected for follow-up experiments. Trypan blue stain (Hyclone), which selectively colors dead cells blue, was used to evaluate cell viability.
Western Blot Analysis
Cells were harvested with Trypsin 0.05% and then lysed with lysis buffer (50 mm Tris (pH 7.5), 150 mm NaCl, and 1% Triton X-100) supplemented with Roche complete protease inhibitor mixture. Lysates were cleared by centrifugation (15,000 × g, 10 min, 4 °C). Protein concentration was determined by MicroBCA assay (Pierce). Endoglycosidase H (endo H) and peptide-N-glycosidase F (New England Biolabs) digestion was performed according to the published procedure (29). Aliquots of cell lysates were separated in an 8% SDS-PAGE gel, and Western blot analysis was performed using the appropriate antibodies. Band intensity was quantified using ImageJ software from the National Institutes of Health.
Immunoprecipitation
Cell lysates (500 μg) were precleared with 30 μl of protein A/G Plus-agarose beads (Santa Cruz Biotechnology) and 1.0 μg of normal mouse IgG for 1 h at 4 °C to remove nonspecific binding proteins. The precleared cell lysates were incubated with 2.0 μg of mouse anti-α1 antibody (clone BD24, Millipore) or normal mouse IgG (negative control for nonspecific binding) for 1 h at 4 °C and then with 30 μl of protein A/G Plus-agarose beads overnight at 4 °C. The beads were collected by centrifugation at 8000 × g for 30 s and washed three times with lysis buffer. The α1 subunit complex was eluted by incubation with 30 μl of SDS loading buffer in the presence of DTT. The immunopurified eluents were separated in an 8% SDS-PAGE gel, and Western blot analysis was performed.
siRNA Transfection
HEK293 cells were seeded at ∼2.5 × 105 cells/well in 6-well plates for siRNA treatment. Cells were allowed to reach ∼70% confluency before transfection. The following siRNA duplexes were obtained from Dharmacon: VCP (catalog nos. J-008727-09 and J-008727-12) and non-targeting siRNA (D-001810-01-20) as a negative control. Cells were transfected with corresponding 50 nm siRNA using HiPerfect transfection reagent (Qiagen) according to the transfection protocol of the manufacturer prior to protein analysis. Forty-eight hours post-transfection, cells were harvested, lysed, and subjected to SDS-PAGE and Western blot analysis.
Cycloheximide Chase Assay
HEK293 cells stably overexpressing α1(A322D)β2γ2 GABAA receptors were seeded at 2.5 × 105 cells/well in 6-well plates and incubated at 37 °C overnight. Cells were then treated with the chemical compounds or dimethyl sulfoxide vehicle control prior to cycloheximide chase. To stop protein translation, cells were treated with 100 μg/ml cycloheximide (Ameresco) for the indicated time before being lysed for protein analysis.
Biotinylation of Cell Surface Proteins
HEK293 and SH-SY5Y cells stably overexpressing α1(A322D)β2γ2 receptors were plated in 10-cm dishes for surface biotinylation experiments according to published procedures (31). Intact cells were washed twice with ice-cold PBS and incubated with the membrane-impermeable biotinylation reagent Sulfo-NHS SS-Biotin (0.5 mg/ml, Pierce) in PBS containing 0.1 mm CaCl2 and 1 mm MgCl2 (PBS + CM) for 30 min at 4 °C to label surface membrane proteins. To quench the reaction, cells were incubated with 10 mm glycine in ice-cold PBS + CM twice for 5 min at 4 °C. Sulfhydryl groups were blocked by incubating the cells with 5 nm N-ethylmaleimide in PBS for 15 min at room temperature. Cells were solubilized for 1 h at 4 °C in lysis buffer (1% Triton X-100, 50 mm Tris-HCl, 150 mm NaCl, and 5 mm EDTA (pH 7.5)) supplemented with Roche complete protease inhibitor mixture and 5 mm N-ethylmaleimide. The lysates were cleared by centrifugation (16,000 × g, 10 min at 4 °C) to pellet cellular debris. The supernatant contained the biotinylated surface proteins. The concentration of the supernatant was measured using a microBCA assay (Pierce). Biotinylated surface proteins were affinity-purified from the above supernatant by incubating for 1 h at 4 °C with 100 μl of immobilized neutravidin-conjugated agarose bead slurry (Pierce) and being subjected to centrifugation (16,000 × g, 10 min, at 4 °C). The beads were washed six times with buffer (0.5% Triton X-100, 50 mm Tris-HCl, 150 mm NaCl, and 5 mm EDTA (pH 7.5)). Surface proteins were eluted from beads by boiling for 5 min with 200 μl of Laemmli sample buffer/urea buffer (2× Laemmli sample buffer with 100 mm DTT and 6 m urea (pH 6.8)) for SDS-PAGE and Western blot analysis.
Whole-cell Patch Clamp Electrophysiology Recording
Whole-cell currents were recorded 48 h post-transfection using HEK293 cells. The glass electrodes were pulled from thin-walled borosilicate capillary glass (Kimble-Chase) and fire-polished on a DMZ universal puller (Zeitz Instruments), having a tip resistance of 3–5 MΩ. The internal solution contained 153 mm KCl, 1 mm MgCl2, 5 mm EGTA, 10 mm HEPES, and 2 mm MgATP (pH 7.3). The external solution contained 142 mm NaCl, 8 mm KCl, 6 mm MgCl2, 1 mm CaCl2, 10 mm glucose, 10 mm HEPES, and 120 nm fenvalerate (pH 7.4). Coverslips containing HEK293 cells were placed in an RC-25 recording chamber (Warner Instruments) on the stage of an Olympus IX-71 inverted fluorescence microscope and perfused with external solution. Fast GABA application was accomplished with a pressure-controlled perfusion system (Warner Instruments) positioned within 50 μm of the cell utilizing a Quartz MicroManifold with 100-μm inner diameter inlet tubes (ALA Scientific). The whole-cell GABA-induced currents were recorded at a holding potential of −20 mV in voltage clamp mode using an Axopatch 200B amplifier (Molecular Devices). The signals were filtered at 2 kHz and detected at 10 kHz using pClamp10 acquisition software.
