

Background: Nuclear SIRT1 and SIRT6 switch monocyte energy sources from glycolysis to fatty acid oxidation during sepsis adaptation.
Results: Sequential actions of nuclear SIRT1 and RELB differentially induce SIRT3 expression and increase mitochondrial biogenesis during sepsis adaptation.
Conclusion: SIRT1 and RELB link nuclear and mitochondrial alterations in bioenergetics during sepsis.
Significance: Communication between nuclear and mitochondrial functions may influence sepsis outcomes.
Keywords: Mitochondrial Respiratory Chain Complex, Monocyte, Reactive Oxygen Species (ROS), Sepsis, Sirtuin 1 (SIRT1), Acute Inflammation, Mitochondrial Bioenergetics, RELB, SIRT3, Nuclear-mitochondrial Communication
Abstract
We reported that NAD+-dependent SIRT1, RELB, and SIRT6 nuclear proteins in monocytes regulate a switch from the glycolysis-dependent acute inflammatory response to fatty acid oxidation-dependent sepsis adaptation. We also found that disrupting SIRT1 activity during adaptation restores immunometabolic homeostasis and rescues septic mice from death. Here, we show that nuclear SIRT1 guides RELB to differentially induce SIRT3 expression and also increases mitochondrial biogenesis, which alters bioenergetics during sepsis adaptation. We constructed this concept using TLR4-stimulated THP1 human promonocytes, a model that mimics the initiation and adaptation stages of sepsis. Following increased expression, mitochondrial SIRT3 deacetylase activates the rate-limiting tricarboxylic acid cycle (TCA) isocitrate dehydrogenase 2 and superoxide dismutase 2, concomitant with increases in citrate synthase activity. Mitochondrial oxygen consumption rate increases early and decreases during adaptation, parallel with modifications to membrane depolarization, ATP generation, and production of mitochondrial superoxide and whole cell hydrogen peroxide. Evidence of SIRT1-RELB induction of mitochondrial biogenesis included increases in mitochondrial mass, mitochondrial-to-nuclear DNA ratios, and both nuclear and mitochondrial encoded proteins. We confirmed the SIRT-RELB-SIRT3 adaptation link to mitochondrial bioenergetics in both TLR4-stimulated normal and sepsis-adapted human blood monocytes and mouse splenocytes. We also found that SIRT1 inhibition ex vivo reversed the sepsis-induced changes in bioenergetics.
Introduction
A successful inflammatory response requires orderly stages of initiation, adaptation, and resolution (1). Failure to progress through these “defend → mend → restore” stages contributes to many diseases with major impact on human health. In chronic inflammatory illnesses, such as obesity with diabetes, atherosclerosis, and Alzheimer dementia, the pro-inflammation stage does not fully shift to adaptation. In contrast, acute inflammatory diseases progress to adaptation, the length of which depends on the magnitude of the initial response and affects outcomes (2).
Among acute systemic inflammatory diseases, sepsis is a major cause of death worldwide, with rising incidence, mortality rates that often exceed 50%, and no available molecular-based treatment (3). With up to 15 million cases worldwide per year, the economic and life costs of sepsis are enormous. In lethal sepsis, the temporal states are dysregulated. Early sepsis mortality occurs soon after initiation, when the magnitude of the acute pro-inflammatory response (cytokine storm) causes cardiovascular collapse and rapid organ failure. Sepsis mortality remains high during the unresolved adaptation stage, which can persist for days or weeks before resolution (2, 4–6), and is clinically associated with immunosuppression and sustained dysfunction of multiple organs (2, 7–10). Understanding what sustains sepsis adaptation and determining how to resolve it are urgent goals. Treating sepsis initiation in humans has uniformly failed (3, 11) probably because it shifts to adaptation by the time anti-inflammatory treatment is initiated. Strategies to both prevent and treat sepsis once it is in the adaptation stage are urgently needed.
Accumulating data show that changes in NAD+ levels and sirtuin activation promote the shift from initiation to adaptation by simultaneously reprogramming immunity and metabolism (8). Seven mammalian sirtuins reside in nuclear (SIRT1, -6, and -7), mitochondrial (SIRT3, -4, and -5), and cytosolic (SIRT2) compartments (12). Nuclear SIRT1 and SIRT6 play a critical role in switching the initial inflammatory response to adaptation. In monocytes and neutrophils, this switch generates silent heterochromatin by inactivating NFκB factor RelA/p65 and activating NFκB RELB transcription factor and other histone and DNA modifiers (2, 13–16). Mechanistically, SIRT1 deacetylates lysine 310 of RelA/p65 and histone protein H1K27 and recruits RELB to promoters of target genes (15). SIRT6 also deacetylates RelA/p65 and histone H3K9 (17, 18) to exert anti-proinflammatory activity.
SIRT1 acts as an inflammation rheostat during sepsis. Increases in NAD+ availability and the cooperative actions of nuclear SIRT1, SIRT6, and RELB are required to move from initiation to adaptation (15, 19). When NAD+ or SIRT1 levels decrease in cell models of sepsis and sepsis mice, the initial acute inflammatory response is amplified by allowing excessive NFκB RelA/p65 activation (19, 20). To adapt to acute inflammation, nuclear SIRT1 guides RELB to generate silent heterochromatin at pro-immune genes like TNF-α and IL-1β (21) and to activate the euchromatin of genes responsible for fatty acid uptake and oxidation. In contrast, SIRT6 represses glycolysis by inhibiting HIF-1α (22). After the immunometabolic switch from initiation to adaptation, sustained increases in NAD+ and SIRT1 protein stabilization maintain the adaptation state.
Because SIRT1 influences the course of acute inflammation and sepsis, we tested whether it might be a therapeutic target for reversing the adaptation state. Remarkably, we found in mice that inhibiting SIRT1 activity after the switch to adaptation improves peripheral circulation, restores immune competence, and markedly improves survival (20). That study, however, did not address whether SIRT1 inhibition rebalanced glucose and fatty acid mitochondrial oxidation or altered mitochondrial bioenergetics. This question is important because changes in mitochondrial function are prominent features of sepsis, and dysregulated bioenergetics may contribute to poor sepsis outcomes (10). For example, sepsis causes pronounced changes in mitochondrial numbers, structure, and function (23). Mitochondria in immune and tissue cells (e.g. muscle and heart) are injured or destroyed by excessive production of reactive oxygen species (ROS).3 In contrast, accelerating mitochondrial biogenesis during early sepsis can increase survival in septic animals (24–26), but sustained mitochondrial dysfunction during adaptation may adversely influence sepsis outcome (27).
This study tested how nuclear SIRT1-dependent immunometabolic reprogramming during sepsis adaptation modifies mitochondrial bioenergetics. Our research approach used THP-1 promonocytes, primary human monocytes, and mouse splenocytes and extended findings to human and mouse sepsis. We found that sequential activation of SIRT1 and RELB altered mitochondrial bioenergetics during sepsis adaptation by differentially inducing SIRT3 expression, increasing mitochondrial biogenesis, and modifying mitochondrial respiration. We also observed that SIRT1 inhibition in adapted cells reversed sepsis-induced changes in mitochondrial bioenergetics.
EXPERIMENTAL PROCEDURES
Human Promonocytic THP-1 Cell Model of the Acute Inflammatory Response
Promonocyte THP-1 cells from the American Type Culture Collection (ATCC) were maintained in complete RPMI 1640 medium (Invitrogen) supplemented with 100 units/ml of penicillin, 100 μg/ml of streptomycin, 2 mm l-glutamine, and 10% fetal bovine serum (FBS, HyClone, Logan, UT) in a humidified incubator with 5% CO2 at 37 °C. Acute inflammatory response was induced by stimulating THP-1 cells with 1 μg/ml of Gram-negative bacteria lipopolysaccharide (LPS, Escherichia coli serotype 0111:B4, Sigma) for the indicated times to generate different inflammatory phenotypes. In this model, the acute pro-inflammatory phenotype was assessed at 4–8 h, and the post-acute adaptation stage was present 24–48 h after LPS stimulation. This adapted response was confirmed by inhibition of the pro-inflammatory gene TNF-α in response to LPS restimulation. In some experiments, cells were treated with 1 μm of the SIRT1 inhibitor EX-527 at 8 h after LPS, when mitochondrial bioenergetics reaches its peak.
