

Background: Lethal mutagenesis is an antiviral strategy.
Results: Nucleoside analogue mutagenicity is correlated to base pair stability and the ability to function as a template during reverse transcription.
Conclusion: Two simple biophysical/biochemical assays predict mutagenicity of nucleoside analogues.
Significance: Mutagenicity of anti-HIV-1 compounds can be reliably predicted in vitro without labor-intensive and costly in cellula tests.
Keywords: Antiviral Agent, Human Immunodeficiency Virus (HIV), Mutagenesis, Nucleoside/Nucleotide Analogue, Reverse Transcription, Lethal
Abstract
Because of their high mutation rates, RNA viruses and retroviruses replicate close to the threshold of viability. Their existence as quasi-species has pioneered the concept of “lethal mutagenesis” that prompted us to synthesize pyrimidine nucleoside analogues with antiviral activity in cell culture consistent with an accumulation of deleterious mutations in the HIV-1 genome. However, testing all potentially mutagenic compounds in cell-based assays is tedious and costly. Here, we describe two simple in vitro biophysical/biochemical assays that allow prediction of the mutagenic potential of deoxyribonucleoside analogues. The first assay compares the thermal stabilities of matched and mismatched base pairs in DNA duplexes containing or not the nucleoside analogues as follows. A promising candidate should display a small destabilization of the matched base pair compared with the natural nucleoside and the smallest gap possible between the stabilities of the matched and mismatched base pairs. From this assay, we predicted that two of our compounds, 5-hydroxymethyl-2′-deoxyuridine and 5-hydroxymethyl-2′-deoxycytidine, should be mutagenic. The second in vitro reverse transcription assay assesses DNA synthesis opposite nucleoside analogues inserted into a template strand and subsequent extension of the newly synthesized base pairs. Once again, only 5-hydroxymethyl-2′-deoxyuridine and 5-hydroxymethyl-2′-deoxycytidine are predicted to be efficient mutagens. The predictive potential of our fast and easy first line screens was confirmed by detailed analysis of the mutation spectrum induced by the compounds in cell culture because only compounds 5-hydroxymethyl-2′-deoxyuridine and 5-hydroxymethyl-2′-deoxycytidine were found to increase the mutation frequency by 3.1- and 3.4-fold, respectively.
Introduction
Three decades after the discovery of the human immunodeficiency virus type 1 (HIV-1) as the causative agent of the acquired immunodeficiency syndrome (AIDS) (1, 2), extensive research on the viral replication mechanisms and their inhibition has provided a panel of 24 currently distinct chemical entities approved by the Food and Drug Administration (aidsinfo.nih.gov). Reverse transcription of the single-stranded HIV-1 RNA genome into double-stranded DNA is a key step of the viral replication cycle. This process is carried out by the viral reverse transcriptase (RT), bearing RNA- and DNA-dependent DNA polymerase activities as well as RNase H activity (3). The first anti-HIV compound was an RT inhibitor (4), and RT still remains, to date, a major therapeutic target, with 19 Food and Drug Administration-approved single and combination drugs (5, 6).
HIV-1 is an actively replicating virus, and RT is a highly error-prone enzyme. Combined with a lack of proofreading activity of RT (7) and the absence of DNA repair mechanisms within RNA/DNA hybrids (8), it results in mutation rates as high as 5–10 mutations per replication cycle (9–16) As a consequence, antiviral therapies rapidly lead to the emergence of viral populations that are resistant to all the drugs used in highly active antiretroviral therapies. The inevitable emergence and transmission of resistance thus drive the need for the constant search for new drug targets and/or new inhibitors against existing targets, possibly acting through new molecular mechanisms, active against resistant viruses, or displaying different resistance patterns.
As a direct consequence of their high mutation propensity, HIV-1 and other viruses that replicate through RNA-dependent RNA polymerases or RT co-exist within an infected individual as “quasi-species,” a viral population of related genomes of different, albeit similar, sequences (17–19). For these viruses, part of the mutant viral population is nonviable, a handicap that is overcome, in the particular case of retroviruses, by a high replication rate (1010 viruses produced per day (20, 21)) and a high rate of genetic recombination (22, 23), which allow the selection of the fittest viruses. The existence of quasi-species is thus based on an equilibrium between mutations and their selection for survival. For most of the viruses with an RNA genome, the quasi-species theory predicts that a small increase in the number of genomic mutations might push the viral population beyond a threshold that results in a sudden loss of viability (24). The lethal accumulation of mutations was named “error catastrophe” in virology (25, 26) and has lead to the concept of “lethal mutagenesis” (26–28) that aims at the ultimate extinction of all viable viral sequences.
Enhanced mutagenesis as an antiviral strategy was first demonstrated on vesicular stomatitis virus and poliovirus (29). The same concept was applied to several other RNA viruses such as foot-and-mouth disease virus (FMDV)6 (30–32), Hantaan virus (33, 34), or hepatitis C virus (35, 36). Notably, the ribonucleoside analogue ribavirin has been used to treat hepatitis C infection since 1998 (37, 38) before its mode of action, by lethal mutagenesis (35, 39, 40), was even elucidated.
The lethal mutagenesis concept was also tested on retroviruses such as spleen necrosis virus (41) and HIV (27, 42). In the latter case, Loeb et al. (27) used the deoxynucleoside analogue 5-hydroxy-2′-deoxycytidine (5-OH-dC), incorporated by HIV-1 RT opposite guanosines in the vRNA template strand. The subsequent incorporation of deoxyadenosine during (+)-strand DNA synthesis resulted in a 2-fold increase in the mutation rate of the HIV-1 genome due to an increase of G to A substitutions and had a strong effect on viral lethality (27). The compound 5,6-dihydro-5-aza-2′-deoxycytidine (KP1212) also efficiently inhibited HIV replication in cell culture (43), and its pro-drug form, KP1461, underwent phase II clinical trials with Koronis Pharmaceuticals. This trial revealed no toxicity nor safety issues, but although the analogue induced mutations, there was no impact on viral replication (44). A recent study revealed that the mutagenic properties of KP1212 were linked to its ability to adopt multiple tautomeric forms that pair with purine bases (A and G) (45). Finally, even though azidothymidine or 5-OH-dC alone failed to completely extinguish HIV-1 viruses that replicated efficiently, the combination of the two drugs was proven to eradicate HIV-1 in cell culture (46).
Even though it has been challenging to get promising mutagenic compounds into the clinic, the validity of lethal mutagenesis as an antiviral strategy was reinforced in the past few years with the understanding of the cellular defense mechanisms against viruses, especially in the case of HIV-1. The host-encoded apolipoprotein B mRNA-editing complex (APOBEC) family of cytidine deaminases, and more precisely the APOBEC3 family, acts against viruses by hypermutagenesis of single-stranded viral DNA. In the case of HIV-1, APOBEC3G/3F lead to G to A hypermutation of the (+)-strand DNA (47–50). The lethal mutagenesis induced by APOBEC3G/3F is so powerful that retroviruses have developed a viral infectivity factor specifically to prevent expression and induce degradation of these restriction factors (51–53). Interestingly, APOBEC3G/3F induce lethal mutagenesis very rapidly, within a single round of replication (54, 55), which has brought back the hope that it should be possible to mimic, in therapy, what cellular evolution has reached (56, 57).
Based on that idea, we have synthesized several pyrimidine nucleoside analogues that are modified exclusively on position 5 of the base moiety by carbamoyl, carbamoylmethyl, hydroxyl or hydroxymethyl groups to remain potential substrates for HIV-1 RT after their conversion into triphosphates, to base pair with at least two natural nucleosides, and ultimately to increase the error rate in the proviral DNA (58). Accordingly, O'Neil et al. (12) established that the contribution of RT in HIV-1 mutagenesis is about 2-fold higher than the contribution of host cell polymerases. As a control, we have also prepared one analogue modified on position 3 (Watson-Crick position) of the base moiety by a carbamoylmethyl group. Interestingly, some of these compounds displayed antiviral activity in cell culture, and two of them displayed antiviral activity in cell culture in a way that was consistent with an accumulation of deleterious mutations in the HIV-1 genome (58).
Nucleoside analogues added to cell culture assays or administered to patients must undergo several steps before exerting their inhibitory effects. 1) They have to be phosphorylated at a significant level by the cellular enzymes. 2) The tri-phosphorylated nucleoside analogues have to be incorporated efficiently by RT into the (−)-strand DNA. 3) The incorporated nucleoside analogues have to be extended during (−)-strand DNA synthesis. 4) During (+)-strand DNA synthesis, nucleoside analogues incorporated into the (−)-strand DNA have to base pair with matched (no mutagenesis) or mismatched (mutagenesis) natural nucleotides. 5) Duplexes have to be extended. 6) Alternatively, nucleotide analogues can be incorporated and extended during (+)-strand DNA synthesis to form mutagenic mismatched base pairing. 7) Incorporated mutagenic nucleotides have to escape DNA repair. If all these steps are completed, mutated RNA genomes, leading to nonviable proteins, can be produced through cellular RNA transcription after provirus integration.
Here, we describe two simple in vitro assays, one based on measuring the stability of mismatched base pairs and the other assessing the efficiency of steps 4 and 5 described above. The predictive potential of our screens was confirmed by detailed analysis of the mutation spectrum in HIV-1 genomes induced by the compounds in cellula.
