

Background: Elevated expression of SHP-1 in diabetic podocytes may potentially interact with nephrin phosphorylation.
Results: Rat nephrin Tyr1127 and Tyr1152 are essential for SHP-1 induced nephrin dephosphorylation under high-glucose conditions.
Conclusion: SHP-1 contributes to nephrin deactivation in podocytes exposed to high glucose levels.
Significance: Reduction of SHP-1 may serve as potential therapeutic target to prevent glomerular pathology in diabetes.
Keywords: Diabetes, Nephrology, Phosphotyrosine, Podocyte, Src Homology 2 domain (SH2 domain), Protein-Tyrosine Phosphatase (Tyrosine Phosphatase), Nephrin
Abstract
Nephrin, a critical podocyte membrane component that is reduced in diabetic nephropathy, has been shown to activate phosphotyrosine signaling pathways in human podocytes. Nephrin signaling is important to reduce cell death induced by apoptotic stimuli. We have shown previously that high glucose level exposure and diabetes increased the expression of SHP-1, causing podocyte apoptosis. SHP-1 possesses two Src homology 2 domains that serve as docking elements to dephosphorylate tyrosine residues of target proteins. However, it remains unknown whether SHP-1 interacts with nephrin and whether its elevated expression affects the nephrin phosphorylation state in diabetes. Here we show that human podocytes exposed to high glucose levels exhibited elevated expression of SHP-1, which was associated with nephrin. Coexpression of nephrin-CD16 and SHP-1 reduced nephrin tyrosine phosphorylation in transfected human embryonic kidney 293 cells. A single tyrosine-to-phenylalanine mutation revealed that rat nephrin Tyr1127 and Tyr1152 are required to allow SHP-1 interaction with nephrin. Overexpression of dominant negative SHP-1 in human podocytes prevented high glucose-induced reduction of nephrin phosphorylation. In vivo, immunoblot analysis demonstrated that nephrin expression and phosphorylation were decreased in glomeruli of type 1 diabetic Akita mice (Ins2+/C96Y) compared with control littermate mice (Ins2+/+), and this was associated with elevated SHP-1 and cleaved caspase-3 expression. Furthermore, immunofluorescence analysis indicated increased colocalization of SHP-1 with nephrin in diabetic mice compared with control littermates. In conclusion, our results demonstrate that high glucose exposure increases SHP-1 interaction with nephrin, causing decreased nephrin phosphorylation, which may, in turn, contribute to diabetic nephropathy.
Introduction
Podocytes are highly specialized epithelial cells that play a key role in glomerular blood filtration by wrapping the blood capillaries with their cytoplasmic projections, called foot processes. The foot processes of adjacent podocytes interdigitate with each other to form a zipper-like structure called the slit diaphragm, which helps in maintaining the link between adjacent podocytes. This structure is critical in maintaining the function of the glomerulus because a dysfunction of the foot process is linked to various nephropathies (1–3). Diabetic nephropathy (DN)2 is the leading cause of end-stage renal disease worldwide and an independent risk factor for all-cause and cardiovascular mortalities in diabetic patients (4, 5). Morphometric observations from kidney biopsies of diabetic patients showed a significant reduction in the number of podocytes in patients with a short duration of diabetes before the apparition of microalbuminuria or other DN markers (6–8). Of all morphological changes, a decreased number of podocytes in the glomeruli is the strongest predictor of DN progression (6). Multiple mechanisms have been proposed to explain podocyte dysfunction and apoptosis in DN. Production of reactive oxygen species by oxidative stress in podocytes exposed to high glucose concentration has been associated with an increase in podocyte apoptosis (9). Activation of protein kinase δ in podocytes has been shown to promote podocyte apoptosis induced by high glucose levels (10). It is becoming increasingly clear that vascular endothelial growth factor (VEGF) and insulin are important factors in podocyte survival and that they are disturbed in DN. VEGF has been found to be critical in maintaining the integrity of the glomerulus and is primarily secreted by the podocyte. In humans, VEGF levels and downstream signaling are increased in early stages of DN but then appear to be reduced below normal with progressive DN (11, 12). Inhibition or deletion of VEGF resulted in podocyte apoptosis, foot process effacement, loss of mesangial and endothelial cell fenestration, and death within the first days of life (13). Insulin action on podocytes has also been found to be critical to their integrity. Mice with specific deletion of the insulin receptor in the podocyte develop a pathology similar to DN, with loss of podocyte morphology, thickening of the glomerular basement membrane, and glomerulosclerosis (14).
The transmembrane protein nephrin has been found to be of utmost importance in maintaining the integrity of the slit diaphragm. Mutation in the gene coding for nephrin causes the most severe form of congenital nephrotic syndrome (Finnish-type congenital syndrome), and affected infants excrete massive amounts of protein in their urine at birth (15). Accumulating evidence suggests that nephrin offers not only a structural contribution to the filtration barrier but that it can also mediate signal transduction from the slit diaphragm in the podocytes. Nephrin consists of a large extracellular portion with eight IgG-like domains, a single fibronectin type 3 motif, and a cytoplasmic domain with eight tyrosine phosphorylation sites, most of which are conserved from zebrafish to humans. Phosphorylation of various tyrosine residues found in the nephrin intracellular domain is involved in the regulation of multiple intracellular signaling pathways. These tyrosine residues, contained in sequences recognized by Src homology 2 (SH2) domain-containing proteins, are phosphorylated by the Src family kinase Fyn (16). Phosphorylation of nephrin is known to regulate actin dynamics in podocytes by interaction with the adaptor protein Nck (17) and inhibition of apoptosis by an interaction with the PI3K/Akt pathway (18). However, much less studied is how nephrin phosphorylation is regulated in diabetic nephropathy.
