

Background: ACTN4 is an actin-binding protein and is associated with kidney diseases.
Results: Nuclear ACTN4 interacts with NF-κB and is recruited to NF-κB-targeted promoters to potentiate its transcription activity.
Conclusion: ACTN4 coactivates NF-κB activity independently of its cytoplasmic activity in cultured human podocytes.
Significance: Nuclear ACTN4 may play an important role in glomerular function.
Keywords: Glucocorticoid Receptor, NF-kappa B (NF-KB), Podocyte, Transcription Regulation, Transcriptional Coactivator, Alpha-Actinin 4, Focal Segmental Glomerulosclerosis, FSGS, ACTN4, Glomerulopathies
Abstract
Glomerular podocytes are highly specialized terminally differentiated cells that act as a filtration barrier in the kidney. Mutations in the actin-binding protein, α-actinin 4 (ACTN4), are linked to focal segmental glomerulosclerosis (FSGS), a chronic kidney disease characterized by proteinuria. Aberrant activation of NF-κB pathway in podocytes is implicated in glomerular diseases including proteinuria. We demonstrate here that stable knockdown of ACTN4 in podocytes significantly reduces TNFα-mediated induction of NF-κB target genes, including IL-1β and NPHS1, and activation of an NF-κB-driven reporter without interfering with p65 nuclear translocation. Overexpression of ACTN4 and an actin binding-defective variant increases the reporter activity. In contrast, an FSGS-linked ACTN4 mutant, K255E, which has increased actin binding activity and is predominantly cytoplasmic, fails to potentiate NF-κB activity. Mechanistically, IκBα blocks the association of ACTN4 and p65 in the cytosol. In response to TNFα, both NF-κB subunits p65 and p50 translocate to the nucleus, where they bind and recruit ACTN4 to their targeted promoters, IL-1β and IL-8. Taken together, our data identify ACTN4 as a novel coactivator for NF-κB transcription factors in podocytes. Importantly, this nuclear function of ACTN4 is independent of its actin binding activity in the cytoplasm.
Introduction
The kidney glomerular podocytes are highly specialized epithelial cells that extend numerous lamellipodia that branch into primary and secondary processes, which further ramify into smaller processes known as foot processes (1). These foot processes are spanned by slit diaphragms containing nephrin that prevent the filtration of larger plasma proteins into the urine. The maintenance of the slit diaphragm is critical for proper glomerular filtration and kidney function.
Mutations in several genes encoding components of the slit diaphragm or the foot processes have been linked to familial forms of FSGS2 (for review, see Refs. 2 and 3), including α-actinin-4 (ACTN4) (4, 5). ACTN4 was originally isolated as a protein that binds F-actin to modulate cytoskeletal organization and cell motility (6), and initial theories of its function in FSGS have centered on this role, postulating that increased actin binding activity contributes to pathogenesis. However, the mechanisms by which FSGS-linked ACTN4 mutations cause glomerular diseases remain elusive.
While investigating histone deacetylase 7 (HDAC7)-interacting proteins, we identified ACTN4 as a transcriptional co-activator for MEF2 transcription factors (7). This finding was recently confirmed by An et al. (8). We also established that ACTN4 has a regulatory function on transcription mediated by nuclear hormone receptors such as estrogen receptor, retinoic acid receptor, peroxisome proliferator-activated receptor, and vitamin D receptor via its N-terminal nuclear receptor-interacting motif, LXXLL (9, 10). Notably, FSGS-linked ACTN4 mutants, which are predominantly cytoplasmic, lose their ability to potentiate hormone-dependent transcriptional activation (11). Based on these observations, we conclude that ACTN4 plays a role in both cytoskeletal dynamics and transcription through nuclear hormone receptor signaling.
NF-κB transcription factors regulate immune responses, cell proliferation, apoptosis, and differentiation through controlling the expression of genes encoding pro-inflammatory cytokines, chemokines, and adhesion molecules (12, 13). NF-κB signaling comprises the canonical pathway that involves heterodimers of RelA/p65 and p105/p50 and the non-canonical pathway that includes heterodimers of RelB and p100/p52. The canonical pathways are activated by pattern recognition receptors and the tumor necrosis family receptors in response to a variety of pro-inflammatory signals including cytokines such as TNFα. Upon stimulation, the IκB kinase complex phosphorylates IκB (the cytoplasmic repressor for the NF-κB p50/p65 heterodimer), which promotes IκB degradation and subsequent p65/p50 heterodimer release and translocation to the nucleus. Upon nuclear translocation, NF-κB transcription factors bind their target genes and recruit chromatin modification enzymes and transcriptional cofactors to regulate specific target gene expression.
Several studies have linked dysregulated activation of NF-κB with human chronic kidney diseases, podocyte function, and nephrin expression levels (14–20). Because of the known interactions between the nuclear hormone glucocorticoid receptor (GR) and NF-κB signaling, and also the importance of glucocorticoids in the treatment of proteinuric kidney diseases, we wished to determine whether ACTN4 participates in transcriptional responses mediated by NF-κB and GR. In this study, we describe a novel role for ACTN4 as a transcriptional coactivator of NF-κB in podocytes. In addition, the previously described FSGS-linked ACTN4 mutant, K255E, fails to potentiate NF-κB activity (11). These transcriptional regulatory functions appear to be independent of cytoskeletal functions associated with actin binding defects of the K255E ACTN4 mutation. Taken together, our findings highlight a previously underappreciated function of ACTN4 in transcriptional activation in glomerular physiology and a novel pathological consequence of the mutations in ACTN4 associated with FSGS.
EXPERIMENTAL PROCEDURES
Cell Culture and Differentiation
HEK293T and HeLa cells were grown in DMEM supplemented with 10% FBS and penicillin-streptomycin (50 units/ml) at 37 °C in 5% CO2. Human immortalized podocytes (HPCs) were cultured in RPMI 1640 (Life Technologies) supplemented with 10% fetal bovine serum, penicillin-streptomycin (50 units/ml), and insulin-transferrin-selenite (Sigma). HPCs were grown at the permissive temperature of 33 °C and transferred to 37 °C to induce differentiation for 3 days prior to treatments. Cells were starved in RPMI 1640 supplemented with 2% fetal bovine serum, penicillin-streptomycin (50 units/ml), and insulin-transferrin-selenite.
Plasmid Construction
CMX-HA-ACTN4, GST-ACTN4 (WT), different ACTN4 mutations expression plasmids, as well as GST-ACTN4 deletion constructs were described previously (9–11). The expression plasmids CMX-HA-p65 were generated by PCR and subcloned into a CMX-1H vector (21). For MCP1-Luc reporter plasmid, 3 kb upstream of MCP-1 transcription start site was subcloned into the CMX-Luc reporter vector.