EXPERIMENTAL PROCEDURES
Melting Temperatures
Undecamer DNA oligodeoxyribonucleotides (oligonucleotides) with the sequence 5′-CGACTXGATC (X standing for compound 1, 2, 4, or 5, Fig. 1) were prepared on solid phase and purified as described previously (59). The oligonucleotides with X = compound 3 or 6 were purchased from Eurogentec (Liège, Belgium) (Table 1). The corresponding unmodified 11-mer control oligonucleotides, as well as the complementary strand, were purchased from Thermo Electron (Courtaboeuf, France). Pairs of complementary oligonucleotides were dissolved at a final concentration of 10 μm in a solution of sodium citrate (0.1 m) and NaCl (1 m) at pH 7 in the presence of 0.25 μm SYBR Green I (Stratagene) and 0.5 μm ROX (Invitrogen). Melting curves were determined by measuring the fluorescence emission at 516 nm (excitation wavelength, 492 nm) on a Stratagene MX-4000 quantitative PCR apparatus. Duplexes were hybridized by heating from 25 to 95 °C at 2 °C/s, cooling to 4 °C at 0.1 °C/s, heating to 70 °C at 0.2 °C/s, and finally cooling to 15 °C at 0.2 °C/s. For melting temperature determination by acquisition of the fluorescence emission, temperature of the solution was increased from 15 to 91 °C at a 0.6 °C/s rate; no difference was observed when heating was performed at 0.2 °C/min. The melting temperature was defined as the maximum of the first derivative of the fluorescence intensity versus temperature. For each sample, Tm was measured twice, and the results are the average of at least three independent experiments. Results are expressed as mean ± S.D.
FIGURE 1.
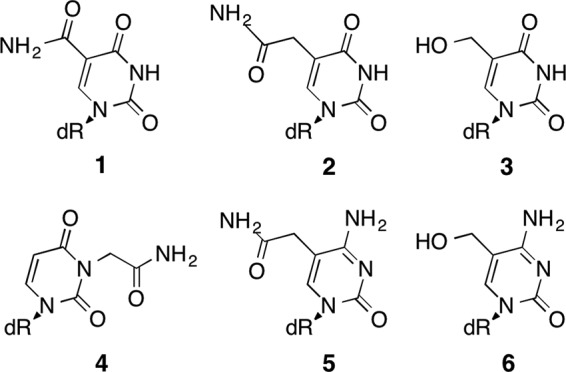
Structure of the nucleoside analogues evaluated in this study. dR stands for deoxyribose. Compound 1, 5-carbamoyl-2′-deoxyuridine also named 5-carbamoyl-dU; compound 2, 5-carbamoylmethyl-2′-deoxyuridine or ncm5-dU; compound 3, 5-hydroxymethyl-2′-deoxyuridine or hm5-dU; compound 4, 3-carbamoylmethyl-2′-deoxyuridine or ncm3-dU; compound 5, 5-carbamoylmethyl-2′-deoxycytidine also named ncm5-dC; and compound 6, 5-hydroxymethyl-2′-deoxycytidine or hm5-dC.
TABLE 1.
Oligodeoxyribonucleotides used for thermal stability measurements
The names and sequences of the oligonucleotides with modified or natural bases in the central position are listed. The numbers in the middle of the sequence refer to the modified nucleotides shown in Fig. 1.
Plus-strand DNA Synthesis
31-Mer DNA oligonucleotides containing the modified 2′-deoxynucleotide analogues 1, 2, 4, or 5 at position 20 from their 5′ end were synthesized on solid phase and purified as described previously (59); those containing compounds 3 and 6 were purchased from Eurogentec (Liège, Belgium). The complementary unmodified 19-mer primer (Thermo Electron, Courtaboeuf, France) was labeled at its 5′ end with [γ-32P]ATP using phage T4 polynucleotide kinase and purified on 8% denaturing polyacrylamide gels. Primer extension assays were performed using the p66/p51 RT bearing the E478Q mutation that abolishes RNase H activity (60), which was expressed and purified according to a method adapted from Ref. 61.
Primer-template complexes were formed by incubating the primer with a 3-fold excess of modified 31-mer oligonucleotide template in water for 2 min at 90 °C, cooling on ice for 2 min, and incubating for 20 min at 50 °C in 100 mm NaCl. Primer-template complexes at a final concentration of 10 nm were preincubated with 30 nm RT (buffer composition: 62.5 mm Tris-HCl, pH 8.3, at 37 °C, 50 mm KCl, 6 mm MgCl2, 1,5 mm dithioerythritol, 3.25% glycerol, 2% polyethylene glycol 6000, 25 mm potassium acetate) at 37 °C for 4 min. Reactions were initiated by the addition of 20 μm of one of the four dNTPs with or without 20 μm ddGTP and stopped at various times with an equal volume of buffer containing formamide. Reaction products were denatured for 2 min at 90 °C prior to separation on a 15% polyacrylamide denaturing gel and quantified using a Fuji FLA-5100 analyzer and the ImageGauge software. Data are the average values of two experiments.
Sequencing of the HIV-1 pol Gene after Treatment with Mutagenic Nucleosides
Serial HIV-1 passage experiments in the presence of compounds 1-6 or in the presence of DMSO (as control) were described previously (58). After the first incubation with the nucleoside analogues (passage 0) and at the end of the procedure (passage 8), total cellular DNA was extracted and purified using the DNeasy tissue kit (Qiagen). Viral DNA of the samples treated with compounds 1-3 and 6 and the DMSO control were amplified using Pfu DNA polymerase using pol forward primer, 5′-ATTAAAGCCAGGAATGGATGGCCCAAAAG-3′ and pol reverse primer, 5′-ATCTCCCTGTTTTCTGCCAGTTCTAGCTCTG-3′, amplifying a sequence of 835 nucleotides (from nucleotides 2611 to 3445) in the polymerase gene of the HIV-1 genome. Amplification consisted of 30 cycles of the following steps (94 °C, 5 min/94 °C, 1 min/63 °C, and 1 min/72 °C, 1 min). The amplified DNAs (four independent PCRs performed on each DNA sample) were ligated into the PCR II-TOPO TA cloning vector (Invitrogen) and sequenced using the adjacent M13 primers. For each PCR, 16 individual clones were selected for sequencing using the Sanger technique (GATC Biotech, Germany).
RESULTS
Compared with classical antiviral nucleoside analogues acting as chain terminators, the biological evaluation of potential mutagenic nucleoside analogues is particularly labor-intensive, as it requires analysis of the effects of the drug candidates over successive viral passages and detailed analysis of the mutations induced by these compounds (35, 62, 63). Therefore, simple biophysical/biochemical tests providing information about the mutagenic potential of nucleoside analogues would be very useful for reducing the number of compounds that will undergo biological evaluation. Among a series of eight nucleoside analogues, we previously showed that two of them, hm5-dU (compound 3) and hm5-dC (compound 6) (Fig. 1), were able to progressively inhibit HIV-1 replication, most likely by causing lethal mutagenesis (58). Here, we compared six of these compounds, including the two mutagenic nucleosides, using thermal denaturation of short DNA duplexes and extension of a primer annealed to a modified template. In addition, we determined the mutation rate and spectrum of the HIV-1 genome upon treatment with compounds 1-3 and 6 in cell culture and compared them with the in vitro data.
Thermal Stability of DNA Duplexes Containing Nucleoside Analogues
Mutagenic nucleotide analogues must be able to form at least two fairly stable base pairs with natural nucleotides, and in the absence of proofreading activity, as is the case for HIV-1 RT, the frequency of misincorporation is expected to be correlated, as was the case for repair enzymes (64), to the stability of mismatched base pairs (65, 66). To estimate the stability of these base pairs, we evaluated the thermal stability of a series of short DNA duplexes in which each nucleotide analogue was base paired with the four natural nucleotides. The stability of DNA duplexes containing pairs of matched or mismatched natural nucleotides at the same position was used as a reference.
For this study, the influence of the base modifications on the duplex thermal stability was evaluated on 11-mer oligonucleotides, as they allowed us to obtain reliable Tm values in the range of 40–70 °C. Their sequences were chosen to avoid self-annealing and formation of intra-molecular hairpin structures (Table 1). In all cases, the modification was introduced at the central position of the oligonucleotides (Table 1, entries 1–6), and the natural oligonucleotides (Table 1, entries 7–10) were used as reference. The complementary (Compl.) strands (Table 1, entries 11–14) with one of the natural bases at the central position were used to form all the possible matched and mismatched duplexes for each of the modified or natural bases. Because establishing the classical UV melting curves of DNA duplexes is time-consuming, we determined the melting temperature by following the fluorescence emission decrease of the SYBR Green I intercalant as temperature increased on a quantitative PCR apparatus. This approach allowed us to determine up to 96 melting curves simultaneously. The Tm values for the 16 possible combinations of dX/Compl. dY hybrids containing unmodified nucleosides are presented in Table 2 (boldface values). We found that dT/dY mismatches destabilized DNA duplexes by 8.5–12.2 °C (compare ΔTm between the mismatched dT/dY and the matched dT/dA pairs (Table 3, dT entry)), and that, as expected from the literature (65), the dT/dG mismatch was the most stable. For dC/dY mismatches, we observed more pronounced destabilization compared with dT/dY mismatches, with Tm values decreased by 16.4–18.4 °C. These results are in agreement with the literature as we found that the dC/dT mismatch was the most stable (Table 3, dC entry). In addition, the base pairs follow the expected order as follows: dC/dG ≫ dA/dT ≫ dT/dG ≅ dT/dT ≅ dG/dG ≅ dA/dG > dA/dA ≅ dT/dC > dA/dC ≅ dC/dC (65). Because the “dX-11-mer” and “Compl. dY-11-mer” oligonucleotides were designed to be fully complementary, except at the central position (depending on the X and Y bases), the Tm of the duplexes should reflect the dX/dY pairing or mispairing abilities. Indeed, the Tm of the dX/Compl. dY duplexes were not significantly different from those of the dY/Compl. dX duplexes (p > 0.05), except for the dA/dC (p = 0.01) and dT/dC duplexes (p = 0.05), indicating that the neighboring bases effect is negligible and that the variation observed in Tm values is mainly due to the modifications in the central position (Table 2, boldface values).