The regulation of protein-tyrosine phosphatases (PTP) is an important mechanism for developmental control and homeostasis of an array of cellular processes, including cell growth, differentiation, mitotic cycle, and oncogenic transformation. Any disruption in the equilibrium between protein-tyrosine kinase activity and protein-tyrosine phosphatase activity will trigger abnormal cell proliferation or death, thereby resulting in various pathophysiological abnormalities. PTP has been shown to be important in maintaining the cytoskeleton integrity of podocytes because the nonspecific PTP inhibitor sodium vanadate causes important pathological changes in cultured murine podocytes (19). We have reported previously that elevated SH2 domain-containing phosphatase 1 (SHP-1) expression caused the inhibition of VEGF and insulin actions, leading to apoptosis in podocytes exposed to high glucose levels (10, 20). Nephrin also contains a binding site for SH2 domains and may interact with SHP-1. Here we explore the regulation of nephrin phosphorylation and its potential interaction with SHP-1 in diabetic nephropathy.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Primary antibodies were obtained from commercial sources as follows, unless stated otherwise. Actin (HRP, catalog no. I-19), SHP-1 (catalog no. C-19), Tyr(P) (catalog no. PY99), and nephrin (catalog no. H300) were from Santa Cruz Biotechnology. Anti-DYKDDDDK (FLAG) was from Cell Signaling Technology (rabbit polyclonal) or Sigma (mouse monoclonal). Nephrin (catalog no. ab58968), human nephrin Tyr(P)1176/1193, and Tyr(P)1217 were from Abcam. EDTA, leupeptin, phenylmethylsulfonyl fluoride, aprotinin, d-glucose, d-mannitol, and Na3VO4 were purchased from Sigma-Aldrich, unless otherwise stated.
Plasmid Construction and Transfection
HEK 293T cells were transfected using Qiagen Effectene reagent according to the protocol of the manufacturer. Construction of nephrin CD16/7 has been described previously (17). The plasmid containing rat Fyn wild-type, rat nephrin wild-type, and tyrosine-to-phenylalanine mutants (rY1127F, rY1152F, rY1171F, rY1194F, rY1204F, and rY1228F) have been described previously (16). Murine SHP-1 cDNA was cloned into the p3XFLAG-CMV-14 expression vector (Sigma-Aldrich) between HindIII and XbaI to express SHP-1 with three FLAG epitopes in the N-terminal end of the protein.
FLP-IN Constitutive Cell Line
HEK 293 cells containing the FRT domain to establish a constitutive stable cell line was a gift from Dr. Gallo-Payet (21). HEK-FR cells were transfected with the wild type and various tyrosine mutants of nephrin to create cells stably and constitutively expressing the various mutants.
Cell Culture
The human podocyte cell line was a gift from Dr. Saleem and cultured as described previously (22). Briefly, cells were grown under permissive conditions at 33 °C in RPMI 1640 medium containing 10% FBS and 100 units/ml of penicillin/streptomycin in a collagen-coated Petri dish. Then cells were allowed to differentiate at 37 °C for 14 days with medium changes on alternate days. After differentiation of podocytes, medium was changed to RPMI 0.1% FBS containing normal glucose (5.6 mmol/liter) or high glucose (25 mmol/liter) up to 96 h. HEK 293T cells were cultured in high-glucose DMEM supplemented with 10% FBS and 100 units/ml of penicillin/streptomycin.
Adenoviral Vector Transfection
Adenoviral vectors containing GFP (Ad-GFP) and dominant negative SHP-1 (Ad-dnSHP-1) in which the catalytic center cysteine (Cys453) was replaced by a serine, causing inactivation of SHP-1 phosphatase activity as described previously (23), were used to infect podocytes, as we reported previously in vascular cells (24) and podocytes (20). Cultured human podocytes were infected with the described adenoviral vectors at a multiplicity of infection of 75 in serum-free medium for 2 h in a 37 °C incubator supplemented with 5% CO2, and adenovirus with the same parental genome carrying the Ad-GFP gene was used as a control. The cells were then grown in medium containing 10% FBS for an additional 24 h in an incubator. Medium of infected cells were removed and replaced with RPMI 0.1% FBS with 25 mmol/liter glucose for 96 h. The infectivity of these adenoviruses was evaluated by the percentage of green light-emitting cells under a fluorescent microscope (Nikon, Avon, MA). The presence of ∼80% of Ad-GFP-positive cells was considered to be a successful infection and used for further experimentation.
Coimmunoprecipitation and Immunoblot Analyses
Coimmunoprecipitation assays were performed as described previously (20). Briefly, 500 μg of lysate proteins was incubated for 90 min with 1 μg of antibodies specific against nephrin, GFP, or FLAG epitope. Then 50 μl of precleaned A/G protein-coated agarose beads was added to the mix and rotated at 4 °C for 30 min. Beads were then pelleted and washed to isolate the protein complex containing the protein of interest. 25 μl of 2× Laemmli was then added to separate the proteins from the beads. For immunoblot analysis, 10–50 μg of lysate proteins was separated by SDS-PAGE and then transferred to a PVDF membrane, which was blocked with 5% skim milk. Proteins were identified by chemiluminescence using ECL (Pierce Thermo Fisher).
Animals and Experimental Design
C57BL/6J and diabetic heterozygous male Ins2+/C96Y (Akita) mice were purchased from The Jackson Laboratory and bred in our animal facility. Throughout the period of study, animals were provided with free access to water and standard rodent chow (Harlan Teklad, Madison, WI). All experiments were conducted in accordance with the Canadian Council of Animal Care and Institutional Guidelines and were approved by the Animal Care and Use Committee of the University of Sherbrooke according to National Institutes of Health guidelines. The blood glucose level of mice was measured by a glucometer (Contour, Bayer).
Blood Glucose, Urinary Albumin Excretion, and Glomerular Pathology
Blood glucose was measured by a glucometer (Contour, Bayer). 24-h urine collections were obtained from mice 1 day prior to sacrifice by housing them in individual mouse metabolic cages (Nalgene, model 650-0311, Nalgene Nunc International, Rochester, NY) with free access to water and rodent mash. Urinary creatinine concentration was measured using alkaline picrate colorimetry on the basis of the Jaffe reaction (The Creatinine Companion, Exocell, Philadelphia, PA), and albumin levels were measured using an indirect competitive ELISA according to the instructions of the manufacturer (Albuwell M, Exocell). Mice were sacrifice at 7 months of age. Left mouse kidneys were harvested for pathological examination, and sections were fixed in 4% paraformaldehyde (Sigma-Aldrich) for 18 h and then transferred to 70% ethanol. The tissue was embedded in paraffin, and 6-μm sections were stained with periodic acid-Schiff stain (Sigma) to evaluate glomerular hypertrophy and mesangial matrix expansion as we described previously (20).
Laser Capture Microdissection and Quantitative PCR
The kidney was removed and immediately whole-mounted in optimal cutting temperature compound. An 8-μm-thick renal cryosection was cut, thaw-mounted onto nonadhesive glass slides, and then placed immediately on dry ice, followed by fixation for 30 s in 75% ethanol, dehydration through a series of alcohol gradients, and clearing for 4 min in xylene (Fluka). Sections were air-dried in a fume hood for 2 min and then subjected to laser microdissection using a PixCell laser capture microscope (Arcturus Engineering, ABI). Sections were stained with isolectin-B4-FITC. Multiple glomeruli were harvested from the sections for SHP-1 and nephrin mRNA expression. Approximately 1 μg of RNA was used to generate cDNA using SuperScript III reverse transcriptase and random hexamers (Invitrogen) at 50 °C for 60 min. GAPDH mRNA expression was used for normalization. PCR products were gel-purified, subcloned using a QIA Quick PCR purification kit (Qiagen), and sequenced in both directions to confirm identity.