Antibodies and Chemicals
The anti-ACTN4 antibodies have been described previously (7). For immunoprecipitation assays, anti-HA antibodies (Santa Cruz Biotechnology Sc-805), anti-p65 antibodies (Santa Cruz Biotechnology Sc-372) were used. For immunostaining, anti-HA (Santa Cruz Biotechnology Sc-805), anti-p65 (Santa Cruz Biotechnology Sc-372), anti-lamin B (Sc-6216), and anti-α-tubulin (T-5168, Sigma-Aldrich) antibodies were used. The secondary antibodies for immunofluorescence were from Life Technologies (anti-mouse or anti-rabbit Alexa Fluor 488,0 or Alexa Fluor 594). TNFα was purchased from Promega (G5241), and dexamethasone (D4902) was purchased from Sigma-Aldrich.
Generation of Stable Knockdown Cells
shRNAs against human ACTN4 (TRCN0000055783, TRCN0000055784, TRCN0000055785, TRCN0000055786, and TRCN0000055787), shRNA against human IκBα (TRCN0000004539, TRCN0000004540, TRCN0000004541, and TRCN0000004542), and shRNA against human HDAC7 (TRCN0000195442) were purchased from Sigma-Aldrich. The packaging plasmids used were pMD2.G and psPAX2, which were kindly provided by Dr. Barbara Bedogni. After cotransfection into the HEK293T packaging cell line with shRNA and packaging plasmid, the supernatants containing the lentivirus were collected and used to infect the HEK293T and HPC cell lines. Cells were stably infected with lentivirus and selected in puromycin-containing medium (1.5 μg/ml) for 2 days. Knockdown of ACTN4 was confirmed by Western blots.
Subcellular Fractionation
Isolation of nuclei and cytoplasm from HPCs and extraction of nuclear and cytosolic proteins were performed according to a published protocol (11). Nuclear and cytoplasmic fractions were resolved on SDS-polyacrylamide gels and followed by Western blotting with the indicated antibodies.
In Vitro Protein-Protein Interaction Assays
GST fusion proteins were expressed in Escherichia coli DH5α strain, affinity-purified, and immobilized on glutathione-Sepharose 4B beads. Immobilized, purified GST, GST-ACTN4, or GST-p65 fusion proteins were incubated with whole cell extracts expressing HA-p65, HA-ACTN4, or HA-p50 followed by extensive washes as described (22). The pulldown fractions were subjected to Western blotting with the HA antibodies. The immobilized GST and GST fusion proteins were visualized by Coomassie Brilliant Blue staining.
Coimmunoprecipitation
HPCs were treated with vehicle or TNFα (20 ng/ml) and harvested, and whole cell lysates were prepared (11). Immunoprecipitation and Western blotting assays were carried out following our published protocol (9).
Glomerular Isolation
Glomerular extracts were prepared following an established protocol (23). Twenty-week-old male C57BL/6 mice (n = 5) were anesthetized. Kidneys were harvested, decapsulated, minced, and digested, and the glomerular RNA were prepared using an RNA spin kit (USB Corp.).
Transient Transfection Reporter Assays
HEK293T cells stably expressing shRNA against human ACTN4 or HDAC7 or overexpressing wild-type or mutant HA-ACTN4 were cultured in 24-well plates. Cells were cotransfected with an NF-κB luciferase reporter (a generous gift from Dr. Bedogni) or MCP-1-Luc reporter and pCMX-β-galactosidase construct using Lipofectamine 2000. The cells were lysed in reporter lysis buffer 8 h after TNFα or 12 h after dexamethasone treatment and 48 h after transfection (Promega). Luciferase and β-gal activities were measured according to the manufacturer's protocol using a luciferase assay system (Promega). Luciferase activity was normalized to β-gal activity. Each reaction was performed in triplicates.
RNA Isolation and qPCR
Total RNA was extracted using the RNeasy isolation kit (Qiagen) and reverse-transcribed using reverse transcription kit (Bio-Rad) according to the manufacturer's instructions. The PCR primers used in this study and their sequences were previously described. The iQ SYBR Green PCR supermix (Bio-Rad and Qiagen) and the CFX96 real-time PCR detection system (Bio-Rad) were used to quantify cDNAs according to the manufacturer's instructions. The 18 S rRNA was used for normalization. The relative mRNA expression was calculated by the 2−(ΔΔCt) method. The PCR primers used in this study and their sequences were previously described (24).
ChIP Assay
ChIP assays were carried out as described with modifications (24). Chromatin was sheared by sonication, and the extracts were precleared by protein-A-coupled beads (Repligen). Immunoprecipitation was performed using anti-ACTN4 and anti-p65 antibodies. The PCR primers used in this study and their sequences were previously described (24).
Immunofluorescence Microscopy
HPCs were grown on a sterile coverslip in 12-well plates, treated with or without TNFα, fixed with 3.7% paraformaldehyde for 30 min at room temperature, and incubated with anti-p65, anti-ACTN4 (Santa Cruz Biotechnology). The secondary antibodies used were Alexa Fluor 488 and Alexa Flour 594 (Invitrogen). DAPI was applied to the samples after the final wash to visualize cell nuclei. Images were visualized using a Leica epifluorescence microscope.
Statistical Analysis
Data were expressed as the mean ± S.D. Analyses were performed with Student's t test using GraphPad Prism6 software. A value of p < 0.05 was considered to be significant. p < 0.05 and p < 0.001 are designated by * and **, respectively.
RESULTS
ACTN4 Is Required for NF-κB Transcriptional Activity
Our previous work has indicated that ACTN4 plays a role in transcriptional regulation by MEF2 transcription factors and nuclear hormone receptors (7). Because of the intersection of the nuclear hormone receptors and NF-κB signaling, we wished to investigate whether ACTN4 is also involved in NF-κB transcriptional regulation. We established several independent HEK293T and HPC cell lines stably expressing a control shRNA (shCtrl) or ACTN4 shRNA (shACTN4) expressing different levels of endogenous ACTN4 protein. We found that knockdown of ACTN4 decreased IL-1β mRNA expression, a known NF-κB target gene (Fig. 1A). These results indicated that ACTN4 positively regulated IL-1β expression in both cell lines and raised the possibility that ACTN4 played an important role in promoting NF-κB signaling. To test whether ACTN4 regulates p65-mediated transcriptional activity, we examined the effect of ACTN4 knockdown on an NF-κB-driven reporter activity and found that knockdown of ACTN4 significantly decreased basal and TNFα-induced NF-κB reporter activity (Fig. 1B). Consistently, overexpression of HA-ACTN4 increased TNFα-induced NF-κB reporter activity (Fig. 1C). We further generated a reporter construct with native MCP-1 promoter, a classical NF-κB target gene, and determined the effect of ACTN4 knockdown on the promoter activity. We found that knockdown of ACTN4 significantly blocked the ability of TNFα to induce MCP-1 promoter activity (Fig. 1D). Taken together, these data indicate that ACTN4 is important for TNFα-mediated transactivation activity of NF-κB.
FIGURE 1.