TABLE 2.
Melting temperatures of matched and mismatched dX/Compl. dY undecamer duplexes
Melting temperatures were measured by fluorescence. Tm was obtained for the matched and mismatched (dX/Compl. dY) duplexes, where X is one of the six modified nucleoside analogues, and Y one of the four natural nucleoside. In boldface type are Tm values obtained for the matched and mismatched natural (dX/Compl. dY) duplexes. Tm (°C) values are the means of three independent experiments ± S.D.
| Compl. dA | Compl. dT | Compl. dC | Compl. dG | |
|---|---|---|---|---|
| dT | 66.4 ± 1.7 | 56.8 ± 1.3 | 54.2 ± 1.4 | 57.9 ± 1 |
| 5-Carbamoyl-dU (1) | 63.7 ± 1 | 54.5 ± 1 | 52.2 ± 1 | 55.2 ± 1 |
| ncm5-dU (2) | 59.6 ± 1.6 | 52.4 ± 1 | 46.2 ± 1 | 55.7 ± 1.2 |
| hm5-dU (3) | 62.3 ± 0.8 | 55.6 ± 1.2 | 53.7 ± 0.4 | 57.8 ± 0.4 |
| ncm3-dU (4) | 50.2 ± 0.5 | 48.6 ± 1 | 48.2 ± 1 | 49 ± 1 |
| dC | 50.9 ± 1.2 | 52.9 ± 1.1 | 52.5 ± 1.3 | 69.3 ± 1.6 |
| ncm5-dC (5) | 47 ± 1 | 46.6 ± 1.3 | 41 ± 1.3 | 62 ± 1.6 |
| hm5-dC (6) | 53.9 ± 0 | 53.6 ± 0.4 | 50.2 ± 0.4 | 66.3 ± 0.6 |
| dA | 54.4 ± 0.7 | 65.8 ± 0.7 | 53 ± 0.6 | 56.2 ± 0.5 |
| dG | 55.1 ± 1.1 | 58.1 ± 1.1 | 68.7 ± 1.5 | 56.3 ± 0.6 |
TABLE 3.
Differences in Tm between different types of duplexes
In each row, the differences in Tm (ΔTm) between the duplex with a central “matched” base pair (see Table 2, 1st column for dU analogues and 4th column for dC analogues) and duplexes with a central “mismatched” base pair (Table 2, 2nd, 3rd, and 4th columns for dU analogues and 1st, 2nd, and 3rd columns for dC analogues) are indicated in black. Differences of Tm between the control matched duplexes (Table 2, 1st column/1st row for dU analogues and/or 4th column/6th row for dC analogues) and the matched duplexes with a central nucleoside analogue (Table 2, 1st column (dU analogues) or and 4th column (dC analogues) values in normal type) are indicated in green. In each column, differences in ΔTm (ΔΔTm) between the duplexes containing a central nucleoside analogue and the control duplexes in this table, 1st row for dU analogues and 6th row for dC analogues), containing only natural unmodified nucleotides, are indicated in red when negative and in blue when positive.
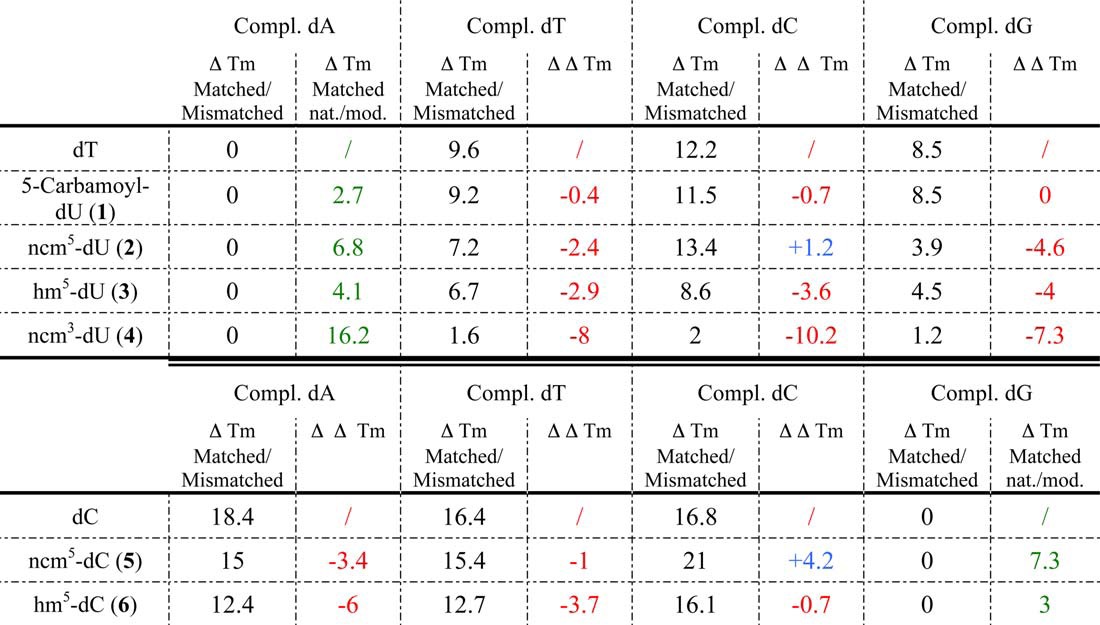
Nucleoside analogues that would decrease the Tm difference (ΔTm, Table 3, values in black) between matched and mismatched base pairs for at least two natural nucleosides should increase the probability of mismatch formation during reverse transcription. Based on this criterion, 5-carbamoyl-dU (compound 1) should be a very poor mutagenic compound as the ΔTm between the duplex with a central matched base pair and the ones with a central mismatched base pair were almost the same as for dT, e.g. ΔΔTm (Table 3, values in red) were close to 0. Compound 4 (ncm3-dU), modified on its Watson-Crick side, is expected to have poor base pairing abilities, and thus it acts as our negative control compound. Accordingly, compound 4 formed base pairs of similar low stability with all unmodified dYs (Tables 2 and 3); ΔTm (matched/mismatched) values were small, and ΔΔTm values were large and negative (Table 3). Thus, if this nucleoside analogue was to be introduced into DNA during (−)-strand DNA synthesis, the efficiencies of incorporation of the four unmodified dY opposite compound 4 during (+)-strand DNA synthesis would be expected to be similar. However, all these base pairs, including the compound 4/dA base pair, were very unstable (Table 2); thus, addition of any dY in front of ncm3-dU is expected to be very inefficient. In addition, as the compound 4/dA base pair was considerably less stable than the dT/dA base pair (Tables 2 and green values of ΔTm (matched natural/matched modified) in Table 3), it is expected that the ncm3-dU-TP (compound 4) analogue would not be incorporated during (−)-strand DNA synthesis, because of the competition with dTTP.
Thus, the two criteria to consider for putative mutagen nucleoside analogues are as follows: (i) a large decrease in the gap between the stability of the matched and mismatched base pairs (large negative ΔΔTm, in red in Table 3) representative of the possibility to form one or more mismatched base pair, and (ii) a small destabilization of the matched base pair involving the nucleoside analogue, as compared with the natural matched base pair (in green in Table 3) for more favorable competition of the modified nucleoside analogue triphosphates with the natural ones. According to these criteria, compound 3 (hm5-dU) appears as the most promising dT analogue as it combines a fairly large ΔΔTm for all three 3/dY mismatches with a moderate destabilization of the matched duplexes (Table 3). In contrast, the ncm5-dU (compound 2) displayed a large negative value for only one mismatch (compound 2/dG) and induced a more pronounced destabilization of the matched duplexes (Table 3). Concerning the dC analogues, compound 6 (hm5-dC) appeared clearly superior to compound 5 (Table 3). Indeed, in this assay, hm5-dC (compound 6) has the most promising properties of all nucleotide analogues we tested, as it has very large (negative) ΔΔTm values for two mismatches, and the hm5-dC/dG is only 3 °C less stable than the natural base pair (Table 3).
Note that, except for our negative control ncm3-dU (compound 4), the matched base pairs formed by the modified nucleotides analogues are more stable than the mismatched pairs of unmodified nucleotides (Table 2). Thus, provided they are efficiently converted into their triphosphate form, they should be incorporated fairly efficiently during (−)-strand DNA synthesis by forming a matched base pair and introduce mutations during (+)-strand DNA strand synthesis (if their ΔΔTm is large and negative). However, mismatches involving nucleotide analogues are usually less stable than those formed by unmodified nucleotides (with the noticeable exception of compound 6/dA and compound 6/dT mismatches); thus the mutational potential of nucleotide analogues is expected to be lower during (−)-DNA strand synthesis than during (+)-DNA strand synthesis. To summarize, measurements of the thermal stability of matched and mismatched base pairs in DNA duplexes allow ranking of the mutagenic potential of our compounds: compound 4 (no stable base pairings, nonmutagenic) < 1 ≈ 2 (only one stable base pairing, nonmutagenic) ≪ 5 (multiple base pairings with medium thermal stabilities, poorly mutagenic) ≪ 3 ≈ 6 (multiple base pairings with good thermal stabilities, mutagenic).