Human Monocyte and Murine Splenocyte Studies
Human primary monocytes purified by negative selection were purchased from Lifeline Cell Technology (Frederick, MD) and maintained in complete RPMI 1640 culture medium for the indicated time in the presence or absence of 100 ng/ml of LPS. Human blood was drawn from septic subjects according to a protocol approved by the Institutional Review Board of Wake Forest University School of Medicine. Sepsis patients were selected according to the diagnostic criteria. Septic human monocytes were isolated by adhesion of peripheral leukocytes for 90 min in a CO2 incubator followed by 3 washes with warm RPMI medium. The resulting adhesive cells were >90% CD14-positive based on flow cytometry analysis and minimally express TNF-α in response to LPS stimulation. Septic monocytes were cultured overnight in the presence or absence of 1 μm EX-527 before assay.
The sublethal animal sepsis model of cecal ligation and puncture (CLP) was generated with C57BL/6 (6–8-week-old) mice or 129Si/SvImJ wild-type and 129Sirt3tm1.1Fwa/J3 SirT3−/− mice according to a protocol approved by the Wake Forest University School of Medicine Institutional Animal Care and Use Committee. All mice were obtained from Jackson Laboratories (Bar Harbor, ME), and the standard CLP procedure (two punctures with 22-gauge needles) was performed under anesthesia as described (20). Animals that received laparotomy without CLP served as sham controls. This model displays the acute inflammatory phenotype within 4–12 h and the post-acute phenotype 18 h after CLP. C57BL/6 animals were given one dose of 10 mg/kg of EX-527 or dimethyl sulfoxide (DMSO) 24 h after CLP. Splenocytes were isolated at 36 h (12 h after EX-527 treatment) by passing mashed spleens through the 40-μm cell strainer. Septic splenocytes are hyporesponsive to LPS stimulation ex vivo. Normal splenocytes were treated with 100 ng/ml of LPS or LPS plus 1 μm EX-527, respectively, for the indicated time before assay. For the survival study, 129Si/SvImJ wild-type and 129Sirt3tm1.1Fwa/J3 Sirt3−/− mice were followed for 7 days after CLP.
Mitochondrial Respiration
Mitochondrial oxidative phosphorylation reactions were assessed by measuring the oxygen consumption rate (OCR) and spare respiratory capacity (SRC) with a cell mito stress kit using an XF-24 Extracellular Flux Analyzer (Seahorse Bioscience). A 24-well XF sensor cartridge containing 500 μl/well of XF calibrate solution was preincubated overnight at 37 °C in a CO2-free incubator.
For a mitochondrial respiratory assay at the whole cell level, a 24-well microplate was pretreated for 20 min with 3.5 μg of Cell-Tak/cm2 of surface area (BD Biosciences) diluted in 0.1 m sodium bicarbonate (pH 8.0) buffer. Excess Cell-Tak was removed by 2 washes with sterile water. THP-1 cells (2 × 105/well), human monocytes or murine splenocytes (3 × 105 per well), in bicarbonate- and Hepes-free RPMI medium (pH 7.4, Invitrogen) supplemented with 2% FBS were preincubated for 1 h at 37 °C in a CO2-free incubator. 10-Fold concentrated compounds (Seahorse Biosciences) of oligomycin (Complex V inhibitor), carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP, electron transport chain uncoupler), or a mixture of rotenone (Complex I inhibitor) and antimycin A (Complex III inhibitor) were loaded into a sensor cartridge to produce final concentrations of 1 μm, 1.5 μm, 100 nm, and 1 μm, respectively.
After a 30-min calibration of the XF sensor with the preincubated sensor cartridge, the cell plate was loaded into the analyzer, and OCR was analyzed under basal conditions. Oxygen use for ATP generation, SRC, and net mitochondrial oxygen consumption were analyzed by sequential injection of the complex inhibitors oligomycin, FCCP, and the mixture of rotenone and antimycin A. SRC was determined as the difference between maximal and basal OCRs. Data were analyzed using XF software (Seahorse Bioscience) and normalized with protein loaded into each well. Four replicates of each sample were analyzed.
To perform the electron transport chain respiration assay on isolated mitochondria, we first extracted mitochondria from THP-1 cells using a method we developed. Briefly, about 10 million cells were suspended in Chappell Perry (CP) I buffer (KCl 100 mm; MOPS 50 mm; EDTA 1 mm; MgSO4 5 mm; ATP 1 mm, pH 7.4) and homogenized using a glass-glass Dounce homogenizer. Further CP II buffer (KCl 100 mm; MOPS 50 mm; EDTA 1 mm; MgSO4 5 mm; ATP 0.2 mm; fatty acid-free BSA 0.50%, pH 7.4) was added to the homogenized cells, mixed, and centrifuged at 600 × g for 10 min at 4 °C. The supernatant was then centrifuged at 10,000 × g for 10 min at 4 °C, and the pellet was washed with CP II buffer followed by CP I buffer. Finally, the pellet was suspended in mitochondrial assay solution (MAS) (sucrose 35 mm; mannitol 110 mm; KH2PO4 2.5 mm; MgCl2 2.5 mm; Hepes 1.0 mm; EGTA 0.5 mm; fatty acid-free BSA 0.10%). Mitochondrial protein concentration was determined using the BCA protein assay kit (Thermo Scientific, Rockford, IL). Ten μg of mitochondrial protein with 10 mm succinate and 2 μm rotenone were loaded into each well in a 24-well microplate. Adenosine diphosphate (ADP, 2 mm), oligomycin (2 μm), FCCP (6 μm), and antimycin A (2 μm) were added in that order to give the final concentration (in parentheses). OCRs were measured by the XF-24 Extracellular Flux Analyzer in buffer-free RPMI medium containing 2% FBS under basal conditions followed by sequential addition of 1 μm oligomycin, 1.5 μm FCCP, and 100 nm rotenone + 1 μm antimycin A (Seahorse Bioscience). Four replicates of each sample were analyzed to determine the mean ± S.E.
Flow Cytometry
Live THP-1 cells in a 24-well cell-culture plate were stained with the indicated mitochondrial fluorescence dyes for 20 min in a CO2 incubator followed by 1 wash in warm cell-culture medium. Cells were resuspended in warm medium and analyzed using an Accuri C6 Flow Cytometer (BD Bioscience). Data collected from 10,000 individual cells were analyzed using BD Bioscience Accuri C6 software.
Fluorescence Microscopy
Aliquots of mitochondrial fluorescence dye-stained live cells from the flow cytometry assay were washed and resuspended in phenol-free RPMI medium. Cells were transferred to an 8-well chambered cover glass and imaged by an LSM 510 microscope with LSM 510 image browser software (Carl Zeiss, Jena, Germany).
Real-time PCR
mRNA levels of human SIRT1, RELB, SIRT3 and murine Sirt1, Relb, Sirt3, and mitochondrial genes were measured by quantitative RT-PCR using gene-specific TaqMan primer/probe sets in an ABI prism 7000 sequence detection system (Applied Biosystems) as described (15). GAPDH mRNA was the internal loading control.
To assess the ratio of mitochondrial DNA to nuclear DNA (nDNA), total cellular DNA was analyzed by PCR using primer pairs (Integrated DNA Technologies) specific for the mitochondrial DNA D-loop and the proximal NFκB binding site of the TNF-α promoter. The primer sequences were: D-loop forward, 5′-CACCCTATTAACCACTCACG-3′, and reverse, 5′-TGAGATTAGTAGTATGGGAG-3′; NFκB forward, 5′-TACCGCTTCCTCCAGATGAG-3′, and reverse, 5′-TGCTGGCTGGGTGTGCCAA-3′.
Transduction and Transfection
For stable RNA interference, SIRT1, RELB, and SIRT3 genes were silenced by infecting the cells with gene-specific lentiviral shRNA according to the manufacturer's protocol (Santa Cruz). A control shRNA lentiviral particle was used as a negative control. Successfully transfected cells were selected using 5 μg/ml of puromycin.
To overexpress SIRT1 or RELB in RELB or SIRT1 gene-silenced cells, 0.5 μg of pcDNA3-HA vector DNA, HA-RELB plasmid (GenScript Corp.) (28), pECE-Flag vector DNA, or pECE-Flag SIRT1 plasmid (Addgene plasmid 1791) (29) were electroporated to cells as described (15, 16). Successful introduction of shRNA and plasmid DNA were confirmed by analyzing gene transcription.