The effect of ACTN4 knockdown on NF-κB-mediated transcriptional activation. A, ACTN4 was stably knocked down by several short hairpin (sh) RNAs (shA1–4) targeting different regions of ACTN4 mRNA in HEK293T cells (lanes 1–5) or HPCs (lanes 6–8). A control shRNA (shCtrl) was used for comparison. The mRNA expression levels of IL-1β of corresponding cells are shown. B, control and ACTN4 stably knockdown HEK293T cells were transiently transfected with a plasmid constitutively expressing β-galactosidase and an NF-κB luciferase (Luc) reporter plasmid. NF-κB BS: NF-κB-binding site. Following transfection, cells were treated with TNFα (20 ng/ml) for 12 h, and luciferase activity was measured. A schematic representation of the reporter construct (top) and the expression of ACTN4 (bottom) are shown. C, HEK293T cells were cotransfected with an NF-κB luciferase reporter plasmid and increasing amounts of HA-ACTN4 expression plasmid followed by treatment with vehicle or TNFα (20 ng/ml) for 12 h, and luciferase activity was measured. B and C, the relative luciferase activity was normalized to β-galactosidase activity (*, p < 0.05). D, the MCP-1 promoter reporter construct and CMX-β-gal expression plasmid were transiently transfected into shCtrl or shA4-1 HEK293T cells, treated with or without TNFα, and the reporter activity was assayed. Veh., vehicle. Error bars indicate mean ± S.D.
Knockdown of ACTN4 Does Not Disrupt TNFα-induced p65 Nuclear Translocation
To dissect the mechanism underlying ACTN4-mediated transcriptional activation of NF-κB target genes in HPCs, control and ACTN4 knockdown HPCs were treated with or without TNFα and NF-κB target gene expression was analyzed by RT-qPCR. We found that knocking down ACTN4 significantly decreased mRNA levels of NF-κB target genes, IL-1β, IL8, and MCP-1 (Fig. 2A). The nephrin gene NPHS1 has been shown to be an NF-κB target gene (25). Indeed, NPHS1 mRNA expression was induced by TNFα, and knockdown of ACTN4 abolished TNFα-mediated induction of NPHS1 mRNA. Nuclear translocation of the NF-κB family of transcription factors is an essential step to regulate the expression of their target genes. A previous study has suggested that overexpression of ACTN4 promoted nuclear translocation of p65 (26). To further dissect the mechanism by which loss of ACTN4 decreased NF-κB reporter activity and its target gene expression, we determined whether knockdown of ACTN4 affects TNFα-induced nuclear translocation of p65. Control and ACTN4 knockdown HPCs were treated with or without TNFα followed by subcellular fractionation and indirect immunofluorescence microscopy. Subcellular fractionation of HPCs indicated that knockdown of ACTN4 had little or no effect on TNFα-induced nuclear translocation of p65 (Fig. 2B). Immunofluorescence microscopy further demonstrated that p65 was capable of translocating to the nucleus in ACTN4 knockdown HPCs (Fig. 2C). Taken together, our data demonstrated that knockdown of ACTN4 significantly inhibited the expression of basal and TNFα-induced NF-κB target genes, but had little or no effect on TNFα-induced nuclear translocation of p65.
FIGURE 2.

The effect of ACTN4 knockdown on TNFα-induced p65 nuclear translocation in HPCs. A, ACTN4 was stably knocked down by shRNAs in HPCs. HPCs were starved overnight and treated with TNFα (20 ng/ml) for 6 h prior to harvest. Total RNA was extracted and quantitative RT-PCR analysis was conducted using gene-specific primers as indicated. Error bars indicate mean ± S.D. B and C, subcellular localization of p65 in ACTN4 knockdown HPCs was analyzed by Western blotting analyses (B) or immunofluorescence microscopy (C). B, control and ACTN4 knockdown HPCs were treated with 20 ng/ml TNFα for 1 h and harvested, and subcellular fractions were prepared followed by Western blotting using anti-ACTN4, anti-p65, and anti-lamin B or anti-α-tubulin as loading controls. The ratio of p65 to lamin B or α-tubulin at dimethyl sulfoxide (DMSO)-treated HPCs was set at 1. The relative p65 intensity is shown. The quantitation data were an average of two Western blots. C, control and ACTN4 knockdown HPCs were treated with TNFα (20 ng/ml) for 30 min followed by immunostaining with ACTN4 and p65 antibodies and fluorescence microscopy.
Glucocorticoid-mediated Transrepression in Podocytes
Liganded GRs can have diverse effects on both glucocorticoid response element-mediated and NF-κB-mediated transcriptional activation, depending on the nature of the ligands. For example, the glucocorticoid dexamethasone (Dex) activates glucocorticoid response element-containing reporter activity while repressing NF-κB reporter activity. Fig. 3A shows that Dex treatment markedly repressed basal and TNFα-mediated induction of IL-1β, IL-8, and MCP-1 mRNAs. We also observed Dex-mediated transrepression of NF-κB activity using a reporter that contains native promoter derived from MCP-1 in HPCs (Fig. 3B) Using HPCs in which ACTN4 was stably knocked down, we further demonstrated that Dex effectively repressed IL-1β and MCP-1 gene expression in ACTN4 knockdown HPCs, but had no effect on IL-8 mRNA expression (Fig. 3C). Taken together, these data indicated that Dex represses NF-κB transcription activity in HPCs and that ACTN4 plays a role in Dex-mediated transrepression of NF-κB activity in a gene-specific manner.
FIGURE 3.

GRα-mediated transrepression of NF-κB target genes in podocytes. A, the mRNA expression levels of NF-κB target genes in TNFα- or Dex-treated HPCs. HPCs were starved overnight and then treated with Dex (100 ng/ml) or TNFα (20 ng/ml) for 6 h. Total RNA was extracted and quantitative RT-PCR analysis was conducted using gene-specific primers as indicated. Veh., vehicle. B, Dex-mediated transrepression of TNFα activity in a reporter construct that contains native MCP-1 promoter. The MCP-1 promoter reporter construct was transiently transfected into HPCs and treated with vehicle, TNFα, Dex, or both TNFα and Dex, and the reporter activity was assayed. C, the expression of NF-κB target genes in control and ACTN4 knockdown HPCs treated with vehicle or Dex. Control and ACTN4 stable knockdown HPCs were treated with Dex (100 ng/ml) for 4 h. Total RNA was extracted and RT-qPCR analysis was conducted using gene-specific primers as indicated. Error bars indicate mean ± S.D.