DNA Synthesis Opposite Nucleoside Analogues Inserted into the Template Strand
To estimate more directly the mutagenic potential of the nucleoside analogues, we introduced the modified nucleosides 1-6 into 31-mer DNA templates and followed the extension of a 19-mer DNA primer by HIV-1 RT in vitro (Fig. 2A). This assay mimics synthesis of the (+)-strand DNA during HIV-1 reverse transcription. Two steps of primer elongation were assessed as follows: we investigated the ability of HIV-1 RT to incorporate matched and mismatched natural dNTPs opposite the modified nucleosides (i.e. at position n + 1, Fig. 2) and also checked whether RT was able to further extend (to position n + 2 or beyond, Fig. 3) the paired or mispaired duplexes involving a modified nucleoside. To these aims, unextended and extended primers were separated by PAGE (Fig. 2, B–G), and the intensities of the bands at each position were quantified as a function of the time.
FIGURE 2.

Primer extension when modified nucleosides are inserted in the template. A, sequences of the template-primer duplexes used in this study. X corresponds to the modified nucleoside (1–6) inserted in the template. B–G, extension of the 19-mer primer with one of the four natural dNTPs, in the presence or absence of ddGTP (the next incoming nucleotide), and quantification of the disappearance of unextended primers as a function of time. Fractions of the reverse transcription reaction mixture were collected at 0, 0.5, 1, 5, 10, 20, and 30 min. The first nucleotide (n + 1) is incorporated opposite the modified nucleoside inserted in the template, and the resulting extended primer is marked by *. When two consecutive nucleotides are incorporated, the extended primer is indicated with **, and the primer extended by three nucleotides is indicated with ***. Unextended primer is marked by −. Quantification of the disappearance of unextended primers was performed in the presence of only one of the four dNTPs. Values are means of two independent experiments.
FIGURE 3.

Primer extension products with templates bearing the modified nucleosides. Extensions were performed with one dNTP alone (dATP, dTTP, dCTP, or dGTP) or with one dNTP associated with the next incoming nucleotide in its dideoxy form (dATP + ddGTP, dTTP + ddGTP, dCTP + ddGTP, or dGTP + ddGTP), for 10 min. For each compound, “n + 1” (in blue) corresponds to the percentage of primer extended by one nucleoside triphosphate; “n + 2” (in orange) corresponds to the percentage of primer extended by two nucleotides, the first nucleoside triphosphate facing the modified nucleoside; “n + 3” (in pink) is the percentage of primer extended by three nucleotides, the first one facing the modified nucleoside. Incorporation of ddGTP only at position n + 1 is represented by the blue bar on the right-hand side of each histogram.
To evaluate the ability of HIV-1 RT to incorporate each of the four natural dNTPs opposite each modified nucleoside, we monitored the disappearance of the 19-mer primer over a 30-min period (Fig. 2, B–G). The 5-carbamoyl-dU (compound 1) and ncm3-dU (compound 4) allowed incorporation of either only the matched natural dNTP in the case of compound 1 (Fig. 2B) or none of the four dNTPs in the case of compound 4 (Fig. 2E), as expected for our negative control nucleoside modified on its Watson-Crick side. This suggests that the latter modifications will not favor mutagenesis. The carbamoylmethyl modification at position 5 of pyrimidine bases, when introduced on dC (compound 5), allowed RT to incorporate dGTP and to a lesser extent the mismatched dATP and dTTP (Fig. 2F), whereas only dATP and dGTP were efficiently and moderately, respectively, incorporated opposite ncm5-dU (2) (Fig. 2C). Finally, our results clearly indicated that 5-hydoxymethyl at position 5 was more conducive that carbamoylmethyl in inducing mispairing. Indeed, HIV-1 RT was able to efficiently incorporate three different natural dNTPs opposite the bases modified with the 5-hydroxymethyl group, the matched dATP and mismatched dCTP or dGTP opposite compound 3 (Fig. 2D) and the matched dGTP and mismatched dATP or dTTP opposite compound 6 (Fig. 2G), suggesting that these two analogues could induce extensive mutagenesis during HIV-1 replication.
Next, we investigated the ability of HIV-1 RT to extend the matched or mismatched base pairs formed between a natural dNTP (incorporated at position +1) and the modified nucleoside inserted into the template. To this end, experiments were performed under the conditions described above except that, together with one of the natural dNTPs, we also added ddGTP expected to be incorporated, as a chain terminator, at position n + 2 (Fig. 2A). Unextended primer and primers extended by one, two, or more nucleotides in some cases were quantified after 10 min of reverse transcription (Figs. 2 and 3).
Unsurprisingly, because n + 1 products were formed at very low yields, we were not able to detect n + 2 products with the ncm3-dU (compound 4) negative control nucleoside present in the template (Figs. 2E and 3). In the case of the uridine analogues 1 and 2, extension of the matched base pairs is clearly detectable. Extension also occurs when both modified dU form an initial mismatched base pair with dGMP (Figs. 2, B and C, and 3).
For compounds 3, 5, and 6 that permitted matched and mismatched primers extensions with more than one natural dNTP (Fig. 2, D, F, and G), our results clearly indicate that the hydroxymethyl modification on dU (compound 3) or dC (compound 6) allows extension of matched but also mismatched base pairs up to position n + 2 (Figs. 2, D and G, and 3) in the presence of dGTP and ddGTP, whereas the carbamoylmethyl modification on dC (compound 5) only sustains extension of the matched n + 1 base pairing (Figs. 2F and 3). Notably, for hm5-dC (compound 6) it was even possible to detect the n + 3 mismatched elongation product with dATP and to a lesser extent with dTTP and dGTP (Figs. 2G and 3), even though the template sequence dictates the incorporation of dCTP (Fig. 2A). These results suggest that our compounds allow mispairing with natural incoming nucleotide triphosphates during the elongation process but also extension of the latter mispaired duplexes. Interestingly, extension of the matched or mismatched duplexes containing the modified base can also be matched or mismatched, thus increasing even further the possibilities of accumulating mutations. In the case of compound 5 (ncm5-dC), even though RT was able to incorporate two successive matched dGTP (n + 1 and n + 2), DNA synthesis was blocked after incorporation of mismatched dATP or dTTP opposite the nucleoside analogue, indicating that this analogue may represent a new class of chain terminators. As a result, dNTP incorporation opposite our modified compounds suggests the following ranking for mutagenicity: 4 (no dNTP incorporated, nonmutagenic) < 1 (only one dNTP incorporated, nonmutagenic) < 2 (matched dNTP is incorporated and only one mismatched dNTP is poorly incorporated, poorly mutagenic), < 5 (three dNTPs are incorporated fairly efficiently but only the matched base pair is extended, poorly mutagenic) ≪ 3 ≈ 6 (three dNTPs are well incorporated and further extended, mutagenic).
Mutation Frequency and Spectrum after Serial Passages of HIV-1 in Cell Culture
Our previous study has shown that serial passages of HIV-1 in the presence of compounds 1-3 and 6 resulted in a progressive loss of viral replication, which was almost complete after eight passages with compounds 2, 3, and 6, and very significant with compound 1 (58). When viruses passaged seven times in the presence of nucleoside analogues were collected and subsequently cultured in the absence of inhibitor, viruses passaged in the presence of compounds 3 and 6 did not replicate, as expected for lethal mutagenesis (58). By contrast, viruses passaged in the presence of compounds 1 and 2 replicated efficiently once the inhibitors were removed, suggesting that these nucleoside analogues do not induce lethal mutagenesis but weakly inhibit HIV-1 replication by a different mechanism. To validate these conclusions and to elucidate the mechanism by which compounds 3 and 6 induce mutagenesis, we recovered the total cellular DNA after the first passage of HIV-1 in cell culture in the presence of the nucleoside analogues 1–3 and 6 or DMSO as a control (passage 0, p0) and at the end of the procedure (passage 8, p8). On each sample, four independent PCRs were performed to amplify an 870-nucleotide fragment of the HIV-1 pol gene. The PCR products were cloned, and 16 clones from each PCR were sequenced to analyze the pattern of mutations introduced by the nucleoside analogues. Sequencing data revealed no significant difference among the mutation frequency and spectrum coming from the four PCRs performed on the same DNA sample. The sequences coming from the four PCRs on each DNA sample were thus pooled together for further analysis (Table 4). In addition, we did not find two sequences with identical mutation sets, indicating that our data reflect the mutation diversity present in the viral DNA samples (data not shown).
TABLE 4.