Quantitative RT-PCR
The relative expression levels of target genes were analyzed using quantitative RT-PCR according to a published procedure (34). Cells were incubated with the chemical compounds at 37 °C for the indicated amount of time before total RNA was extracted from the cells using an RNeasy mini kit (Qiagen, catalog no. 74104). cDNA was synthesized from 500 ng of total RNA using a QuantiTect reverse transcription kit (Qiagen, catalog no. 205311). Quantitative PCR reactions (45 cycles of 15 s at 94 °C, 30 s at 57 °C, and 30 s at 72 °C) were performed using cDNA, a QuantiTect SYBR Green PCR kit (Qiagen, catalog no. 204143), and corresponding primers in the StepOnePlus system (Applied Biosystems) and analyzed using StepOne v2.2 software (Applied Biosystems). The forward and reverse primers for CHOP and GAPDH (housekeeping gene control) were as follows: CHOP forward (5′ > 3′) GGAAACAGAGTGGTCATTCCC and reverse (5′ > 3′) CTGCTTGAGCCGTTCATTCTC and GAPDH forward (5′ > 3′) GTCGGAGTCAACGGATT and reverse (5′ > 3′) AAGCTTCCCGTTCTCAG. The threshold cycle (CT) was extracted from the PCR amplification plot, and the ΔCT value was defined as ΔCT = CT (target gene) − CT (housekeeping gene). The relative mRNA expression level of target genes of the chemical compound-treated cells was normalized to that of untreated cells as follows: relative mRNA expression level = 2 exp [− (ΔCT (treated cells) − ΔCT (untreated cells))]. Each data point was evaluated in triplicate and measured three times.
RT-PCR Analysis of XBP-1 Splicing
After reverse transcription, PCR reactions were performed using cDNA, TaqDNA polymerase, and primers against XBP-1 and GAPDH as follows: XBP-1 forward (5′ > 3′) TTACGAGAGAAAACTCATGGC and reverse (5′ > 3′) GGGTCCAAGTTGTCCAGAATGC and GAPDH forward (5′ > 3′) GTCGGAGTCAACGGATT and reverse (5′ > 3′) AAGCTTCCCGTTCTCAG. PCR products were separated on a 2.5% agarose gel. Unspliced XBP-1 yielded a 289-bp amplicon, and spliced XBP-1 yielded a 263-bp amplicon.
Statistical Analysis
All data are presented as mean ± S.E., and any statistical significance was calculated using two-tailed Student's t test.
RESULTS
VCP Interacts Stronger with Misfolding-prone α1(A322D) Subunits Than with WT α1 Subunits
It has been reported that the A322D mutation in the α1 subunit resulted in rapid degradation of the α1(A322D) subunit mainly by ERAD (29). We confirmed this result by adding lactacystin (5 μm, 24 h), a potent proteasome inhibitor, to HEK293 cells overexpressing WT or α1(A322D)β2γ2 GABAA receptors. Lactacystin treatment clearly increased the total protein level for the WT α1 subunit (Fig. 1B, cf. lane 4 with lane 1) and the α1(A322D) subunit (Fig. 1B, cf. lane 8 with lane 6), indicating that proteasome inhibition led to the accumulation of total α1 subunits in cells. To determine whether lactacystin enhanced the maturation of the α1(A322D) subunit, we used an endo H enzyme digestion assay to monitor its folding and trafficking. The endo H enzyme selectively cleaves after N-acetylglucosamine (GlcNAc) in the N-linked glycans incorporated on the α1 subunit in the ER. After the high-mannose form is remodeled in the Golgi, endo H is not able to remove the oligosaccharide chain. Therefore, endo H-resistant α1 subunits represent properly folded, post-ER α1 subunit glycoforms that traffic at least to the Golgi compartment. The peptide-N-glycosidase F enzyme cleaves between the innermost GlcNAc and asparagine residues from N-linked glycoproteins, serving as a control for unglycosylated α1 subunits (Fig. 1B, lane 3). Lactacystin treatment only increased the immature endo H-sensitive ER glycoforms of the α1(A322D) subunit (Fig. 1B, cf. lane 9 with lane 7), indicating that proteasome inhibition itself did not enhance the folding and trafficking of the α1(A322D) subunit.
Presumably, VCP extracts misfolded α1(A322D) subunits, a well established ERAD substrate (3, 29), from the ER membrane to the cytosol. To confirm a cellular interaction between VCP and α1 subunits, HEK293 cells expressing WT or α1(A322D)β2γ2 GABAA receptors were immunoprecipitated using an anti-α1 antibody and blotted for VCP. Clearly, VCP was pulled down with WT α1 and α1(A322D) subunits (Fig. 1C, lanes 6 and 7), indicating that both WT α1 and α1(A322D) subunits interact with VCP directly or indirectly. Furthermore, the ratio of immunoprecipitated VCP/α1 is 2.7-fold higher in cells expressing α1(A322D) subunits than that in cells expressing WT subunits (Fig. 1C, cf. lane 7 with lane 6, see Fig. 1D for quantification), indicating that VCP interacts stronger with misfolding-prone α1(A322D) subunits than with WT α1 subunits.
Silencing VCP Increases the Total Protein Level and Trafficking Efficiency of the α1(A322D) Subunit
Because of the critical role of VCP in extracting misfolded α1 subunits from the ER to the cytosol for degradation, we hypothesized that silencing VCP increased the total α1(A322D) protein level. Knockdown of VCP using siRNA (69% knockdown efficiency) increased the total protein level of α1(A322D) subunits 3.5-fold in HEK293 cells expressing α1(A322D)β2γ2 GABAA receptors, whereas knockdown of VCP using siRNA (60% knockdown efficiency) increased the total protein level of α1 subunits 1.6-fold in HEK293 cells expressing WT GABAA receptors (Fig. 2A, quantification is shown in the right panel). In addition, we tested a second VCP siRNA to avoid a potential siRNA off-target effect. As shown in Fig. 2B, VCP depletion using this siRNA also significantly increased both WT and mutant α1 subunit total protein levels, although less increase was observed than with the application of the first VCP siRNA. In both VCP siRNA treatment cases, a more dramatic increase of the total protein was found in cells expressing α1(A322D) subunits than in those expressing WT α1 subunits.