ACTN4 Physically Interacts with p65 in HPCs
We further dissected the mechanism by which ACTN4 potentiates NF-κB transcription activity by examining whether ACTN4 and p65 interact. First, we demonstrated that endogenous ACTN4 was coprecipitated with exogenously expressed HA-p65 in HEK293T cells (Fig. 4A). We further confirmed that this interaction occurs for endogenous ACTN4 and p65 (Fig. 4B). In the absence of TNFα, ACTN4 is distributed throughout the cells, whereas p65 is primarily in the cytoplasm (Fig. 2). In response to TNFα, a significant fraction of p65 translocates into the nucleus, whereas subcellular localization of ACTN4 remains relatively unchanged. These observations raise an intriguing question. Do ACTN4 and p65 interact in the cytoplasm or in the nucleus? Indeed, we observed that ACTN4 and p65 coimmunoprecipitated in the nuclear extracts prepared from TNFα-treated HEK293T cells (Fig. 4C). Similarly, we observed that p65 only interacts nuclear, but not cytoplasmic ACTN4 in TNFα-treated HPCs (Fig. 4D). Lastly, we isolated glomeruli from mice and prepared total cellular extracts. Immunoprecipitation with anti-p65 antibodies indicated that ACTN4 and p65 interact in mouse glomeruli (Fig. 4E). To further confirm this association, we examined whether endogenous ACTN4 interacts with exogenously transfected HA-p65. In summary, these data indicate that p65 and ACTN4 interact in glomeruli, HPCs, and HEK293T cells.
FIGURE 4.
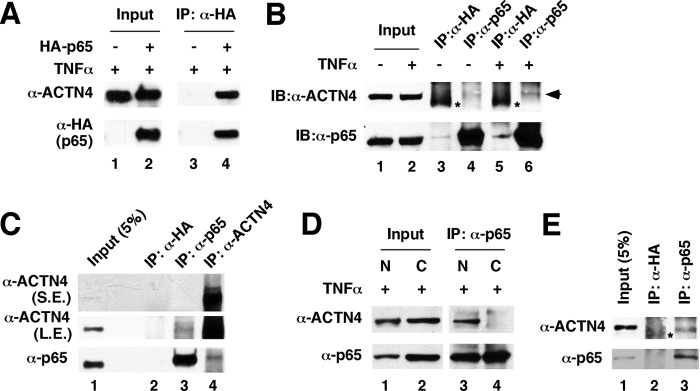
ACTN4 and p65 interact in glomeruli and in mammalian kidney cells. A, exogenous p65 and endogenous ACTN4 interact in TNFα-treated HEK293T cells. HEK293T cells were transiently transfected with a control or a HA-p65 expression plasmid and treated with TNFα, and immunoprecipitation (IP) was performed with anti-HA antibodies followed by immunoblotting with anti-HA and anti-ACTN4 antibodies. B, HEK293T cells were treated with or without TNFα (20 ng/ml) for 1 h, and whole cell extracts were prepared and immunoprecipitated with anti-p65 antibodies and anti-HA antibodies as control. Proteins were detected by Western blotting (IB) using anti-ACTN4 and anti-p65 antibodies as indicated. The asterisks show nonspecific background signals. C, HEK293T cells were treated with TNFα (20 ng/ml) for 1 h, and nuclear extracts were prepared and immunoprecipitated with anti-HA (control), anti-p65, or anti-ACTN4 antibodies. The immunopellets were subjected to Western blotting with anti-p65 and anti-ACTN4 antibodies. D, nuclear ACTN4 and p65 interact in HPCs. HPCs were treated with TNFα, and nuclear and cytoplasmic fractions were prepared. Coimmunoprecipitation was carried out using anti-p65 antibodies followed by Western blotting with the indicated antibodies. E, mouse glomerular extracts were prepared and immunoprecipitated with anti-p65 antibodies or anti-HA antibodies followed by Western blotting with anti-ACTN4 and p65 antibodies. The asterisks E represent nonspecific background signals. S.E., short exposure; L.E., longer exposure.
The Role of Cytoplasmic IκBα in ACTN4-p65 Interaction
Because cytoplasmic NF-κB is bound by IκB, the inability of ACTN4 to bind NF-κB in the cytoplasm could be due to the masking of the ACTN4-interacting surface in p65 by cytoplasmic IκBα. Alternatively, it is possible that nuclear and cytoplasmic p65 and p50 have distinct biochemical properties such as differential posttranslational modification. p65 and p50 are subjected to intensive posttranslational modification such as phosphorylation, acetylation, methylation, glycosylation, and ubiquitination (27). To distinguish these two possibilities, we first tested whether bacterially expressed GST-p65 is capable of pulling down HA-ACTN4 expressed in mammalian cells. Indeed, GST-p65 was capable of pulling down HA-ACTN4, suggesting that posttranslational modification of p65 is not required for interacting with ACTN4 (Fig. 5A). We further mapped ACTN4 interaction domain in p65 to the first 313 amino acids (Fig. 5B), the same region where IκBα binds. Lastly, we demonstrated that depletion of IκBα in HPCs significantly enhances the interaction between ACTN4 and p65 in the cytoplasm. In summary, these data support a model in which cytoplasmic IκBα masks the ACTN4 interaction domain, thus blocking ACTN4 from binding p65.
FIGURE 5.

IκBα blocks ACTN4 from interacting with p65 in the cytosol. A, purified, immobilized GST-p65 was incubated with cell extracts expressing HA-ACTN4 or HA-p50. Pulldown fractions were subject to Western blotting with anti-HA antibodies. B, purified, immobilized GST-ACTN4 was incubated with cell extracts expressing HA-p65 (full-length), HA-p65 (1–313), or HA-p65 (314–551) followed by Western blotting with anti-HA antibodies. C, knockdown of IκBα results in an association between ACTN4 and p65. HPCs were transiently transfected with a control or IκBα shRNA, and nuclear (N) and cytoplasmic (C) fractions were prepared, immunoprecipitated (IP) with anti-p65 antibodies, and Western blotted with the indicated antibodies.
Association of ACTN4 (WT) and Its Variant with NF-κB Subunits
We have previously described a spliced variant of ACTN4 (Fig. 6A), ACTN4 (Iso), which binds and potentiates transcriptional activity by nuclear receptors (9, 10) and MEF2C (7). We also demonstrated that the LXXLL nuclear interacting motif is essential for nuclear receptor binding and transcriptional activation (9, 11). We next addressed whether p65 binds ACTN4 variants by GST pulldown assays and whether the LXXLL motif is critical for the interaction between ACTN4 and p65. We found that mutation of the LXXLL motif, LXXAA (AA), had little effect on p65 binding activity (Fig. 6B, lanes 3 and 4) and that GST-ACTN4 (Iso) also binds p65, but less robustly than the full-length protein (lanes 3 and 5). We have previously shown that the FSGS-linked ACTN4 mutant, K255E, is defective in interacting with nuclear receptors (11). However, ACTN4 (K255E) interacts with p65 similarly to the wild-type protein. Interestingly, the p65 heterodimeric partner, p50, also binds ACTN4 and its variants (Fig. 6C), although p50 binds ACTN4 (Iso) better than the full-length protein (lanes 3 and 5). Furthermore, ACTN4 (K255E) exhibited a weaker binding to p50. We further determined the ability of these ACTN4 variants to potentiate NF-κB reporter activity (Fig. 6D). We observed a higher reporter activity in ACTN4 (Iso) and ACTN4 (Iso, AA) transfected cells, although their expression levels were significantly lower than the full-length ACTN4 (Fig. 6D, right panel). In contrast, the FSGS-linked mutant, K255E, significantly lost its ability to activate NF-κB reporter activity. Taken together, these data indicate that the p65 and p50 binding surfaces in ACTN4 do not overlap with those of nuclear receptors and that ACTN4 (Iso) possess intrinsically more potent coactivation activity.
