

Background: The mechanism by which ginsenoside Rg5 regulates vascular function remains unclear.
Results: Rg5 increases angiogenesis and vasorelxation by activating multiple signal transduction pathways downstream of insulin-like growth fastor-1 receptor (IGF-1R).
Conclusion: Rg5 promotes endothelial cell function through activation of IGF-1R.
Significance: These findings reveal a mechanism for the positive regulation of vascular function by Rg5-mediated IGF-1R activation.
Keywords: Angiogenesis, Endothelial Cell, Insulin-like Growth Factor (IGF), Nitric Oxide, Vascular Biology, Insulin-like Growth Factor-1 Receptor, Vascular Function
Abstract
Ginsenoside Rg5 is a compound newly synthesized during the steaming process of ginseng; however, its biological activity has not been elucidated with regard to endothelial function. We found that Rg5 stimulated in vitro angiogenesis of human endothelial cells, consistent with increased neovascularization and blood perfusion in a mouse hind limb ischemia model. Rg5 also evoked vasorelaxation in aortic rings isolated from wild type and high cholesterol-fed ApoE−/− mice but not from endothelial nitric-oxide synthase (eNOS) knock-out mice. Angiogenic activity of Rg5 was highly associated with a specific increase in insulin-like growth factor-1 receptor (IGF-1R) phosphorylation and subsequent activation of multiple angiogenic signals, including ERK, FAK, Akt/eNOS/NO, and Gi-mediated phospholipase C/Ca2+/eNOS dimerization pathways. The vasodilative activity of Rg5 was mediated by the eNOS/NO/cGMP axis. IGF-1R knockdown suppressed Rg5-induced angiogenesis and vasorelaxation by inhibiting key angiogenic signaling and NO/cGMP pathways. In silico docking analysis showed that Rg5 bound with high affinity to IGF-1R at the same binding site of IGF. Rg5 blocked binding of IGF-1 to its receptor with an IC50 of ∼90 nmol/liter. However, Rg5 did not induce vascular inflammation and permeability. These data suggest that Rg5 plays a novel role as an IGF-1R agonist, promoting therapeutic angiogenesis and improving hypertension without adverse effects in the vasculature.
Introduction
Endothelial cells line blood vessels, which play an important role in vascular homeostasis and functions such as angiogenesis, vasorelaxation, and vascular inflammation and remodeling. Angiogenesis is a tightly controlled biological process characterized by changes in endothelial cell (EC)4 behavior that leads to increased growth, migration, and assembly into capillary structures (1). This process is accompanied by vasodilation and hyperemia of pre-existing capillaries (2). Angiogenesis is a pivotal process not only in embryonic development but also in the progression of a variety of pathologic conditions such as ischemic heart disease and wound healing (1). A large number of bioactive molecules, including vascular endothelial growth factor (VEGF), promote angiogenesis via stimulation of ECs to elicit multiple signal pathways (3, 4) and improve tissue and organ function in ischemia has been caused by defective blood circulation.
ECs constitutively express the endothelial isoform of endothelial nitric-oxide synthase (eNOS), in which catalytic activity is regulated by two extinct posttranslational modifications, specifically Ca2+-dependent dimerization and phosphorylation at Ser1177, leading to elevated endothelial NO production (5, 6). EC-derived NO plays a critical role in vasodilation via soluble guanylyl cyclase-mediated cGMP production and angiogenesis via promotion of EC survival (2). Impairments in endothelium-dependent vasodilation and angiogenesis are largely mediated by a reduction in the bioavailability of endothelium-derived NO. Indeed, mice lacking eNOS induce hypertension (7) and decrease VEGF-induced angiogenesis (2). Endogenous or exogenous molecules that regulate eNOS activity are suggested to improve hypertension (7).
Insulin-like growth factor-1 receptor (IGF-1R) signaling initiated by ligand binding mediates many crucial cell responses including angiogenesis (9). IGF-1R ligation has been shown to induce angiogenesis and endothelial proliferation in a retina model (9). Moreover, IGF-1R has also been shown to mediate neovascularization in human lung development and zebrafish cardiovascular development (10, 11) and promote vasculogenesis via differentiation of embryonic stem cells into endothelial cells (12). Moreover, a recent study determined that mouse overexpression of human IGF-1R in the endothelium enhances endothelium regeneration after denuding arterial injury and vasorelaxation (13), suggesting that IGF-1R activation is a useful target for treating vascular repair and hypertension disorders. Thus, IGF-1R agonists can be used as a beneficial means to treat human diseases with vascular dysfunction.
Although some ginsenosides present in ginseng are shown to regulate angiogenesis and vasodilation (14, 15), the beneficial effect of ginsenoside Rg5, a compound newly synthesized during the steaming process of Panax ginseng, on vascular function have not yet been studied. In the present study, we extensively investigated the pharmacological effect of Rg5 on angiogenesis and vasorelaxation as well as its cellular targets. We demonstrated that Rg5 regulates neovascularization and hypertension via action as a novel, nonbiological, IGF-1R agonist.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Rg5 was purified by a previously reported method (16), and its purity was >98%. Rg5 solutions (20 and 200 mm) were prepared in dimethyl sulfoxide as a stock solution. All cell culture media and reagents were purchased from Invitrogen. VEGF and basic fibroblast growth factor was purchased from Upstate Biotechnology (Lake Placid, NY). Antibodies used in this study were obtained from Cell Signaling Technology (Beverly, MA), R&D Systems (Minneapolis, MN), Santa Cruz Biotechnology (Santa Cruz, CA), and BD Transduction Laboratories (San Diego, CA). Recombinant human IGF-1 and [125I]IGF were obtained from R&D Systems and PerkinElmer Life Sciences, respectively. PD98059, LY294002, pertussis toxin (PTX), BAPTA-AM, U73122, PP2, NG-nitro-l-arginine methyl ester (l-NAME), and 1H-[1,2,4]oxadiazole[4,3-a]quinoxalin-1-one (ODQ) were from Calbiochem. Fluo-4-acetoxymethyl (Fluo-4 AM) ester and 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) diacetate were obtained from Molecular Probes (Eugene, OR). A human phospho-receptor tyrosine assay kit was purchased from R&D Systems. The siRNAs for human and mouse IGF-1R, human phospholipase C-γ1 (PLC-γ1), and scrambled control were from Santa Cruz Biotechnology. The primers specific for human VEGF, 5′-GTGGACATCTTCCAGGAGTA-3′ (sense) and 5′-GCGAGTCTGTGTTTTTGC-3′ (antisense), were purchased from Bioneer (Daejeon, Korea). All other chemicals were obtained from Sigma unless indicated otherwise.
Cell Culture
Human umbilical cords were obtained after full-term normal deliveries under protocols approved by the Institutional Review Board at Kangwon National University Hospital, and informed consent was obtained from all patients. The investigation conforms to the principles outlined in the Declaration of Helsinki. Human umbilical vein endothelial cells (HUVECs) were isolated and grown in M199 as described previously (17), and only passages 2–7 were used. Cells were grown in M199 media supplemented with 20% fetal bovine serum (FBS), 100 units/ml penicillin, 100 ng/ml streptomycin, 3 ng/ml basic fibroblast growth factor, and 5 units/ml heparin at 37 °C under 5% CO2/95% air.
Animals
Seven-week-old male mice (C57BL/6J, eNOS−/−, and ApoE−/−) and Sprague-Dawley rats were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained on a standard (normal) chow diet ad libitum in a laminar airflow cabinet under specific pathogen-free conditions. Some ApoE−/− mice were fed a high cholesterol diet (D12108C, Research Diet Inc., New Brunswick, NJ) for 8 weeks. Animal experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Ethics Committee of Kangwon National University. Moreover, this investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (38).