FIGURE 2.

Silencing VCP enhances the trafficking of the α1(A322D) subunit in HEK293 cells stably expressing α1(A322D)β2γ2 GABAA receptors. A and B, silencing VCP using siRNA1 (A) or siRNA2 (B) increases the total protein level of the α1 subunit in HEK293 cells stably expressing WT α1β2γ2 or α1(A322D)β2γ2 GABAA receptors. Cells were transfected with 50 nm siRNA1 or siRNA2 for VCP using transfection reagent prior to protein analysis. Forty-eight hours post-transfection, cells were harvested, lysed, and subjected to SDS-PAGE and Western blot analysis (n = 3). The intensity of α1 subunit was quantified, normalized to that of the WT, and is shown in the right panels. IB, immunoblot. C and D, knockdown of VCP using siRNA1 (C) or siRNA2 (D) increases the endo H-resistant post-ER glycoform of the α1(A322D) subunit. Forty-eight hours post-siRNA transfection, cells were harvested, lysed, digested by endo H enzyme, and subjected to SDS-PAGE and Western blot analysis (n = 3). After endo H digestion, subunits with a molecular weight equal to unglycosylated α1 subunits were considered endo H-sensitive (bottom arrow), whereas those with a higher molecular weight were considered endo H-resistant (top arrow) (lanes 2 and 4). The curly brackets indicate the top endo H-resistant bands in lanes 2 and 4. There are two endo H-resistant bands because the α1 subunit has two glycosylation sites (Asn-38 and Asn-138) in the ER. The ratio of endo H-resistant to total α1(A322D) serves as a measure of trafficking efficiency of the α1(A322D) subunit. Quantification of the ratio of endo H-resistant/total α1(A322D) subunit bands is shown in the right panels. E, silencing VCP using siRNA1 increases the BiP protein level. Forty-eight hours post-siRNA transfection, cells were harvested, lysed, and subjected to SDS-PAGE and immunoblotting (n = 2). *, p < 0.05; **, p < 0.01.
To determine whether the increased total α1(A322D) subunit protein after VCP depletion folded properly in the ER, we used an endo H enzyme digestion assay. Knockdown of VCP using siRNA (69% knockdown efficiency) clearly increased the upper two endo H-resistant α1(A322D) subunit bands in HEK293 cells (Fig. 2C, cf. lane 4 with lane 2), indicating that silencing VCP increased properly folded, post-ER glycoforms of the α1(A322D) subunit. The ratio of endo H-resistant/total α1(A322D) serves as a measure of trafficking efficiency of the α1(A322D) subunit. VCP knockdown significantly increased the ratio of endo H-resistant/total α1(A322D) (Fig. 2C, cf. lane 4 with lane 2, quantification is shown in the right panel), indicating that VCP knockdown increased the trafficking efficiency of the α1(A322D) subunit. Consistently, the application of a second VCP siRNA also yielded a significant increase in α1(A322D) subunit trafficking efficiency (Fig. 2D).
Silencing VCP increased the protein level of BiP but not Hsp70, Hsp90, or Grp94 in HEK293 cells expressing α1(A322D)β2γ2 receptors (Fig. 2E). We showed previously that BiP overexpression promotes the folding and trafficking of the α1(A322D) subunit (33). Therefore, an increase in BiP protein level after VCP depletion could partially contribute to the enhanced trafficking of the α1(A322D) subunit.
However, VCP depletion (69% knockdown efficiency) caused cell toxicity (data not shown). Therefore, in the following experiments, we used a VCP inhibitor under non-toxic conditions and determined its effect on α1(A322D) subunit maturation.
Inhibiting VCP Using Eeyarestatin I Increases the Total Protein Level and Trafficking Efficiency of the α1(A322D) Subunit, and Coapplication of SAHA Enhances This Effect without an Induction of the Unfolded Protein Response
EerI is a potent VCP inhibitor (see Fig. 3A for its chemical structure) (35). No apparent cell toxicity was observed for 5 μm EerI treatment up to 6 h in HEK293 cells expressing α1(A322D)β2γ2 GABAA receptors according to trypan blue staining (data not shown). Therefore, we used this condition for our following experiments.
FIGURE 3.

Inhibiting VCP increases the total protein level and trafficking of the α1(A322D) subunit in HEK293 cells, and coapplication of SAHA enhances this effect without an induction of the Unfolded protein response. A, chemical structures of EerI (left panel) and SAHA (right panel). B, effect of EerI and SAHA on the total protein levels of the α1(A322D), β2, and γ2 subunits in HEK293 cells stably expressing α1(A322D)β2γ2 GABAA receptors. EerI (5 μm) was added to the cell culture medium for the last 6 h, and SAHA (2.5 μm) was added for 24 h before cell lysis and Western blot analysis (n = 3). The dosage regimen is shown at the top. The intensity of the α1(A322D), β2, or γ2 subunits was quantified, normalized to that of dimethyl sulfoxide (DMSO) vehicle control, and is shown in the right panel. IB, immunoblot. C and D, effect of EerI and SAHA on endo H-resistant α1 subunits in HEK293 cells expressing α1(A322D)β2γ2 (C) and WT α1β2γ2 receptors (D) (n = 3). The dosage regimen is the same as that in B. The curly brackets indicate the top endo H-resistant bands in lanes 2, 4, and 6 (C) and in lanes 2 and 4 (D). Quantification of the ratio of endo H-resistant/total α1 subunit bands is shown in the right panels. NT, non-targeting; Ctl, no enzyme digestion control; Eh, endo H. E, effect of EerI and SAHA on XBP1 splicing using RT-PCR analysis. HEK293 cells stably expressing α1(A322D)β2γ2 receptors were incubated with EerI (5 μm, 6 h) in the absence or presence of SAHA (2.5 μm, 24 h) prior to RNA extraction and RT-PCR analysis (n = 2). Unspliced XBP1 yielded a 289-bp amplicon, whereas spliced XBP1-s yielded a 263-bp amplicon. Thapsigargin (TG, 1 μm, 6 h) was used as a positive control to induce XBP1 splicing (lane 5, bottom band). GAPDH was used as a loading control. F, effect of EerI and SAHA on CHOP mRNA levels using quantitative RT-PCR analysis. After chemical compound incubations as in E, HEK293 cells were subjected to mRNA extraction and quantitative RT-PCR analysis. The experiments were done three times with triplicates each time. Thapsigargin (1 μm, 6 h) was used as a positive control to induce CHOP expression. G, EerI treatment (5 μm, 6 h) does not alter the protein levels of major chaperones in the ER in HEK293 cells stably expressing α1(A322D)β2γ2 GABAA receptors (n = 2). Each data point in B–D and F is reported as mean ± S.E. *, p < 0.05; **, p < 0.01.