Immunofluorescence
Antigen retrieval was performed on kidney sections for 20 min at 95 °C in 10 mmol/liter citrate buffer (0.05% Tween (pH 6)). Sections were then blocked in PBS containing 10% goat serum for 1 h, exposed in sequence to primary antibodies (nephrin, catalog no. GP-N2, PROGEN Biotechnik, 1:50; SHP-1, catalog no. SC-287, Santa Cruz Biotechnology, 1:100) overnight, followed by incubation with secondary antibodies, Alexa Fluor 488-conjugated anti-guinea pig IgG (Jackson ImmunoResearch Laboratories, 1:400), and Alexa Fluor 546-conjugated anti-rabbit IgG (Invitrogen, 1:400). Mounting media (Vectashield, Vector Laboratories Inc., Burlingame, CA) was added to mount the coverslip. Sections were examined with an Plan Apo ×60 oil immersion objective, numerical aperture 1.42, on an inverted spectral scanning confocal microscope (FV1000, Olympus, Tokyo, Japan). Specimens were laser-excited at 488 nm (40 milliwatt, blue argon laser) and 543.5 nm (1.0 milliwatt, green helium neon laser). To avoid the cross-talk between the emitted Alexa Fluor 488 and Alexa Fluor 543, fluorescence was collected sequentially at wavelengths of 500–530 nm and >560 nm, respectively. Serial horizontal optical sections of 512 × 512 pixels with 2× line averaging were taken at 0.4-μm intervals through the entire thickness of the sections (optical resolution: lateral, 0.2 μm; axial, 0.8 μm). Images of one experiment were taken at the same time under identical settings and handled in Adobe Photoshop similarly across all images.
Statistical Analyses
Data are shown as mean ± S.D. for each group. Statistical analysis was made using unpaired Student's t test. Results were considered statistically significant at p < 0.05.
RESULTS
SHP-1 Is Increased by High Glucose in Human Podocytes and Is Associated with Nephrin
We have demonstrated previously that high glucose exposure increased SHP-1 expression in mouse podocytes (20). We confirmed that human podocytes exposed to high glucose levels for 96 h exhibited significantly elevated SHP-1 expression by 120% (p = 0.0385) (Fig. 1A). Human podocytes express higher levels of nephrin compared with mouse podocytes. We chose human podocytes to evaluate a potential nephrin/SHP-1 interaction by immunoprecipitation assays under high-glucose conditions. As shown in Fig. 1B, the association of SHP-1 and nephrin was increased significantly by 235% (p = 0.0002) in human podocytes exposed to 25 mmol/liter of glucose compared with normal glucose levels.
FIGURE 1.
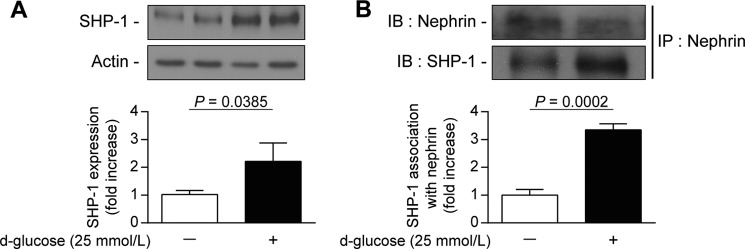
Increased expression of SHP-1 by high glucose levels is associated with nephrin in podocytes. Shown are protein expression of SHP-1 (A) and coimmunoprecipitation with nephrin (B) following an immunoblot of SHP-1 and nephrin in podocytes exposed to high glucose levels (25 mmol/liters) for 96 h. Protein expression was detected by Western blot, and densitometric quantitation was measured. Results are shown as mean ± S.D. of three independent experiments. IB, immunoblot; IP, immunoprecipitation.
SHP-1 Reduces Nephrin Tyrosine Phosphorylation
We demonstrated that SHP-1 can be associated with nephrin. However, it remains unknown whether this interaction leads to reduced nephrin tyrosine phosphorylation. To investigate this, we used HEK cells transfected with nephrin-CD16 that allowed us to induce nephrin phosphorylation following clustering with CD16 antibody, as reported previously (25). Cells were transfected to express both nephrin-CD16 and a SHP-1/FLAG tag. A coimmunoprecipitation assay using anti-FLAG antibody indicated that nephrin-CD16 was associated with SHP-1, suggesting an interaction between both proteins (Fig. 2A). This interaction was not observed in cells transfected with SHP-1/FLAG or nephrin-CD16 alone. We next evaluated the nephrin phosphorylation status in this setting. Cells were transfected to express nephrin-CD16 or nephrin-CD16 with SHP-1/FLAG and treated with antibody against CD16. In cells treated with anti-CD16 antibody, a 1.9-fold increase of nephrin tyrosine phosphorylation was observed compared with untreated cells (Fig. 2B). However, coexpression of SHP-1/FLAG with nephrin-CD16 significantly reduced anti-CD16-mediated nephrin phosphorylation. These results indicate that SHP-1 association with nephrin can lead to a reduction of nephrin tyrosine phosphorylation.
FIGURE 2.
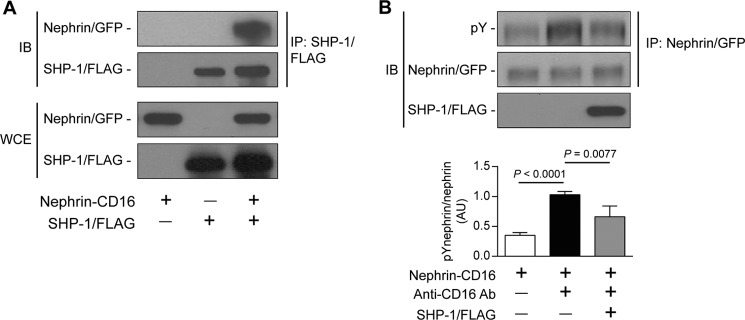
Interaction of SHP-1 and nephrin caused the reduction of nephrin phosphorylation. Shown are coimmunoprecipitation of SHP-1/FLAG with nephrin was performed in HEK cells transfected with nephrin/CD16/GFP and SHP-1/FLAG (A) and stimulated with anti-CD16 antibody (B). Protein expression of nephrin and SHP-1 and tyrosine phosphorylation of nephrin was detected by Western blot, and densitometric quantitation was measured. Results are shown as mean ± S.D. of three independent experiments. AU, arbitrary units; IB, immunoblot; IP, immunoprecipitation; WCE, whole cell extract.