In Vitro Angiogenesis Assay
Angiogenic activity was determined by measuring cell proliferation, migration, and tube formation as described previously (18). Cell proliferation was determined by a [3H]thymidine incorporation assay. HUVECs were pretreated with various inhibitors for 30 min and stimulated with the indicated concentrations, 20 μm Rg5 or 10 ng/ml VEGF, for 30 h followed by the addition of 0.5 μCi/ml of [3H]thymidine (Amersham Biosciences) for 6 h. 3H-Labeled high molecular DNAs were determined using a liquid scintillation counter. A chemotactic migration assay was performed using Transwell plates with 6.5-mm-diameter polycarbonate filters (8-μm pore size). The lower surface of the filter was coated with 10 μg of gelatin. The fresh M199 medium (1% FBS) containing the indicated concentrations, 20 μm Rg5 or 10 ng/ml VEGF was placed in the lower wells. HUVECs (1 × 104 cells/μl) were loaded into each of the upper wells. The chamber was incubated at 37 °C for 4 h. Migrated cells were stained with H&E and quantified using a phase-contrast microscope (×100). Tube formation was determined after culturing the HUVECs on a layer of growth factor-reduced Matrigel. Briefly, HUVECs treated with the indicated concentrations, 20 μm Rg5 or 10 ng/ml VEGF were plated onto the layer of Matrigel at a density of 2 × 105 cells/well. After 20 h, tube formation was observed by an inverted phase-contrast microscope (×40) and quantified using Image-Pro Plus, version 4.5 (Media Cybernetics, San Diego).
Ex Vivo and in Vivo Angiogenesis Assay
An aortic ring sprouting assay was performed using a modified method based on a previous report (19). Sprague-Dawley (7-week-old male) were deeply anesthetized with inhaled halothane (5%) and then humanely sacrificed. Dorsal aortas were isolated and carefully cut into 1-mm rings. The aortic rings were placed in the 48-well plates precoated with 120 μl of Matrigel, sealed in place with an overlay of 50 μl of Matrigel, and incubated with Rg5 (40 μm) or VEGF (20 ng/ml) in serum-free medium. On day 8, newly formed vessels were fixed and stained with FITC-labeled isolectin B4. The assay was scored from 0 (least positive) to 5 (most positive) in a double-blinded manner. A Matrigel plug assay was performed as described previously (20). C57BL/6J mice were injected subcutaneously with 400 μl of Matrigel containing 10 units of heparin combined with either 200 nmol of Rg5 or 100 ng of VEGF under anesthesia with pentobarbital (50 mg/kg, intraperitoneal injection). After 7 days, mice were sacrificed by cervical dislocation, and Matrigel plugs were carefully removed and photographed. Hemoglobin was measured using Drabkin's reagent (Sigma-Aldrich) for quantification of blood vessel formation. Neovascularization was determined by intravital fluorescence microscopy as described previously (18). C57BL/6J mice were anesthetized by inhalation of 1.5% isoflurane and O2-N2O using a vaporizer (Surgivet, Waukesha, WI), and titanium-based imaging windows were surgically implanted between the skin and abdominal wall of t5he mice. Matrigel (100 μl) containing Rg5 or VEGF was applied to the inner space of the window, which was surgically implanted between the skin and abdominal wall of the mice. After 4 days, neovascularization was recorded by a Zeiss Axiovert 200M microscope (Carl Zeiss) after intravenous injection of 50 μl of 25 mg/ml FITC-labeled dextran (molecular mass, 250,000 Da) via the tail vein.
Mouse Model of Hind Limb Ischemia
Rg5 was delivered into the right gastrocnemius muscle of C57BL/6J mice by three injections (total, 300 μmol of Rg5/100 μl/mouse) following double ligation of the superficial femoral artery proximal to the deep femoral artery and distal femoral artery under anesthesia (100 mg/kg ketamine mixed with 2 mg/kg xylazine delivered by intraperitoneal injection). Sham-operated control animals were subjected to the same surgical protocol, but the femoral artery was not ligated. Pain caused by the surgical procedures was managed preoperatively and 1 and 2 days after surgery by intramuscular injection with buprenorphine (0.1 mg/kg). At days 7, 14, and 21 post-surgery, the mice were anesthetized with inhaled isoflurane (2%), and blood flow in both hind limbs was determined by laser-Doppler perfusion imaging (Moor Instruments, Axminster, UK). The flow ratios of the occluded/non-occluded leg were compared among experimental groups. Following sacrifice of the mice by overdose of isoflurane via inhalation, the gastrocnemius muscles were surgically removed to determine vascular density.
Aortic Vascular Tension Assay
Male C57BL/6J wild type and eNOS−/− mice were anesthetized in a closed chamber ventilated with 2.5% isoflurane for 3–5 min, and the thoracic aorta was rapidly removed. The aortas were placed in ice-cold oxygenated Krebs-Ringer bicarbonate solution and cleared of adherent connective tissues. The mouse aortas were cut into 1.5-mm rings and incubated for 30 min in DMEM containing Rg5 (20 μm) in the presence or absence of l-NAME (1 mm) or ODQ (10 μm). Some aortic rings were transfected with IGF-1R or scrambled siRNA using Lipofectamine 2000 reagent followed by incubation with Rg5. The aortic rings were suspended between two wire stirrups (150 μm) in a multiwire myograph system (DMT-USA Inc., Ann Arbor, MI) in 10 ml Krebs-Ringer buffer (95% O2, 5% CO2, pH 7.4, at 37 °C). One stirrup was connected to a three-dimensional micromanipulator, and the other was attached to a force transducer. The rings were passively stretched at 10-min intervals in increments of 200 mg to reach optimal tone (600 mg), and the response to a maximal dose of KCl was used to normalize the responses to agonist across vessel rings. Dose responses to the vasodilators acetylcholine (Ach) (10−9-10−5 m) were performed after preconstriction with U46619 (10−8 m) or phenylephrine (1 μm). At the end of the experiments, NO-dependent vasorelaxation activity was confirmed by adding the guanylyl cyclase inhibitor ODQ. Data were collected online using a MacLab system and analyzed using dose response software (AD Instruments).
Monocyte-Endothelial Cell Interaction
Monocytes (U937 cells) were labeled with 5 μm calcein-AM in RPMI 1640 containing 10% FBS at 37 °C for 1 h and washed twice with PBS by centrifugation. HUVECs were stimulated with Rg5 (20 μm) or VEGF (10 ng/ml) in 24-well plates for 8 h and then incubated with labeled monocytes (1 × 106 cells/ml) at 37 °C for 30 min. Non-adherent cells were removed by washing with RPMI 1640, and plates were photographed by fluorescence microscopy.
Endothelial Cell and Vascular Permeability Assays
[14C]Sucrose permeability in HUVECs was determined using Transwell plates (18). Confluent HUVECs in the upper compartment of Transwell plates were treated with Rg5 (40 μm) or VEGF (20 ng/ml) for 1 h. [14C]Sucrose (50 μl at 0.8 μCi/ml) was added to the upper compartment. The amount of radioactivity that diffused into the lower compartment after 30 min was determined using a liquid scintillation counter. For assaying Miles vascular permeability (18), Evans blue dye (100 μl of a 1% solution in 0.9% NaCl) was injected into the tail vein of C57BL/6J mice. After 10 min, 10 μl of Rg5 (100 μm) or VEGF (50 ng) was injected intradermally into the shaved back skin of mice. After 30 min, the animals were euthanized, and an area of skin (1 × 1 cm) that included the blue spot resulting from leakage of the dye was removed. Evans blue dye was extracted from the skin by incubation with formamide for 4 days at room temperature, and the absorbance of the extracted dye was measured at 620 nm with a spectrophotometer.