Immunoprecipitation and Immunoblot
To detect acetylated human isocitrate dehydrogenase 2 (IDH2) and superoxide dismutase 2 (SOD2), THP-1 cells were stimulated for the indicated times with 1 μg/ml of LPS. Whole cell lysates were precleared by incubation for 1 h with anti-rabbit IgG trueblot beads (Rockland) followed by incubation with primary rabbit anti-human SIRT1 or -SOD2 antibodies (Genetex) for 1 h on ice. The immunocomplexes were then precipitated by anti-rabbit IgG trueblot beads for 1 h on a rocking platform. The pelleted beads were washed 3 times with 500 μl of lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40), resuspended in Laemmli buffer, and heated to 100 °C for 10 min. The IgG bead-free supernatants were loaded onto the SDS-PAGE gel, probed with rabbit anti-acetyl lysine antibody, and developed with rabbit IgG trueblot.
Cellular protein levels were analyzed by Western blot as described (15). Primary antibodies were purchased from Santa Cruz (SIRT1, SIRT3, RELB, and β-actin), Genetex (IDH2, SOD2), and Abcam (voltage-dependent anion channel (VDAC)).
In Vitro Assessments of ATP and Enzyme Activities
Colorimetric assay kits were used to biochemically assess the ATP production and enzymatic activity of citrate synthase (Biovision) and SOD2 (Dojindo Molecular Technologies) according to the manufacturers' instructions. To specifically assess SOD2 activity, 1 mmol of potassium cyanide was used to inactivate Cu, Zn-SOD and extracellular SOD activity. Data were normalized with loaded protein.
IDH2 activities were measured by reducing NADP+ to NADPH at 340 nm using a Bio-Rad spectrophotometer according to the protocols (30, 31). In brief, 20 μl of sonicated cell lysate were added to each well of a 96-well plate followed by 180 μl of a reaction mixture containing 33 mm KH2PO4·K2HPO4, 3.3 mm MgCl2, 167 μm NADP+, and 167 μm (+)-potassium Ds-threo-isocitrate monobasic (Sigma). The absorbance at 340 nm was immediately read at 30-s intervals for 7 min. All samples were run in triplicate at room temperature. A blank control was set with all reagents except substrate isocitrate. Enzyme activity (micromoles/min/mg of protein) in the sample was calculated using an NADPH extinction coefficient of 6.22 mm/cm.
Data Analysis
Data were analyzed by one-way ANOVA to compare the different treatments or time points and multiple unpaired t test to compare the treated and untreated groups using GraphPad Prism version 6 (San Diego, CA). The area under the curve (AUC) was analyzed by XF software (Seahorse Bioscience). The survival curve was statistically analyzed by log-rank (Mantel-Cox) test. p values less than 0.05 were considered significant.
RESULTS
Biphasic Shifts in Mitochondrial Bioenergetics Accompany Inflammation Adaptation
We reported that between 4 and 8 h after TLR4 activation, THP-1 promonocytic cells switch their dominant metabolic profile from an acute-stage Warburg-like glycolysis to a post-acute adaptation stage of increased fatty acid β-oxidation in a SIRT1- and SIRT6-dependent process (15, 19). A similar switch was observed in adapted blood leukocytes isolated from septic humans and septic mouse splenocytes. We refer to these shifts as immunometabolic adaptation.
Here, we hypothesized that the change in metabolism between the initiation and adaptation stages of the acute inflammatory response modifies mitochondrial bioenergetics. To test it, we used our THP-1 cell model of the TLR4-dependent (1 μg/ml of LPS) acute inflammatory response (32) to time the phase of oxidative phosphorylation as the cells transition between the two stages. First, we measured the mitochondrial OCR using a Seahorse XF-24 oxygen and proton flux analyzer. At 4 h, TLR4 stimulation had significantly increased OCR, which peaked by 8 h. During that time, the metabolic fuel switched from glycolysis to fatty acid oxidation (19), and by 24 h OCR had declined (Fig. 1A). The biphasic changes in OCR were paralleled by modifications in ATP production estimated by the difference in OCR between basal and oligomycin treatment (Fig. 1B) or in vitro biochemical measurement (Fig. 1C). At the same time, mitochondrial membrane potential changed, as determined using MitoTracker Red either with fluorescence microscopy analysis (Fig. 1D, left panel) or flow cytometry (Fig. 1D, right panel).
FIGURE 1.

Biphasic shifts in mitochondrial bioenergetics accompany inflammation adaptation. THP-1 cells were stimulated with 1 μg/ml of LPS for the indicated times, spanning the phenotype shift from acute initiation (0–8 h) to postacute adaptation (8–24 h). A, acute inflammatory response-induced dynamic changes in mitochondrial OCR analyzed using a Cell Mito Stress kit. B, ATP generation calculated by subtracting OCR after oligomycin treatment from basal OCR. C, kinetics of mitochondrial ATP generation evaluated by the biochemical assay in vitro. D, modifications in mitochondrial membrane potential analyzed by confocal microscopy (left panel) and flow cytometry (right panel; data normalized on 0 h) using MitoTracker Red dye. E, mitochondrial superoxide production assessed by confocal microscopy (left panel) and flow cytometry (right panel; data normalized on 0 h) using Mito-Sox Red dye. F, fluorescence microscopy study of the production of mitochondrial superoxide and cellular hydrogen peroxide stained by dichlorofluorescein diacetate. G, mitochondrial SRC identified by differences between the basal and maximal OCR (8 h after LPS) after adding ETC-uncoupler FCCP. H, dynamic changes in ETC respiration in purified mitochondria over the course of TRL4 response. Mitochondria were isolated from THP-1 cells treated with or without LPS for 8 h. Bioenergetics was analyzed in an equal amount (10 μg) of mitochondrial protein as described under “Experimental Procedures.” The inset shows purity of mitochondrial preparations. The data in A, B, C, G, and H are mean ± S.E. of quadruplicate wells from one of three separate experiments. The flow cytometry data in C and D are mean ± S.E. from three independent experiments. The one-way ANOVA p value in A–E depicts the significance across time points; subsequent unpaired t tests determined the significance between 0 h and other time points. The AUC in F and G is analyzed by XF software. *, p < 0.05; **, p < 0.01; ***, p < 0.001; #, p > 0.05; ∧ indicates p > 0.05 in comparison to OCR at 4 h after LPS.
Increases in mitochondrial respiration with ATP production often parallel accumulation of electron transport chain (ETC)-dependent superoxide anions (O2⨪). As a ROS, they can prompt beneficial signal transduction pathways by specific redox protein modifications or become autotoxic by disrupting protein structure and activating cell death pathways (33). Analyzing mitochondrial O2⨪ using fluorescence microscopy (Fig. 1E, left panel) and flow cytometry (Fig. 1E, right panel) with Mito-Sox dye, we found that levels rapidly increased to a peak at 4 h and decreased by 24 h. We then assessed whole cell hydrogen peroxide (H2O2) by dichlorofluorescein fluorescence microscopy; it peaked at 8 h and substantially decreased at 24 h (Fig. 1F).
SRC is another important parameter of mitochondrial bioenergetics. It varies among cell types and subtypes; for example, it is minimal in neutrophils but much higher in T memory cells that have transitioned from T effector cells. They gain extra mitochondrial capacity if they are restimulated through the antigen-specific receptor (34). We compared the SRC of the initiation and adaptation phenotypes, using the mitochondrial uncoupler FCCP, and found substantial increases at 8 h when OCR peaks (Fig. 1G).
The observed shifts in mitochondrial bioenergetics at the whole cell level could reflect changes in mitochondrial mass and/or qualitative modifications in ETC. To address these possibilities, we purified mitochondria from LPS-treated or untreated THP-1 cells and compared their ETC respiration by loading equal amounts (10 μg/well) of mitochondrial protein. Results reproduced the TLR4-mediated increase in OCR; however, the SRC increase at the whole cell level is normalized by mitochondrial protein, suggesting changes in mitochondrial mass and/or quantity (Fig. 1H).
Together, these data indicate that during the acute inflammatory response, (a) as the primary energy nutrients of THP-1 monocytes switch from glycolysis to fatty acid oxidation, mitochondrial oxidative phosphorylation, ROS, and SRC switch from initiation to adaptation (19); and (b) ROS decrease in THP-1 cells when NAD+ levels and fatty acid β-oxidation remain elevated, as reported (15, 19). Both suggest the activation of antioxidant pathways.