EerI treatment (5 μm, 6 h) significantly increased the total protein level of the α1(A322D) subunit, but not the β2 or γ2 subunits, in HEK293 cells expressing α1(A322D)β2γ2 GABAA receptors (Fig. 3B, cf. lane 2 with lane 1). Coapplication of SAHA (2.5 μm, 24 h) with EerI (5 μm, 6 h) further increased the α1(A322D) subunit protein level compared with EerI treatment alone (Fig. 3B, cf. lane 3 with lane 2), and this combination increased the β2 or γ2 subunit protein level because of the effect of SAHA (Fig. 3B) (33).
Furthermore, EerI treatment (5 μm, 6 h) increased both the intensity of the endo H-resistant α1(A322D) subunit bands and the ratio of endo H-resistant/total α1(A322D) subunits (Fig. 3C, cf. lane 4 with lane 2, see the right panel for quantification), indicating that this treatment facilitated the trafficking of the α1(A322D) subunits. Moreover, coapplication of SAHA (2.5 μm, 24 h) with EerI (5 μm, 6 h) further promoted the trafficking of the α1(A322D) subunits (Fig. 3C, cf. lane 6 with lane 4, see the right panel for quantification). Interestingly, this combination also increased the trafficking efficiency of WT α1 subunits (Fig. 3D, cf. lane 4 with lane 2, see the right panel for quantification), presumably because certain amounts of WT subunits are also targeted to ERAD (Fig. 1B). However, the effect of this coapplication to increase the trafficking efficiency is more dramatic for α1(A322D) subunits than WT α1 subunits (cf. Fig. 3C with Fig. 3D).
We next evaluated whether such treatments induced the unfolded protein response (UPR). The UPR regulates ER proteostasis by enhancing the ER folding capacity through transcriptional up-regulation of ER chaperones as well as decreasing the ER folding load (36–38). If ER proteostasis cannot be reestablished during prolonged ER stress, then the UPR can lead to apoptosis. In mammals, three ER stress sensor proteins, IRE1, ATF6, and PERK, have been established. PERK activation eventually leads to apoptosis during sustained ER stress. We tested whether our treatments activated the IRE1 or PERK pathways. IRE1 responds to ER stress by oligomerization, resulting in trans-autophosphorylation that activates its cytosolic endonuclease function, which precisely splices the mRNA that encodes the transcription factor XBP1. RT-PCR analysis was used to detect the spliced form of XBP-1 (XBP1-s) in HEK293 cells expressing α1(A322D)β2γ2 receptors. Thapsigargin (1 μm, 6 h), a potent UPR inducer, was used as a positive control (Fig. 3E, lane 5). EerI treatment (5 μm, 6 h) with or without SAHA (2.5 μm, 24 h) did not lead to the detection of XBP1-s, indicating that the IRE1 arm was not activated (Fig. 3E, lanes 3 and 4). The transcription factor CCAAT/enhancer-binding protein homologous protein (CHOP, also known as DDIT3/GADD153) operates as a downstream component of PERK activation. Quantitative RT-PCR analysis showed that EerI treatment (5 μm, 6 h) with or without SAHA (2.5 μm, 24 h) only slightly increased CHOP mRNA levels, but not significantly (Fig. 3F), indicating that such treatments did not result in substantial CHOP induction or proapoptotic PERK activation. Consistently, EerI treatment (5 μm, 6 h) did not increase the protein levels of BiP and calnexin, which are UPR downstream targets, in HEK293 cells expressing α1(A322D) subunits (Fig. 3G).
EerI Treatment Reduces the ERAD of the α1(A322D) Subunit, and Coapplication of SAHA Further Attenuates Its ERAD without an Apparent Effect on Its Endocytosis
We next clarified whether the small molecule treatments decreased the degradation rate of α1(A322D) subunits using a cycloheximide (CHX) chase assay. The α1(A322D) subunit had a half-life of 45 min when fitted to a single exponential function (Fig. 4A), consistent with the reported [35S]methionine chase result (39, 40). EerI treatment (5 μm, 6 h) increased the half-life of the α1(A322D) subunit from 45 to 82 min, and, remarkably, coapplication of SAHA (2.5 μm, 24 h) further extended its half-life to 130 min (Fig. 4A). Moreover, EerI treatment (5 μm, 6 h) decreased the ubiquitin signal relative to α1(A322D) subunits, and coapplication of SAHA (2.5 μm, 24 h) further reduced this ubiquitin modification on the α1(A322D) subunits (Fig. 4B), indicating that EerI treatment reduced and SAHA treatment further attenuated the ERAD of α1(A322D) subunits.
FIGURE 4.