FIGURE 6.

The association of ACTN4 variants with p65 and p50. A, schematic representations of ACTN4 and its spliced variant, Iso. CH, calponin homology domain; SR, spectrin repeat, CaM, calmodulin. The nuclear receptor-interacting motif, LXXLL (amino acids 84–88), is N-terminal to the splicing junction such that both the full-length form and the isoform share the LXXLL motif with distinct C-terminal sequences to the motif (9). The FSGS-linked K255E mutant is indicated. The C-terminal tandem solid black boxes are EF hand calcium-binding domains. B, HeLa cells were transiently transfected with an HA-p65 expression plasmid. Whole cell lysates were incubated with immobilized GST or GST-ACTN4 (WT) or its variants. The pulldown fractions were analyzed by Western blotting with anti-HA antibodies. The expressions of GST or GST-ACTN4 were visualized by Coomassie Blue staining. C, the experiments were similar to that in B except that HA-p50-expressed HeLa extracts were used for pulldown. D, HEK293T cells were cotransfected with an NF-κB luciferase reporter construct and HA-ACTN4 (WT) or the variants Iso, Iso with AA mutation in the LXXLL motif (Iso-AA), or the K255E FSGS mutant (K-E) and treated with TNFα (20 ng/ml) for 12 h, and luciferase activity was measured. The luciferase activity was normalized to β-galactosidase activity. The expression of ACTN4 and variants are shown by Western blot on the right panel. Veh., vehicle. Error bars indicate mean ± S.D.
The HDAC7-interacting Domain Is Important for ACTN4-mediated Transcriptional Activation
We have previously demonstrated that the ability of ACTN4 to potentiate MEF2- and estrogen receptor α (ERα)-mediated transactivation requires its HDAC7-interacting domain (7, 9). We therefore determined whether HDAC7 plays a role in ACTN4-mediated potentiation of NF-κB activity. Indeed, the ACTN4 mutants defective in interacting with HDAC7 significantly lost their ability to potentiate NF-κB-driven reporter activity (Fig. 7A). Furthermore, knockdown of HDAC7 potentiates NF-κB-driven reporter activity (Fig. 7B) and endogenous IL-1β mRNA expression (Fig. 7C). Based on these data, we conclude that HDAC7 plays a role in ACTN4-mediated potentiation of NF-κB transactivation activity.
FIGURE 7.

The role of HDAC7 in ACTN4-mediated potentiation in NF-κB transactivation activity. A, HEK293T cells were transiently transfected with ACTN4 (FL), ACTN4 (FL, Δ 831–869), ACTN4 (Iso), or ACTN4 (Iso, Δ 441–479) and an NF-κB reporter and treated with or without TNFα, and reporter activity was measured. Veh., vehicle. B, HEK293T cells were infected with a shCtrl or shHDAC7 expression viral vector and an NF-κB reporter and treated with or without TNFα, and reporter activity was determined. C, HPCs were transiently infected with a shCtrl or shHDAC7 expression viral vector and treated with or without TNFα, total RNA was isolated, and RT-qPCR was performed with gene-specific primers. Error bars indicate mean ± S.D.
ACTN4 Is Recruited to NF-κB Target Gene Promoters
We further determined the p65 interaction domain in ACTN4. Immobilized, purified GST-ACTN4 (WT) was incubated with nuclear and cytoplasm extracts expressing HA-p65 (Fig. 8A) or HA-p50 (Fig. 8B) followed by Western blotting with anti-HA antibodies. In agreement with coimmunoprecipitation experiments, GST-ACTN4 was capable of pulling down nuclear p65 (Fig. 8A, lane 5) and p50 (Fig. 8B, lane 5) in vitro. In contrast, no interaction was observed when cytoplasmic extracts were used for pulldown assays. To map the domain in ACTN4 that is required for interacting with nuclear p65, we used GST fusion proteins with full-length ACTN4 (full-length), isoform ACTN4 (Iso), and truncated ACTN4 (Iso) at amino acids 2–102 and 95–521 for pulldown assays. The C-terminal deletion mutant, ACTN4 (2–102), contains part of CH1 (calponin homology 1) domain, whereas ACTN4 (95–521) contains the remaining C terminus of ACTN4 (Iso) that includes part of Spectrin repeat 2 (SR2), SR3, SR4, and the calmodulin-like domain (Fig. 5A). Fig. 8, A and B, show that amino acids 2–102 are sufficient to bind both HA-p65 and HA-p50 (lane 10). However, the C terminus did not interact with HA-p65 (Fig. 8A, lane 12) and interacted poorly with p50 (Fig. 8B, lane 12).
FIGURE 8.

Association of p65 and ACTN4 with IL1-β and IL-8 promoters in HPCs. A, HeLa cells were transiently transfected with an HA-p65 expression plasmid. Nuclear (N) and cytoplasmic (C) lysates were incubated with immobilized GST or GST-ACTN4 (WT) (lanes 1–6). Lanes 7–12, pulldown assays were performed with nuclear extracts expressing HA-P65 and immobilized GST, GST-ACTN4, and its variants. Pulldown fractions were analyzed by Western blotting with anti-HA antibodies. The expressions of GST fusion proteins were visualized by Coomassie Blue staining. The asterisk shows contaminated bacterial proteins copurified with GST proteins. B, the experiments were similar to that in A except that HA-p50-expressed HeLa nuclear extracts were used for pulldown assays. C–E, HPCs were treated with vehicle or TNFα for 1 h followed by ChIP assays with anti-p65 and anti-ACTN4 antibodies. Immunoprecipitated chromatin was analyzed by qPCR using the primers flanking p65-binding sites on IL-1β (C) and IL-8 (D) promoters. TNFα-induced association of p65 and ACTN4 with the promoters is shown as relative promoter occupancy. E, HPCs stably expressing shCtrl or shACTN4 were used for ChIP assays as described in C and D. The relative recruitment of ACTN4 to the IL-1β promoter was determined by normalizing the PCR products from shACTN4 cells to that of the shCtrl cells. Error bars indicate mean ± S.D.