Mitochondrial Biogenesis and Enzyme Activation Increase with Inflammation Adaptation
Increases in OCR, ROS production, and SRC suggest that mitochondrial biogenesis and enzyme activation increase after TLR4 stimulation, when initiation shifts to adaptation. To test this hypothesis, we first assessed mitochondrial mass by flow cytometry using MitoTracker Green dye and found it had increased by 4 h, peaked at 8 h, and decreased at 24 h (Fig. 2A). The increase was supported by a higher mitochondrial to nuclear DNA ratio (Fig. 2B) and increases in the nuclear DNA-encoded mitochondrial enzyme proteins IDH2 and SOD2 and the structural protein, VDAC (Fig. 2C). Each of these proteins had substantially increased by 4 h and remained elevated at 24 h, when adaptation was fully established. Mitochondrial DNA-encoded (NADH dehydrogenase subunit 6) and nuclear-encoded (Cox IV) ETC proteins also increased during the TLR4 response (Fig. 2D). These data mirrored the increased expression of ETC complexes, as assessed in whole cell lysates (Fig. 2E, left panel) and purified mitochondrial lysates (Fig. 2E, right panel). Increases in mitochondrial protein expression correlated with elevated mRNA levels of mitochondrial functional proteins; 24 h after TLR4 activation, IDH2, SOD2, SIRT3, and VDAC had increased 2.07-, 9.3-, 2.22-, and 3.77-fold, respectively (Fig. 2F).
FIGURE 2.

Mitochondrial biogenesis and enzyme activation increase with inflammation adaptation. THP-1 cells were stimulated with 1 μg/ml of LPS for the indicated times. A, flow cytometry analysis of mitochondrial mass determined by MitoTracker Green dye at 0, 4, 8, and 24 h after TLR4 activation. B, LPS-induced changes in the mitochondrial-to-nuclear DNA ratio. C, immunoblots of nuclear DNA-encoded IDH2, SOD2, and structural protein VDAC probed at time points between 0 and 24 h. D, LPS-induced expression of mitochondrial DNA-encoded NADH dehydrogenase subunit 6 and nuclear DNA-encoded complex IV. E, dynamic changes of ETC complex proteins in whole cell lysates (left panel) and purified mitochondria (right panel) over the course of TLR4 response. F, LPS-induced gene expression of IDH2, SOD2, and VDAC analyzed by RT-PCR. LPS-induced changes in IDH2 (G), SOD2 (H), and citrate synthase (I) activities were assessed biochemically in vitro. Total cell lysates were analyzed in C, D, and the left panel of E with β-actin as loading control; isolated mitochondrial proteins were analyzed with Cox-IV as loading control (E, right panel). One of three experiments is shown in A-C; one of two experiments is shown in D, E, G, H, and I; one of five experiments is shown in F. One-way ANOVA p values are shown to determine the significance across time points. The significance between 0 h and other time points is determined by subsequent unpaired t tests, *, p < 0.05; **, p < 0.01; ***, p < 0.001; #, p > 0.05.
We next determined whether enzyme activation accompanied mitochondrial biogenesis. We selected IDH2, the rate-limiting enzyme in the tricarboxylic acid (TCA) cycle, and mitochondrial SOD2, the rate-limiting enzyme in the antioxidant defense pathway. Both IDH2 and SOD2 activity significantly increased after LPS stimulation (Fig. 2, G and H). We also measured citrate synthase activity, which catalyzes the first step of the TCA cycle after acetyl-CoA generation from pyruvate or fatty acids. Citrate synthase activity increased during adaptation (Fig. 2I). Together, these data indicate that during the postacute adaptation response, increased mitochondrial biogenesis is associated with activation of mitochondrial bioenergy regulators.
SIRT3 Expression and Activation Increase with Inflammation Adaptation
Because SIRT3 is a major activator of mitochondrial bioenergetics (35, 36), the increases in mitochondrial enzymatic activities and biphasic shifts in oxidative phosphorylation suggest that bioenergetics changes during inflammation adaptation. To examine this question, we first assessed gene expression of SIRT3 by measuring mRNA and protein. We found that both increased by 4 h and remained elevated 24 h after TLR4 stimulation (Fig. 3, A and B). To further test the role of SIRT3 during the adaptive process, we assessed acetylation states (inactive form) of IDH2 and SOD2, two important rate-limiting enzymes and SIRT3 targets. Because site-specific IDH2 and SOD2 acetylation antibodies are unavailable, we immunoprecipitated total protein followed by Western blot analysis with an anti-pan acetyl antibody. Levels of the acetylated inactive form of IDH2 decreased by 4 h and remained low at 24 h. Acetylated SOD2 levels decreased slightly at 4 h but diminished dramatically by 24 h after TLR4 stimulation (Fig. 3C). These data indicate that SIRT3 plays a pivotal role in the postacute inflammation adaptation response.
FIGURE 3.
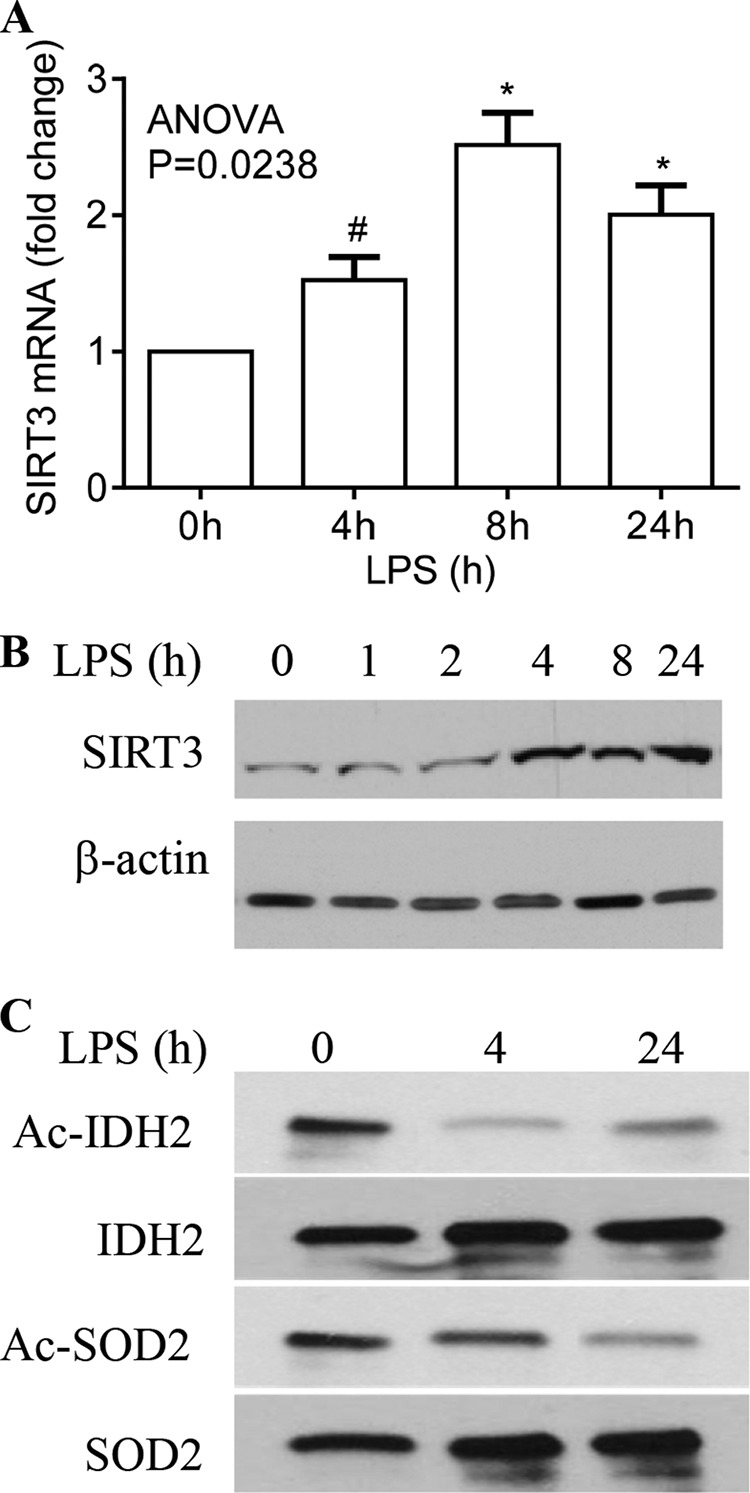
SIRT3 expression and activation increased with inflammation adaptation. THP-1 cells were stimulated with or without 1 μg/ml of LPS for the indicated times. A, LPS-induced SIRT3 gene transcription. B, LPS-induced changes in SIRT3 protein were analyzed by Western blot. C, deacetylation activation of representative mitochondrial rate-limiting enzymes followed at baseline, 4, and 24 h post-LPS using anti-acetyl lysine antibody developed by rabbit IgG trueblot. Total cellular IDH2 and SOD2 were co-immunoprecipitated with anti-IDH2 or anti-SOD2 antibodies. Data in A are shown as mean ± S.E. of five experiments. One of five experiments is shown in B, and one of three experiments is shown in C. The one-way ANOVA p value in A determines the significant changes in SIRT3 mRNA across time points. The significant changes in SIRT3 mRNA between 0 h and other time points were analyzed by subsequent unpaired t tests, *, p < 0.05; #, p > 0.05.
