

Background: Focal adhesion kinase (FAK) is enriched at focal adhesions through its focal adhesion targeting (FAT) domain, a four-helix bundle.
Results: Mutations that facilitate or prevent opening of the first helix have profound consequences on the biochemical properties of FAK and its function in cells.
Conclusion: The ability of FAT to open and close is essential for FAK function.
Significance: This provides evidence for the functional importance of the conformational dynamics of FAT.
Keywords: Conformational Change, Focal Adhesions, Protein Kinase, Protein Structure, Tyrosine-Protein Kinase (Tyrosine Kinase)
Abstract
Focal adhesion (FA) kinase (FAK) regulates cell survival and motility by transducing signals from membrane receptors. The C-terminal FA targeting (FAT) domain of FAK fulfils multiple functions, including recruitment to FAs through paxillin binding. Phosphorylation of FAT on Tyr925 facilitates FA disassembly and connects to the MAPK pathway through Grb2 association, but requires dissociation of the first helix (H1) of the four-helix bundle of FAT. We investigated the importance of H1 opening in cells by comparing the properties of FAK molecules containing wild-type or mutated FAT with impaired or facilitated H1 openings. These mutations did not alter the activation of FAK, but selectively affected its cellular functions, including self-association, Tyr925 phosphorylation, paxillin binding, and FA targeting and turnover. Phosphorylation of Tyr861, located between the kinase and FAT domains, was also enhanced by the mutation that opened the FAT bundle. Similarly phosphorylation of Ser910 by ERK in response to bombesin was increased by FAT opening. Although FAK molecules with the mutation favoring FAT opening were poorly recruited at FAs, they efficiently restored FA turnover and cell shape in FAK-deficient cells. In contrast, the mutation preventing H1 opening markedly impaired FAK function. Our data support the biological importance of conformational dynamics of the FAT domain and its functional interactions with other parts of the molecule.
Introduction
Focal adhesion kinase (FAK)5 is a non-receptor tyrosine kinase enriched at focal adhesions (FAs) (1–3). FAK plays a major role in transducing signals downstream from integrins and other membrane receptors (4–6) and it is involved in the control of cell growth, survival, and migration (7, 8). Embryonic lethality of the FAK null mutation before midgestation underlines its biological importance (9). The overexpression of FAK observed in many tumors correlates with increased tumor invasiveness and metastases, making FAK an attractive therapeutic target (10).
Following integrin engagement by extracellular matrix proteins, FAK is recruited to FAs. The C-terminal focal adhesion targeting (FAT) domain mediates this recruitment by association with paxillin (11, 12) through two binding sites for paxillin LD-motifs, formed by helices H1/H4 and H2/H3 on opposite sides of the molecule (13–16). FAT also interacts with talin, which may contribute to recruitment of either partner to FAs (17, 18). The molecular basis of the FAK-talin interaction is not known but it seems not to require the structural integrity of FAT (17, 19).
Enrichment of FAK at FAs favors its transient dimerization, which triggers trans-autophosphorylation of Tyr397 (20). This is essential for FAK function, as shown by the embryonic lethality of mutant mice selectively lacking the autophosphorylation site (21). Phosphorylation of Tyr397, which is located in the linker region between the N-terminal FERM (4.1, ezrin, radixin, moesin) (22) and the central kinase domains, allows binding and activation of Src-family kinases (SFKs) (23). Subsequent phosphorylation of Tyr925 by SFKs creates a binding site for the SH2 domain of Grb2, linking FAK to the Ras/extracellular signal-regulated kinase (ERK) pathway (24, 25) and facilitating the release of FAK from FAs (26). Tyr925, positioned within H1 of the four-helix bundle, is not accessible for phosphorylation and Grb2 binding (19, 27, 28). This apparent contradiction suggested that the FAT domain can undergo conformational rearrangement in cells. In support of this hypothesis, H1-swapped dimeric FAT was observed in crystallographic studies, suggesting that H1 has the capacity to dissociate from the rest of the bundle (27). Opening of H1 is likely to result from a strain introduced by the Pro944-APP motif that connects H1 and H2 (27), because a similar PXP motif triggers domain swapping in p13suc1 (29–31). H1 opening has subsequently been observed in several studies (15, 32, 33). The conformational strain in this hinge region was experimentally apparent (34, 35) and relieved in the open or H1-swapped dimeric conformation of Pro944-APP (27, 35). The open state would allow Tyr925 phosphorylation and subsequent binding to Grb2. Intriguingly, despite the strain in the H1-H2 hinge region, only 0.1% FAT Y925E molecules were in the open conformation (33) and only 2.5% of total FAT were stable, presumably arm-exchanged dimers in size exclusion chromatography analysis (15). The low probability of this transition is consistent with the observation that Tyr925 in the native FAT domain is a much poorer substrate for SFK phosphorylation than the unstructured peptide mimics of the region around Tyr925 or FAT domains with destabilized cores (27, 34, 35). Moreover, by severely destabilizing the FAT bundle structure, H1 opening is also predicted to affect other FAT functions, such as paxillin binding and FAK dimerization. Thus, although these data establish that the opening of H1 occurs occasionally in vitro and hence might provide a functional switch for FAT, its biological role remains highly questionable.