We next performed ChIP assays to determine whether ACTN4 is recruited to the promoters of NF-κB-targeted promoters. Indeed, TNFα increased the recruitment of p65 and ACTN4 to IL-1β (Fig. 8C) and IL-8 promoters (Fig. 8D). As a control, knockdown of ACTN4 markedly reduced the association of ACTN4 with the IL-1β promoter regardless of the presence of TNFα (Fig. 8E). Interestingly, knockdown of ACTN4 slightly reduced the binding of p65 to the IL-1β promoter. Taken together, we conclude that ACTN4 and p65 are recruited to IL-1β and IL-8 promoters in response to TNFα in HPCs.
DISCUSSION
We and others have previously demonstrated that in addition to its role in the cytoplasm, ACTN4 is capable of potentiating the activity of several transcription factors that include nuclear receptors such as the estrogen receptor, peroxisome proliferator-activated receptors, retinoic acid receptor, vitamin D receptor (9–11), and MEF2s (7, 8). These observations raise a mechanistically important question regarding the role of ACTN4 in podocyte function and kidney disease. Is the known cytoplasmic actin binding activity of ACTN4 required for its ability to coactivate transcription factors?
We previously reported that ACTN4 is recruited to the promoter of estrogen receptor target gene, pS2 (9). When tethered to the Gal4 DNA-binding domain (DBD), Gal DBD-ACTN4 potently activates Gal4 reporter activity, likely through its interaction with transcriptional coactivators such as p160 coactivators and the histone acetyltransferase activity p300/CBP-associated factor (PCAF) (10). Our current study provides strong evidence demonstrating that 1) ACTN4 is essential for NF-κB-mediated transcriptional activation, 2) ACTN4 interacts with p65 and p50 in the nucleus and is recruited to the promoters of NF-κB target genes, IL-1β and IL-8, 3) loss of ACTN4 does not affect TNFα-induced nuclear translocation of p65, and 4) the spliced variant, ACTN4 (Iso), does not contain an intact actin-binding domain and is a much more potent transcriptional coactivator than the full-length ACTN4. Moreover, although the FSGS-linked mutant K255E is capable of interacting with p65, it significantly lost its ability to potentiate NF-κB reporter activity. We reason that because K255E is predominantly cytoplasmic (11), it is therefore unable to potentiate the activity of transcription factors in the nucleus. This is consistent with recent studies indicating that in response to myoblast differentiation signal, ACTN4 translocates into the nucleus and coactivates MEF2 transcription activity and myogenic gene expression (8). In contrast to hormone-dependent interaction between ACTN4 and nuclear receptors, this finding provides an alternative mechanism by which ACTN4 potentiates transcriptional activity. A previous study has suggested a role of ACTN4 in activating NF-κB activity (28). However, it was concluded that ACTN4 and NF-κB interact in the cytoplasm, and it is unclear how cytoplasmic ACTN4 potentiates NF-κB activity. However, the current study and previous reports support the notion that although ACTN4 is found in the nucleus and cytoplasm, nuclear ACTN4 binds and potentiates transcription factor activity in the nucleus, independently of its cytoplasmic actin binding activity (9–11).
We noted that although ACTN4 (Iso) binds p65 similarly to the wild-type protein, ACTN4 (Iso) has a more potent transactivation activity than the wild-type protein (Fig. 6D). This is consistent with our previous report that Gal-ACTN4 (Iso) has a higher transactivation activity than the wild-type protein, likely due to a better binding activity of ACTN4 (Iso) to p160 coactivators and the histone acetyltransferase activity PCAF than the ACTN4 (FL) (10).
Several lines of evidence suggest that cytoplasmic IκBα masks and prevents ACTN4 from binding p65. 1) GST-ACTN4 pulls down nuclear, but not cytoplasmic p65 or p50, 2) endogenous ACTN4 only associated with p65 when IκBα was knocked down, and 3) the interaction domain between ACTN4 and p65 overlapped the interaction domain between IκBα and p65. In sum, these data indicate that a compartment-dependent interaction between ACTN4 and p65 regulated by signal-dependent nuclear translocation of p65 and p50 is indispensable for the activation of NF-κB activity (Fig. 9).
FIGURE 9.

A model in which ACTN4 binds and coactivates NF-κB transactivation activity in the nucleus. We propose that in the absence of TNFα, NF-κB transcription factor is sequestered by IκBα in the cytosol, thereby blocking ACTN4 from binding to NF-κB (left panel). However, knockdown of IκBα induces the association between ACTN4 and NF-κB in the cytosol (center panel). In response to TNFα, NF-κB translocates into the nucleus, where it binds and recruits ACTN4 to its targeted promoters (right panel). RHD, Rel homology domain.
Glucocorticoid receptor is a member of the nuclear receptor family that regulates target gene expression through one of several mechanisms that involve activation, repression, and transrepression (29). Glucocorticoid-mediated transrepression of NF-κB activity is a critical mechanism underlying its anti-inflammatory activity (29). In proteinuric kidney diseases, the glucocorticoids are a longstanding treatment strategy, although their exact mechanism of action is not fully understood. Although anti-inflammatory effects are likely involved, in vitro studies have suggested a direct beneficial effect on podocyte function (30) including stabilizing podocyte actin filaments (31). Our data indicated that ACTN4 may play a role in Dex-mediated transrepression of NF-κB activity in a gene-specific manner. Further investigation is necessary to dissect the mechanism by which ACTN4 regulates GR-mediated transrepression of NF-κB target genes.
As an actin-binding protein that coordinates cytoskeletal dynamics of the foot processes in podocytes, ACTN4 mutations that alter its actin binding activity are likely disrupting slit diaphragm and causing podocyte injury. However, although some FSGS-associated ACTN4 mutants acquire increased actin binding activity, other FSGS-associated ACTN4 mutants bind actin similarly to the wild-type protein (32). Importantly, the FSGS-linked ACTN4 mutant, K255E, not only acquires increased actin binding activity, but also exhibits predominantly cytoplasmic distribution (11) and accelerated turnover rate (33). Furthermore, FSGS-linked ACTN4 mutants are defective in ligand-dependent interaction with nuclear receptors and decrease ligand-dependent activation of nuclear reporter activity (11). In this study, we found that although K255E binds NF-κB in vitro, it fails to potentiate NF-κB reporter activity, likely due to nuclear exclusion of this mutant. Based on these observations, we hypothesize that ACTN4 (K255E) is both gain-of-function in acquiring increased actin binding activity and loss-of-function in losing nuclear localization, reducing protein abundance, and therefore is defective in transcriptional coactivation. Interestingly, ACTN4 deficiency is also found in multiple human primary glomerulopathies including sporadic FSGS, minimal change disease, and IgA nephropathy (34–37). Recent studies further indicated that the abundance of ACTN4 protein is significantly reduced in experimental glomerular diseases (38, 39). Indeed, Actn4 knock-out mice do not survive the perinatal period, and the remaining mice that survive exhibit severe glomerular defects, podocyte injury, and proteinuria (40). Collectively, these observations suggest that expression levels of normal, not just mutant ACTN4, play an important role in many forms of glomerular diseases independent of a genetic alteration in F-actin binding.