Substitution rates after 0 and 8 HIV-1 passages in cell culture in the presence of compounds 1–3 and 6
DMSO was used as a control, because the nucleotide stock solutions were prepared in DMSO. For compounds 3 and 6, substitution rate values that are more than twice the control value are indicated in boldface.
| Compound | Passage | Global substitution rate (×105) | Individual substitution rates/nucleotide (×105) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A → |
T → |
C → |
G → |
|||||||||||
| T | C | G | A | C | G | A | T | G | A | T | C | |||
| DMSO | 0 | 93 | 0 | 0 | 32 | 18 | 45 | 0 | 0 | 211 | 0 | 142 | 0 | 10 |
| 8 | 91 | 0 | 5 | 27 | 0 | 65 | 0 | 0 | 94 | 0 | 219 | 0 | 10 | |
| 1 | 0 | 85 | 0 | 0 | 5 | 9 | 17 | 9 | 0 | 175 | 0 | 222 | 0 | 0 |
| 8 | 81 | 0 | 0 | 0 | 7 | 49 | 0 | 10 | 171 | 0 | 180 | 8 | 0 | |
| 2 | 0 | 71 | 2 | 2 | 18 | 0 | 35 | 0 | 5 | 163 | 0 | 127 | 0 | 0 |
| 8 | 31 | 0 | 0 | 0 | 0 | 87 | 0 | 0 | 0 | 0 | 49 | 0 | 0 | |
| 3 | 0 | 242 | 5 | 0 | 149 | 0 | 48 | 0 | 13 | 275 | 0 | 517 | 0 | 52 |
| 8 | 284 | 0 | 0 | 283 | 61 | 183 | 0 | 87 | 346 | 0 | 126 | 14 | 70 | |
| 6 | 0 | 81 | 16 | 0 | 37 | 0 | 9 | 0 | 13 | 173 | 0 | 133 | 0 | 0 |
| 8 | 313 | 0 | 0 | 0 | 0 | 15 | 0 | 0 | 212 | 0 | 779 | 554 | 0 | |
As expected, in the absence of nucleoside analogues, similar mutation rates were observed at p0 and p8, and transitions (especially G→A and C→T) were more frequent than transversions (Table 4). The global mutation rates of the viruses treated with compounds 1 and 2 are similar to the control, and no alteration of the mutation spectra was observed with these nucleoside analogues. By contrast, the overall mutation rate of viruses replicated in the presence of compound 3 was increased by 2.6- and 3.1-fold at p0 and p8, respectively, compared with the control viruses treated with DMSO alone (Table 4). Strangely enough, the HIV-1 mutation rate was not affected by compound 6 at p0, but it increased by 3.4-fold at p8. Despite the fact that a relatively large number of nucleotides were sequenced (619,562 for the whole study), the limited number of clones sequenced (64/compound/passage) does not allow detection of statistically significant hot spots of mutations when viruses were treated with compounds 3 or 6. However, with the latter compounds, we noticed that the number of mutated positions decreased between p0 and p8, which likely reflects the elimination of viruses bearing deleterious mutation(s).
These two compounds also affected the mutation spectrum in different manners (Table 4). Virus treated with compound 3 displayed significant increases of A→G and G→C mutations both at p0 and p8. The 5–10-fold increase of A→G mutations might result from Watson-Crick base pairing of compound 3-TP with A during (−)-strand DNA synthesis, followed by incorporation of dGMP in front of compound 3-MP during (+)-strand DNA synthesis (Fig. 4). The 5–7-fold increase in G→C mutations requires two non-Watson-Crick base pairing: incorporation of compound 3-MP in front of G-MP during (−)-strand DNA synthesis, followed by incorporation of dC-MP in front of compound 3-MP during (+)-strand DNA synthesis (Fig. 4). Two types of mutations were only increased at p8. On the one hand, the 3-fold increase of T→C mutations might result from base pairing of compound 3-TP with U-MP in the RNA template, followed by base pairing of dC-TP in front of compound 3-MP during (+)-strand DNA synthesis (Fig. 4). On the other hand, C→A mutations might be the consequence of base pairing of compound 3-TP with dC-MP during (−)-strand DNA synthesis, followed by Watson-Crick base pairing during (+)-strand DNA synthesis (Fig. 4). Finally, and surprisingly, an excess of G→A mutations was observed at p0 but not at p8. They might result from base pairing of compound 3-MP with G during (−)-strand DNA synthesis (Fig. 4). Only the following two types of mutation were increased in HIV-1 treated with compound 6 and only at p8 (Table 4): G→A and G→T. Both types of mutation types involve classical Watson-Crick of compound 6-TP with G during (−)-strand DNA synthesis, followed by mispairing of 6-MP with either dA-TP or dT-TP during (+)-strand DNA, respectively (Fig. 4).
FIGURE 4.
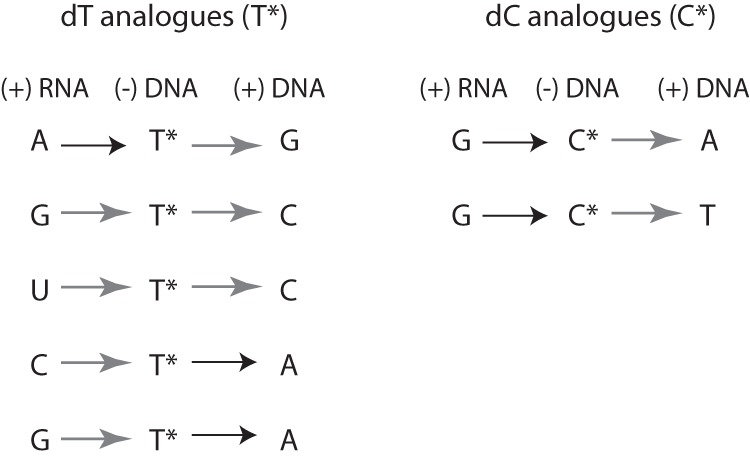
Possible mutagenic pathways. Proposed base pairings coherent with the nucleoside substitutions observed after 0 and 8 passages of HIV-1 in cell culture, in the presence of the mutagenic compounds 1–3 and 6. The 1st column corresponds to the sequence of the RNA substrate. The 2nd column corresponds to possible base pairings of the nucleoside triphosphate analogues with the RNA template during (−)-strand DNA synthesis. The 3rd column corresponds with possible base pairings of natural dNTPs with the nucleoside analogue monophosphate, during (+)-strand DNA synthesis. Black and gray arrows correspond to Watson-Crick and non-Watson-Crick base pairings, respectively.
DISCUSSION
Because HIV-1 systematically develops resistance to escape antiviral strategies, there is a constant urge to expand the therapeutic options. We investigated the use of nucleoside analogues that act through unconventional mechanisms, and we have previously reported the synthesis and antiviral activity of a series of pyrimidine nucleoside analogues modified at position 5 of the aglycone moiety but unmodified on the sugar part (58). Testing the potential mutagenic activity of nucleoside analogues in cell-based assays is labor-intensive and costly. We therefore implemented here two simple in vitro assays to serve as first screens to predict the mutagenic potential of nucleotides analogues before the lead compounds are taken into cell culture and in vivo assays.
Mutagenic compounds must form reasonably stable base pairs with at least two natural nucleotides, the matched one and one or several mismatched ones. We postulated that their frequency of incorporation by HIV-1 RT must therefore correlate with the thermal stability of the matched and mismatched duplexes that contain the analogue. The combination of a relatively small destabilization of the matched duplex involving the nucleoside analogue compared with the one containing the natural dNTP (Table 2) and a gap as small as possible between the thermal stabilities of the mismatched duplexes containing a nucleoside analogue and the corresponding control duplexes containing the natural nucleoside (Table 3, (ΔΔTm)) led to the prediction that compounds 3 and 6 should be the most effective mutagens.
These predictions were challenged in in vitro incorporation assays, using HIV-1 RT, of matched or mismatched natural dNTP opposite the modified analogues, inserted beforehand in a template strand, as well as extension assays of the newly formed matched or mismatched duplexes. Consistently with the thermal stability measurements, we found that dATP, dCTP, and dGTP as well as dGTP, dATP, and dCTP were readily incorporated opposite compounds 3 and 6, respectively (Fig. 2, D and G). Compound 1, which displayed favorable properties in terms of the stability of the matched duplex, but unfavorable properties when the ΔΔTm values were considered, behaves in the in vitro polymerization assay according to the predictions, only matched dATP was found to be incorporated (Fig. 2B). The same was true for compound 2 (Fig. 2C), where melting temperatures predicted that only (compound 2)/dA formed a stable base pair (Table 2). As expected from the low thermal stabilities of all the duplexes involving the negative control compound 4, no dNTP incorporation was observed opposite this modified analogue. Conversely, the mispairing abilities observed in vitro for ncm5-dC (compound 5, Fig. 2F) in the presence of dATP and dTTP were not recalled from the hybridization study. This discrepancy might be explained by the formation of a twisted structure where the nucleotide opposite the modification induces a distortion in the oligomer. As a result, dATP or dTTP can be incorporated at position n + 1, but the incorporation of the next incoming nucleotide is impaired, raising the possibility that this compound might act as a mismatched-induced chain terminator. A similar effect, although less pronounced, could be occurring with compounds 1 and 2, for which the efficiency of mismatch extension is poor. Interestingly, Dapp et al. (67) recently reported ribonucleoside analogues that increase HIV-1 mutational loads. Among their compounds, some, like our compound 5 and to a lesser extend compounds 1 and 2, induce premature termination of viral DNA synthesis. Accordingly, the lower thermal stabilities observed for duplexes containing compound 5 could be due to the fact that the base pair flanking the ncm5-dC/dA pair cannot form or is distorted.