Here, we investigated the biological importance of FAT dynamics using mutant forms of the isolated FAT domain and full-length FAK in which the FAT H1-H2 hinge region was modified to increase or decrease its propensity to open (Fig. 1A). Our results demonstrate that these mutations have profound consequences on specific FAK functions in vitro and in vivo. Comparative analysis of the wild-type (WT) and mutant phenotypes strongly supports that the conformational dynamics of FAT are an essential regulator for the cellular function of wt FAK at FAs.
FIGURE 1.

Mutations in the H1-H2 hinge of FAT alter its self-association. A, mutations designed to relax (R) or increase (T) the tension in the HI-H2 FAT hinge region. B, pull-down assays with immobilized GST-FAT or GST and purified FAT domain: WT-FAT (lanes 2 and 3), R-FAT (lane 4), and T-FAT (lane 5). Input (lane 1): 1 μg of soluble FAT equivalent to the total amount used in the pull-down assays. Molecular mass markers positions are indicated in kDa. C, quantification of bound FAT in three independent pull-down experiments (mean ± S.E., a.u., arbitrary units). One-way ANOVA, F2,6 = 37.7, p = 0.0004, Tukey's test **, p < 0.01; ***, p < 0.001; ns, not significant. D, representative surface plasmon resonance sensorgrams showing the binding curves of purified FAT domains to a sensor chip covalently coated with purified GST-FAT. RU, resonance units. Differential response was obtained by subtracting the signal in the blank channel from that in the experimental channel. Estimated Kd: FAT, 23 ± 8.6 μm; T-FAT, 1.3 ± 0.8 μm; R-FAT, 24 ± 5.5 μm (mean ± S.E., n = 3).
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Rabbit polyclonal antibodies were from the following sources: anti-FAK A17, Santa Cruz Biotechnology (1:2000); anti-phospho-Tyr397-FAK (Tyr(P)397-FAK), BIOSOURCE (1:2000); anti-Tyr(P)925-FAK, Cell Signaling (1:2000); and anti-Tyr(P)861-FAK or Tyr(P)576-FAK, Invitrogen (both 1:1000). Affinity-purified anti-VSV rabbit polyclonal antibodies (1:2000) were a generous gift from M. Arpin (Curie Institute, Paris). A homemade anti-VSV rabbit serum was used for FAK-paxillin immunoprecipitation. Mouse monoclonal antibodies were from the following sources: FAK 4.47, Millipore (1:1000); Fyn (1/1000), Transduction Laboratories; talin, clone 8D4 (1:1000), and anti-VSV, Sigma (1:1000); paxillin, Zymed Laboratories Inc. (1:1000). All other reagents were from Sigma unless otherwise noted.
DNA Constructs and Mutagenesis
The N-terminal tagging of full-length rat FAK (AF 020777) by VSV and HA was realized as follows: first a 300-nucleotide fragment containing a SacII site in the 3′ untranslated region of FAK was eliminated by digestion with BamHI-SmaI, filled by T4 DNA polymerase and self-ligated. Then, a new SacII site was introduced immediately downstream from the FAK ATG start codon, without affecting the primary sequence. Synthetic phosphorylated oligonucleotides corresponding to VSV and HA epitopes flanked by semi-SacII sites were introduced in-frame with the FAK sequence in the newly created SacII site, generating the pBKCMV-VSV-FAK and pBKCMV-HA-FAK plasmids. All constructs were verified by DNA sequencing. We used rat FAK° “standard” isoform, without an additional exon (36).