SIRT1, RELB, and SIRT3 Interactions Regulate Mitochondrial Bioenergetics
Because we demonstrated that NAD+ redox state sensor SIRT1 requires RELB expression and promoter loading to epigenetically coordinate acute inflammation and metabolism (8, 15, 19), we reasoned that SIRT1 and RELB might also link to SIRT3 expression and mitochondrial biogenesis. To test this idea, we used gene-specific shRNA to stably silence gene expression of SIRT1, RELB, or SIRT3 before TLR4 stimulation.
Flow cytometry analysis showed that reducing SIRT1 or RELB attenuated TLR4-mediated changes in MitoTracker Green intensity (biogenesis). In contrast, reducing SIRT3 did not alter biogenesis (Fig. 4A). This observation was confirmed by fluorescence microscopy (Fig. 4B). We next determined the effect of gene silencing on ETC respiration. As expected, SIRT3 knockdown diminished TLR4-induced increases in OCR, and OCR was similarly reduced after knockdown of either SIRT1 or RELB (Fig. 4C). These gene silence-mediated OCR changes paralleled reductions in SRC (Fig. 4D). In support of bioenergetics changes, acetylated levels (inactive form) of IDH2 and SOD2 were dramatically decreased in LPS-treated cells at 24 h compared with untreated cells (at 0 h). In contrast, individual knockdown of SIRT1, RELB, or SIRT3 attenuated LPS-mediated deacetylation of both proteins (0 versus 24 h of LPS treatment) (Fig. 4E).
FIGURE 4.

SIRT1, RELB, and SIRT3 regulate mitochondrial bioenergetics. To test distinct functions, SIRT1, RELB, or SIRT3 genes were stably silenced in THP-1 cells by infecting the cells with control or gene-specific lentiviral shRNA. Cells were then stimulated with 1 μg/ml of LPS for the indicated times. A, changes in mitochondrial mass defined by mean fluorescence intensity of MitoTracker Green dye at 0, 4, 8, and 24 h. B, fluorescence microscopy study of mitochondrial mass stained by MitoTracker Green. C, OCR changes in control versus gene-specific shRNA cells assessed at 0 and 24 h. D, gene silencing effects on bioenergetics as determined by XF-24 Extracellular Flux Analyzer. E, acetylated IDH2 and SOD2 were determined using anti-acetylated lysine antibody at 0 and 24 h (left panel) and densitometry assay of Ac-IDH2 and Ac-SOD2 normalized against total immunoprecipitated proteins (right panel). F, mitochondrial superoxide O2⨪ production determined by flow cytometry using Mito-Sox dye. G, SOD2 activity assessed biochemically in vitro. Data in A are shown as mean ± S.E. (n = 3). Data in C, D, F, and G are mean ± S.E. of triplicate wells (representative of n = 3). A representative experiment of two is shown in B and E. One-way ANOVA p values in A determined the significance across the time points for each RNAi; results of unpaired t tests are shown in C, F, and G. AUCs between medium and LPS treatments in D are analyzed by XF software. *, p < 0.05; ***, p < 0.001; #, p > 0.05. CTRL: control.
Next, we examined the effect of gene silencing on O2⨪ accumulation. Reducing SIRT1 or RELB but not SIRT3 levels prevented LPS induction of O2⨪ levels (Fig. 4F). We expected the increase in O2⨪ accumulation in SIRT3 knock-out because of reduced SOD2 activity (Fig. 4G), which is controlled by SIRT3. Taken together, the SIRT1, RELB, and SIRT3 knockdown data suggested that SIRT1 and RELB nuclear activation are proximal signals for differential induction of both SIRT3 expression and mitochondrial biogenesis, but note that SIRT3 does not induce biogenesis.
SIRT1, RELB, and SIRT3 Act Sequentially
To determine whether SIRT1, RELB, and SIRT3 act in sequence to modify mitochondrial bioenergetics, we used shRNA to stably silence each of them (Fig. 5A). As shown in Fig. 5B, TLR4 activation significantly stimulated expression of SIRT1, RELB, and SIRT3 mRNA in control RNAi THP-1 cells. In contrast, knockdown of SIRT1 reduced basal transcription of RELB and SIRT3 and significantly prevented induction of their mRNA during LPS stimulation. Reducing RELB diminished the basal and LPS-induced SIRT3 mRNA but did not affect SIRT1 mRNA. In contrast, reducing SIRT3 had no effect on basal and LPS-induced SIRT1 or RELB gene expression. These data show that SIRT1 regulates RELB gene expression as reported (15) and that RELB supports increased SIRT3 expression. This sequential gene transcription was confirmed by Western blot analysis of protein levels (Fig. 5C); furthermore, reducing SIRT1 or RELB but not SIRT3 prevented LPS-induced IDH2 and SOD2 levels, showing that SIRT3 does not induce mitochondrial biogenesis.
FIGURE 5.

SIRT1, RELB, and SIRT3 sequentially control mitochondrial bioenergetics. THP-1 cells with and without individual RNAi reductions were stimulated with 1 μg/ml of LPS for 24 h. A, stable knockdown efficiency determined by Western blot. B, effects of gene silencing on mRNA expression of the 3 genes analyzed by quantitative PCR. LPS induces mRNAs of SIRT1, RELB, and SIRT3 in control knockdown cells; reducing SIRT1 attenuates LPS-induced mRNAs of RELB and SIRT3; reducing RELB minimizes LPS-induced SIRT3 but not SIRT1 mRNAs; reducing SIRT3 has no effect on LPS-induced SIRT1 and RELB mRNAs. C, effects of gene silencing on LPS induction of SIRT1, RELB, SIRT3, IDH2, and VDAC proteins. D, compensation of SIRT1 or RELB deficiency in LPS-induced OCR changes with overexpression of the opposite gene. RELB knock-in overcame SIRT1 KD reduction in OCR, but SIRT1 knock-in had little effect on RELB KD reduction of OCR. Inset shows knock-in efficiency. Data in B and D are shown as mean ± S.E. (n = 3). One of three experiments is shown in C. One-way ANOVA p values are shown in B and D. The subsequent unpaired t analysis in B (*, p < 0.05; **, p < 0.01) indicates the significance of mRNA changes between medium and LPS treatment in control KD cells, and (∧, p < 0.05; ∧, p < 0.01; triple ∧, p < 0.001) compare the LPS-induced mRNA in each gene-specific silenced cell with that in control RNAi. t tests in D compare the OCR in other groups to basal OCR, ***, p < 0.001; #, p > 0.05. Med, medium; KD, knockdown; KI, knock-in.
To further assess the sequence of SIRT1 and RELB action, we determined TLR4-mediated OCR changes in SIRT1 or RELB RNAi cells by overexpressing (genetically inducing) the opposite gene. Overexpression of RELB overcame SIRT1 RNAi-dependent reductions in OCR. However, increased expression of SIRT1 did not overcome RELB RNAi-induced reductions in OCR (Fig. 5D). The inset in Fig. 5D shows gene knock-in efficiency. Together, these data suggest that SIRT1, RELB, and SIRT3 act sequentially to shift bioenergetics during the adapted acute inflammatory response.