EerI treatment reduces the ERAD of the α1(A322D) subunit in HEK293 cells, and coapplication of SAHA further attenuates its ERAD without an apparent effect on its endocytosis. A, effect of EerI and SAHA on the degradation of the α1(A322D) subunit using CHX chase analysis. HEK293 cells stably expressing α1(A322D)β2γ2 receptors were treated with EerI (5 μm, 6 h) in the absence or presence of SAHA (2.5 μm, 24 h), followed by a chase with CHX (100 μg/ml), which inhibits protein synthesis, for the indicated time. Cells were harvested, lysed, and subjected to 8% SDS-PAGE gel and immunoblot (IB) analysis (n = 3). The dosage regimen is shown at the top. Degradation kinetics were plotted by quantifying α1(A322D) intensity against time after CHX addition. Data were normalized to the α1(A322D) intensity at t = 0 after CHX addition (right panel). DMSO, dimethyl sulfoxide. B, effect of EerI and SAHA on ubiquitinated α1(A322D) subunits. HEK293 cells stably expressing α1(A322D)β2γ2 receptors were treated with EerI (5 μm, 6 h) in the absence or presence of SAHA (2.5 μm, 24 h). Then the cells were lysed, immunoprecipitated using an anti-α1 antibody, and blotted for ubiquitin. The dosage regimen is shown at the top. The ubiquitin band intensity relative to α1(A322D) subunits post-immunoprecipitation (IP) was quantified and is shown in the right panel (n = 2). C, effect of EerI (5 μm, 6 h) and SAHA (2.5 μm, 24 h) on the cell surface α1(A322D) subunits in the absence or presence of a potent dynamin-1 inhibitor, dynole 34-2 (2.5 μm, 24 h). HEK293 cells stably expressing α1(A322D)β2γ2 receptors were treated with chemical compounds as in the dosage regimen. Then the surface α1(A322D) subunit was measured using a cell surface biotinylation assay. Quantification of the surface α1(A322D) subunit intensity is shown in the right panel (n = 3). Each data point in A, B, and C is reported as mean ± S.E. *, p < 0.05; **, p < 0.01.
Because GABAA receptors are known to undergo dynamin-1-dependent endocytosis on the plasma membrane (39, 41), we next examined whether the small molecule treatments inhibit this process. Surface biotinylation assay was used to quantify the cell surface α1(A322D) subunit level in HEK293 cells. Treatment with dynole 34-2 (2.5 μm, 24 h), a specific, potent dynamin-1 inhibitor (42), significantly increased the surface α1(A322D) subunit (Fig. 4C, cf. lane 4 with lane 1), consistent with our prior report (33). Coapplication of EerI and dynole 34-2 produced at least an additive effect to increase the surface α1(A322D) subunit (Fig. 4C, cf. lane 5 with lanes 2 and 4, see quantification in the right panel), indicating that EerI did not influence the dynamin-1-dependent endocytosis of the α1(A322D) subunit. Our prior study revealed that the effect of SAHA on α1(A322D) subunit maturation does not rely on dynamin-1-dependent endocytosis (33). Therefore, it appears that EerI, SAHA, and dynole 34-2 use different pathways to increase α1(A322D) subunit surface levels. Strikingly, coadministration of these three chemical compounds increased the cell surface α1(A322D) subunit level by 9.0-fold (Fig. 4C, lane 6), the best rescue we achieved for α1(A322D) subunits.
Combining EerI and SAHA Treatment Additively Increases the Cell Surface α1(A322D) Subunits
To determine whether properly folded α1(A322D) subunits after chemical compound treatments were successfully trafficked to the plasma membrane, we quantified the cell surface α1 subunit level using a cell surface biotinylation assay. EerI treatment (5 μm, 6 h) significantly increased the surface α1(A322D) subunits in HEK293 cells and neuronal SH-SY5Y cells (Fig. 5, A and B, cf. lane 2 with lane 1). Coapplication of EerI (5 μm, 6 h) and SAHA (2.5 μm, 24 h) yielded an additive effect to increase the cell surface α1(A322D) subunits in HEK293 cells and SH-SY5Y cells (Fig. 5, A and B, cf. lane 4 with lanes 2 and 3).
FIGURE 5.

EerI and SAHA yield an additive restoration of surface protein expression of the α1(A322D) subunit in HEK293 cells and neuronal SH-SY5Y cells. A and B, effect of EerI treatment, SAHA treatment, or coapplication on the surface α1(A322D) subunits in HEK293 cells (A) or neuronal SH-SY5Y cells (B) stably expressing α1(A322D)β2γ2 GABAA receptors. The cells were treated for 6 h with 5 μm EerI only and 24 h with 2.5 μm SAHA only, or cotreated with EerI (5 μm, 6 h) and SAHA (2.5 μm, 24 h). Then the cells were subjected to surface biotinylation experiments and analyzed using Western blot (n = 2). The dosage regimen is shown at the top. The intensity of the surface α1(A322D) protein was quantified using ImageJ software, normalized to the dimethyl sulfoxide vehicle control treatment, and is shown in the right panels. Ctl, control; IB, immunoblot. Each data point is reported as mean ± S.E. *, p < 0.05; **, p < 0.01.
Combining EerI and SAHA Treatment Additively Increases GABA-induced Currents
To study the functional consequence of increased cell surface levels of α1(A322D) subunits, we used whole-cell patch clamp electrophysiological experiments to record GABA-induced chloride currents. Compared with the WT, the A322D mutation in the α1 subunit resulted in significantly lowered GABA-induced currents. The peak current was 1.0 ± 0.4 picoampere (pA) in response to GABA (3 mm) in HEK293 cells expressing α1(A322D)β2γ2 receptors, whereas it was 138 ± 12 pA in response to GABA (1 mm) in HEK293 cells expressing WT α1β2γ2 receptors (Fig. 6A). EerI (5 μm, 6 h) treatment slightly increased the peak current to 4.3 ± 0.3 pA in HEK293 cells expressing α1(A322D)β2γ2 receptors (Fig. 6A, see Fig. 6B for quantification). Acute application of EerI (5 μm, 1 min) in the external perfusion-recording medium to HEK293 cells expressing GABAA receptors during the whole-cell patch clamp experiment did not significantly change the GABA-induced peak current (data not shown), indicating that EerI did not act as an antagonist of GABAA receptors. Furthermore, coapplication of EerI (5 μm, 6 h) and SAHA (2.5 μm, 24 h) increased the peak current to 33.6 ± 3.0 pA in HEK293 cells expressing α1(A322D)β2γ2 receptors, amounting to 24% of that in cells expressing WT receptors (Fig. 6, A and B). This combination significantly rescued function of the misfolding-prone GABAA receptors.