Mutations in the FAT region of FAK were produced using an XhoI (nucleotide 2950)-SacI (nucleotide 3430) fragment of FAK subcloned in pBlueScript.SK+ (Stratagene) as a template, corresponding pairs of oligonucleotides designed with the desired mutation, and the QuikChange mutagenesis kit were from Stratagene. Mutations included replacement of several prolines in the hinge between the first two helices of the FAT domain (Pro944, Pro946, and Pro947) by glycine residues, producing “relaxed” mutant (R-FAK), or deletion of the two residues, producing “tense” mutant -P-PP (T-FAK). After validation of every mutation by sequencing, each mutated version of FAT (fragment XhoI (2950)-SacI (3430)) was cloned back to full-length VSV- or HA-tagged FAK. A recombinant N-terminal His6-tagged FAK (His6-FAK) was produced in a baculovirus-based expression system. The rat cDNA FAK sequence was amplified by PCR from pCMV2-FAK° using a forward primer introducing a BamHI site and a reverse primer bearing a KpnI site and cloned in the similarly digested pFastBac HT B donor plasmid (Invitrogen).
Determination of Improved FAT Structure
FAT(892–1052) was produced and crystallized as reported previously (27). Data were collected at the European Synchrotron Radiation Facility, Grenoble, France, at beamline ID14-2, using a wavelength of 0.97984 Å. However, differently from our previous FAT crystal structures, these crystals were cryo-protected in paraffin oil (Hampton Research). Data integration, molecular replacement, and structure refinement were carried out as described (37) (Table 1). Values shown in Table 1 indicate that the quality of this structure was within the expected range for a crystal structure of this resolution (see Structure Validation section of the deposited PDB file 3S9O).
TABLE 1.
Crystallographic data collection and refinement statistics for FAT
| Data collection | FAT(892–1052) |
|---|---|
| Space group | C2221 |
| Cell dimensions: a, b, c (Å) | 89.92, 223.98, 98.01 |
| Resolution (Å) | 30.43–2.60 (2.67–2.60)a |
| Rmerge (%)b | 10.7 (48.7) |
| I/σI | 11.0 (1.4) |
| Completeness (%) | 84.1 (54.0) |
| Redundancy | 3.2 (2.1) |
| Refinement | |
| Resolution (Å) | 30.43–2.6 |
| No. reflections used | 25973 |
| Rwork/Rfree (%)c,d | 23.3/28.7 |
| No. atoms: protein/solvent | 3271/36 |
| Mean B-factors (Å2): refinement/Wilson plot | 64.3/63.0 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.021 |
| Bond angles (°) | 2.008 |
| Ramachandran plot (%): most favored/outliers | 91.6/1.2 |
a Values in parentheses are for the highest-resolution shell.
b Rmerge = ΣhklΣi|Ii(hkl) − 〈I(hkl)〉|/Σhkl Σi Ii(hkl), where Ii(hkl) and 〈I(hkl)〉 are the observed individual and mean intensities of a reflection with the indices hkl, respectively; Σi is the sum over the individual measurements of a reflection with indices hkl; and Σhkl is the sum over all reflections.
c Rwork = Σh‖Foh| − | Fch‖/Σh|Foh.
d All measured reflections were included in the refinement, except the 2.1% (546 reflections) that were used to calculate Rfree.
GST Fusion Proteins, Pull-downs, and in Vitro Kinase Assays
The FAT domain of FAK (rat FAK(892–1053)) bearing the mutations in the hinge region were generated by PCR amplification from full-length mutated FAK plasmids and cloned through the BamHI site into the pGEX-6P2 expression vector (Novagen). The recombinant proteins were purified on glutathione-Sepharose 4B columns and cleaved when indicated with 3C protease (GE Healthcare) as described (27). For pull-down assays, 5 μg of GST-FAT on the beads were incubated with 1 μg of cleaved FAT. After incubation at 4 °C in 10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm EDTA on a rotator for 2 h, the complexes were washed three times with the same buffer before elution with SDS-PAGE sample buffer at 100 °C.
For in vitro phosphorylation assays, active human GST-Src (Sigma) was used at 0.1 μg/ml to phosphorylate 5 μg of GST-FAT (15 min at 30 °C) according to the manufacturer's indications. The reaction was stopped by adding SDS-PAGE sample buffer at 100 °C. Then, after separation by SDS-PAGE and transfer to nitrocellulose (GE Healthcare), the phosphorylated proteins were detected by immunoblotting with a phospho-specific anti-Tyr(P)925 antibody.