IGF-1 Binding Assay
HUVECs were cultured in 24-well plates overnight. The cells were changed to serum-free M199 and incubated for 1 h. The medium was removed, and cells were incubated with fresh serum-free medium containing 1 × 10−7-5 × 10−2 m Rg5 at 37 °C for 20 min followed by the addition of 50 μl (1 μCi) of [125I]IGF-1 and then further incubated for 10 min. The medium was decanted, and cell plates were washed twice with serum-free medium. Cells were lysed in 300 μl of 0.1 n NaOH solution containing 0.1% SDS, transferred to scintillation vials, and mixed with 1 ml of Ultima GoldTM mixture solution (PerkinElmer Life Sciences). Cell-associated [125I]IGF-1 was analyzed in a scintillation counter. The nonspecific binding was determined by co-incubation with unlabeled IGF-1 (50 nm).
Measurement of [Ca2+]i, NO, and cGMP
The changes of [Ca2+]i were monitored by a confocal microscope (Olympus FV-300) according to a previously described method (21). Briefly, cells grown on round coverslips were incubated with the Ca2+ probe Fluo-4 AM ester (2 μm) in M199 containing 1% FBS for 30 min and treated with 20 μm Rg5 in the presence or absence of several inhibitors. Then, [Ca2+]i was monitored using a confocal microscope. The levels of cellular NO production were measured in situ by using DAF-FM diacetate. Briefly, after treatment with or without 20 μm Rg5 and 1 mm l-NAME for 1 h, HUVECs were incubated with 5 μm DAF-FM diacetate for 30 min at 37 °C. After the excess probe was removed, the relative levels of intracellular NO were determined from the fluorescence intensity of DAF-FM by confocal microscopy. cGMP levels in the cell extract and vascular ring extracts were measured using a commercial ELISA kit.
Docking Simulations
Blind docking of Rg5 to IGF-1R (PDB code 1IGR) was performed using Autodock 4.2 with a Lamarckian genetic algorithm (22). A grid box of 0.2 Å spacing was set up to encompass the whole IGF-1R molecule. The number of energy evaluations was increased to 50 million to account for the large number (17) of rotatable bonds in Rg5. Chimera software was used for the graphic presentation (23).
Other Analytical Methods
Western blotting and immunoprecipitation were performed (17–19), and dimer and monomer of eNOS were separated using low temperature SDS-PAGE as described previously (24). All blots are representative of two to three independent experiments. The level of VEGF mRNA was determined by RT-PCR with primers specific for human VEGF. HUVECs were also transfected with 100 nm IGF-1R or scrambled siRNA using microporation (NanoEntek, Seoul, Korea) according to the manufacturer's protocol. For the promoter assay, HUVECs were transiently transfected with a VEGF promoter (2.6 kb)/luciferase reporter construct by a Lipofectamine method. After 24 h, cells were treated with Rg5 (20 μm) or TNF-α (10 ng/ml) for 12 h. Promoter activity was measured in cell lysates by assaying the luciferase activity.
Statistical Analysis
Data are presented as means ± S.D. of at least three independent experiments. Statistical significance was determined using Student's t test for unpaired observations between two groups or by analysis of variance with the Bonferroni correction for multiple group comparisons. p values of <0.05 were considered significant.
RESULTS
Rg5 Increases Angiogenesis in Vitro, ex Vivo, and in Vivo
When the angiogenic activities of 11 ginsenosides isolated from ginseng were examined in an EC culture system, Rg5 exhibited the strongest cell proliferation activity as compared with other ginsenosides (Fig. 1). Rg5 increased EC proliferation in a dose-dependent manner (Fig. 2A) as well as promoting chemotactic migration and tube formation (Fig, 2, B and C). The responses of ECs to 20 μm Rg5 were higher than the responses to 10 ng/ml VEGF (Fig. 2, A–C), suggesting that Rg5 stimulates the in vitro angiogenic behavior of ECs. We next examined whether Rg5 regulates angiogenesis ex vivo and in vivo. Rg5 significantly stimulated vessel sprouting in the cut edge of ex vivo explanted rat aortic rings, as compared with control (Fig. 2D). The Matrigel plug assay in mice was performed to evaluate the in vivo angiogenic activity of Rg5. Matrigel plugs containing Rg5 appeared dark red in color in the vessels, with increased hemoglobin inside the plugs, as compared with control plugs, indicating that Rg5 promotes extensive neovascularization (Fig, 2E). Moreover, intravital microscopic analysis showed that Rg5 increased angiogenic characteristics, such as capillary sprouting and bud formation, in Matrigel implanted under the abdominal skin of mice, which was not observed in control mice (Fig. 2F). These results indicate that Rg5 is capable of promoting neovessel formation.
FIGURE 1.
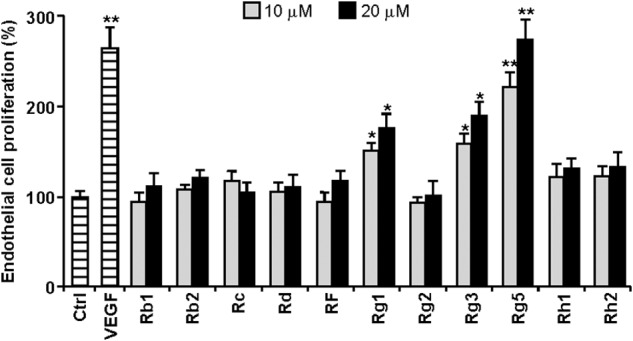
Effects of ginsenosides on HUVEC proliferation. Cells were treated with 10 ng/ml VEGF or with each ginsenoside at 10 and 20 μm for 30 h followed by the addition of 0.5 μCi/ml of [3H]thymidine for 6 h. 3H-Labeled high molecular DNAs were determined by a liquid scintillation counter (n = 3). *, p < 0.05, and **, p < 0.01 versus control.
FIGURE 2.

Rg5 induces angiogenesis in vitro, ex vivo, and in vivo. A, HUVECs were treated with the indicated concentrations of Rg5 or with 10 ng/ml VEGF for 30 h, and cell proliferation was determined by a [3H]thymidine incorporation assay (n = 3). B and C, HUVECs were treated with 20 μm Rg5 or 10 μg/ml VEGF, and chemotactic motility (B) and tube formation (C) were determined. D, rat aortic segments were treated with Rg5 (40 μm) or VEGF (20 ng/ml) for 8 days and stained with FITC-conjugated isolectin B4. Sprouting activity was assessed (n = 8 aortic rings/group, with two aortic rings prepared from each mouse). E, mice were injected with 400 μl of Matrigel containing 10 units of heparin combined with either 200 nmol of Rg5 or 100 ng of VEGF. After 7 days, Matrigel plugs were excised and photographed. Hemoglobin levels were determined in Matrigel plugs (n = 4). F, Matrigel (100 μl) containing 200 nmol of Rg5 or 100 ng of VEGF was put into abdominal windows that were surgically implanted in mice. After 4 days, neovascularization was recorded by intravital fluorescence microscopy, and neovessel formation was quantitated (n = 5). *, p < 0.05, and **, p < 0.01 versus control.