Perturbed activation of NF-κB is implicated in various nephrotic diseases. Constitutive activation of NF-κB is associated with FSGS in HIV-associated nephropathy as well as passive Heymann nephritis and contributes to proteinuria (41). However, inhibition of NF-κB is thought to contribute to TGFβ-mediated apoptosis of murine podocytes (42), and down-regulation of NF-κB is found in podocytes in c-MIP elevated idiopathic nephrotic syndrome (43). A better elucidation of the role of NF-κB, its target genes, and regulation in podocytes will help understand the pathogenesis of podocytopathies. Our finding that ACTN4 is essential for NF-κB-mediated transcriptional activation opens a new avenue for future study involving anti-inflammatory treatment effects and podocyte function in FSGS.
This work was supported, in whole or in part, by National Institutes of Health Grants HL093269, DK078965 (to H. Y. K.).
- FSGS
- focal segmental glomerulosclerosis
- GR
- glucocorticoid receptor
- HPC
- human immortalized podocyte
- Dex
- dexamethasone
- qPCR
- quantitative PCR
- CBP
- CREB-binding protein
- CREB
- cAMP-response element-binding protein
- FL
- full-length
- Iso
- isoform
- AA
- LXXAA motif.
REFERENCES
- 1. Mundel P., Shankland S. J. (2002) Podocyte biology and response to injury. J. Am. Soc. Nephrol. 13, 3005–3015 [DOI] [PubMed] [Google Scholar]
- 2. Rood I. M., Deegens J. K., Wetzels J. F. (2012) Genetic causes of focal segmental glomerulosclerosis: implications for clinical practice. Nephrol. Dial. Transplant. 27, 882–890 [DOI] [PubMed] [Google Scholar]
- 3. Schell C., Huber T. B. (2012) New players in the pathogenesis of focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 27, 3406–3412 [DOI] [PubMed] [Google Scholar]
- 4. Kaplan J. M., Kim S. H., North K. N., Rennke H., Correia L. A., Tong H. Q., Mathis B. J., Rodríguez-Pérez J. C., Allen P. G., Beggs A. H., Pollak M. R. (2000) Mutations in ACTN4, encoding α-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 24, 251–256 [DOI] [PubMed] [Google Scholar]
- 5. Choi H. J., Lee B. H., Cho H. Y., Moon K. C., Ha I. S., Nagata M., Choi Y., Cheong H. I. (2008) Familial focal segmental glomerulosclerosis associated with an ACTN4 mutation and paternal germline mosaicism. Am. J. Kidney Dis. 51, 834–838 [DOI] [PubMed] [Google Scholar]
- 6. Honda K., Yamada T., Endo R., Ino Y., Gotoh M., Tsuda H., Yamada Y., Chiba H., Hirohashi S. (1998) Actinin-4, a novel actin-bundling protein associated with cell motility and cancer invasion. J. Cell Biol. 140, 1383–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chakraborty S., Reineke E. L., Lam M., Li X., Liu Y., Gao C., Khurana S., Kao H. Y. (2006) Alpha-actinin 4 potentiates myocyte enhancer factor-2 transcription activity by antagonizing histone deacetylase 7. J. Biol. Chem. 281, 35070–35080 [DOI] [PubMed] [Google Scholar]
- 8. An H. T., Kim J., Yoo S., Ko J. (2014) Small leucine zipper protein (sLZIP) negatively regulates skeletal muscle differentiation via interaction with α-actinin-4. J. Biol. Chem. 289, 4969–4979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khurana S., Chakraborty S., Cheng X., Su Y. T., Kao H. Y. (2011) The actin-binding protein, actinin α 4 (ACTN4), is a nuclear receptor coactivator that promotes proliferation of MCF-7 breast cancer cells. J. Biol. Chem. 286, 1850–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Khurana S., Chakraborty S., Zhao X., Liu Y., Guan D., Lam M., Huang W., Yang S., Kao H. Y. (2012) Identification of a novel LXXLL motif in α-actinin 4-spliced isoform that is critical for its interaction with estrogen receptor α and co-activators. J. Biol. Chem. 287, 35418–35429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khurana S., Chakraborty S., Lam M., Liu Y., Su Y. T., Zhao X., Saleem M. A., Mathieson P. W., Bruggeman L. A., Kao H. Y. (2012) Familial focal segmental glomerulosclerosis (FSGS)-linked α-actinin 4 (ACTN4) protein mutants lose ability to activate transcription by nuclear hormone receptors. J. Biol. Chem. 287, 12027–12035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DiDonato J. A., Mercurio F., Karin M. (2012) NF-κB and the link between inflammation and cancer. Immunol. Rev. 246, 379–400 [DOI] [PubMed] [Google Scholar]
- 13. Tornatore L., Thotakura A. K., Bennett J., Moretti M., Franzoso G. (2012) The nuclear factor κB signaling pathway: integrating metabolism with inflammation. Trends Cell Biol. 22, 557–566 [DOI] [PubMed] [Google Scholar]
- 14. Martinka S., Bruggeman L. A. (2006) Persistent NF-κB activation in renal epithelial cells in a mouse model of HIV-associated nephropathy. Am. J. Physiol. Renal Physiol. 290, F657–F665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ross M. J., Martinka S., D'Agati V. D., Bruggeman L. A. (2005) NF-κB regulates Fas-mediated apoptosis in HIV-associated nephropathy. J. Am. Soc. Nephrol. 16, 2403–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brähler S., Ising C., Hagmann H., Rasmus M., Hoehne M., Kurschat C., Kisner T., Goebel H., Shankland S., Addicks K., Thaiss F., Schermer B., Pasparakis M., Benzing T., Brinkkoetter P. T. (2012) Intrinsic proinflammatory signaling in podocytes contributes to podocyte damage and prolonged proteinuria. Am. J. Physiol. Renal Physiol. 303, F1473–F1485 [DOI] [PubMed] [Google Scholar]
- 17. Bruggeman L. A., Drawz P. E., Kahoud N., Lin K., Barisoni L., Nelson P. J. (2011) TNFR2 interposes the proliferative and NF-κB-mediated inflammatory response by podocytes to TNF-α. Lab. Invest. 91, 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hussain S., Romio L., Saleem M., Mathieson P., Serrano M., Moscat J., Diaz-Meco M., Scambler P., Koziell A. (2009) Nephrin deficiency activates NF-κB and promotes glomerular injury. J. Am. Soc. Nephrol. 20, 1733–1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yamashita M., Millward C. A., Inoshita H., Saikia P., Chattopadhyay S., Sen G. C., Emancipator S. N. (2013) Antiviral innate immunity disturbs podocyte cell function. J. Innate Immun. 5, 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu R., Zhong Y., Li X., Chen H., Jim B., Zhou M. M., Chuang P. Y., He J. C. (2014) Role of transcription factor acetylation in diabetic kidney disease. Diabetes 63, 2440–2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kao H. Y., Lee C. H., Komarov A., Han C. C., Evans R. M. (2002) Isolation and characterization of mammalian HDAC10, a novel histone deacetylase. J. Biol. Chem. 277, 187–193 [DOI] [PubMed] [Google Scholar]