SHP-1 Dephosphorylates Nephrin Specifically on Tyr1176/1193 and Tyr1217 Residues
Phosphorylation of specific tyrosine residues of nephrin can regulate different intracellular pathways, notably PI3K and Nck (17, 18). Because SHP-1 induced nephrin tyrosine dephosphorylation, we evaluated which tyrosine residues were dephosphorylated by SHP-1. Immunoblot analysis using antibodies specific against Tyr1176/1193 or Tyr1217 of nephrin was performed in HEK cells transfected with a plasmid of nephrin-CD16 only or nephrin-CD16 and SHP-1/FLAG. Cotransfection of SHP-1 and nephrin-CD16 decreased the phosphorylation levels of Tyr1176/1193 by 44% compared with cells expressing nephrin-CD16 alone and stimulated with anti-CD16 antibody (Fig. 3A). Similar results were observed with the phosphorylation levels of Tyr1217, with a 35% reduction of Tyr1217 phosphorylation level when SHP-1/FLAG was coexpressed with nephrin-CD16 (Fig. 3B). These results show that SHP-1 dephosphorylated nephrin specifically on Tyr1176/1193 and Tyr1217.
FIGURE 3.
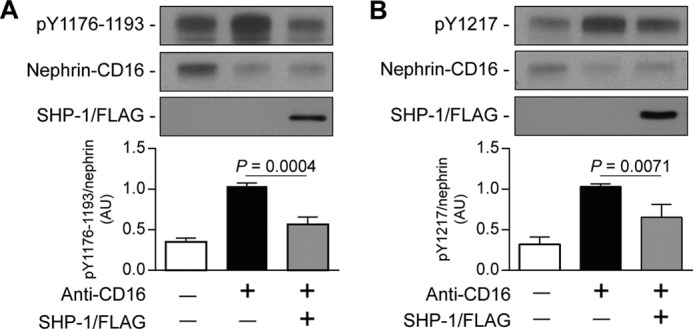
SHP-1 reduced Tyr1176/1193 and Tyr1217 phosphorylation of nephrin. Shown is nephrin phosphorylation of Tyr1176/1193 (A) and Tyr1217 (B) in HEK cells transfected with nephrin/CD16/GFP and SHP-1/FLAG and then stimulated with anti-CD16 antibody. Protein expression was detected by Western blot, and densitometric quantitation was measured. Results are shown as mean ± S.D. of three to four independent experiments. AU, arbitrary units.
Rat Nephrin Tyr1127 and Tyr1152 Are Required for SHP-1 Action
Although SHP-1 reduced Tyr1176/1193 and Tyr1217 phosphorylation, other tyrosine residues could be dephosphorylated and/or involved in binding of the N-terminal and C-terminal SH2 domains of SHP-1. To specifically identify which tyrosine residues are responsible for SHP-1 association with nephrin, we used rat nephrin constructs that possessed tyrosine-to-phenylalanine mutations (rY1127F, rY1152F, rY1171F, rY1194F, rY1204F, and rY1228F), as described previously (16). As shown in Fig. 4, mutation of rY1171F, rY1194F, rY1204F, and rY1228F did not prevent SHP-1-induced inhibition of nephrin phosphorylation. In contrast, both rY1127F and rY1152F significantly impaired the capability of SHP-1 capability to blunt nephrin phosphorylation, suggesting that these two nephrin tyrosine residues are required for SHP-1 actions.
FIGURE 4.

Nephrin mutation of Tyr1127 or Tyr1152 is required for SHP-1-induced nephrin dephosphorylation. Expression of SHP-1 and nephrin and phosphorylation of nephrin in HEK cells transfected with rat Fyn, nephrin, and SHP-1. Protein expression was detected by Western blot, and densitometric quantitation was measured. Results are shown as mean ± S.D. of three to four independent experiments. AU, arbitrary units.
Absence of Rat Nephrin Tyr1127 and Tyr1152 Prevents SHP-1 Interaction with Nephrin
To evaluate whether rat nephrin Tyr1127 and Tyr1152 are essential for the SHP-1 binding interaction, we performed coimmunoprecipitation assays with SHP-1 and nephrin mutants. Our results confirmed that Tyr1127 and Tyr1152 of rat nephrin are needed for the interaction of SHP-1 with nephrin. In cells transfected with SHP-1 and nephrin wild-type or nephrin rY1194F, SHP-1 was able to bind with nephrin. In contrast, the capacity of SHP-1 to interact with nephrin was lost when cotransfected with mutated nephrin rY1127F and rY1152F (Fig. 5).
FIGURE 5.
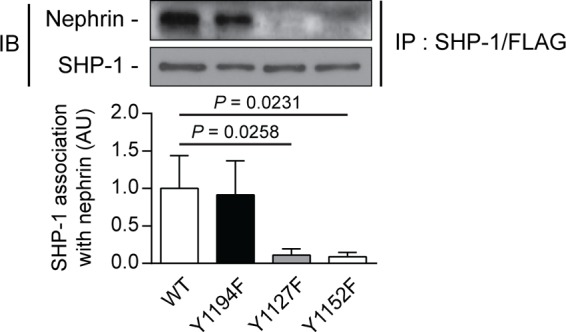
Loss of nephrin Tyr1127 or Tyr1152 prevented SHP-1 binding with nephrin. Shown is coimmunoprecipitation of SHP-1 with either nephrin WT, Y1194F-, Y1127F-, and Y1152F-transfected HEK293T cells. Protein expression was detected by Western blot, and densitometric quantitation was measured. Results are shown as mean ± S.D. of four independent experiments. AU, arbitrary units; IB, immunoblot; IP, immunoprecipitation.
Inhibition of SHP-1 in Podocytes Exposed to High Glucose Concentrations Restores Nephrin Tyrosine Phosphorylation Levels
A previous study has reported that another protein-tyrosine phosphatase (PTP1B) can dephosphorylate nephrin. To demonstrate that nephrin dephosphorylation is associated with SHP-1 expression under high-glucose conditions, we used an adenoviral vector expressing dominant negative SHP-1. Human podocytes were transduced with either GFP or dominant negative SHP-1 adenovirus and then exposed to 25 mmol/liter of glucose for 96 h. In GFP-transduced cells, high glucose decreased nephrin tyrosine phosphorylation by 60% (p = 0.0202) (Fig. 6). In contrast, overexpression of SHP-1 dominant negative in human podocytes prevented the high glucose-induced reduction of nephrin tyrosine phosphorylation (Fig. 6). The results suggest that SHP-1 up-regulation by high glucose mediates nephrin dephosphorylation.