To demonstrate the predictive potential of the above-described in vitro assays, we analyzed the mutation spectrum induced by compounds 1–3 and 6 at p0, the first viral supernatant after infection, and after eight serial passages of HIV-1 in cell culture at p8. In the literature, this type of analysis has been performed after 11 (62) and 16 passages (27). However, analysis at p0 is probably the only step that provides a good approximation of the frequency of mutations during one round of viral replication, before the less fit viruses are eliminated in the following passages. Compounds 1 and 2 did not, as predicted (Tables 2 and 3; Figs. 2 and 3), alter the mutation spectrum as compared with the control (Table 4). Compounds 3 and 6 were, as predicted, the best mutagens. In the case of compound 3, the mutation rates are high and almost similar at p0 and p8, suggesting that viruses treated with this nucleoside analogue acquired the highest level of bearable mutations very quickly compared with those treated with compound 6. Notably, among all the observed base pairings, only the “hm5-dU (3)-T” one was not predictable from the in vitro assays (Tables 2 and 3; Figs. 2 and 3). Hence, we conclude that the prediction potential of the in vitro assays was supported by the detailed analysis of the mutation spectra induced by the lead compounds. The in vitro assays thus constitute relevant first screens for potential mutagenic compounds. The study also further demonstrates that molecules that bear modifications on position 5 of the base moiety and inhibit HIV-1 replication (58) are promising mutagenic nucleoside analogues. From a mechanistic point of view, we have demonstrated that they allow mismatch formation and, importantly, that the extension of these mismatches, to form again either matched or mismatched base pairs, further extends their mutagenic potential.
The global increase of the mutation rate in our study (between 2.6- and 3.4-fold) is comparable with the ones measured previously with viruses treated with KP1212 (62) or 5-OH-dC (27). In the case of KP1212, a general increase of 1.44-fold in the mutation rate was linked with mainly A→G and G→A transitions, even though T→C transversions were also observed (62). In the case of 5-OH-dC, after 16 passages, there was a 5.6-fold increase in G→A substitutions (27). In our case, we observe a mixture of transition and transversion mutations, which are fully compatible with the measured thermal stabilities of the matched and mismatched duplexes involving the analogues (Table 4).
Notably, 5-hydroxymethylcytosine (hm5-C), the heterocyclic base of our compound 6, is increasingly being recognized as the sixth base in mammalian DNA (68, 69), present at high frequency in CpG dinucleotides. Oxidative damage could lead to the oxidation of 5-methylcytosine (5mC) to hm5-C (70), but the conversion also occurs via the Ten-Eleven-Translocation (TET) proteins (71). The formation of hm5-C leads to the demethylation of DNA, thus modulating the 5mC-dependent gene regulation. The hm5-C could thus be an intermediate in a possible active DNA demethylation pathway involving a DNA repair mechanism. Recently, the DNA methyltransferases DNMT3A and DNMT3B were proven to function bi-directionally, both as DNA methyltransferases and as dehydroxymethylases (72). These recent developments raise of course the question of a potential repair of the proviral genome treated with compound 6 as a mutagen, but this repair could only take place in the nucleus, after the nucleoside analogue has exerted its mutagenic effects.
The usage of lethal mutagenesis as a new intervention strategy (73) faces several challenges. First, lethal mutagenesis requires the usage of compounds that display no, or at least limited, toxicity to the cell. This undeniably precludes the use of chemical mutagens. The mutagen KP1212 has a proven record of low genotoxicity and mitochondrial toxicity (62), and 5-OH-dC was also shown to inhibit viral replication at concentrations that displayed minimal toxicity to the human cells (27). In the future, knowledge of the three-dimensional structures of viral and cellular polymerases and that of ternary primer-template-dNTP complexes will increase our understanding of the basis of nucleotide selection. This might enable the rational design of nucleoside analogues that specifically target viral enzymes. Second, emergence of drug resistance is a fundamental issue. Interestingly, viral strains resistant to classical RT inhibitors might be sensitive to mutagens, as documented for KP1212 (62). The emergence of drug-resistant viruses has been documented in the case of hepatitis C virus treatment with the mutagenic ribonucleoside ribavirin, where resistance has occurred, either in cell culture (74) or in patients (75). Resistance were also detected during FMDV treatment with mutagenic nucleosides in combination with conventional drugs (76). By analogy with ribavirin treatment (74), resistance mutations against our mutagenic nucleoside analogues should decrease the viral genome variability and thus disfavor the accumulation of resistance mutations against other anti-HIV-1 drugs, suggesting that our compounds, and more generally mutagenic nucleosides, should be useful in combination therapy. Finally, if resistance was to occur, it is also likely to be delayed rather than immediate, with a delayed selection of growth advantage compared with other inhibitors. In the case of the known nucleoside reverse transcriptase inhibitors and non-nucleoside reverse transcriptase inhibitors, resistance mutations result in improved replication capacities of the virus in the presence of the drug. In the case of mutagenic nucleoside analogues, the mutations introduced remain from one passage to the other and take a while to be cleared. In our case, sequencing of the p51 RT region in the pol gene (Table 4) did not indicate the selection of any resistance mutations after eight passages of HIV-1 in the presence of the mutagenic analogues, showing that resistance mutations are probably genuinely difficult to acquire. In the case of poliovirus (77) and human enterovirus 71 (78) treated with ribavirin, a mutation in the finger subdomain of the polymerase conferred high replication fidelity (77). Ribavirin-resistant mutation in the FMDV polymerase was shown to result in impaired RNA binding, polymerization, and ribavirin-monophosphate incorporation (79), through a complex network of interactions that reach the polymerase active site. From the crystal structure of RT involved in quaternary complexes with primer-template and incoming nucleotide (80), no amino acid side chain enters in close contact with position 5 of pyrimidines. As a consequence, the possibilities of steric hindrance to prevent incorporation of pyrimidine analogues modified on position 5 of the base moiety are strongly reduced, and the pathways to generate resistance should therefore be more sophisticated and involve several amino acids. Finally, mutagenic analogues need to compete with the pool of intracellular natural dNTPs, present at high concentrations, for incorporation by HIV-1 RT. However, one of the advantages of the strategy lies in the cumulative effect of mutations over multiple rounds of replication, thus making the issue of high concentrations of mutagenic analogues less crucial.
Below a basal mutation rate, complementing interactions in a viral population dominate and are responsible for its gain of fitness. When the mutation rate increases, interfering interactions gradually take over complementation (81–84). Thus, a possible outcome from a mutagenic therapy could be the effective suppression of viral load, rather than complete virus eradication, which would then give the immune system the opportunity to clear the virus. To achieve that, a subtle interplay between mutagenesis and inhibition is required with a combination of mutagens and inhibitors, administered in the right way. In cell culture, extinction of HIV-1 was achieved by the combination of 5-OH-dC and AZT, added at the same time (46). However, treatment of FMDV has proven that a sequential administration of inhibitor followed by a mutagenic agent is more effective than the administration of both drugs at the same time (82). Indeed, the latter protocol, allowing for mutagen-induced errors, most likely increases the probability of selecting inhibitor escape mutants that will in turn increase the viral load. Furthermore, in the case of HIV-1, but also other viruses, inhibitor-resistant strains with lower replicative capacities might be more susceptible to treatment with mutagens. The challenges for the future are thus to design effective, nontoxic mutagenic agents and new antiviral protocols that successfully combine mutagens and inhibitors to achieve eradication of HIV-1.
Acknowledgments
We thank Delphine Richer for technical help. The CEM-SS cells were obtained from P. Nara through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health.
This work was supported by CNRS, the Agence Nationale de Recherche sur le SIDA et les Hépatites Virales, SIDACTION, and the Strasbourg University. Funding for open access charges: Centre National de la Recherche Scientifique.
- FMDV
- foot-and-mouth disease virus
- 5-OH-dC
- 5-hydroxy-2′-deoxycytidine
- pol
- polymerase
- p0
- passage 0
- p8
- passage 8
- MP
- monophosphate
- TP
- triphosphate.