Rg5 Does Not Induce Vascular Inflammation and Permeability
The angiogenic activity of some ginsenosides may be the result of either direct action on ECs or induction of pro-angiogenic genes, including VEGF (15, 25). We determined whether Rg5 regulates VEGF expression in ECs. Rg5 did not increase the mRNA level and promoter activity of VEGF, whereas TNF-α, a known inducer of VEGF, markedly increased its transcript and promoter activity (Fig. 3, A and B). To reconfirm these results, we examined whether a VEGF-neutralizing antibody inhibits the angiogenic activity of Rg5. Treatment with an antibody for VEGF did not affect Rg5-induced EC proliferation, but it effectively reduced VEGF-induced cell proliferation (Fig. 3C). We next examined the effects of Rg5 on the expression of adhesion molecules (ICAM-1 and VCAM-1) and VE-cadherin phosphorylation-mediated disruption of adherens junction, which are known adverse effects induced during angiogenic therapy using VEGF (25). Rg5 did not affect ICAM-1 and VCAM-1 expression in ECs and adhesion of monocytes to ECs, whereas VEGF promoted these vascular inflammatory events (Fig, 3, D and E). Moreover, Rg5 treatment did not affect VE-cadherin phosphorylation, endothelial adherens junction, and transendothelial permeability in HUVEC monolayer cultures, whereas VEGF significantly promoted these adverse events (Fig. 3, F–H). Moreover, Rg5 did not elevate vascular permeability in a mouse model as compared with VEGF (Fig. 3I). These results suggest that Rg5 directly stimulates angiogenesis without increasing VEGF expression, vascular inflammation, and vascular permeability.
FIGURE 3.

Rg5 does not induce VEGF expression and vascular inflammation and permeability. A, HUVECs were stimulated with Rg5 (20 μm) or TNF-α (10 ng/ml) for 8 h, and VEGF expression was determined by RT-PCR. B, VEGF promoter activity was assessed following treatment with Rg5 (20 μm) or TNF-α (10 ng/ml) (n = 3). C, HUVECs were incubated with Rg5 (20 μm) or VEGF (10 ng/ml) in the presence or absence of a neutralizing VEGF antibody (α-VEGF, 1 mg/ml) for 30 h. Cell proliferation was determined by a [3H]thymidine incorporation assay (n = 3). D, HUVECs were treated with Rg5 (20 μm) or VEGF (10 ng/ml) for 10 h, and expression levels of ICAM-1 and VCAM-1 were determined by immunoblotting. E, HUVECs pretreated with Rg5 (20 μm) or VEGF (10 ng/ml) for 8 h were co-cultured with fluorescence-labeled monocytic U937 cells. Adherent monocytes were photographed by fluorescence microscopy. F, HUVECs were treated with Rg5 (20 μm) or VEGF (10 ng/ml) for 40 min. Tyrosine phosphorylation of VE-cadherin was determined by Western blotting. G, HUVECs were treated with Rg5 (20 μm) or VEGF (10 ng/ml) for 1 h and immunostained with a VE-cadherin antibody. H, HUVECs were treated with Rg5 (40 μm) or VEGF (20 ng/ml) in Transwell plates for 1 h. Endothelial permeability of [14C]sucrose was determined by a liquid scintillation counter (n = 3). I, Miles vascular permeability was performed in a mouse model following intradermal injection with 10 μl of Rg5 (100 μm) or VEGF (50 ng), and extravasated Evans blue dye was determined in mouse skin by spectrophotometry (n = 3). **, p < 0.01 versus control.
Rg5 Activates Multiple Angiogenic Signaling Pathways
Various angiogenic factors induce multiple signal pathways responsible for the survival, proliferation, migration, and tube formation of ECs (8, 25). We investigated whether Rg5 regulates the activation of angiogenic signal pathways. Treatment with Rg5 increased phosphorylation-dependent activation of ERK, Akt, eNOS, Src, FAK, and paxillin in a time-dependent manner (Fig. 4A). Activation of ERK, Akt, and Src was observed at 10 min; however, eNOS, FAK, and paxillin phosphorylation occurred 20 min after Rg5 stimulation. Because phosphorylated eNOS increases NO production, which is involved in angiogenesis (2, 3, 25), we examined the effect of Rg5 on eNOS-dependent NO production. Treatment with Rg5 promoted an increase in NO production, the effect of which was abrogated by co-treatment with the NOS inhibitor l-NAME (Fig. 4B). We next examined the functional involvement of these signal mediators in Rg5-induced angiogenesis using several inhibitors. Rg5-mediated activation of ERK, Akt, and FAK was suppressed by the inhibitors of MEK (PD98059), PI3K (LY294002), and Src (PP2), respectively (Fig. 4C). Similarly, proliferation of ECs by Rg5 was inhibited by PD98059, LY294002, and l-NAME, and the endothelial cell migration was suppressed by LY294002, PP2, and l-NAME. In addition, Rg5-mediated tube formation of ECs was significantly decreased by all of these inhibitor (Fig. 4D). These results suggest that Rg5 promotes angiogenesis by activating multiple signaling pathways, such as PI3K/Akt/eNOS, MEK/ERK, and Src/FAK/paxillin.
FIGURE 4.
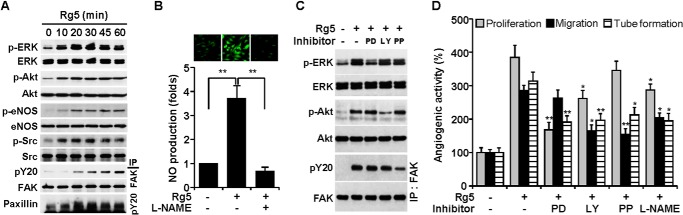
Rg5 induces angiogenesis through the activation of the multiple angiogenic signaling pathways. A, HUVECs were treated with Rg5 (20 μm) for the indicated time periods. Phosphorylation of angiogenic signal mediators was determined by immunoblotting. B, HUVECs were treated with Rg5 (20 μm) or in combination with l-NAME (1 mm) for 1 h. The intracellular levels of NO were assessed by confocal microscopy using DAF-FM and evaluated quantitatively (n = 3). **, p < 0.01. C, HUVECs were treated with Rg5 (20 μm) in the presence or absence of PD98059 (PD) (25 μm), LY294002 (LY) (20 μm), and PP2 (PP) (10 μm) for 30 min, and phosphorylation of target proteins was determined by immunoblotting. D, HUVECs were treated with Rg5 (20 μm) in the presence or absence of the indicated inhibitors, and cell proliferation, migration, and tube formation were determined (n = 3). *, p < 0.05, and **, p < 0.01 versus Rg5 alone.
Rg5-induced Angiogenesis Requires Gi Protein, PLC, and Ca2+ Mobilization
Some ginsenosides promote angiogenesis via activation of the glucocorticoid receptor or Gi protein-mediated signaling (15, 26). We determined whether these mediators were involved in Rg5-induced angiogenesis. Rg5-induced activation of ERK and FAK (but not Akt and eNOS) was blocked by treatment with the Gi protein inhibitor PTX but not by the glucocorticoid receptor inhibitor RU486 (Fig. 5A). Activation of the Gi protein complex stimulates PLC activation and Ca2+ mobilization, resulting in promotion of cell growth and migration (27, 28). We next examined whether Rg5 regulates the Gi protein-mediated PLC/Ca2+ pathway. Stimulation of ECs with Rg5 resulted in a significant increase in [Ca2+]i, which was inhibited by PTX, siRNA-mediated PLC-γ1 knockdown, and the Ca2+ chelator BAPTA-AM (Fig. 5B). In addition, Rg5-induced ERK activation was inhibited by the PLC inhibitor U73122, but not by PP2 and BAPTA-AM. However, all of these inhibitors suppressed Rg5-induced activation of FAK, but not Akt (Fig. 5C). Because [Ca2+]i plays an important role in the active dimer formation of eNOS, we assessed the effects of Rg5 on eNOS dimerization and NO production. Rg5 increased the eNOS dimer/monomer ratio and intracellular NO level, which were suppressed by PTX, U73122, and BAPTA-AM (Fig. 5D). Furthermore, these inhibitors and PLC-γ1 knockdown significantly suppressed proliferation, migration, and tube formation of ECs stimulated with Rg5, with the exception of the non-inhibitory effect of PP2 on Rg5-mediated EC proliferation (Fig. 5E). These results suggest that Rg5 activates the Gi protein-dependent and -independent pathways, which are likely responsible for Rg5-induced angiogenesis.