SIRT1 Inhibition during Early Adaptation Reverses Bioenergetics Modifications
To confirm the indispensable role of SIRT1 in SIRT1-RELB-SIRT3 signaling, we examined whether inhibiting it would reverse established mitochondrial reprogramming. The SIRT1-specific inhibitor EX-527 reversed SIRT1-dependent epigenetic silencing of the TNF-α gene and restored the immunocompetent monocyte phenotype (15). THP-1 cells were treated with or without EX-527 at the OCR peak time of 8 h after LPS stimulation and incubated for an additional 16 h before analysis at 24 h. We found that EX-527 treatment reversed TLR4-mediated increases in basal OCR and SRC (Fig. 6A) and gene expression of RELB, SIRT3, IDH2, and VDAC. Surprisingly, SOD2 mRNA levels remained high (Fig. 6B). EX-527 treatment also significantly reduced RELB and SIRT3 but not VDAC levels (Fig. 6C). These data indicate that SIRT1 is the proximal regulator of nuclear-mitochondrial signaling, and SIRT-dependent bioenergy shifts during adaptation are reversible.
FIGURE 6.

SIRT1 inhibition during early adaptation reverses bioenergetics modifications. THP-1 cells were treated with 1 μg/ml of LPS for 24 h. One set of cells in culture was treated with 1 μm EX-527 at 8 h after LPS. A, SIRT1 inhibition by EX-527 reverses LPS-mediated modifications in OCR and SRC. Inset shows experiment design. B, SIRT1 inhibition by EX-527 affects gene expression of RELB, SIRT3, IDH2, VDAC, and SOD2 as analyzed by RT-PCR. Data are shown as mean ± S.E. of five separate experiments. C, effect of inhibiting SIRT1 by EX-527 on levels of RELB, SIRT3, IDH2, VDAC, and SOD2. One of three experiments is shown in A and C. AUCs between treatments and medium in A are analyzed by XF software. Unpaired t tests compare the significant differences between LPS and LPS + EX-527 treatments in B, *, p < 0.05; **, p < 0.01; ***, p < 0.001.
SIRT1 Controls Bioenergy Shifts in Normal and Sepsis-adapted Human Monocytes
Normal Human Monocytes
To test whether shifts in mitochondrial bioenergetics identified in THP-1 cells during acute inflammation also occur in human primary monocytes, we used normal human blood monocytes in culture. Primary monocytes from healthy humans responded to ex vivo TLR4 stimulation by increasing mRNA expression of SIRT1, RELB, SIRT3, SOD2, and VDAC (Fig. 7A); gene induction peaked at 8 h and decreased by 24 h. We were unable to determine protein levels due to an insufficient number of isolated cells.
FIGURE 7.

SIRT1, RELB, and SIRT3 regulate mitochondrial bioenergetics in normal and sepsis-adapted monocytes. A, modifications in mRNA expression of SIRT1, RELB, SIRT3, IDH2, SOD2, and VDAC in normal human monocytes purified by negative selection and stimulated with 100 ng/ml of LPS for 0, 8, and 24 h. Data are shown as mean ± S.E. of four experiments. B, effects of SIRT1 inhibition by EX-527 administered at 8 h post-LPS on LPS-induced mRNA expression in normal primary monocytes at 24 h. Data are shown as mean ± S.E. (n = 3). C, mitochondrial bioenergetics changes in LPS-stimulated primary monocytes between 0 and 24 h and reverses with the EX-527 SIRT1 inhibitor added at 8 h after LPS (representative of four experiments). D, TNF-α expression in healthy and septic monocytes in response to 100 ng/ml of LPS stimulation. Data are shown as mean ± S.E. (n = 5). E, mRNA expression in adapted (LPS-tolerant) human sepsis monocytes, treated or untreated with SIRT1 inhibitor EX-527 for 16 h. Responses of untreated septic monocytes are set as 1. Data are shown as mean ± S.E. (n = 5). F, effects of EX-527 SIRT1 inhibitor on changes in mitochondrial bioenergetics in adapted septic monocytes treated with SIRT1 inhibitor EX-527 for 16 h. A representative experiment of five is shown. The one-way ANOVA p values for individual gene expression across the time points are shown in A, and the subsequent unpaired t tests determined the significance at 0 h and the other time points. Unpaired p values (*, p < 0.05; **, p < 0.01) are shown in B, D, and E. AUC statistics are shown in C and F, *, p < 0.05; ***, p < 0.001, in comparison with medium.
We also used the SIRT-specific inhibitor EX-527 (8, 15, 20) to test whether SIRT1 activation is indispensable for reprogramming mitochondrial bioenergetics in TLR4-adapted primary human monocytes. Adding EX-527 ex vivo 8 h after LPS stimulation decreased the mRNA level of all genes except SOD2 after normalizing against untreated cells (Fig. 7B). We also found that EX-527 treatment could reverse the increased OCR and SRC we observed in the adaptation phenotype of normal monocytes (Fig. 7C).
Septic Human Monocytes
We previously reported that increased SIRT1 and RELB levels in sepsis-adapted human leukocytes, assessed by tolerance to LPS stimulation of TNF-α or IL-1β, sustained increased mitochondrial fatty acid oxidation and decreased glucose oxidation (19). To determine whether monocyte mitochondrial bioenergetics is altered during the human sepsis adaptation state, we isolated CD14+ monocytes from venous blood obtained from patients with sepsis. First, we confirmed that the cells were in the adaptation state by demonstrating repressed TNF-α transcription after LPS stimulation (LPS tolerance, Fig. 7D). To determine the dependence and reversibility of shifts in mitochondrial bioenergetics in sepsis monocytes, we treated them ex vivo with EX-527 overnight. We found significantly repressed mRNA levels of RELB, SIRT3, IDH2, SOD2, and VDAC after normalizing against cells not treated with EX-527 (Fig. 7E). We also observed that EX-527 restored the increases in basal OCR and SRC levels (Fig. 7F) in sepsis-adapted monocytes to near normal (Fig. 7C).
Thus, as in the THP1 sepsis cell model, SIRT1, RELB, and SIRT3 regulate shifts in mitochondrial bioenergetics in LPS-stimulated normal and sepsis-adapted blood monocytes. Furthermore, SIRT1 is indispensable in controlling these shifts.
SIRT1 Controls Bioenergy Shifts in Normal and Sepsis-adapted Mouse Splenocytes
Normal Mouse Splenocytes
We next explored the Sirt1-Relb-Sirt3 nuclear-to-mitochondrial link using normal mouse splenocytes because this total population of mixed immune cells reflects the adaptation phenotype (19). Taking the same approach as for primary human monocytes, we found that LPS-adapted splenocytes have a gene expression pattern similar to that of THP-1 cells and normal human monocytes. LPS significantly induced normal splenocyte Sirt1, Relb, Sirt3, Idh2, Sod2, and Vdac mRNA levels (Fig. 8A), and when added to the cells during early adaptation (8 h), EX-527 reversed these changes (Fig. 8B). In further support of the indispensable contribution of SIRT1, we found that inhibiting it reversed OCR and SRC increases in LPS-adapted normal splenocytes (Fig. 8C).
FIGURE 8.

Sirt1, Relb, and Sirt3 regulate mitochondrial bioenergetics in normal and sepsis-adapted mouse splenocytes. A–C, normal splenocytes were isolated from C57/BL6 mice and stimulated with 100 ng/ml of LPS for the indicated times. Data collected from five (A and B) or three (C) animals are shown. A, mRNA levels of Sirt1, Relb, Sirt3, Idh2, Sod2, and Vdac at 0, 8, and 24 h after LPS stimulation. B, effects of SIRT1 inhibition with EX-527 added 8 h post-LPS on LPS-induced changes in mRNA expression at 24 h in splenocytes. C, changes in basal OCR in normal splenocytes treated or untreated with EX-527 Sirt1 inhibitor added at 8 h after LPS stimulation and assessed at 24 h. D, mRNA changes in Relb, Sirt3, Idh2, Sod2, and Vdac in sepsis splenocytes isolated from mice treated with vehicle DMSO or EX-527 24 h after CLP and assessed ex vivo at 36 h (12-h post EX-527 treatment). Data are shown as mean ± S.E. for three animals. E, effects of EX-527 Sirt1 inhibitor on OCR in septic splenocytes treated with Sirt1 inhibitor EX-527 for 16 h. Error bars represent mean ± S.E. of quadruplicate wells. Data are shown as mean ± S.E. for three animals. F, survival curve of wild-type and SIRT3 knock-out 129-mice after CLP-induced sepsis (7 animals per group). G, scheme showing the sequential actions of SIRT1-RELB-SIRT3 that regulate nuclear-mitochondrial communication and remodel mitochondrial bioenergetics during adaptation to the acute inflammatory response. One-way ANOVA p values of a gene across the time points are shown in A. The subsequent unpaired t test (*, p < 0.05; **, p < 0.01) depicts the significant changes in gene expression compared with 0 h. Unpaired t test p values shown in B–E compare the difference between EX-527 treated and untreated cells. The log-rank (Mantel-Cox) test p value in F compares the survival rates of wild-type and Sirt3 knock-out 129 mice.