In Vitro Interactions with Full-length Proteins
To explore direct binding of mutated FAK proteins, we used recombinant His6-FAK produced in a baculovirus system. Sf9 cells were cultured in Insect-XPRESSTM medium (Cambrex BioScience). Infection was done using a His6-FAK recombinant baculovirus. Cells were harvested 48 h after infection and recombinant proteins were extracted and affinity purified under native conditions on a nickel-chelating resin (nickel-resin, ProBond system, Invitrogen) according to the manufacturer's protocol. After purification, His6-FAK-containing fractions were pooled and dialyzed for 24 h at 4 °C against the “native” buffer (50 mm NaH2PO4, pH 8, 0.5 m NaCl) and then immobilized on nickel-resin. In the binding assays, transfected cells were lysed in an immune precipitate wash buffer (38) and the lysates containing wild-type or mutated FAK were pre-cleared with nickel-resin for 1 h at 4 °C. Pre-cleared lysates (500 μl) were incubated with 1 μg of recombinant His6-FAK-immobilized on nickel beads for 2 h at 4 °C with constant rocking. Finally, samples were washed three times with the immune precipitate wash buffer and the bound proteins were eluted by boiling in a Laemmli sample buffer and analyzed by SDS-PAGE followed by immunoblotting with anti-tag antibodies. An unrelated protein, the splicing factor His6-U2AF protein (a gift from A. Maucuer, Institut du Fer à Moulin, Paris) was used in parallel as a specificity control.
Surface Plasmon Resonance
Binding experiments were performed at 25 °C in 0.01 m HEPES buffer at pH 7.4 containing 0.15 m NaCl, 3 mm EDTA, and 0.005% Surfactant P20 (GE Healthcare), using a Biacore 3000 Instrument (IFR 83 platform, Paris). GST-FAT was covalently coupled to a CM5 sensor chip (GE Healthcare) via its primary amine groups. The analytes (isolated WT FAT, T-FAT, and R-FAT) were injected at various concentrations at a flow rate of 5 μl/min.
Immunoprecipitation
Forty-eight hours after transfection, COS7 cells were homogenized in modified radioimmunoprecipitation assay buffer (1% Triton X-100 (v/v), 5 g/liter of sodium deoxycholate, 1 g/liter of SDS, 50 mm Tris, pH 7.4, 150 mm NaCl, 1 mm sodium orthovanadate) and Complete® (Boehringer) protease inhibitors as described (39). For FAK-paxillin-talin co-immunoprecipitations, cells were lysed in FAK-paxillin buffer (40) (1% (v/v) Triton X-100, 150 mm NaCl, 20 mm Tris base, 0.05% (v/v) Tween 20, 1 mm NaF, 1 mm Na3VO4, supplemented with Complete). Lysates were pre-cleared by incubation for 1–3 h at 4 °C with 100 μl of a mixture (50% each (v/v) of Sephacryl and protein A- or G-Sepharose beads (GE Healthcare) saturated with 25 g/liter of BSA). Immunoprecipitation was carried out overnight at 4 °C with 40 μl of rabbit anti-VSV serum, or 5 μl of monoclonal anti-VSV or anti-HA antibodies. The beads were washed three times with lysis buffer, resuspended in Laemmli loading buffer, heated at 100 °C for 2 min, and subjected to SDS-PAGE. Separated proteins were transferred to a nitrocellulose membrane, immunoblotted with the appropriate antibodies, and visualized either with horseradish peroxidase-conjugated anti-mouse/anti-rabbit antibodies (1:4000) and the ECL detection system (GE Healthcare) or with IR-Dye 800 CW donkey anti-mouse/anti-rabbit IgG antibodies (Rockland) and detection by infrared fluorescence with an Odyssey Li-Cor scanner.
Cell Culture and Transfection
Mouse embryonic FAK−/− (Ptk2−/−) fibroblasts (MEFs) (9) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) plus 0.001% (v/v) β-mercaptoethanol. COS7 cells were cultured in DMEM supplemented with 10% (v/v) FCS. MEFs cells were transfected with 1.3 μg of DNA per 35-mm diameter culture dish with Lipofectamine 2000 (Invitrogen), and COS7 cells were transfected with and 8 μg of DNA/100-mm diameter culture dish in the presence of polyethyleneimine as described previously (39). Total DNA quantity was kept constant with empty pcDNA3 plasmid.