FIGURE 5.

Rg5-induced angiogenesis requires Gi protein, PLC, and Ca2+ mobilization. A–E, HUVECs were treated with Rg5 (20 μm) in the presence or absence of RU486 (RU) (10 μm), pertussis toxin (PT) (100 ng/ml), U73122 (U7) (5 μm), or BAPTA-AM (BA) (10 μm). Endothelial cells were also transfected with scramble (SCR) or PLC-γ1 siRNA (siPLC) (100 nm). A, after 30 min, phosphorylation of angiogenic signal mediators was determined by immunoblotting. B, the changes in [Ca2+]i were monitored by confocal microscopy using Fluo-4 AM. Cellular Ca2+ images are representative of three independent experiments. The arrowhead indicates the application of Rg5. Immunoblotting was performed to determine PLC-1γ level in cells transfected with its siRNA. C, phosphorylation of signal mediators was determined by immunoblotting. D, eNOS dimerization was determined in HUVECs treated with Rg5 for 1 h by low temperature SDS-PAGE analysis, and intracellular NO levels were determined by confocal microscopy using DAF-FM. Numerical data are the average of three independent experiments. E, endothelial cell proliferation, migration, and tube formation were determined (n = 3). IP, immunoprecipitation. *, p < 0.05, and **, p < 0.01 versus Rg5 alone.
Rg5 Promotes Angiogenesis via IGF-1R Activation
We investigated an angiogenesis-associated membrane receptor, which can be activated by Rg5 using a human phospho-receptor tyrosine kinase array kit. Treatment of HUVECs with Rg5 resulted in a 4.3-fold increase in IGF-1R phosphorylation and non-significant increases in phosphorylation of insulin receptor and VEGF receptor-2 (Fig. 6A). However, Rg5 did not alter the phosphorylation of other receptors compared with untreated controls (Fig. 6A). We further confirmed that Rg5 elicited increases in IGF-1R and IRS-1 phosphorylation in HUVECs, which were not altered by a neutralizing human IGF-1 antibody (Fig. 6B). These data indicate that Rg5 stimulates IRS-1 phosphorylation via IGF-1R activation. To investigate the possible interaction of Rg5 with IGF-1R, a docking analysis was performed. Docking results showed that Rg5 binds strongly at two sites, A and B, with Kd values of 20 and 27 nm, respectively, to the cysteine-rich domain of IGF-1R (Fig. 6C, left). At the stronger binding site A, the hydrophobic moiety of Rg5 was anchored in a hydrophobic patch formed by Phe241, Phe251, Ile255, and Phe266, and the sugar moiety formed hydrogen bonds with Lys80, Glu242, and Glu272 (Fig. 6C, top right). At the other binding site B, only Ile282 interacted hydrophobically with Rg5, and the hydrogen-bonding network between Rg5 and the IGF-1R residues, such as Gln321, Gly322, Glu345, Asn346, and Gly349, made a major contribution to the tight binding (Fig. 6C, bottom right). We next examined whether IGF-1R is critically involved in Rg5-mediated angiogenic signal cascades using a siRNA-mediated IGF-1R knockdown method. Specific knockdown of IGF-1R suppressed Rg5-mediated activation of angiogenic signal mediators ERK, Akt, eNOS, and FAK (Fig. 6D) as well as inhibiting Rg5-induced NO production and cGMP generation (Fig. 6, E and F). In addition, IGF-1R knockdown significantly suppressed proliferation, migration, and proliferation of ECs stimulated with Rg5 (Fig. 6G). Pretreatment with Rg5 blocked the binding of radiolabeled IGF-1 to HUVECs with an IC50 value of ∼90 μmol/liter (Fig. 6H), which was greater than an IC50 value of ∼1.4 nmol/liter for unlabeled IGF-1 (data not shown). These results suggest that Rg5 promotes angiogenesis via activation of the angiogenic pathways, probably by directly binding to IGF-1R.
FIGURE 6.

Rg5-induced angiogenesis is mediated by IGF-1R activation. A, lysates of HUVECs stimulated with or without Rg5 (20 μm) for 30 min were applied to phospho-receptor tyrosine array kit. Phosphorylated receptors were determined, and the indicated boxes are compared in the lower panel. B, cells were stimulated with Rg5 (20 μm) alone or in combination with an anti-neutralizing IGF-1 antibody (α-IGF-1, 1 mg/ml), and levels of phospho-IGF-1R and IRS-1 were determined by immunoprecipitation (IP) and immunoblotting. C, in silico molecular docking analysis of Rg5 and IGF-1R was performed using Autodock 4.2 with a Lamarckian genetic algorithm. D–G, HUVECs were transfected with 100 nm scrambled (S) or IGF-1R siRNA (I) and stimulated with Rg5 (20 μm). D, after 30 min, the levels of target proteins were determined by immunoblotting. E, after 1 h, the intracellular levels of NO were determined by confocal laser microscopy using DAF-FM diacetate (n = 3). F, after 4 h, cellular cGMP levels were assessed by ELISA (n = 3). G, cell proliferation, migration, and tube formation were determined as described in the legend for Fig. 2 (n = 3). H, HUVECs were treated with 1 × 10−7-5 × 10−2 m Rg5 for 20 min followed by incubation with 125I-labeled IGF-1 for 30 min. Cell-bound radiolabeled IGF-1 was counted by a scintillation counter (n = 3). *, p < 0.05, and **, p < 0.01.
Rg5 Promotes Angiogenesis in Murine Model of Hindlimb Ischemia
We examined the angiogenic effect of Rg5 in a mouse model of experimental hind limb ischemia. Mice treated with Rg5 demonstrated significantly improved blood flow in the ischemic hind limb, compared with control animals (Fig. 7, A and B). Consistent with improved perfusion recovery, ischemic muscle from Rg5-treated mice showed higher capillary density than in control mice (Fig, 7C). These data suggest that Rg5 restores blood perfusion via neovascularization in ischemic tissues.
FIGURE 7.
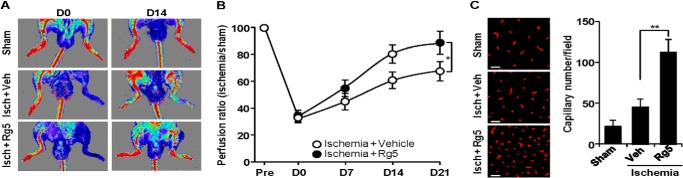
Rg5 promotes angiogenesis and local blood flow recovery after mouse hind limb ischemia. A, mice were subjected to femoral artery ligation followed by intramuscular injection with Rg5 (300 μmol of Rg5/100 μl/mouse). Representative laser-Doppler perfusion imaging of mice at days 0 and 14 after hind limb ischemia (Isch) is shown. Veh, vehicle. B, quantification of blood flow recovery was calculated from five mice/group. C, representative images show CD31-positive capillaries in transverse sections of gastrocnemius muscles from ischemic mouse hind limbs. Capillary density was quantitated from five mice/group. Scale bar: 50 μm. *, p < 0.05, and **, p < 0.01.