FIGURE 6.

EerI and SAHA yield an additive increase of the peak amplitude of GABA-induced chloride currents in HEK293 cells expressing α1(A322D)β2γ2 GABAA receptors. A, representative whole-cell patch clamp recording traces in HEK293 cells expressing WT or α1(A322D)β2γ2 GABAA receptors. HEK293 cells transiently expressing α1(A322D)β2γ2 GABAA receptors were treated with EerI (5 μm, 6 h) and/or SAHA (2.5 μm, 24 h) before whole-cell patch clamp recording with a holding potential of −20 mV. The dosage regimen is shown at the top. B, quantification of the peak currents (Imax). The number of patched cells in each group is shown at the top of the bar. AD, A322D. Each point in B is reported as mean ± S.E. **, p < 0.01.
DISCUSSION
Here we showed that VCP inhibition reduces the ERAD of α1(A322D) subunits without an apparent effect on their dynamin-1-dependent endocytosis. The α1(A322D) subunits cannot efficiently form the TM3 α helix in the ER membrane, resulting in its misfolding (40). Misfolded α1(A322D) subunits are recognized by the ER quality control machinery, polyubiquitinated, extracted from the ER membrane to the cytosol, and targeted to the proteasome for degradation. The major players in each ERAD step (15) for GABAA receptors have not yet been established. Our findings strongly support the critical role of VCP in extracting misfolded α1(A322D) subunits from the ER membrane to the cytosol for degradation. As a result, VCP inhibition stabilizes α1(A322D) subunits in the ER so that more proteins can engage the trafficking machinery for transport to the plasma membrane.
Furthermore, we demonstrated an additive rescue of mutant α1(A322D) subunit function in patch clamp experiments using both EerI and SAHA. EerI treatment inhibits the VCP-mediated ERAD and accumulates available mutant α1 subunits in the ER. Our prior study revealed that SAHA enhances the folding of α1(A322D) subunits posttranslationally by promoting calnexin and BiP-assisted folding. Therefore, increased mutant α1 subunits in the ER by EerI treatment can fold properly after SAHA treatment, yielding an additive rescue of mutant α1 subunit folding, trafficking, and, therefore, function (Fig. 7).
FIGURE 7.
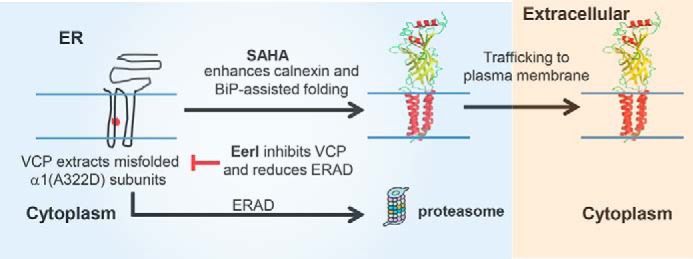
Proposed model for an additive rescue of the function of α1(A322D) subunits using both EerI and SAHA. VCP extracts misfolded α1(A322D) subunits from the ER membrane to the cytosol for degradation through ERAD. SAHA treatment promotes the folding and trafficking of mutant subunits posttranslationally by enhancing calnexin and BiP-assisted folding. EerI treatment inhibits VCP-mediated ERAD and accumulates more mutant subunits in the ER, which can be further rescued by SAHA treatment. Therefore, coapplication of SAHA and EerI yields an additive restoration of the function of mutant subunits. Red asterisk, A322D mutation in the α1 subunit. The folded subunit schematics are constructed using the crystal structure of GABAA receptor β3 subunits (PDB code 4COF) (48).
It is desirable to test the effect of SAHA and EerI in vivo. There are at least 19 GABAA receptor subunit isoforms in humans: six α subunits (α1–6), three β subunits (β1–3), three γ subunits (γ1–3), one δ subunit, one ϵ subunit, one θ subunit, one π subunit, and three ρ subunits (ρ1–3). Many knockout or knockin mice have been generated for various subunits of GABAA receptors (43), including α1 knockout mice (44–46). Recent studies showed that deleting α1 subunits can produce absence-like epilepsy (44). Because the A322D mutation leads to exceedingly low surface expression of α1 subunits and has a dominant negative effect (47), it is highly possible that the α1(A322D) knockin mice recapitulate epilepsy phenotypes. Therefore, it is of great interest to generate α1(A322D) knockin mice and examine the in vivo effect of SAHA and EerI in future studies. Because epilepsy is a threshold event, the significant functional rescue using this combination strategy might be clinical relevant and, therefore, holds great promise to be further developed for the treatment of idiopathic epilepsy resulting from GABAA receptor misfolding.
Acknowledgment
We thank Dr. Ya-Juan Wang (Case Western Reserve University) for critical input.
This work was supported by the Epilepsy Foundation of America (225243).
- ERAD
- endoplasmic reticulum-associated degradation
- VCP
- valosin-containing protein
- ER
- endoplasmic reticulum
- TM
- transmembrane
- SAHA
- suberanilohydroxamic acid
- EerI
- Eeyarestatin I
- endo H
- endoglycosidase H
- UPR
- unfolded protein response
- IRE1
- inositol-requiring kinase 1
- ATF6
- activating transcription factor 6
- PERK
- protein kinase RNA-like endoplasmic reticulum kinase
- CHOP
- CCAAT/enhancer-binding protein homologous protein
- CHX
- cycloheximide
- pA
- picoampere.