Septic Mouse Splenocytes
Because we found that EX-527 SIRT1 inhibition reverses immune suppression and rescues septic mice from death, we tested whether changes in mitochondrial bioenergetics occur in the splenocytes of septic mice, and if so, whether they can be reversed by Sirt1 inhibition. Using our established model of murine sepsis (cecal ligation and puncture) (20), we found that treating sepsis splenocytes ex vivo for 24 h with the SIRT1 inhibitor EX-527 significantly decreased mRNA levels of Relb, Sirt3, Idh2, Sod2, and Vdac (Fig. 8D). Similar treatment reversed OCR elevation in septic splenocytes (Fig. 8E).
These data show the important sequential actions of Sirt1, Relb, and Sirt3 in regulating inflammation adaptation. Our previous study showed that inhibiting Sirt1 broke sustained adaptation and markedly decreased mortality (20). We assessed sepsis survival rates in Sirt3 knock-out mice to determine whether the effects of Sirt1 on sepsis outcome were due to its link with SIRT3. Although Sirt3 knock-out was done in 129S1/SvImJ background rather than C57 BL/6 mice, survival rates between wild-type and Sirt3-deficient mice did not differ statistically (Fig. 8F). This result suggests that the beneficial effect of Sirt1 inhibition is not due solely to its control of Sirt3 expression and activation. Fig. 8G summarizes the sequence of SIRT1, RELB, and SIRT3 in regulating mitochondrial bioenergetics during inflammation adaptation.
DISCUSSION
The major discovery of this study, linking NAD+-dependent nuclear SIRT1 function to mitochondrial bioenergetics through the induction of SIRT3 expression and increased biogenesis, clarifies sepsis adaptation in monocytes. In addition, the relationship between the functions of nuclear SIRT1 and mitochondrial SIRT3 suggests that NAD+ generation in the two subcellular compartments is coordinated during sepsis adaptation. This study also expands the importance of the nuclear SIRT1-RELB connection in regulating sepsis adaptation (15) by showing its influence on mitochondrial bioenergetics as well as immune and metabolic reprogramming (8). Thus, three major phenotypic changes identified in sepsis-adapted monocytes, repressing innate immunity, shifting mitochondrial fueling from glucose to fatty acids, and modifying mitochondrial bioenergetics, are controlled, at least in part, by nuclear SIRT1 and RELB, suggesting that an integrated epigenetic process directs the course of sepsis.
The proximal control of SIRT1 over RELB function and its downstream effects on mitochondrial bioenergetics explain why specifically inhibiting nuclear Sirt1 during the adaptation state of mouse sepsis might rebalance the innate immune response, metabolism, and bioenergetics to promote survival (20). However, the relative role of each in influencing sepsis outcomes and how their programming is coordinated must still be determined. Our finding that the sepsis survival rate in Sirt3 knock-out mice did not differ significantly from that in wild-type control mice supports the possibility that persistently dysregulated mitochondrial fueling sources or alterations in quality control (e.g. mitophagy; fission and fusion; biogenesis) might affect survival, rather than direct changes in bioenergetics.
Another new finding describes how ETC activity shifts during sepsis adaptation. Biphasic changes in OCR, ATP levels, membrane depolarization, and ROS levels between initiation and adaption in THP1 promonocytes peaked 6–8 h after initiation and subsided somewhat by 24 h. However, OCR remained above the normal basal state in all of the sepsis cell models. Strikingly, blocking SIRT1 ex vivo significantly decreased the basal OCR in sepsis-adapted human monocytes, mouse splenocytes, and adapted THP1 cells. These findings, along with persistent increases in Sirt1, Relb, Sirt3, and Vdac expression and their reduction with SIRT1 inhibition, suggest that mitochondria are stressed or dysregulated during prolonged adaptation. Several observations support this conclusion. In this study, SIRT1 inhibition ex vivo reversed the shift in mitochondrial bioenergetics in our sepsis cell model and in human and mouse sepsis leukocytes. In our published report, Sirt1 inhibition in vivo during sepsis adaptation improved survival concomitant with reversing immune repression and stabilizing vascular function (20). We also previously reported that persistent shifts from glucose to fatty acid oxidation in sepsis-adapted human blood monocytes and mouse splenocytes could be reversed ex vivo by inhibiting SIRT1 (19).
Taken together, these results indicate that SIRT1 broadly influences homeostasis, which is dysregulated during sepsis adaptation. What causes this dysregulation? Does metabolic inflexibility during sepsis adaptation imbalance both mitochondrial bioenergetics and immune function by sustaining fatty acid over glucose oxidation (37)? In support of this concept, others found that short and medium chain carnitine fatty acids accumulate in the plasma of septic humans and highly predict poor survival rates 28 days after sepsis onset (38). Another study reports that transferring normal mitochondria into pulmonary alveoli protects animals against acute endotoxin-induced lung injury (39). We favor the concept that increases in NAD+ with persistent activation of SIRT1 delays sepsis resolution, at least in part, by dysregulating mitochondrial fuel sources and altering bioenergetics.
In summary, we show that the sequential actions of nuclear SIRT1, RELB, and mitochondrial SIRT3 link nuclear and mitochondrial functions during sepsis adaptation. These findings indicate that SIRT1 may provide a new treatment target to promote homeostasis and improve survival during sepsis adaptation.
Acknowledgments
We thank Drs. Barbara Yoza, Candice M. Brown, and Linda McPhail for the study discussion. We thank Dr. Richard Loeser for critical suggestions, and Jean Hu, Sue Cousart, and Young Choi for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants R01AI079144, R01-AI065791, and R01GM102497 (to C. E. M.), and R01GM099807 (to V. V.).
- ROS
- reactive oxygen species
- CLP
- cecal ligation and puncture
- OCR
- oxygen consumption rate
- SRC
- spare respiratory capacity
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- IDH2
- isocitrate dehydrogenase 2
- SOD2
- superoxide dismutase 2
- AUC
- area under the curve
- ANOVA
- analysis of variance
- ETC
- electron transport chain
- VDAC
- voltage-dependent anion channel.