Rg5 Promotes NO-dependent Vasorelaxation via IGF-1R
Because eNOS-mediated NO production appears to play a critical role in vascular relaxation via cGMP production, we examined whether Rg5 regulates cGMP production in ex vivo cultured mouse aortic rings. Mouse aortic rings incubated with Rg5 increased cGMP production, which was abrogated by co-treatment with l-NAME and transfection with IGF-1R siRNA but not with scrambled siRNA (Fig. 8A). As expected, stimulation with Rg5 potentiated the vasorelaxant response to Ach as compared with control, and this response was significantly inhibited by l-NAME (Fig, 8B). Transfection of aortic rings with IGF-1R siRNA exerted a significant inhibitory effect on Rg5-mediated vasorelaxation as compared with scrambled siRNA (Fig. 8C). Interestingly, the aortic rings from ApoE−/− mice fed a high cholesterol diet showed reduced vasodilatory responses to Ach compared with those from normal diet-fed ApoE−/− mice, and this reduced vasorelaxation was significantly potentiated by treatment with Rg5 (Fig. 8D). In addition, aortic rings from wild type mice were more sensitive to the Rg5-induced vasodilatory response than those from eNOS−/− mice, and this response was significantly abrogated by l-NAME and ODQ (Fig. 8E). These data suggest that Rg5 reduces blood pressure via vasodilation.
FIGURE 8.

Rg5 promotes NO-mediated vasodilation. A, aortic rings were treated with Rg5 (20 μm) for 2 h following transfection with 100 nm scrambled (S) or IGF-1R (I) siRNA or pretreatment with l-NAME (1 mm). cGMP levels in vascular ring extracts were measured by ELISA (n = 6 aortic rings/ group, with two aortic rings prepared from each mouse). B and C, aortic rings from normal mice were pretreated with l-NAME or transfected with scrambled or IGF-1R siRNA followed by incubation with Rg5. Concentration response curves to Ach were performed (n = 8 aortic rings/group, with two aortic rings prepared from each mouse). Immunoblot was performed to determine IGF-1R level in cells transfected with its siRNA. D, aortic rings from normal diet (ND)- or high cholesterol diet (HCD)-fed ApoE−/− mice were incubated with Rg5 in the presence or absence of l-NAME. Ach-dependent vasorelaxation response was determined (n = 8 aortic rings/group, with two aortic rings prepared from each mouse). E, aortic rings from wild type and eNOS−/− mice were incubated with Rg5 (20 μm) in the presence or absence of l-NAME (1 mm) or ODQ (10 μm). Vasodilatory responses were determined (n = 6 aortic rings/group, with two aortic rings prepared from each mouse). *, p < 0.05, and **, p < 0.01. ns, no significance.
DISCUSSION
Some angiogenic inducers, including VEGF, evoke angiogenesis mainly by activating multiple signal pathways, such as MEK/ERK, Akt/eNOS, and Src/FAK, through binding their receptors (20, 25). However, other factors indirectly activate angiogenic signaling by inducing angiogenic factor expression (15, 29). In the present study, Rg5 increased angiogenesis and blood perfusion in a mouse hind limb ischemia model by activating the angiogenic signal pathways without VEGF expression. These data suggest that Rg5 is a direct angiogenic inducer. In addition, this compound promoted vasodilation by activating the eNOS/NO/cGMP pathway. However, Rg5 did not increase vascular inflammation and permeability, which are known adverse effects induced during VEGF-based angiogenic therapy. These results suggest that Rg5 promotes therapeutic angiogenesis via activation of common angiogenic signal cascades and vasodilation via eNOS-derived increases in NO production without inducing harmful side effects on vascular function.
Although some angiogenic compounds elicit the common angiogenic signal pathways of MEK/ERK, Src/FAK, and Akt/eNOS, their apical targets or receptors have not been identified as yet (25, 29). We found here that Rg5 specifically activated IGF-1R, which is associated with regulation of the EC function. Silencing of IGF-1R inhibited Rg5-induced angiogenic events and signaling pathways, indicating that Rg5 promotes angiogenesis by directly binding to IGR-1R. This possibility was confirmed by in silico docking analysis that showed significant interactions of Rg5 with the cysteine-rich domain of IGF-1R. Indeed, we performed 1000 blind docking simulations from which 11 conformations were obtained with a Kd value less than 100 nm and found that all of these were docked at two sites with Kd values of 20 and 27 nm. The higher affinity binding site “A” is exactly the same as the interaction for the C-domain of IGF. This site of interaction occurs with the cysteine-rich domain of IGF-1R at the cluster of residues Phe241, Phe251, and Phe266, which has been demonstrated using monoclonal antibodies and site-directed mutagenesis (30). This information provides insights into the future design of IGF-1R agonists for drug discovery. Indeed, we found that Rg5 bound to IGF-1R with an IC50 value of ∼90 and its angiogenic activity was inhibited by IGF-1R knockdown. Although Rg5 can inhibit interaction of LPS to TLR4 in macrophages and suppressed NF-κB-dependent inflammatory gene expression (16), this possibility is not likely linked to its pro-angiogenic activity in endothelial cells. As the purity of Rg5 used in this study was >98%, we could not completely rule out the possibility of a contaminant in Rg5 preparation. However, we found that Rg5 of the 11 ginsenosides that are most often found in steam-processed ginseng showed the highest angiogenic activity. These data suggest that Rg5, rather than a contaminant, is the major natural product responsible for Rg5-induced angiogenesis. These results also suggest that Rg5 can bind directly to IGF-1R at the binding site of IGF and trigger the IGF-1R-mediated angiogenic signal pathway.
Although IGF-1R is known as a receptor tyrosine kinase, its biological activity also requires PTX-sensitive Gi protein (28, 32), indicating that this receptor evokes dual signaling mechanisms, such as receptor tyrosine kinase and Gi-mediated PLC pathways. Signal cascades induced by IGF-1 mediate many cellular responses, including proliferation, differentiation, and angiogenesis (8, 33), and are also involved in tumor angiogenesis and developmental neovascularization in human and zebrafish (12, 13, 34). A recent study demonstrated that IGF-1R activation by IGF-1 promotes differentiation of stem cells into mesoderm and endothelial progenitor cells into mature ECs, indicating IGF-1 as a potential drug for enabling vascular regeneration (12). Moreover, endothelium-targeted IGF-1R transgenic mice enhance endothelial remodeling and vasorelaxation (13), suggesting that an activator of IGF-1R may be a useful therapeutic strategy to treat vascular repair and hypertension disorders. In this study, we found that Rg5 activated common angiogenic signal pathways via IGF-1R. These signal mediators were independently blocked by their own upstream inhibitors of PI3K, MEK, and Src, respectively, without cross-inhibition of each other. Such data indicate that these signal pathways are independently activated following the activation of IGF-1R by Rg5. Moreover, Rg5 promoted Ca2+-dependent eNOS dimerization and NO production, which were mediated by the Gi-mediated PLC pathway. These data are consistent with previous results in which IGF-1R activated Gi-mediated PLC activation and Ca2+ mobilization (27, 28). Inhibitors of these signal mediators suppressed Rg5-mediated FAK activation and angiogenesis, suggesting that Gi protein-mediated elevation of [Ca2+]i is responsible for FAK phosphorylation (35). Thus, Rg5 directly promotes in vivo angiogenesis by activating both receptor tyrosine kinase and Gi-mediated PLC pathways via IGF-1R.