REFERENCES
- 1. Barré-Sinoussi F., Chermann J. C., Rey F., Nugeyre M. T., Chamaret S., Gruest J., Dauguet C., Axler-Blin C., Vézinet-Brun F., Rouzioux C., Rozenbaum W., Montagnier L. (1983) Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220, 868–871 [DOI] [PubMed] [Google Scholar]
- 2. Gallo R. C., Sarin P. S., Gelmann E. P., Robert-Guroff M., Richardson E., Kalyanaraman V. S., Mann D., Sidhu G. D., Stahl R. E., Zolla-Pazner S., Leibowitch J., Popovic M. (1983) Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS). Science 220, 865–867 [DOI] [PubMed] [Google Scholar]
- 3. Hu W.-S., Hughes S. H. (2012) HIV-1 reverse transcription. Cold Spring Harbor Perspect. Med. 2012; 2:a006882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mitsuya H., Weinhold K. J., Furman P. A., St Clair M. H., Lehrman S. N., Gallo R. C., Bolognesi D., Barry D. W., Broder S. (1985) 3′-Azido-3′-deoxythymidine (BW A509U): An antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc. Natl. Acad. Sci. U.S.A. 82, 7096–7100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. El Safadi Y., Vivet-Boudou V., Marquet R. (2007) HIV-1 reverse transcriptase inhibitors. Appl. Microbiol. Biotechnol. 75, 723–737 [DOI] [PubMed] [Google Scholar]
- 6. Waters L., John L., Nelson M. (2007) Non-nucleoside reverse transcriptase inhibitors: a review. Int. J. Clin. Pract. 61, 105–118 [DOI] [PubMed] [Google Scholar]
- 7. Kohlstaedt L. A., Wang J., Friedman J. M., Rice P. A., Steitz T. A. (1992) Crystal structure at 3.5Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 256, 1783–1790 [DOI] [PubMed] [Google Scholar]
- 8. Modrich P., Lahue R. (1996) Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 65, 101–133 [DOI] [PubMed] [Google Scholar]
- 9. Preston B. D., Poiesz B. J., Loeb L. A. (1988) Fidelity of HIV-1 reverse transcriptase. Science 242, 1168–1171 [DOI] [PubMed] [Google Scholar]
- 10. Roberts J. D., Bebenek K., Kunkel T. A. (1988) The accuracy of reverse transcriptase from HIV-1. Science 242, 1171–1173 [DOI] [PubMed] [Google Scholar]
- 11. Mansky L. M., Temin H. M. (1995) Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69, 5087–5094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O'Neil P. K., Sun G., Yu H., Ron Y., Dougherty J. P., Preston B. D. (2002) Mutational analysis of HIV-1 long terminal repeats to explore the relative contribution of reverse transcriptase and RNA polymerase II to viral mutagenesis. J. Biol. Chem. 277, 38053–38061 [DOI] [PubMed] [Google Scholar]
- 13. Abram M. E., Ferris A. L., Shao W., Alvord W. G., Hughes S. H. (2010) Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 84, 9864–9878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Menéndez-Arias L. (2002) Molecular basis of fidelity of DNA synthesis and nucleotide specificity of retroviral reverse transcriptase. Prog. Nucleic Acid Res. Mol. Biol. 71, 91–147 [DOI] [PubMed] [Google Scholar]
- 15. Menéndez-Arias L. (2009) Mutation rates and intrinsic fidelity of retroviral reverse transcriptases. Viruses 1, 1137–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Menendez-Arias L. (2013) in Human Immunodeficiency Virus Reverse Transcriptase. A Bench-to-Bedside Success (LeGrice S., Gotte M. eds) pp. 225–252, Springer, New York [Google Scholar]
- 17. Eigen M., McCaskill J., Schuster P. (1988) Molecular quasi-species. J. Phys. Chem. 92, 6881–6891 [Google Scholar]
- 18. Holland J. J., De la Torre J. C., Steinhauer D. A. (1992) RNA virus populations as quasispecies. Curr. Top. Microbiol. Immunol. 176, 1–20 [DOI] [PubMed] [Google Scholar]
- 19. Smyth R. P., Davenport M. P., Mak J. (2012) The origin of genetic diversity in HIV-1. Virus Res. 169, 415–429 [DOI] [PubMed] [Google Scholar]
- 20. Ho D. D., Neumann A. U., Perelson A. S., Chen W., Leonard J. M., Markowitz M. (1995) Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373, 123–126 [DOI] [PubMed] [Google Scholar]
- 21. Wei X., Ghosh S. K., Taylor M. E., Johnson V. A., Emini E. A., Deutsch P., Lifson J. D., Bonhoeffer S., Nowak M. A., Hahn B. H. (1995) Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373, 117–122 [DOI] [PubMed] [Google Scholar]
- 22. Mikkelsen J. G., Pedersen F. S. (2000) Genetic reassortment and patch repair by recombinaison in retroviruses. J. Biomed. Sci. 7, 77–99 [DOI] [PubMed] [Google Scholar]
- 23. Smyth R. P., Schlub T. E., Grimm A. J., Waugh C., Ellenberg P., Chopra A., Mallal S., Cromer D., Mak J., Davenport M. P. (2014) Identifying recombination hot spots in the HIV-1 genome. J. Virol. 88, 2891–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eigen M., Schuster P. (1977) The hypercycle. A principle of natural self-organization. Part A: Emergence of the hypercycle. Naturwissenschaften 64, 541–565 [DOI] [PubMed] [Google Scholar]
- 25. Biebricher C. K., Eigen M. (2005) The error threshold. Virus Res. 107, 117–127 [DOI] [PubMed] [Google Scholar]
- 26. Eigen M. (2002) Error catastrophe and antiviral strategy. Proc. Natl. Acad. Sci. U.S.A. 99, 13374–13376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Loeb L. A., Essigmann J. M., Kazazi F., Zhang J., Rose K. D., Mullins J. I. (1999) Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. U.S.A. 96, 1492–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perales C., Iranzo J., Manrubia S. C., Domingo E. (2012) The impact of quasispecies dynamics on the use of therapeutics. Trends Microbiol. 20, 595–603 [DOI] [PubMed] [Google Scholar]
- 29. Holland J. J., Domingo E., de la Torre J. C., Steinhauer D. A. (1990) Mutation frequencies at defined single codon sites in vesicular stomatitis virus and poliovirus can be increased only slightly by chemical mutagenesis. J. Virol. 64, 3960–3962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sierra S., Dávila M., Lowenstein P. R., Domingo E. (2000) Response of foot-and-mouth disease virus to increased mutagenesis: influence of viral load and fitness in loss infectivity. J. Virol. 74, 8316–8323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Airaksinen A., Pariente N., Menéndez-Arias L., Domingo E. (2003) Curing of foot-and-mouth disease virus from persistently infected cells by ribavirin involves enhanced mutagenesis. Virology 311, 339–349 [DOI] [PubMed] [Google Scholar]
- 32. Agudo R., Arias A., Pariente N., Perales C., Escarmís C., Jorge A., Marina A., Domingo E. (2008) Molecular characterization of a dual inhibitory and mutagenic activity of 5-fluorouridine triphosphate on viral RNA synthesis. Implications for lethal mutagenesis. J. Mol. Biol. 382, 652–666 [DOI] [PubMed] [Google Scholar]
- 33. Severson W. E., Schmaljohn C. S., Javadian A., Jonsson C. B. (2003) Ribavirin causes error catastrophe during hantaan virus replication. J. Virol. 77, 481–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chung D.-H., Sun Y., Parker W. B., Arterburn J. B., Bartolucci A., Jonsson C. B. (2007) Ribavirin reveals a lethal threshold of allowable mutation frequency for Hantaan virus. J. Virol. 81, 11722–11729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Contreras A. M., Hiasa Y., He W., Terella A., Schmidt E. V., Chung R. T. (2002) Viral RNA mutations are region specific and increased by ribavirin in a full-length hepatitis C virus replication system. J. Virol. 76, 8505–8517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou S., Liu R., Baroudy B. M., Malcolm B. A., Reyes G. R. (2003) The effect of ribavirin and IMPDH inhibitors on hepatitis C virus subgenomic replicon RNA. Virology 310, 333–342 [DOI] [PubMed] [Google Scholar]
- 37. Davis G. L., Esteban-Mur R., Rustgi V., Hoefs J., Gordon S. C., Trepo C., Shiffman M. L., Zeuzem S., Craxi A., Ling M. H., Albrecht J. (1998) Interferon α-2b alone or in combination with ribavirin for the treatment of relapse of chronic hepatitis C. International Hepatitis Interventional Therapy Group. N. Engl. J. Med. 339, 1493–1499 [DOI] [PubMed] [Google Scholar]
- 38. McHutchison J. G., Gordon S. C., Schiff E. R., Shiffman M. L., Lee W. M., Rustgi V. K., Goodman Z. D., Ling M. H., Cort S., Albrecht J. K. (1998) Interferon α-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. N. Engl. J. Med. 339, 1485–1492 [DOI] [PubMed] [Google Scholar]
- 39. Crotty S., Maag D., Arnold J. J., Zhong W., Lau J. Y., Hong Z., Andino R., Cameron C. E. (2000) The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 6, 1375–1379 [DOI] [PubMed] [Google Scholar]
- 40. Crotty S., Cameron C. E., Andino R. (2001) RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. U.S.A. 98, 6895–6900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pathak V. K., Temin H. M. (1992) 5-Azacytidine and RNA secondary structure increase the retrovirus mutation rate. J. Virol. 66, 3093–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Loeb L. A., Mullins J. I. (2000) Lethal mutagenesis of HIV by mutagenic ribonucleoside analogs. AIDS Res. Hum. Retroviruses 16, 1–3 [DOI] [PubMed] [Google Scholar]
- 43. Daifuku R. (2003) Stealth nucleosides: mode of action and potential use in the treatment of viral diseases. BioDrugs 17, 169–177 [DOI] [PubMed] [Google Scholar]