REFERENCES
- 1. Balch W. E., Morimoto R. I., Dillin A., Kelly J. W. (2008) Adapting proteostasis for disease intervention. Science 319, 916–919 [DOI] [PubMed] [Google Scholar]
- 2. Kim Y. E., Hipp M. S., Bracher A., Hayer-Hartl M., Hartl F. U. (2013) Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355 [DOI] [PubMed] [Google Scholar]
- 3. Guerriero C. J., Brodsky J. L. (2012) The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 92, 537–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gidalevitz T., Prahlad V., Morimoto R. I. (2011) The stress of protein misfolding: from single cells to multicellular organisms. Cold Spring Harb. Perspect. Biol. 3, a009704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vilchez D., Simic M. S., Dillin A. (2014) Proteostasis and aging of stem cells. Trends Cell Biol. 24, 161–170 [DOI] [PubMed] [Google Scholar]
- 6. Franz A., Ackermann L., Hoppe T. (2014) Create and preserve: proteostasis in development and aging is governed by Cdc48/p97/VCP. Biochim. Biophys. Acta 1843, 205–215 [DOI] [PubMed] [Google Scholar]
- 7. Zhang X., Shaw A., Bates P. A., Newman R. H., Gowen B., Orlova E., Gorman M. A., Kondo H., Dokurno P., Lally J., Leonard G., Meyer H., van Heel M., Freemont P. S. (2000) Structure of the AAA ATPase p97. Mol. Cell 6, 1473–1484 [DOI] [PubMed] [Google Scholar]
- 8. Meyer H., Bug M., Bremer S. (2012) Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123 [DOI] [PubMed] [Google Scholar]
- 9. Yamanaka K., Sasagawa Y., Ogura T. (2012) Recent advances in p97/VCP/Cdc48 cellular functions. Biochim. Biophys. Acta 1823, 130–137 [DOI] [PubMed] [Google Scholar]
- 10. Wolf D. H., Stolz A. (2012) The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim. Biophys. Acta 1823, 117–124 [DOI] [PubMed] [Google Scholar]
- 11. Olzmann J. A., Kopito R. R., Christianson J. C. (2013) The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol. 5, a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leitman J., Ron E., Ogen-Shtern N., Lederkremer G. Z. (2013) Compartmentalization of endoplasmic reticulum quality control and ER-associated degradation factors. DNA Cell Biol. 32, 2–7 [DOI] [PubMed] [Google Scholar]
- 13. Claessen J. H., Kundrat L., Ploegh H. L. (2012) Protein quality control in the ER: balancing the ubiquitin checkbook. Trends Cell Biol. 22, 22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brodsky J. L. (2012) Cleaning up: ER-associated degradation to the rescue. Cell 151, 1163–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vembar S. S., Brodsky J. L. (2008) One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vij N., Fang S., Zeitlin P. L. (2006) Selective inhibition of endoplasmic reticulum-associated degradation rescues ΔF508-cystic fibrosis transmembrane regulator and suppresses interleukin-8 levels: therapeutic implications. J. Biol. Chem. 281, 17369–17378 [DOI] [PubMed] [Google Scholar]
- 17. Wang F., Song W., Brancati G., Segatori L. (2011) Inhibition of endoplasmic reticulum-associated degradation rescues native folding in loss of function protein misfolding diseases. J. Biol. Chem. 286, 43454–43464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wójcik C., Rowicka M., Kudlicki A., Nowis D., McConnell E., Kujawa M., DeMartino G. N. (2006) Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol. Biol. Cell 17, 4606–4618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Macdonald R. L., Olsen R. W. (1994) GABAA receptor channels. Annu. Rev. Neurosci. 17, 569–602 [DOI] [PubMed] [Google Scholar]
- 20. Dougherty D. A. (2008) Cys-loop neuroreceptors: structure to the rescue? Chem. Rev. 108, 1642–1653 [DOI] [PubMed] [Google Scholar]
- 21. Lester H. A., Dibas M. I., Dahan D. S., Leite J. F., Dougherty D. A. (2004) Cys-loop receptors: new twists and turns. Trends Neurosci. 27, 329–336 [DOI] [PubMed] [Google Scholar]
- 22. Mowrey D. D., Kinde M. N., Xu Y., Tang P. (2015) Atomistic insights into human Cys-loop receptors by solution NMR. Biochim. Biophys. Acta Biomembranes 1848, 307–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen A. S., Berkovic S. F., Cossette P., Delanty N., Dlugos D., Eichler E. E., Epstein M. P., Glauser T., Goldstein D. B., Han Y., Heinzen E. L., Hitomi Y., Howell K. B., Johnson M. R., Kuzniecky R., Lowenstein D. H., Lu Y. F., Madou M. R., Marson A. G., Mefford H. C., Esmaeeli Nieh S., O'Brien T. J., Ottman R., Petrovski S., Poduri A., Ruzzo E. K., Scheffer I. E., Sherr E. H., Yuskaitis C. J., Abou-Khalil B., Alldredge B. K., Bautista J. F., Berkovic S. F., Boro A., Cascino G. D., Consalvo D., Crumrine P., Devinsky O., Dlugos D., Epstein M. P., Fiol M., Fountain N. B., French J., Friedman D., Geller E. B., Glauser T., Glynn S., Haut S. R., Hayward J., Helmers S. L., Joshi S., Kanner A., Kirsch H. E., Knowlton R. C., Kossoff E. H., Kuperman R., Kuzniecky R., Lowenstein D. H., McGuire S. M., Motika P. V., Novotny E. J., Ottman R., Paolicchi J. M., Parent J. M., Park K., Poduri A., Scheffer I. E., Shellhaas R. A., Sherr E. H., Shih J. J., Singh R., Sirven J., Smith M. C., Sullivan J., Lin Thio L., Venkat A., Vining E. P., Von Allmen G. K., Weisenberg J. L., Widdess-Walsh P., Winawer M. R. (2013) De novo mutations in epileptic encephalopathies. Nature 501, 217–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Noebels J. L. (2003) The biology of epilepsy genes. Annu. Rev. Neurosci. 26, 599–625 [DOI] [PubMed] [Google Scholar]
- 25. Steinlein O. K. (2004) Genetic mechanisms that underlie epilepsy. Nat. Rev. Neurosci. 5, 400–408 [DOI] [PubMed] [Google Scholar]