In the vasculature, NO is produced predominantly by constitutive, Ca2+-dependent eNOS expressed in ECs and acts as a key determinant of vascular relaxation and angiogenesis (2, 7). The functional involvement of EC-derived NO in angiogenesis is strongly evidenced from results showing that eNOS mediates the angiogenic activity of VEGF (3) and from the observation of reduced angiogenesis in eNOS−/− mice (2). On the other hand, NO released by the endothelium diffuses into vascular smooth muscle cells and causes vasodilatation by producing cGMP via activation of soluble guanylyl cyclase. In fact, eNOS−/− mice are hypertensive and lack NO-mediated, endothelium-dependent vasorelaxation (7). Our previous studies show that natural compounds elevate eNOS-mediated NO production and promote angiogenesis and vasorelaxation (17, 25). In addition, several studies effectively demonstrate that the IFG-1R/IGF-1 axis activates the PI3K/Akt/eNOS pathway in endothelial cells and promotes angiogenesis in human and animal development and endothelium-dependent vasorelaxation (9–13, 34, 36, 37,). Based on this evidence and our present data, we concluded that the responses of ECs to Rg5 are directly associated with IGR-1R activation. Additionally, l-NAME or eNOS deficiency suppressed the ability of Rg5 to increase NO production and cGMP synthesis or to induce angiogenesis and vasorelaxation, suggesting that eNOS-derived NO is an important axis for Rg5 for exertion of its vascular effects. We also found that the relaxation-response curves for Rg5 are significantly shifted to the left in the vascular rings from ApoE−/− mice fed a high fat diet, which decreased the eNOS activity (31). This indicates that Rg5 improves vascular endothelial function in cardiovascular diseases associated with reduced eNOS-mediated NO production. Based on the current data, we concluded that Rg5 regulates vascular homeostasis by activating the eNOS/NO pathway via IGF-1R activation.
The regulation of eNOS activity involves a range of posttranscriptional mechanisms. Among them, [Ca2+]i-dependent eNOS dimerization and Akt-mediated eNOS phosphorylation at Ser1177 play a predominant role (4, 6). In line with these reports, our data show that Rg5 phosphorylates eNOS at Ser1177 via IGF-1R-dependent activation of the PI3K/Akt pathway, as well as enhancing eNOS dimerization via IGF-1R-mediated increases in Ca2+ mobilization, resulting in elevated NO production. Both mechanisms of action are directly linked to the promotion of angiogenesis and vasorelaxation. These findings suggest that Rg5 increases eNOS-derived NO production via two distinct posttranslational modifications following direct activation of IGF-1R, specifically Akt-dependent eNOS phosphorylation and Ca2+-mediated eNOS dimerization.
Although IGF-1 was considered a pathogenic mediator of vascular disease in native arteries, increasing evidence indicates that the IGF-1/IGF-1R system protects against endothelial dysfunction, atherosclerotic development, and ischemic myocardial damage (31). Some of these effects are related to the induction of eNOS-derived NO production and the activation of multiple signal pathways. Because IGF-1 circulates almost entirely (>99%) bound to six IGF-1-binding proteins (IGFBP-1–6), the therapeutic effectiveness of exogenous IGF-1 is very limited. Thus, chemical IGF-1R agonists, which do not bind to IGFBPs, need to be developed for therapeutic propose. We found that Rg5 did not elicit any different angiogenic activity in serum-free versus 20% serum-supplemented media (data not shown). These data suggest that the angiogenic ability of Rg5 is not affected by serum components, including IGFBPs. Thus, this compound could be useful for treating vascular disease.
In summary, this work reports for the first time that Rg5 promotes neovascularization, local blood perfusion, and vasorelaxation via direct activation of IGF-1R, suggesting that Rg5 is a novel natural agonist IGF-1R. The present findings also offer a mechanistic explanation of the beneficial effect of Rg5 as a non-biological compound on neovascularization and endothelial function under pathological conditions, namely ischemic vascular diseases and hypertension. Our findings provide a rationale for novel therapeutic approaches, without affecting vascular inflammation and permeability, utilizing Rg5 for cardiovascular diseases caused by endothelial dysfunction such as ischemia and hypertension.
Acknowledgment
We thank Dr. Elaine Por for helpful comments and critical reading of this manuscript.
This work was supported by Grants NRF-2011-0028790 and NRF-2013M3A9B6046563 from the National Research Foundation of Korea, funded by the Korean government (Ministry of Science, ICT and Future Planning), and a research grant from Kangwon National University.
- EC
- endothelial cell
- Ach
- acetylcholine
- BAPTA-AM
- 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)
- DAF-FM
- 4-amino-5-methylamino-2′,7′-difluorofluorescein
- eNOS
- endothelial nitric-oxide synthase
- Fluo-4 AM
- fluo-4-acetoxymethyl
- IGF
- insulin-like growth factor
- IGF-1R
- insulin-like growth fastor-1 receptor
- IGFBP
- insulin-like growth factor-binding protein
- HUVEC
- human umbilical vein endothelial cell
- l-NAME
- NG-nitro-l-arginine methyl ester
- ODQ
- 1H-[1,2,4]oxadiazole[4,3-a]quinoxalin-1-one
- PLC
- phospholipase C
- PTX
- pertussis toxin
- VE
- vascular endothelial.
REFERENCES
- 1. Folkman J. (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1, 27–31 [DOI] [PubMed] [Google Scholar]
- 2. Fukumura D., Gohongi T., Kadambi A., Izumi Y., Ang J., Yun C. O., Buerk D. G., Huang P. L., Jain R. K. (2001) Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. U.S.A. 98, 2604–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Papapetropoulos A., García-Cardeña G., Madri J. A., Sessa W. C. (1997) Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Invest. 100, 3131–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Isner J. M., Asahara T. (1999) Angiogenesis and vasculogenesis as therapeutic strategies for postnatal neovascularization. J. Clin. Invest. 103, 1231–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fulton D., Gratton J. P., McCabe T. J., Fontana J., Fujio Y., Walsh K., Franke T. F., Papapetropoulos A., Sessa W. C. (1999) Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399, 597–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hellermann G. R., Solomonson L. P. (1997) Calmodulin promotes dimerization of the oxygenase domain of human endothelial nitric-oxide synthase. J. Biol. Chem. 272, 12030–12034 [DOI] [PubMed] [Google Scholar]
- 7. Huang P. L., Huang Z., Mashimo H., Bloch K. D., Moskowitz M. A., Bevan J. A., Fishman M. C. (1995) Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 377, 239–242 [DOI] [PubMed] [Google Scholar]
- 8. Deleted in proof.