REFERENCES
- 1. Medzhitov R. (2010) Inflammation 2010: new adventures of an old flame. Cell 140, 771–776 [DOI] [PubMed] [Google Scholar]
- 2. McCall C. E., Yoza B., Liu T., El Gazzar M. (2010) Gene-specific epigenetic regulation in serious infections with systemic inflammation. J. Innate Immun. 2, 395–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boomer J. S., To K., Chang K. C., Takasu O., Osborne D. F., Walton A. H., Bricker T. L., Jarman S. D., 2nd, Kreisel D., Krupnick A. S., Srivastava A., Swanson P. E., Green J. M., Hotchkiss R. S. (2011) Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 306, 2594–2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Belikoff B. G., Hatfield S., Georgiev P., Ohta A., Lukashev D., Buras J. A., Remick D. G., Sitkovsky M. (2011) A2B adenosine receptor blockade enhances macrophage-mediated bacterial phagocytosis and improves polymicrobial sepsis survival in mice. J. Immunol. 186, 2444–2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hotchkiss R. S., Karl I. E. (2003) The pathophysiology and treatment of sepsis. N. Engl. J. Med. 348, 138–150 [DOI] [PubMed] [Google Scholar]
- 6. Hotchkiss R. S., Monneret G., Payen D. (2013) Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect. Dis. 13, 260–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carré J. E., Singer M. (2008) Cellular energetic metabolism in sepsis: the need for a systems approach. Biochim. Biophys. Acta 1777, 763–771 [DOI] [PubMed] [Google Scholar]
- 8. Liu T. F., Brown C. M., El Gazzar M., McPhail L., Millet P., Rao A., Vachharajani V. T., Yoza B. K., McCall C. E. (2012) Fueling the flame: bioenergy couples metabolism and inflammation. J. Leukoc. Biol. 92, 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Piantadosi C. A., Suliman H. B. (2012) Transcriptional control of mitochondrial biogenesis and its interface with inflammatory processes. Biochim. Biophys. Acta 1820, 532–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singer M. (2008) Cellular dysfunction in sepsis. Clin. Chest. Med. 29, 655–660 [DOI] [PubMed] [Google Scholar]
- 11. Williams S. C. (2012) After Xigris, researchers look to new targets to combat sepsis. Nat. Med. 18, 1001. [DOI] [PubMed] [Google Scholar]
- 12. Haigis M. C., Guarente L. P. (2006) Mammalian sirtuins: emerging roles in physiology, aging, and calorie restriction. Genes Dev. 20, 2913–2921 [DOI] [PubMed] [Google Scholar]
- 13. El Gazzar M., Yoza B. K., Chen X., Hu J., Hawkins G. A., McCall C. E. (2008) G9a and HP1 couple histone and DNA methylation to TNFα transcription silencing during endotoxin tolerance. J. Biol. Chem. 283, 32198–32208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Foster S. L., Hargreaves D. C., Medzhitov R. (2007) Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447, 972–978 [DOI] [PubMed] [Google Scholar]
- 15. Liu T. F., Yoza B. K., El Gazzar M., Vachharajani V. T., McCall C. E. (2011) NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J. Biol. Chem. 286, 9856–9864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yoza B. K., Hu J. Y., Cousart S. L., Forrest L. M., McCall C. E. (2006) Induction of RelB participates in endotoxin tolerance. J. Immunol. 177, 4080–4085 [DOI] [PubMed] [Google Scholar]
- 17. Gil R., Barth S., Kanfi Y., Cohen H. Y. (2013) SIRT6 exhibits nucleosome-dependent deacetylase activity. Nucleic Acids Res. 41, 8537–8545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kawahara T. L., Michishita E., Adler A. S., Damian M., Berber E., Lin M., McCord R. A., Ongaigui K. C., Boxer L. D., Chang H. Y., Chua K. F. (2009) SIRT6 links histone H3 lysine 9 deacetylation to NF-κB-dependent gene expression and organismal life span. Cell 136, 62–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu T. F., Vachharajani V. T., Yoza B. K., McCall C. E. (2012) NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J. Biol. Chem. 287, 25758–25769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vachharajani V. T., Liu T., Brown C. M., Wang X., Buechler N. L., Wells J. D., Yoza B. K., McCall C. E. (2014) SIRT1 inhibition during the hypoinflammatory phenotype of sepsis enhances immunity and improves outcome. J. Leukoc. Biol. 96, 785–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. El Gazzar M., Yoza B. K., Hu J. Y., Cousart S. L., McCall C. E. (2007) Epigenetic silencing of tumor necrosis factor α during endotoxin tolerance. J. Biol. Chem. 282, 26857–26864 [DOI] [PubMed] [Google Scholar]
- 22. Zhong L., D'Urso A., Toiber D., Sebastian C., Henry R. E., Vadysirisack D. D., Guimaraes A., Marinelli B., Wikstrom J. D., Nir T., Clish C. B., Vaitheesvaran B., Iliopoulos O., Kurland I., Dor Y., Weissleder R., Shirihai O. S., Ellisen L. W., Espinosa J. M., Mostoslavsky R. (2010) The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1α. Cell 140, 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haden D. W., Suliman H. B., Carraway M. S., Welty-Wolf K. E., Ali A. S., Shitara H., Yonekawa H., Piantadosi C. A. (2007) Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. Am. J. Respir. Crit. Care Med. 176, 768–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carré J. E., Orban J. C., Re L., Felsmann K., Iffert W., Bauer M., Suliman H. B., Piantadosi C. A., Mayhew T. M., Breen P., Stotz M., Singer M. (2010) Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am. J. Respir. Crit. Care Med. 182, 745–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suliman H. B., Welty-Wolf K. E., Carraway M., Tatro L., Piantadosi C. A. (2004) Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc. Res. 64, 279–288 [DOI] [PubMed] [Google Scholar]
- 26. Suliman H. B., Welty-Wolf K. E., Carraway M. S., Schwartz D. A., Hollingsworth J. W., Piantadosi C. A. (2005) Toll-like receptor 4 mediates mitochondrial DNA damage and biogenic responses after heat-inactivated E. coli. FASEB J. 19, 1531–1533 [DOI] [PubMed] [Google Scholar]
- 27. Singer M. (2014) The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence 5, 66–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen X., El Gazzar M., Yoza B. K., McCall C. E. (2009) The NF-κB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J. Biol. Chem. 284, 27857–27865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brunet A., Sweeney L. B., Sturgill J. F., Chua K. F., Greer P. L., Lin Y., Tran H., Ross S. E., Mostoslavsky R., Cohen H. Y., Hu L. S., Cheng H. L., Jedrychowski M. P., Gygi S. P., Sinclair D. A., Alt F. W., Greenberg M. E. (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015 [DOI] [PubMed] [Google Scholar]
- 30. Andersson A. K., Miller D. W., Lynch J. A., Lemoff A. S., Cai Z., Pounds S. B., Radtke I., Yan B., Schuetz J. D., Rubnitz J. E., Rubnitz J. E., Ribeiro R. C., Raimondi S. C., Zhang J., Mullighan C. G., Shurtleff S. A., Schulman B. A., Downing J. R. (2011) IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia 25, 1570–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Someya S., Yu W., Hallows W. C., Xu J., Vann J. M., Leeuwenburgh C., Tanokura M., Denu J. M., Prolla T. A. (2010) Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McCall C. E., El Gazzar M., Liu T., Vachharajani V., Yoza B. (2011) Epigenetics, bioenergetics, and microRNA coordinate gene-specific reprogramming during acute systemic inflammation. J. Leukoc. Biol. 90, 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. West A. P., Shadel G. S., Ghosh S. (2011) Mitochondria in innate immune responses. Nat. Rev. Immunol. 11, 389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van der Windt G. J., Everts B., Chang C. H., Curtis J. D., Freitas T. C., Amiel E., Pearce E. J., Pearce E. L. (2012) Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36, 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hebert A. S., Dittenhafer-Reed K. E., Yu W., Bailey D. J., Selen E. S., Boersma M. D., Carson J. J., Tonelli M., Balloon A. J., Higbee A. J., Westphall M. S., Pagliarini D. J., Prolla T. A., Assadi-Porter F., Roy S., Denu J. M., Coon J. J. (2013) Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 49, 186–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lombard D. B., Alt F. W., Cheng H. L., Bunkenborg J., Streeper R. S., Mostoslavsky R., Kim J., Yancopoulos G., Valenzuela D., Murphy A., Yang Y., Chen Y., Hirschey M. D., Bronson R. T., Haigis M., Guarente L. P., Farese R. V., Jr., Weissman S., Verdin E., Schwer B. (2007) Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27, 8807–8814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Muoio D. M., Neufer P. D. (2012) Lipid-induced mitochondrial stress and insulin action in muscle. Cell Metab. 15, 595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Langley R. J., Tsalik E. L., van Velkinburgh J. C., Glickman S. W., Rice B. J., Wang C., Chen B., Carin L., Suarez A., Mohney R. P., Freeman D. H., Wang M., You J., Wulff J., Thompson J. W., Moseley M. A., Reisinger S., Edmonds B. T., Grinnell B., Nelson D. R., Dinwiddie D. L., Miller N. A., Saunders C. J., Soden S. S., Rogers A. J., Gazourian L., Fredenburgh L. E., Massaro A. F., Baron R. M., Choi A. M., Corey G. R., Ginsburg G. S., Cairns C. B., Otero R. M., Fowler V. G., Jr., Rivers E. P., Woods C. W., Kingsmore S. F. (2013) An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci. Transl. Med. 5, 195ra95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Islam M. N., Das S. R., Emin M. T., Wei M., Sun L., Westphalen K., Rowlands D. J., Quadri S. K., Bhattacharya S., Bhattacharya J. (2012) Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 18, 759–765 [DOI] [PMC free article] [PubMed] [Google Scholar]