Immunofluorescence Microscopy
Cells were fixed 48 h after transfection in 40 g/liter of paraformaldehyde for 10 min at room temperature, and then permeabilized by incubation for 20 min in PBS containing 0.2% Triton X-100. After saturation with 50 g/liter of BSA for 30 min at room temperature, the coverslips were incubated overnight at 4 °C with a 1:500 dilution of monoclonal anti-FAK antibody 4.47, washed in PBS containing 1 g/liter of BSA, and incubated with a 1:1000 dilution of goat anti-mouse antibody conjugated to CY3. The coverslips were washed four times with PBS and mounted in Vectashield (Vector Abcys). The cells were examined at the Institut du Fer à Moulin cell imaging facility, with a DM-6000 upright microscope equipped with a SP5 confocal laser scanning device and an argon/krypton laser (Leica Microsystems, Heidelberg, Germany). Circularity was measured with a Fiji plugin: 4 π × area/perimeter2. A value of 1.0 indicates a perfect circle; values between 1.0 and 0.0 indicate an increasingly elongated shape.
Live Cell Imaging and Fluorescence Recovery after Photobleaching (FRAP) Experiments
The 24-h transfected cells were plated at low density on 10 μg/ml of fibronectin-coated glass-bottom μ-dishes (35-mm diameter, Biovalley). Twenty-four hours later, the cells were placed in Hanks' balanced salt solution at 37 °C and imaged with a Leica DMI 4000 inverted microscope (Leica Microsystems) equipped with a confocal spinning disk head (CSU 22; Yokogawa) and a 491-nm laser (MAG Biosystems). The images were acquired with a ×63 HCX PL APO (1.4 NA) objective every 5 min on an EMCCD camera (QuantEM 512 SC, Photometrics) and analyzed using Metamorph software (Molecular Devices). Up to five different fields were sequentially recorded during each experiment using a Märzhäuser (Wetzlar) automated stage piloted by Metamorph software. Ratios of fluorescence at the FA at each time point to that of the same FA at time 0 were calculated.
In the FRAP experiments, the cells were placed in Hanks' balanced salt solution medium at 32 °C and imaged by a Leica SP5 II upright microscope (Leica Microsystems). The images were acquired with a ×40 HCX APO (0.80 NA) water immersion objective and the FRAP experiment performed with the FRAP Wizard software from Leica Microsystems. Five images where taken at low laser intensity (∼5%) before bleaching at the rate of 1 Hz for measuring the basal fluorescence intensity. Photobleaching was achieved with 100% of the 488-nm laser line with four iterations. Recovery was followed with the same laser power as in the pre-bleached session at the same rate of imaging for 60 s. For each time point, the intensity of the bleached area was normalized to the pre-bleached intensity. Data were corrected for photobleaching due to repeated imaging using parallel measurements in a different region of the cell. FRAP recovery curves and analysis were generated using Igor Pro software (WaveMatrics). To avoid possible artifacts from overexpression, only cells expressing a low but detectable amount of paxillin-GFP were chosen for further analysis.
Statistical Analyses
The data are presented as mean ± S.E. for the indicated number of points. Statistical analysis was done with GraphPad Prism 6.0. Comparisons between two sets of means data were done with Student's t test; for three sets or more with one-way ANOVA; and between crossed variables with two-way ANOVA, using Tukey's and Sidak's tests, respectively, for post hoc comparisons. Values for t, F, and p are indicated in the figure legends.