FIGURE 6.
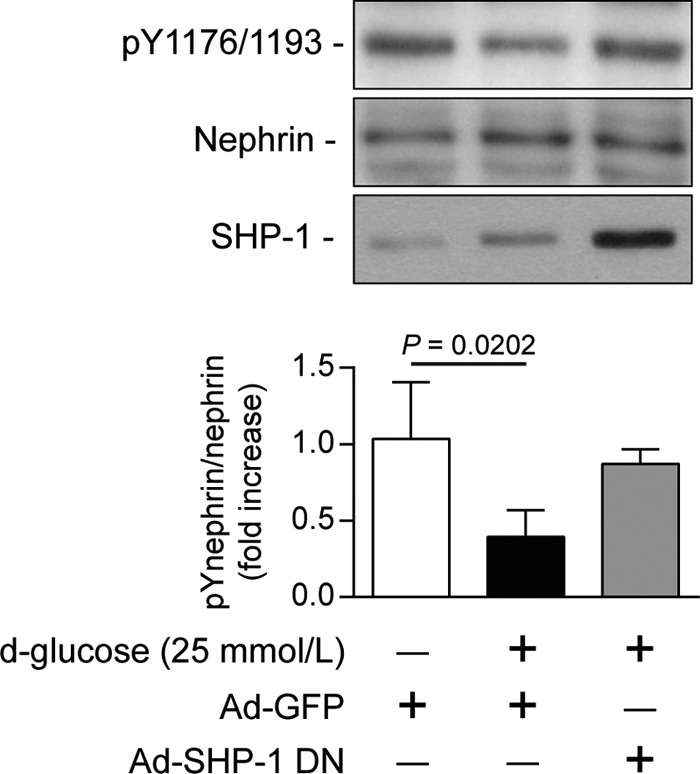
SHP-1 inhibition preserved nephrin phosphorylation during high glucose level exposure. Human podocytes were transduced with either Ad-GFP or Ad-dnSHP-1 and then incubated with normal (5.6 mmol/liter) or high glucose (25 mmol/liter) for 96 h. Protein expression of SHP-1 nephrin and Tyr(P)1176/1193 of nephrin were detected by Western blot, and densitometric quantitation was measured. Results are shown as mean ± S.D. of four independent experiments.
SHP-1 Expression Induced by Hyperglycemia Reduces Nephrin Tyrosine Phosphorylation in Type 1 Diabetic Mice
A previous study has shown that nephrin is phosphorylated in the adult mouse kidney and reduced during puromycin aminonucleoside-induced podocyte injury and proteinuria (25). To examine whether diabetes can affect nephrin tyrosine phosphorylation, the renal cortices of 7-month-old non-diabetic (Ins2+/+) and diabetic (Ins2+/C96Y) mice were harvested for histological analysis, and proteins were extracted for immunoblot analysis. As expected, diabetic Ins2+/C96Y mice exhibited lower body weight, higher levels of blood glucose, and elevated albuminuria and glomerular pathology (hypertrophy and mesangial cell expansion) (Fig. 7). Immunoblot analysis demonstrated that expression of SHP-1 and cleaved caspase-3, a proapoptotic marker, was increased in renal cortices of diabetic compared with non-diabetic littermate control mice (Fig. 8A). Elevated expression of SHP-1 was associated with decreased expression of nephrin and Tyr1217 phosphorylation in diabetic mice (Fig. 8A). The expression of SHP-1 and nephrin mRNA was measured in isolated glomeruli using laser capture microdissection. Our data confirmed elevated SHP-1 expression and reduced nephrin expression in diabetic mice compared with control littermates (Fig. 8B). Coimmunoprecipitation assays and immunofluorescence analysis demonstrated that diabetes increased the interaction of nephrin with SHP-1, which was correlated with a decline of nephrin phosphorylation (Fig. 8, C and D). These in vivo data support the notion that diabetes increased SHP-1 expression and was associated with nephrin, reducing its tyrosine phosphorylation.
FIGURE 7.
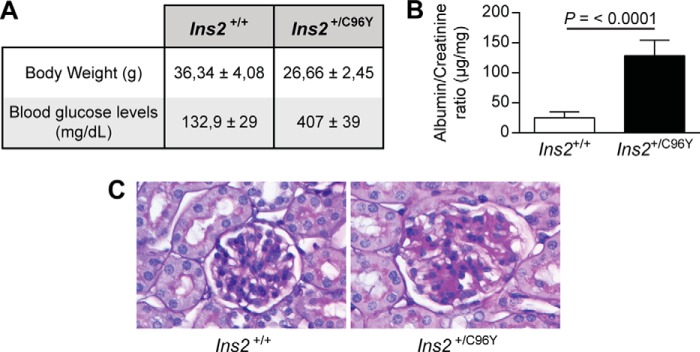
Blood glucose, renal function, and pathology. Shown are body weight and fasting blood glucose levels (A), urine albumin/creatinine ratio (B), and renal glomeruli stained with periodic acid Schiff for renal pathology (C) of Ins2+/+ and Ins2+/C96Y mice. Results are shown as mean ± S.D. of five animals.
FIGURE 8.

Expression of SHP-1 and nephrin is increased markedly in glomeruli of diabetic mice. A, nephrin, Tyr(P)1217 of nephrin, SHP-1, cleaved caspase-3, and actin protein expression from the renal cortex of Ins2+/+ and Ins2+/C96Y mice. B, mRNA expression of SHP-1 and nephrin in glomeruli using laser capture microdissection. C, immunoprecipitation (IP) assays of nephrin, Tyr(P)1176/1193 of nephrin, and SHP-1 from the renal cortex of Ins2+/+ and Ins2+/C96Y mice. Protein expression was detected by Western blot, and densitometric quantitation was measured. D, immunofluorescence labeling of podocytes (green, nephrin) and SHP-1 (red) within glomeruli of Ins2+/+ and Ins2+/C96Y mice. Results are shown as mean ± S.D. of five animals. Scale bar = 20 μm. AU, arbitrary units.