- 44. Mullins J. I., Heath L., Hughes J. P., Kicha J., Styrchak S., Wong K. G., Rao U., Hansen A., Harris K. S., Laurent J.-P., Li D., Simpson J. H., Essigmann J. M., Loeb L. A., Parkins J. (2011) Mutation of HIV-1 genomes in a clinical population treated with the mutagenic nucleoside KP1461. PLoS One 6, e15135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li D., Fedeles B. I., Singh V., Peng C. S., Silvestre K. J., Simi A. K., Simpson J. H., Tokmakoff A., Essigmann J. M. (2014) Tautomerism provides a molecular explanation for the mutagenic properties of the anti-HIV nucleoside 5-aza-5,6-dihydro-2′-deoxycytidine. Proc. Natl. Acad. Sci. U.S.A. 111, E3252–E3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tapia N., Fernàndez G., Parera M., Gómez-Mariano G., Clotet B., Quiñones-Mateu M., Domingo E., Martínez M. A. (2005) Combination of a mutagenic agent with a reverse transcriptase inhibitor results in systematic inhibition of HIV-1 infection. Virology 338, 1–8 [DOI] [PubMed] [Google Scholar]
- 47. Harris R. S., Bishop K. N., Sheehy A. M., Craig H. M., Petersen-Mahrt S. K., Watt I. N., Neuberger M. S., Malim M. H. (2003) DNA deamination mediates innate immunity to retroviral infection. Cell 113, 803–809 [DOI] [PubMed] [Google Scholar]
- 48. Mangeat B., Turelli P., Caron G., Friedli M., Perrin L., Trono D. (2003) Broad antiretroviral defense by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424, 99–103 [DOI] [PubMed] [Google Scholar]
- 49. Zhang H., Yang B., Pomerantz R. J., Zhang C., Arunachalam S. C., Gao L. (2003) The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424, 94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Henriet S., Mercenne G., Bernacchi S., Paillart J.-C., Marquet R. (2009) Tumultuous relationship between the human immunodeficiency virus type 1 viral infectivity factor (Vif) and the human APOBEC-3G and APOBEC-3F restriction factors. Microbiol. Mol. Biol. Rev. 73, 211–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sheehy A. M., Gaddis N. C., Malim M. H. (2003) The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9, 1404–1407 [DOI] [PubMed] [Google Scholar]
- 52. Yu X., Yu Y., Liu B., Luo K., Kong W., Mao P., Yu X.-F. (2003) Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF Complex. Science 302, 1056–1060 [DOI] [PubMed] [Google Scholar]
- 53. Mercenne G., Bernacchi S., Richer D., Bec G., Henriet S., Paillart J.-C., Marquet R. (2010) HIV-1 Vif binds to APOBEC3G mRNA and inhibits its translation. Nucleic Acids Res. 38, 633–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chelico L., Pham P., Goodman M. F. (2009) Mechanisms of APOBEC3G-catalyzed processive deamination of deoxycytidine on single-stranded DNA. Nat. Struct. Mol. Biol. 16, 454–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Armitage A. E., Deforche K., Chang C.-H., Wee E., Kramer B., Welch J. J., Gerstoft J., Fugger L., McMichael A., Rambaut A., Iversen A. K. (2012) APOBEC3G-induced hypermutation of human immunodeficiency virus type-1 is typically a discrete “all or nothing” phenomenon. PLoS Genet. 8, e1002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rosenberg B. R., Papavasiliou F. N. (2007) Beyond SHM and CSR: AID and related cytidine deaminases in the host response to viral infection. Adv. Immunol. 94, 215–244 [DOI] [PubMed] [Google Scholar]
- 57. Albin J. S., Harris R. S. (2010) Interactions of host APOBEC3 restriction factors with HIV-1 in vivo: implications for therapeutics. Expert Rev. Mol. Med. 12, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. El Safadi Y., Paillart J.-C., Laumond G., Aubertin A.-M., Burger A., Marquet R., Vivet-Boudou V. (2010) 5-Modified-2′-dU and 2′-dC as mutagenic anti HIV-1 proliferation agents: synthesis and activity. J. Med. Chem. 53, 1534–1545 [DOI] [PubMed] [Google Scholar]
- 59. Vivet-Boudou V., Isel C., Sleiman M., Smyth R., Ben Gaied N., Barhoum P., Laumond G., Bec G., Götte M., Mak J., Aubertin A.-M., Burger A., Marquet R. (2011) 8-Modified-2′-deoxyadenosine analogues induce delayed polymerization arrest during HIV-1 reverse transcription. PLoS One 6, e27456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mizrahi V., Brooksbank R. L., Nkabinde N. C. (1994) Mutagenesis of the conserved aspartic acid 443, glutamic acid 478, asparagine 494, and aspartic acid 498 residues in the ribonuclease H domain of p66/p51 human immunodeficiency virus type I reverse transcriptase. Expression and biochemical analysis. J. Biol. Chem. 269, 19245–19249 [PubMed] [Google Scholar]
- 61. Lindberg J., Sigurdsson S., Löwgren S., Andersson H. O., Sahlberg C., Noréen R., Fridborg K., Zhang H., Unge T. (2002) Structural basis for inhibitory efficacy of efavirenz (DMP-266), MSC194 and PNU142721 towards the HIV-1 RT K103N mutant. Eur. J. Biochem. 269, 1670–1677 [DOI] [PubMed] [Google Scholar]
- 62. Harris K. S., Brabant W., Styrchak S., Gall A., Daifuku R. (2005) KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antiviral Res. 67, 1–9 [DOI] [PubMed] [Google Scholar]
- 63. Dapp M. J., Clouser C. L., Patterson S., Mansky L. M. (2009) 5-Azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. J. Virol. 83, 11950–11958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Werntges H., Steger G., Riesner D., Fritz H. J. (1986) Mismatches in DNA double strands: thermodynamic parameters and their correlation to repair efficiencies. Nucleic Acids Res. 14, 3773–3790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aboul-ela F., Koh D., Tinoco I., Jr., Martin F. H. (1985) Base-base mismatches. Thermodynamics of double helix formation for dCA3XA3G + dCT3YT3G (X, Y = A,C,G,T). Nucleic Acids Res. 13, 4811–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gaffney B. L., Jones R. A. (1989) Thermodynamic comparison of the base pairs formed by the carcinogenic lesion O6-methylguanine with reference both to Watson-Crick pairs and to mismatched pairs. Biochemistry 28, 5881–5889 [DOI] [PubMed] [Google Scholar]
- 67. Dapp M. J., Bonnac L., Patterson S. E., Mansky L. M. (2014) Discovery of novel ribonucleoside analogs with activity against human immunodeficiency virus type1. J. Virol. 88, 354–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dahl C., Grønbæk K., Guldberg P. (2011) Advances in DNA methylation: 5-hydroxymethylcytosine revisited. Clin. Chim. Acta 412, 831–836 [DOI] [PubMed] [Google Scholar]
- 69. Kriukienė E., Liutkevičiūtė Z., Klimašauskas S. (2012) 5-Hydroxymethylcytosine–the elusive epigenetic mark in mammalian DNA. Chem. Soc. Rev. 41, 6916–6930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kriaucionis S., Heintz N. (2009) The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tahiliani M., Koh K. P., Shen Y., Pastor W. A., Bandukwala H., Brudno Y., Agarwal S., Iyer L. M., Liu D. R., Aravind L., Rao A. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen C.-C., Wang K.-Y., Shen C.-K. (2012) The mammalian de novo DNA methyltransferases Dnmt3a and Dnmt3b are also DNA 5-hydroxymethyl cytosine dehydroxymethylases. J. Biol. Chem. 287, 33116–33121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Perales C., Martín V., Domingo E. (2011) Lethal mutagenesis of viruses. Curr. Opin. Virol. 1, 419–422 [DOI] [PubMed] [Google Scholar]
- 74. Pfeiffer J. K., Kirkegaard K. (2005) Ribavirin resistance in hepatitis C virus replicon-containing cell lines conferred by changes in the cell line or mutations in the replicon RNA. J. Virol. 79, 2346–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Young K. C., Lindsay K. L., Lee K. J., Liu W. C., He J. W., Milstein S. L., Lai M. M. (2003) Identification of ribavirin-resistant NS5B mutation of hepatitis C virus during ribavirin monotherapy. Hepatology 38, 869–878 [DOI] [PubMed] [Google Scholar]
- 76. Pariente N., Airaksinen A., Domingo E. (2003) Mutagenesis versus inhibition in the efficiency of extinction of foot-and-mouth disease virus. J. Virol. 77, 7131–7138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pfeiffer J. K., Kirkegaard K. (2003) A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. U.S.A. 100, 7289–7294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sadeghipour S., Bek E. J., McMinn P. C. (2013) Ribavirin-resistant mutants of human enterovirus 71 express a high replication fidelity phenotype during growth in cell culture. J. Virol. 87, 1759–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ferrer-Orta C., Sierra M., Agudo R., de la Higuera I., Arias A., Pérez-Luque R., Escarmís C., Domingo E., Verdaguer N. (2010) Structure of foot-and-mouth disease virus mutant polymerases with reduced sensitivity to ribavirin. J. Virol. 84, 6188–6199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Huang H., Chopra R., Verdine G. L., Harrison S. C. (1998) Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282, 1669–1675 [DOI] [PubMed] [Google Scholar]
- 81. Grande-Pérez A., Lázaro E., Lowenstein P., Domingo E., Manrubia S. C. (2005) Suppression of viral infectivity through lethal defection. Proc. Natl. Acad. Sci. U.S.A. 102, 4448–4452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Perales C., Agudo R., Tejero H., Manrubia S. C., Domingo E. (2009) Potential benefits of sequential inhibitor-mutagen treatments of RNA virus infections. PLoS Pathog. 5, e1000658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Perales C., Lorenzo-Redondo R., López-Galíndez C., Martínez M. A., Domingo E. (2010) Mutant spectra in virus behavior. Future Virol. 5, 679–698 [Google Scholar]
- 84. Perales C., Agudo R., Manrubia S. C., Domingo E. (2011) Influence of mutagenesis and viral load on the sustained, low-level replication of an RNA virus. J. Mol. Biol. 407, 60–78 [DOI] [PubMed] [Google Scholar]