RESULTS
A Mutation Designed to Open the H1-H2 Hinge Region Increases FAT Self-association
We reasoned that if H1 opening of the FAT domain is relevant for selected FAK functions, then these functions would be enhanced by a FAT mutant with enhanced propensity to open H1, and abrogated by a FAT mutant with defective H1 opening, whereas wild-type FAK would show an intermediate phenotype. Conversely, if the conformational dynamics of FAT were irrelevant for FAK function, then WT FAK would behave like the FAK mutant with abrogated H1 opening. To produce these FAT mutants, we followed an approach previously used to manipulate the domain-opening dynamics of p13suc1 (31). To relieve the strain generated by the prolines in the H1-H2 hinge (Pro944-APP), we replaced the hinge prolines with three flexible glycine residues, generating a relaxed mutant of FAT (R-FAT, Fig. 1A), in which the closed conformation should be stabilized. Conversely, to facilitate FAT opening, we produced a tense mutant of FAT (T-FAT) by deleting the two non-proline residues in the proline-rich motif (Fig. 1A). Because H1 opening promotes FAT dimerization through H1 exchange in vitro (27, 35), we performed FAT self-association experiments to probe H1 release. WT or mutated FAT was used as bait in pull-down assays with GST-FAT (Fig. 1B). Although the 3 forms of FAT were able to bind GST-FAT, T-FAT interacted most strongly (Fig. 1, B and C). We then studied FAT-FAT interactions by surface plasmon resonance with immobilized GST-FAT on the chip (ligand). No binding of GST was detected, whereas all FAT molecules interacted with GST-FAT. As expected, the strongest interaction was observed with T-FAT (Fig. 1D). These results from two different approaches show that FAT interacts with itself and that this interaction is increased by a mutation designed to favor FAT bundle opening. At least two mechanisms could account for the FAT-FAT interactions: swapping of helix H1, as observed in some previously published crystals (27) (Fig. 2A), and binding of an N-terminal extension of one FAT molecule (residues 895–915) to the paxillin binding site on H1/H4 of another FAT molecule (27) (Fig. 2, B and C). The improved crystallographic data presented here (Table 1, Fig. 2, B and C) allowed us to ascertain that this second interaction can occur in cis (Fig. 2B) and in trans (Fig. 2C). However, deletion of residues 895–915 did not prevent the FAT-FAT interaction in pull-down or surface plasmon resonance experiments (data not shown), suggesting that the observed dimerization was due to H1 swapping, as expected from the mutant design.
FIGURE 2.
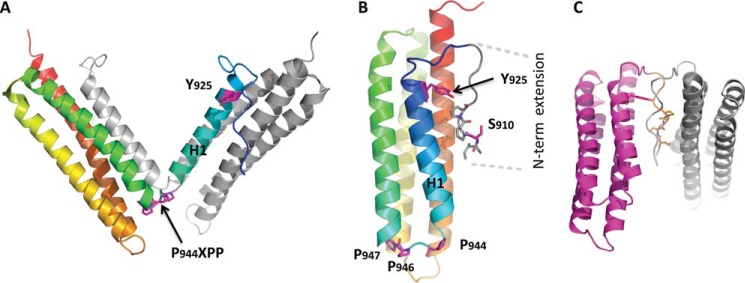
Structural analysis of FAT predicts several possible modes of dimerization. A, crystallographic model for the H1-swapped FAT dimer (PDB accession 1K04). One protomer is shown in gray; the other one is color-ramped from the N terminus (blue) to the C terminus (red). B, interaction in cis between the N-terminal extension (residues 908–915) and FAT H1/H4 observed in the FAT(892–1052) crystal structure determined to a resolution of 2.6 Å for this study (Table 1, PDB accession 3S9O, molecule A is shown). The secondary structure representation is color-ramped as in A. Side chains of relevant residues are shown. C, crystallographic model for a potential FAT-FAT interaction via swapping of the N-terminal extensions (residues 908–915). This arrangement is also taken from PDB accession 3S9O, molecule C, and symmetry-related molecule C.
T-FAK Mutation Promotes Interaction between Full-length FAK Molecules in Vitro
To investigate whether FAT mutations could also modify the interaction between full-length FAK molecules, recombinant His6-FAK bound to nickel-resin was incubated with transfected COS7 cells lysates containing similar levels of VSV-tagged WT FAK, R-FAK, or T-FAK (Fig. 3A). Retained proteins were detected by immunoblot with anti-VSV antibodies (Fig. 3B). Binding of WT FAK and R-FAK to His6-FAK was similar, whereas binding of T-FAK was consistently stronger (Fig. 3, B and C). In the same pull-down assay, T-FAK did not bind to an unrelated His6-tagged protein on nickel-resin, demonstrating the specificity of the interaction with His6-FAK (Fig. 3B, right lane). Thus, full-length FAK can interact with itself and this interaction is facilitated by the tense mutation of the FAT H1-H2 hinge region. It should be emphasized that FAK can self-associate through several mechanisms, including FERM-FERM and FAT-FERM interactions with itself (20).
FIGURE 3.