- 22. Guan D., Factor D., Liu Y., Wang Z., Kao H. Y. (2013) The epigenetic regulator UHRF1 promotes ubiquitination-mediated degradation of the tumor-suppressor protein promyelocytic leukemia protein. Oncogene 32, 3819–3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schlondorff D. (1990) Preparation and study of isolated glomeruli. Methods Enzymol. Methods Enzymol. 191, 130–140 [DOI] [PubMed] [Google Scholar]
- 24. Hsu K. S., Kao H. Y. (2013) β-Transducin repeat-containing protein 1 (β-TrCP1)-mediated silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) protein degradation promotes tumor necrosis factor α (TNFα)-induced inflammatory gene expression. J. Biol. Chem. 288, 25375–25386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ristola M., Arpiainen S., Saleem M. A., Holthöfer H., Lehtonen S. (2012) Transcription of nephrin-Neph3 gene pair is synergistically activated by WT1 and NF-κB and silenced by DNA methylation. Nephrol. Dial. Transplant. 27, 1737–1745 [DOI] [PubMed] [Google Scholar]
- 26. Babakov V. N., Petukhova O. A., Turoverova L. V., Kropacheva I. V., Tentler D. G., Bolshakova A. V., Podolskaya E. P., Magnusson K. E., Pinaev G. P. (2008) RelA/NF-κB transcription factor associates with α-actinin-4. Exp. Cell Res. 314, 1030–1038 [DOI] [PubMed] [Google Scholar]
- 27. Chaturvedi M. M., Sung B., Yadav V. R., Kannappan R., Aggarwal B. B. (2011) NF-κB addiction and its role in cancer: ‘one size does not fit all’. Oncogene 30, 1615–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aksenova V., Turoverova L., Khotin M., Magnusson K. E., Tulchinsky E., Melino G., Pinaev G. P., Barlev N., Tentler D. (2013) Actin-binding protein α-actinin 4 (ACTN4) is a transcriptional co-activator of RelA/p65 sub-unit of NF-κB. Oncotarget 4, 362–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Glass C. K., Saijo K. (2010) Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat. Rev. Immunol. 10, 365–376 [DOI] [PubMed] [Google Scholar]
- 30. Xing C. Y., Saleem M. A., Coward R. J., Ni L., Witherden I. R., Mathieson P. W. (2006) Direct effects of dexamethasone on human podocytes. Kidney Int. 70, 1038–1045 [DOI] [PubMed] [Google Scholar]
- 31. Ransom R. F., Lam N. G., Hallett M. A., Atkinson S. J., Smoyer W. E. (2005) Glucocorticoids protect and enhance recovery of cultured murine podocytes via actin filament stabilization. Kidney Int. 68, 2473–2483 [DOI] [PubMed] [Google Scholar]
- 32. Weins A., Kenlan P., Herbert S., Le T. C., Villegas I., Kaplan B. S., Appel G. B., Pollak M. R. (2005) Mutational and biological analysis of α-actinin-4 in focal segmental glomerulosclerosis. J. Am. Soc. Nephrol. 16, 3694–3701 [DOI] [PubMed] [Google Scholar]
- 33. Yao J., Le T. C., Kos C. H., Henderson J. M., Allen P. G., Denker B. M., Pollak M. R. (2004) α-Actinin-4-mediated FSGS: an inherited kidney disease caused by an aggregated and rapidly degraded cytoskeletal protein. PLoS Biol. 2, e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kimura M., Toyoda M., Kato M., Kobayashi K., Abe M., Kobayashi T., Miyauchi M., Yamamoto N., Umezono T., Suzuki D. (2008) Expression of α-actinin-4 in human diabetic nephropathy. Intern. Med. 47, 1099–1106 [DOI] [PubMed] [Google Scholar]
- 35. Dai S., Wang Z., Pan X., Chen X., Wang W., Ren H., Feng Q., He J. C., Han B., Chen N. (2009) ACTN4 gene mutations and single nucleotide polymorphisms in idiopathic focal segmental glomerulosclerosis. Nephron Clin. Pract. 111, c87–94 [DOI] [PubMed] [Google Scholar]
- 36. Dai S., Wang Z., Pan X., Wang W., Chen X., Ren H., Hao C., Han B., Chen N. (2010) Functional analysis of promoter mutations in the ACTN4 and SYNPO genes in focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 25, 824–835 [DOI] [PubMed] [Google Scholar]
- 37. Liu Z., Blattner S. M., Tu Y., Tisherman R., Wang J. H., Rastaldi M. P., Kretzler M., Wu C. (2011) α-Actinin-4 and CLP36 protein deficiencies contribute to podocyte defects in multiple human glomerulopathies. J. Biol. Chem. 286, 30795–30805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kimura J., Ichii O., Otsuka S., Sasaki H., Hashimoto Y., Kon Y. (2013) Close relations between podocyte injuries and membranous proliferative glomerulonephritis in autoimmune murine models. Am. J. Nephrol. 38, 27–38 [DOI] [PubMed] [Google Scholar]
- 39. Yasuno K., Araki S., Sakashita H., Kobayashi R., Baba T., Kawakami H., Kamiie J., Ogihara K., Shirota K. (2013) Development of podocyte injuries in Osborne-Mendel rats is accompanied by reduced expression of podocyte proteins. J Comp. Pathol. 149, 280–290 [DOI] [PubMed] [Google Scholar]
- 40. Kos C. H., Le T. C., Sinha S., Henderson J. M., Kim S. H., Sugimoto H., Kalluri R., Gerszten R. E., Pollak M. R. (2003) Mice deficient in α-actinin-4 have severe glomerular disease. J. Clin. Invest. 111, 1683–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mudge S. J., Paizis K., Auwardt R. B., Thomas R. J., Power D. A. (2001) Activation of nuclear factor-κB by podocytes in the autologous phase of passive Heymann nephritis. Kidney Int. 59, 923–931 [DOI] [PubMed] [Google Scholar]
- 42. Schiffer M., Bitzer M., Roberts I. S., Kopp J. B., ten Dijke P., Mundel P., Böttinger E. P. (2001) Apoptosis in podocytes induced by TGF-β and Smad7. J. Clin. Invest. 108, 807–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ory V., Fan Q., Hamdaoui N., Zhang S. Y., Desvaux D., Audard V., Candelier M., Noel L. H., Lang P., Guellaën G., Pawlak A., Sahali D. (2012) c-mip down-regulates NF-κB activity and promotes apoptosis in podocytes. Am. J. Pathol. 180, 2284–2292 [DOI] [PubMed] [Google Scholar]