- 26. Macdonald R. L., Kang J. Q., Gallagher M. J. (2010) Mutations in GABAA receptor subunits associated with genetic epilepsies. J. Physiol. 588, 1861–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Steinlein O. K. (2012) Ion channel mutations in neuronal diseases: a genetics perspective. Chem. Rev. 112, 6334–6352 [DOI] [PubMed] [Google Scholar]
- 28. Cossette P., Liu L., Brisebois K., Dong H., Lortie A., Vanasse M., Saint-Hilaire J. M., Carmant L., Verner A., Lu W. Y., Wang Y. T., Rouleau G. A. (2002) Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat. Genet. 31, 184–189 [DOI] [PubMed] [Google Scholar]
- 29. Gallagher M. J., Shen W., Song L., Macdonald R. L. (2005) Endoplasmic reticulum retention and associated degradation of a GABAA receptor epilepsy mutation that inserts an aspartate in the M3 transmembrane segment of the α 1 subunit. J. Biol. Chem. 280, 37995–38004 [DOI] [PubMed] [Google Scholar]
- 30. Krampfl K., Maljevic S., Cossette P., Ziegler E., Rouleau G. A., Lerche H., Bufler J. (2005) Molecular analysis of the A322D mutation in the GABAA receptor α(1)-subunit causing juvenile myoclonic epilepsy. Eur. J. Neurosci. 22, 10–20 [DOI] [PubMed] [Google Scholar]
- 31. Lachance-Touchette P., Brown P., Meloche C., Kinirons P., Lapointe L., Lacasse H., Lortie A., Carmant L., Bedford F., Bowie D., Cossette P. (2011) Novel α 1 and γ 2 GABA(A) receptor subunit mutations in families with idiopathic generalized epilepsy. Eur. J. Neurosci. 34, 237–249 [DOI] [PubMed] [Google Scholar]
- 32. Wang Y. J., Han D. Y., Tabib T., Yates J. R., 3rd, Mu T. W. (2013) Identification of GABAC receptor protein homeostasis network components from three tandem mass spectrometry proteomics approaches. J. Proteome Res. 12, 5570–5586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Di X. J., Han D. Y., Wang Y. J., Chance M. R., Mu T. W. (2013) SAHA enhances proteostasis of epilepsy-associated α1(A322D)β2γ2 GABAA receptors. Chem. Biol. 20, 1456–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mu T. W., Ong D. S., Wang Y. J., Balch W. E., Yates J. R., 3rd, Segatori L., Kelly J. W. (2008) Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell 134, 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Q., Li L., Ye Y. (2008) Inhibition of p97-dependent protein degradation by Eeyarestatin I. J. Biol. Chem. 283, 7445–7454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gardner B. M., Pincus D., Gotthardt K., Gallagher C. M., Walter P. (2013) Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 5, a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang S., Kaufman R. J. (2012) The impact of the unfolded protein response on human disease. J. Cell Biol. 197, 857–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pavitt G. D., Ron D. (2012) New insights into translational regulation in the endoplasmic reticulum unfolded protein response. Cold Spring Harb. Perspect. Biol. 4, a012278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bradley C. A., Taghibiglou C., Collingridge G. L., Wang Y. T. (2008) Mechanisms involved in the reduction of GABAA receptor α 1-subunit expression caused by the epilepsy mutation A322D in the trafficking-competent receptor. J. Biol. Chem. 283, 22043–22050 [DOI] [PubMed] [Google Scholar]
- 40. Gallagher M. J., Ding L., Maheshwari A., Macdonald R. L. (2007) The GABAA receptor α 1 subunit epilepsy mutation A322D inhibits transmembrane helix formation and causes proteasomal degradation. Proc. Natl. Acad. Sci. U.S.A. 104, 12999–13004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Herring D., Huang R., Singh M., Robinson L. C., Dillon G. H., Leidenheimer N. J. (2003) Constitutive GABAA receptor endocytosis is dynamin-mediated and dependent on a dileucine AP2 adaptin-binding motif within the β 2 subunit of the receptor. J. Biol. Chem. 278, 24046–24052 [DOI] [PubMed] [Google Scholar]
- 42. Hill T. A., Gordon C. P., McGeachie A. B., Venn-Brown B., Odell L. R., Chau N., Quan A., Mariana A., Sakoff J. A., Chircop M., Robinson P. J., McCluskey A. (2009) Inhibition of dynamin mediated endocytosis by the dynoles: synthesis and functional activity of a family of indoles. J. Med. Chem. 52, 3762–3773 [DOI] [PubMed] [Google Scholar]
- 43. Rudolph U., Möhler H. (2004) Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annu. Rev. Pharmacol. Toxicol. 44, 475–498 [DOI] [PubMed] [Google Scholar]
- 44. Arain F. M., Boyd K. L., Gallagher M. J. (2012) Decreased viability and absence-like epilepsy in mice lacking or deficient in the GABAA receptor α1 subunit. Epilepsia 53, e161–e165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kralic J. E., Korpi E. R., O'Buckley T. K., Homanics G. E., Morrow A. L. (2002) Molecular and pharmacological characterization of GABAA receptor α1 subunit knockout mice. J. Pharmacol. Exp. Ther. 302, 1037–1045 [DOI] [PubMed] [Google Scholar]
- 46. Vicini S., Ferguson C., Prybylowski K., Kralic J., Morrow A. L., Homanics G. E. (2001) GABA(A) receptor α1 subunit deletion prevents developmental changes of inhibitory synaptic currents in cerebellar neurons. J. Neurosci. 21, 3009–3016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ding L., Feng H.-J., Macdonald R. L., Botzolakis E. J., Hu N., Gallagher M. J. (2010) GABAA receptor α 1 subunit mutation A322D associated with autosomal dominant juvenile myoclonic epilepsy reduces the expression and alters the composition of wild type GABAA receptors. J. Biol. Chem. 285, 26390–26405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Miller P. S., Aricescu A. R. (2014) Crystal structure of a human GABAA receptor. Nature 512, 270–275 [DOI] [PMC free article] [PubMed] [Google Scholar]