- 9. Grant M. B., Mames R. N., Fitzgerald C., Ellis E. A., Caballero S., Chegini N., Guy J. (1993) Insulin-like growth factor 1 as and angiogenic agent: in vivo and in vitro studies. Ann. N.Y. Acad. Sci. 692, 230–242 [DOI] [PubMed] [Google Scholar]
- 10. Han R. N., Post M., Tanswell A. K., Lye S. J. (2003) Insulin-like growth factor-I receptor-mediated vasculogenesis/angiogenesis in human lung development. Am. J. Respir. Cell Mol. Biol. 28, 159–169 [DOI] [PubMed] [Google Scholar]
- 11. Hartnett L., Glynn C., Nolan C. M., Grealy M., Byrnes L. (2010) Insulin-like growth factor-2 regulates early neural and cardiovascular system development in zebrafish embryos. Int. J. Dev. Biol. 54, 573–583 [DOI] [PubMed] [Google Scholar]
- 12. Piecewicz S. M., Pandey A., Roy B., Xiang S. H., Zetter B. R., Sengupta S. (2012) Insulin-like growth factors promote vasculogenesis in embryonic stem cells. PLoS One 7, e32191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Imrie H., Viswambharan H., Sukumar P., Abbas A., Cubbon R. M., Yuldasheva N. (2012) Novel role of the IGF-1 receptor in endothelial function and repair: studies in endothelium-targeted IGF-1 receptor transgenic mice. Diabetes 61, 2359–2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sengupta S., Toh S. A., Sellers L. A., Skepper J. N., Koolwijk P., Leung H. W., Yeung H. W., Wong R. N., Sasisekharan R., Fan T. P. (2004) Modulating angiogenesis: the yin and the yang in ginseng. Circulation 110, 1219–1225 [DOI] [PubMed] [Google Scholar]
- 15. Cheung L. W., Leung K. W., Wong C. K., Wong R. N., Wong A. S. (2011) Ginsenoside-Rg1 induces angiogenesis via non-genomic crosstalk of glucocorticoid receptor and fibroblast growth factor receptor-1. Cardiovasc. Res. 89, 419–425 [DOI] [PubMed] [Google Scholar]
- 16. Kim T. W., Joh E. H., Kim B., Kim D. H. (2012) Ginsenoside Rg5 ameliorates lung inflammation in mice by inhibiting the binding of LPS to toll-like receptor-4 on macrophages. Int. Immunopharmacol. 12, 110–116 [DOI] [PubMed] [Google Scholar]
- 17. Chung B. H., Kim S., Kim J. D., Lee J. J., Baek Y. Y., Jeoung D., Lee H., Choe J., Ha K. S., Won M. H., Kwon Y. G., Kim Y. M. (2012) Syringaresinol causes vasorelaxation by elevating nitric oxide production through the phosphorylation and dimerization of endothelial nitric oxide synthase. Exp. Mol. Med. 44, 191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baek Y. Y., Cho D. H., Choe J., Lee H., Jeoung D., Ha K. S., Won M. H., Kwon Y. G., Kim Y. M. (2012) Extracellular taurine induces angiogenesis by activating ERK-, Akt-, and FAK-dependent signal pathways. Eur. J. Pharmacol. 674, 188–199 [DOI] [PubMed] [Google Scholar]
- 19. Kim C. K., Choi Y. K., Lee H., Ha K. S., Won M. H., Kwon Y. G., Kim Y. M. (2010) The farnesyltransferase inhibitor LB42708 suppresses vascular endothelial growth factor-induced angiogenesis by inhibiting Ras-dependent mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signal pathways. Mol. Pharmacol. 78, 142–150 [DOI] [PubMed] [Google Scholar]
- 20. Pyun B. J., Choi S., Lee Y., Kim T. W., Min J. K., Kim Y., Kim B. D., Kim J. H., Kim T. Y., Kim Y. M., Kwon Y. G. (2008) Capsiate, a nonpungent capsaicin-like compound, inhibits angiogenesis and vascular permeability via a direct inhibition of Src kinase activity. Cancer Res. 68, 227–235 [DOI] [PubMed] [Google Scholar]
- 21. Lee Z. W., Kweon S. M., Kim S. J., Kim J. H., Cheong C., Park Y. M., Ha K. S. (2000) The essential role of H2O2 in the regulation of intracellular Ca2+ by epidermal growth factor in rat-2 fibroblasts. Cell Signal 12, 91–98 [DOI] [PubMed] [Google Scholar]
- 22. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. (2009) Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 24. Lee K. S., Lee D. K., Jeoung D., Lee H., Choe J., Ha K. S., Won M. H., Kwon Y. G., Kim Y. M. (2012) Differential effects of substrate-analogue inhibitors on nitric oxide synthase dimerization. Biochem. Biophys. Res. Commun. 418, 49–55 [DOI] [PubMed] [Google Scholar]
- 25. Chung B. H., Lee J. J., Kim J. D., Jeoung D., Lee H., Choe J., Ha K. S., Kwon Y. G., Kim Y. M. (2010) Angiogenic activity of sesamin through the activation of multiple signal pathways. Biochem. Biophys. Res. Commun. 391, 254–260 [DOI] [PubMed] [Google Scholar]
- 26. Hwang Y. P., Jeong H. G. (2010) Ginsenoside Rb1 protects against 6-hydroxydopamine-induced oxidative stress by increasing heme oxygenase-1 expression through an estrogen receptor-related PI3K/Akt/Nrf2-dependent pathway in human dopaminergic cells. Toxicol. Appl. Pharmacol. 242, 18–28 [DOI] [PubMed] [Google Scholar]
- 27. Poiraudeau S., Lieberherr M., Kergosie N., Corvol M. T. (1997) Different mechanisms are involved in intracellular calcium increase by insulin-like growth factors 1 and 2 in articular chondrocytes: voltage-gated calcium channels, and/or phospholipase C coupled to a pertussis-sensitive G-protein. J. Cell. Biochem. 64, 414–422 [PubMed] [Google Scholar]
- 28. Lyons-Darden T., Daaka Y. (2004) Requirement for G proteins in insulin-like growth factor-I-induced growth of prostate cells. J. Mol. Endocrinol. 33, 165–173 [DOI] [PubMed] [Google Scholar]
- 29. Choi Y. K., Kim C. K., Lee H., Jeoung D., Ha K. S., Kwon Y. G., Kim K. W., Kim Y. M. (2010) Carbon monoxide promotes VEGF expression by increasing HIF-1α protein level via two distinct mechanisms, translational activation and stabilization of HIF-1α protein. J. Biol. Chem. 285, 32116–32125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Keyhanfar M., Booker G. W., Whittaker J., Wallace J. C., Forbes B. E. (2007) Precise mapping of an IGF-I-binding site on the IGF-1R. Biochem. J. 401, 269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Handa P., Tateya S., Rizzo N. O., Cheng A. M., Morgan-Stevenson V., Han C. Y., Clowes A. W., Daum G., O'Brien K. D., Schwartz M. W., Chait A., Kim F. (2011) Reduced vascular nitric oxide-cGMP signaling contributes to adipose tissue inflammation during high-fat feeding. Arterioscler. Thromb. Vasc. Biol. 31, 2827–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Perrault R., Zahradka P. (2013) Identification of novel signalling roles and targets for Gα and Gβγ downstream of the insulin-like growth factor 1 receptor in vascular smooth muscle cells. Biochem. J. 450, 209–219 [DOI] [PubMed] [Google Scholar]
- 33. Aghdam S. Y., Eming S. A., Willenborg S., Neuhaus B., Niessen C. M., Partridge L., Krieg T., Bruning J. C. (2012) Vascular endothelial insulin/IGF-1 signaling controls skin wound vascularization. Biochem. Biophys. Res. Commun. 421, 197–202 [DOI] [PubMed] [Google Scholar]
- 34. Moser C., Schachtschneider P., Lang S. A., Gaumann A., Mori A., Zimmermann J., Schlitt H. J, Geissler E. K., Stoeltzing O. (2008) Inhibition of insulin-like growth factor-I receptor (IGF-IR) using NVP-AEW541, a small molecule kinase inhibitor, reduces orthotopic pancreatic cancer growth and angiogenesis. Eur. J. Cancer 44, 1577–1586 [DOI] [PubMed] [Google Scholar]
- 35. Fan R. S., Jácamo R. O., Jiang X., Sinnett-Smith J., Rozengurt E. (2005) G protein-coupled receptor activation rapidly stimulates focal adhesion kinase phosphorylation at Ser-843: mediation by Ca2+, calmodulin, and Ca2+/calmodulin-dependent kinase II. J. Biol. Chem. 280, 24212–24220 [DOI] [PubMed] [Google Scholar]
- 36. Sukhanov S., Higashi Y., Shai S. Y., Vaughn C., Mohler J., Li Y., Song Y. H., Titterington J., Delafontaine P. (2007) IGF-1 reduces inflammatory responses, suppresses oxidative stress, and decreases atherosclerosis progression in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 27, 2684–2690 [DOI] [PubMed] [Google Scholar]
- 37. Conti E., Carrozza C., Capoluongo E., Volpe M., Crea F., Zuppi C., Andreotti F. (2004) Insulin-like growth factor-1 as a vascular protective factor. Circulation 110, 2260–2265 [DOI] [PubMed] [Google Scholar]
- 38. Committee for the Update of the Guide for the Care and Use of Laboratory Animals and the National Research Council (2011) Guide for the Care and Use of Laboratory Animals, 8th Ed., National Academies Press, Washington, D. C. [Google Scholar]