DISCUSSION
DN is a recognized microvascular complication that can ultimately lead to progressive renal failure. The glomerular podocyte plays a central and critical role in the structural and functional integrity of the glomerular filtration barrier and maintenance of normal renal function. The loss of podocytes is thought to be a key event in DN. It has been reported previously that mice and humans with diabetes exhibit a disruption of nephrin expression (27, 28). Furthermore, a previous study showed that podocytes expressing a mutated form of nephrin displayed increased apoptosis in response to serum starvation (29), suggesting a relationship with the absence of nephrin signaling actions. In our study, we demonstrated that nephrin expression and phosphorylation are reduced significantly in human podocytes exposed to high glucose levels and in the glomeruli of type 1 diabetic mice. The reduction of nephrin phosphorylation is, in part, due to an increase in expression of SHP-1, which binds to rat nephrin Tyr1127 and Tyr1152 residues (Fig. 9).
FIGURE 9.

Schematic of the SHP-1 interaction with nephrin tyrosine residues.
Despite the fact that the nonspecific PTP inhibitor sodium vanadate is able to cause dramatic morphological changes of cultured mouse podocytes, little has been known of the potential role of PTPs in podocyte function. GLEPP1 is the major PTP in the glomerulus and localizes in the apical membrane of podocytes (facing the Bowman capsule). Mice depleted of GLEPP1 display abnormal podocyte morphology and elevated blood pressure after uninephrectomy (30). Other studies have indicated that SHP2, PTP1B, PTP-PEST, and PTPD2 mRNA are expressed in cultured mouse podocytes (19). We have reported recently that another PTP, SHP-1, is also present in mouse podocytes and that its expression is elevated in cells exposed to high glucose levels (20), and we now confirm that high glucose concentrations enhanced SHP-1 protein expression in human podocytes. Besides the SHP-1 interaction with insulin and VEGF receptors (10, 20), our study indicates that SHP-1 binds to nephrin, affecting its phosphorylation state in podocytes. Recently, it has been shown that PTP1B, which is up-regulated in the rat model of puromycin aminonucleoside nephrosis, binds directly to and dephosphorylates nephrin (31). However, mouse podocytes exposed to high glucose levels did not exhibit elevated PTP1B expression (20), suggesting that PTP expression is triggered by different podocyte injury signals.
Nephrin induces a number of cell signaling pathways, including members of the MAP kinase family and PI3K/Akt (18), which are essential to reduce cell death induced by apoptotic stimuli. Additionally, nephrin tyrosine phosphorylation appears to regulate podocyte gene expression as well as the association of nephrin with podocin (16, 18). Until recently, the influence of nephrin tyrosine phosphorylation on the development of foot processes or glomerular injury and repair was uncertain. The only indication of nephrin signaling involvement in glomerular pathology came from the observation of Fyn kinase knockout mice that develop massive proteinuria and foot process effacement and, eventually, podocyte loss (32). Since then, studies by multiple investigators have clearly demonstrated the importance of the nephrin signaling pathway in podocyte function and injury. Nck, an adaptor protein with three SH3 domains and one SH2 domain, have been shown to bind to the phosphorylated Tyr1176/1193 and Tyr1217 residues of nephrin, causing the recruitment of signal transduction molecules such as focal adhesion kinase, p21-activated kinase, and Wiskott-Aldrich syndrome protein and the polymerization of actin (28, 33, 34). The physiological significance of this signal cascade in podocytes in vivo has been recognized using podocyte-specific knockout of Nck1 and Nck2, which exhibited foot process effacement and proteinuria (17, 35). We published previously that SHP-1, which is critical in abating cell response to growth factors, causes podocyte apoptosis (20). This study demonstrates that elevated expression of SHP-1 induced by high glucose levels reduces nephrin phosphorylation in vivo, which could contribute to podocyte dysfunction in diabetic nephropathy. Moreover, our results show that inhibition of SHP-1 by overexpressing the dominant negative form of SHP-1 prevented the high glucose-induced reduction of nephrin phosphorylation. Although the presence of phosphorylation in the normal state and the decrease in phosphorylation in a podocyte injury model clearly suggest that nephrin phosphorylation is important for maintaining the normal structure and function of podocytes, elevated nephrin phosphorylation has been reported in the pathogenesis of certain types of nephritis, such as in the rat Heymann nephritis model (16). Therefore, further animal and human studies are required to fill the gaps regarding the role of nephrin phosphorylation in kidney diseases.
Nephrin has suggested previously to aid podocyte survival because animals deficient in the nephrin-anchoring protein CD2AP have increased podocyte apoptosis and reduced cell survival (18). A recent study revealed that Nck directly regulates the Fyn-mediated phosphorylation of nephrin, which, in turn, potentiates activation of PI3K signaling pathways known to activate Akt involved in podocyte survival (36). Disruption of these signaling pathways has been correlated with the development of renal disease. Decreased Akt phosphorylation on Ser473 has been reported during the onset of proteinuria in a PAN-induced glomerular injury rat model (37). Furthermore, podocyte-specific Akt2-deficient mice (not Akt1) showed podocyte apoptosis and foot process effacement and developed glomerulosclerosis and albuminuria (38). Therefore, the PI3K/Akt signaling pathways through which nephrin acts play a key role in maintaining podocyte function and survival in addition to actin reorganization and barrier permeability. It is also known that SHP-1 binds to the p85 subunit of PI3K and prevents subsequent activation of Akt (39). Using a type 1 diabetic mouse model, we have shown that Akt phosphorylation was blunted by SHP-1 in podocytes (20). However, further studies are required to elucidate whether increased SHP-1 expression in the diabetic glomeruli could reduce Akt activity indirectly via decreased nephrin phosphorylation, leading to podocyte dysfunction and apoptosis in diabetic nephropathy.
SHP-1 and SHP-2 have two SH2 domains. Both N-terminal and C-terminal domains are critical for the localization and regulation of phosphatase activity. A splicing mutation in the N-terminal SH2 domain in mice causes the motheaten (me/me) phenotype, resulting in no detectable SHP-1 protein expression, severe hematopoietic cell disorder, and death within 2–3 weeks of age (26). In our study, we showed that the capability of SHP-1 to bind to nephrin was completely blunted when either the rat nephrin Tyr1127 or Tyr1152 were mutated to phenylalanine, suggesting that each tyrosine residue is essential for SHP-1 action on nephrin. In contrast, mutation of the Nck binding site, Y1194F, did not affect the ability of SHP-1 to interact with nephrin. Moreover, SHP-1-induced nephrin dephosphorylation was impaired when Tyr1127 and Tyr1152 were mutated. Taken together, our data suggest that these tyrosine residues are essential for the SHP-1/nephrin interaction and SHP-1-mediated nephrin dephosphorylation.