T-FAT mutation increases full-length FAK dimerization and interaction with FAT. A–C, mutant and WT FAK proteins with N-terminal VSV tags were expressed by transfection in COS7 cells. A, immunoblotting of FAK in 20% of the amount of cell lysates used for each pull-down. Molecular mass marker positions are in kDa. B, cell lysates were loaded onto Ni2+ beads coated with recombinant His6-FAK (left lanes) or a His6-tagged 65-kDa unrelated protein (right lane). After extensive washing, bound proteins were eluted and the presence of VSV-tagged protein was assessed by immunoblotting. Ponceau red staining of the membrane shows equivalent amounts of His6-FAK bait. C, quantification of the pull-down experiment performed in triplicate (mean ± S.E., au, arbitrary units). One-way ANOVA, F(2,6) = 9.8, p = 0.013, Tukey's test; *, p < 0.05; ns, not significant. In these experiments, full-length FAK interactions are likely to involve FERM-FERM or FAT-FERM interactions, in addition to the FAT-FAT interactions. D, COS7 cell lysates containing identical amounts of VSV-tagged mutant or WT FAK proteins were added to beads coated with GST (lanes 1–4), GST-FERM (lanes 5–8), and GST-FAT (lanes 9–12). After washing and elution, bound FAK was visualized by immunoblotting with VSV antibodies. Equivalent quantities of the three different baits used (GST, GST-FERM, and GST-FAT) were revealed by immunoblotting with a GST antibody. Input (20% of the lysate amount used in the pull-down) is shown on either side of the VSV blot. E and F, quantification of the amount of VSV-FAK retained by GST-FERM (B) and GST-FAT (C) in three independent experiments (mean ± S.E., au, arbitrary units). One-way ANOVA GST-FERM, no significant difference, GST-FAT, F(2,6) = 43, p < 0.001, Tukey's test; ***, p < 0.001; ns, not significant. The T mutation in full-length FAK did not modify its binding to GST-FERM but increased its interaction with GST-FAT. A, B, and D, molecular mass marker positions are indicated in kDa.
To distinguish FERM-FAT from FAT-FAT interactions, we carried out pull-down assays using GST alone or fused to FERM or FAT. Equivalent quantities of GST-proteins bound to glutathione-Sepharose beads were used as “bait” and loaded with calibrated amounts of cell lysates from transfected COS7 cells expressing WT or mutant VSV-FAK (Fig. 3D). No binding to GST alone was observed (Fig. 3D, lanes 1–4). In contrast, FAK-FERM (Fig. 3, D, lanes 6–8, and E) and FAK-FAT (Fig. 3, D, lanes 10–12, and F) interactions were observed. The T-FAK mutation did not alter the interaction with GST-FERM, whereas it increased the interaction with GST-FAT (Fig. 3, D–F). Thus, the T-mutation appeared to selectively alter the FAT-FAT interactions, without interfering with the other types of FAK self-association.
Mutations of the H1-H2 Hinge Alter Phosphorylation of Tyr925 in Solution and in Transfected Cells
Currently available in vitro data suggest that H1 unfolding and dissociation from the FAT core is required for Tyr925 phosphorylation by SFKs and subsequent Grb2 SH2 binding (25, 27, 35). We examined whether mutation of the FAT H1-H2 hinge directly altered phosphorylation of Tyr925, using purified FAT domains (892–1052) as substrates for Src in vitro (Fig. 4, A and B). Tyr925 was more phosphorylated in T-FAT and slightly less in R-FAT than in WT FAT (Fig. 4, A and B), providing evidence that the T mutation increased accessibility of Tyr925 to SFKs, whereas the R mutation decreased it.
FIGURE 4.

Mutations of the FAT H1-H2 hinge alter phosphorylation of Tyr925. A, purified GST-tagged WT FAT domain (lane 1), R-FAT (lane 2, R-FAT), and T-FAT (lane 3) were incubated with Src (0.1 μg/ml) in the presence of ATP for 15 min at 30 °C. Immunoblotting was carried out with Tyr925 phospho-specific antibody (pY925, upper panel) or a FAK C-terminal antibody (FAK, lower panel). The GST-FAT position is indicated by an arrowhead. Note that phosphorylation of a breakdown product with a lower molecular weight was also affected in the same direction as full-length FAT-GST by the hinge mutations. B, quantification of three experiments for Tyr(P)925 as in A, corrected for the amount of FAK (mean ± S.E.). One-way ANOVA, F(2,6) = 105, p < 0.0001, Tukey's test; *, p < 0.05; ***, p < 0.001; ****, p < 0.0001. C, COS7 cells were transfected with WT or mutated VSV-tagged FAK and pY925 and FAK were analyzed by immunoblotting. D, quantification of results in C. One-way ANOVA, F(2,6) = 6.43, p < 0.05. E and F, same as in C and D, except that cells were treated for 16 h with 50 μm orthovanadate before lysis. One-way ANOVA, F(2,9) = 7.28, p < 0.05. G and H, same as in C and D, except that FAK was cotransfected with B-Fyn. One-way ANOVA, F(2,6) = 19.9, p = 0.002. D, F, and H, Tukey's test, *, p < 0.05; **, p < 0.01. A, C, E, and G, molecular mass markers positions are indicated in kDa. C, E, and G) the samples were run on the same gel and blot but intervening lanes were deleted as indicated by a vertical line.