In summary, our work reveals a new mechanism by which SHP-1 deregulates nephrin activity, contributing to the apparition and progression of podocyte dysfunction in diabetic nephropathy. Besides regulating insulin and VEGF actions, SHP-1 decreased nephrin phosphorylation under hyperglycemic conditions. Our results emphasize the emerging hypothesis that, in podocytes, the inhibition of the signaling pathways of phosphorylated survival factors such as nephrin, insulin, and VEGF is an important contributor to the structure and biological function of podocytes. Therefore, the diabetic milieu may represent a vicious cycle of hyperglycemia, nephrin dysfunction, and podocyte insulin resistance, resulting in a severe diabetic nephropathy phenotype.
Acknowledgment
We thank Anne Vezina for assistance with immunofluorescence techniques.
This work was supported by Canadian Institute of Health Research Grant 201209PNI (to P. G.) and Juvenile Diabetes Research Foundation Grant 27-2012-531 (to P. G.). This work was performed at the Centre Hospitalier Universitaire de Sherbrooke Research Center funded by the Fonds de Recherche du Québec—Santé.
- DN
- diabetic nephropathy
- SH
- Src homology
- PTP
- protein-tyrosine phosphatase
- Ad
- adenovirus
- FRT
- flp recombination target.
REFERENCES
- 1. Greka A., Mundel P. (2012) Cell biology and pathology of podocytes. Annu. Rev. Physiol. 74, 299–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kerjaschki D. (2001) Caught flat-footed: podocyte damage and the molecular bases of focal glomerulosclerosis. J. Clin. Invest. 108, 1583–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaplan J. M., Kim S. H., North K. N., Rennke H., Correia L. A., Tong H. Q., Mathis B. J., Rodríguez-Pérez J. C., Allen P. G., Beggs A. H., Pollak M. R. (2000) Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 24, 251–256 [DOI] [PubMed] [Google Scholar]
- 4. Collins A. J., Kasiske B., Herzog C., Chavers B., Foley R., Gilbertson D., Grimm R., Liu J., Louis T., Manning W., McBean M., Murray A., St Peter W., Xue J., Fan Q., Guo H., Li Q., Li S., Qiu Y., Li S., Roberts T., Skeans M., Snyder J., Solid C., Wang C., Weinhandl E., Zhang R., Arko C., Chen S.-C. C., Dalleska F., Daniels F., Dunning S., Ebben J., Frazier E., Hanzlik C., Johnson R., Sheets D., Wang X., Forrest B., Berrini D., Constantini E., Everson S., Eggers P., Agodoa L. (2006) Excerpts from the United States Renal Data System 2006 Annual Data Report. Am. J. Kidney diseases 49, S1–S296 [DOI] [PubMed] [Google Scholar]
- 5. Sarnak M. J., Levey A. S., Schoolwerth A. C., Coresh J., Culleton B., Hamm L. L., McCullough P. A., Kasiske B. L., Kelepouris E., Klag M. J., Parfrey P., Pfeffer M., Raij L., Spinosa D. J., Wilson P. W., American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. (2003) Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 108, 2154–2169 [DOI] [PubMed] [Google Scholar]
- 6. Meyer T. W., Bennett P. H., Nelson R. G. (1999) Podocyte number predicts long-term urinary albumin excretion in Pima Indians with type II diabetes and microalbuminuria. Diabetologia 42, 1341–1344 [DOI] [PubMed] [Google Scholar]
- 7. Pagtalunan M. E., Miller P. L., Jumping-Eagle S., Nelson R. G., Myers B. D., Rennke H. G., Coplon N. S., Sun L., Meyer T. W. (1997) Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Invest. 99, 342–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. White K. E., Bilous R. W., Marshall S. M., El Nahas M., Remuzzi G., Piras G., De Cosmo S., Viberti G. (2002) Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes 51, 3083–3089 [DOI] [PubMed] [Google Scholar]
- 9. Eid A. A., Gorin Y., Fagg B. M., Maalouf R., Barnes J. L., Block K., Abboud H. E. (2009) Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes 58, 1201–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mima A., Kitada M., Geraldes P., Li Q., Matsumoto M., Mizutani K., Qi W., Li C., Leitges M., Rask-Madsen C., King G. L. (2012) Glomerular VEGF resistance induced by PKCδ/SHP-1 activation and contribution to diabetic nephropathy. FASEB J. 26, 2963–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baelde H. J., Eikmans M., Lappin D. W., Doran P. P., Hohenadel D., Brinkkoetter P. T., van der Woude F. J., Waldherr R., Rabelink T. J., de Heer E., Bruijn J. A. (2007) Reduction of VEGF-A and CTGF expression in diabetic nephropathy is associated with podocyte loss. Kidney Int. 71, 637–645 [DOI] [PubMed] [Google Scholar]
- 12. Hohenstein B., Hausknecht B., Boehmer K., Riess R., Brekken R. A., Hugo C. P. (2006) Local VEGF activity but not VEGF expression is tightly regulated during diabetic nephropathy in man. Kidney Int. 69, 1654–1661 [DOI] [PubMed] [Google Scholar]
- 13. Eremina V., Jefferson J. A., Kowalewska J., Hochster H., Haas M., Weisstuch J., Richardson C., Kopp J. B., Kabir M. G., Backx P. H., Gerber H. P., Ferrara N., Barisoni L., Alpers C. E., Quaggin S. E. (2008) VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 358, 1129–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coward R. J., Welsh G. I., Yang J., Tasman C., Lennon R., Koziell A., Satchell S., Holman G. D., Kerjaschki D., Tavaré J. M., Mathieson P. W., Saleem M. A. (2005) The human glomerular podocyte is a novel target for insulin action. Diabetes 54, 3095–3102 [DOI] [PubMed] [Google Scholar]
- 15. Kestila M., Lenkkeri U., Mannikko M., Lamerdin J., McCready P., Putaala H., Ruotsalainen V., Morita T., Nissinen M., Herva R., Kashtan C. E., Peltonen L., Holmberg C., Olsen A., Tryggvason K. (1998) Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol. Cell 1, 575–582 [DOI] [PubMed] [Google Scholar]
- 16. Li H., Lemay S., Aoudjit L., Kawachi H., Takano T. (2004) SRC-family kinase Fyn phosphorylates the cytoplasmic domain of nephrin and modulates its interaction with podocin. J. Am. Soc. Nephrol. 15, 3006–3015 [DOI] [PubMed] [Google Scholar]
- 17. Jones N., Blasutig I. M., Eremina V., Ruston J. M., Bladt F., Li H., Huang H., Larose L., Li S. S., Takano T., Quaggin S. E., Pawson T. (2006) Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 440, 818–823 [DOI] [PubMed] [Google Scholar]
- 18. Huber T. B., Hartleben B., Kim J., Schmidts M., Schermer B., Keil A., Egger L., Lecha R. L., Borner C., Pavenstädt H., Shaw A. S., Walz G., Benzing T. (2003) Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol. Cell. Biol. 23, 4917–4928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reiser J., Pixley F. J., Hug A., Kriz W., Smoyer W. E., Stanley E. R., Mundel P. (2000) Regulation of mouse podocyte process dynamics by protein tyrosine phosphatases rapid communication. Kidney Int. 57, 2035–2042 [DOI] [PubMed] [Google Scholar]
- 20. Drapeau N., Lizotte F., Denhez B., Guay A., Kennedy C. R., Geraldes P. (2013) Expression of SHP-1 induced by hyperglycemia prevents insulin actions in podocytes. Am. J. Physiol. Endocrinol. Metab. 304, E1188–E1198 [DOI] [PubMed] [Google Scholar]
- 21. Roy S., Perron B., Gallo-Payet N. (2010) Role of asparagine-linked glycosylation in cell surface expression and function of the human adrenocorticotropin receptor (melanocortin 2 receptor) in 293/FRT cells. Endocrinology 151, 660–670 [DOI] [PubMed] [Google Scholar]
- 22. Saleem M. A., O'Hare M. J., Reiser J., Coward R. J., Inward C. D., Farren T., Xing C. Y., Ni L., Mathieson P. W., Mundel P. (2002) A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J. Am. Soc. Nephrol. 13, 630–638 [DOI] [PubMed] [Google Scholar]
- 23. Dubois M.-J., Bergeron S., Kim H.-J., Dombrowski L., Perreault M., Fournès B., Faure R., Olivier M., Beauchemin N., Shulman G. I., Siminovitch K. A., Kim J. K., Marette A. (2006) The SHP-1 protein tyrosine phosphatase negatively modulates glucose homeostasis. Nat. Med. 12, 549–556 [DOI] [PubMed] [Google Scholar]
- 24. Geraldes P., Hiraoka-Yamamoto J., Matsumoto M., Clermont A., Leitges M., Marette A., Aiello L. P., Kern T. S., King G. L. (2009) Activation of PKC-δ and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat. Med. 15, 1298–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones N., New L. A., Fortino M. A., Eremina V., Ruston J., Blasutig I. M., Aoudjit L., Zou Y., Liu X., Yu G.-L., Takano T., Quaggin S. E., Pawson T. (2009) Nck proteins maintain the adult glomerular filtration barrier. J. Am. Soc. Nephrol. 20, 1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsui H. W., Siminovitch K. A., de Souza L., Tsui F. W. (1993) Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat. Genet. 4, 124–129 [DOI] [PubMed] [Google Scholar]
- 27. Kim J.-J., Li J. J., Jung D.-S., Kwak S.-J., Ryu D.-R., Yoo T.-H., Han S. H., Choi H. Y., Kim H. J., Han D. S., Kang S.-W. (2007) Differential expression of nephrin according to glomerular size in early diabetic kidney disease. J. Am. Soc. Nephrol. 18, 2303–2310 [DOI] [PubMed] [Google Scholar]
- 28. Welsh G. I., Saleem M. A. (2010) Nephrin-signature molecule of the glomerular podocyte? J. Pathol. 220, 328–337 [DOI] [PubMed] [Google Scholar]
- 29. Foster R. R., Saleem M. A., Mathieson P. W., Bates D. O., Harper S. J. (2005) Vascular endothelial growth factor and nephrin interact and reduce apoptosis in human podocytes. Am. J. Physiol. 288, F48–F57 [DOI] [PubMed] [Google Scholar]
- 30. Wharram B. L., Goyal M., Gillespie P. J., Wiggins J. E., Kershaw D. B., Holzman L. B., Dysko R. C., Saunders T. L., Samuelson L. C., Wiggins R. C. (2000) Altered podocyte structure in GLEPP1 (Ptpro)-deficient mice associated with hypertension and low glomerular filtration rate. J. Clin. Invest. 106, 1281–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aoudjit L., Jiang R., Lee T. H., New L. A., Jones N., Takano T. (2011) Podocyte protein, Nephrin, is a substrate of protein tyrosine phosphatase 1B. J. Signal Transduct. 10.1155/2011/376543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu C. C., Yen T. S., Lowell C. A., DeFranco A. L. (2001) Lupus-like kidney disease in mice deficient in the Src family tyrosine kinases Lyn and Fyn. Curr. Biol. 11, 34–38 [DOI] [PubMed] [Google Scholar]
- 33. Rohatgi R., Nollau P., Ho H. Y., Kirschner M. W., Mayer B. J. (2001) Nck and phosphatidylinositol 4,5-bisphosphate synergistically activate actin polymerization through the N-WASP-Arp2/3 pathway. J. Biol. Chem. 276, 26448–26452 [DOI] [PubMed] [Google Scholar]
- 34. Lu W., Katz S., Gupta R., Mayer B. J. (1997) Activation of Pak by membrane localization mediated by an SH3 domain from the adaptor protein Nck. Curr. Biol. 7, 85–94 [DOI] [PubMed] [Google Scholar]
- 35. Verma R., Kovari I., Soofi A., Nihalani D., Patrie K., Holzman L. B. (2006) Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J. Clin. Invest. 116, 1346–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. New L. A., Keyvani Chahi A., Jones N. (2013) Direct regulation of nephrin tyrosine phosphorylation by Nck adaptor proteins. J. Biol. Chem. 288, 1500–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhu J., Sun N., Aoudjit L., Li H., Kawachi H., Lemay S., Takano T. (2008) Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int. 73, 556–566 [DOI] [PubMed] [Google Scholar]
- 38. Canaud G., Bienaimé F., Viau A., Treins C., Baron W., Nguyen C., Burtin M., Berissi S., Giannakakis K., Muda A. O., Zschiedrich S., Huber T. B., Friedlander G., Legendre C., Pontoglio M., Pende M., Terzi F. (2013) AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat. Med. 19, 1288–1296 [DOI] [PubMed] [Google Scholar]
- 39. Yu Z., Su L., Hoglinger O., Jaramillo M. L., Banville D., Shen S. H. (1998) SHP-1 associates with both platelet-derived growth factor receptor and the p85 subunit of phosphatidylinositol 3-kinase. J. Biol. Chem. 273, 3687–3694 [DOI] [PubMed] [Google Scholar]