We then examined the role of the FAT hinge region in modulating phosphorylation of Tyr925 in full-length FAK in transfected COS7 cells. Cells were grown for 48 h in the absence (Fig. 4, C and D) or presence of 50 μm orthovanadate (Fig. 4, E and F). Tyr925 phosphorylation was slightly decreased in R-FAT and markedly increased in T-FAK (Fig. 4, C–F). FAT mutants were also co-transfected with B-Fyn to determine their potential to serve as substrates for SFKs in intact cells (Fig. 4, G and H). T-FAK was markedly phosphorylated by B-Fyn on Tyr925, whereas R-FAK phosphorylation was less pronounced (Fig. 4, G and H). These results confirmed in intact cells the facilitation of Tyr925 phosphorylation by H1 opening in T-FAK.
T-FAK Mutation Increases Phosphorylation of FAK on Tyr861 but Not Tyr397 or Tyr576 in Transfected Cells
We then examined whether FAT mutations modified phosphorylation of other key residues in FAK in cells. Using a specific Tyr(P)397 antibody, which recognizes the FAK autophosphorylation site (23), we found no difference between mutated and WT FAK in transfected COS7 cells (Fig. 5, A and B). This indicated that the mutations did not alter Tyr397 autophosphorylation or phosphorylation by other kinases. Phosphorylation of Tyr576, a residue in the FAK activation loop phosphorylated by SFKs (41), was also unaltered by FAT hinge mutations (Fig. 5, A and C). In contrast, phosphorylation of Tyr861, a residue crucial in oncogenic cell transformation by H-Ras (42), was strongly increased in T-FAK as compared with WT or R-FAK (Fig. 5, A and C). The lack of change in Tyr397 or Tyr576 phosphorylation provided strong evidence that increased phosphorylation of Tyr925 and Tyr861 is a consequence of altered FAT dynamics and is not mediated by an increased activation of FAK autophosphorylation and/or SFK recruitment.
FIGURE 5.
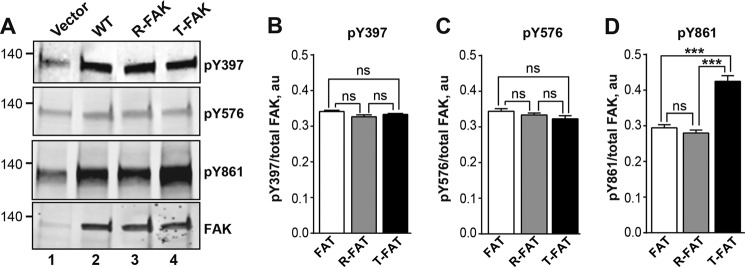
Mutation of the FAT H1-H2 hinge alters phosphorylation of Tyr861 but not Tyr397 or Tyr576. A, COS7 cells were transfected with WT FAK, R-FAK, or T-FAK. Representative blots show phosphorylation of Tyr397, Tyr576, and Tyr861 using phospho-specific antibodies (pY397, pY576, and pY861, respectively). Levels of FAK protein were assessed with 4.47 monoclonal antibody (FAK). Molecular mass markers positions are indicated in kDa. B–D, quantification of three experiments as in A, for Tyr(P)397 (B), Tyr(P)576 (C), and Tyr(P)861 (D), corrected for the amount of FAK (mean ± S.E.). Statistical analysis, one-way ANOVA: Tyr(P)397, F(2,6) = 3.65, p = 0.09; Tyr(P)576, F(2,6) = 2.82, p = 0.14; Tyr(P)861, F(6,2) = 48.1, p = 0.0002, Tukey's post hoc test; ***, p < 0.001; ns, not significant.
FAT H1-H2 Hinge Mutations Alter Phosphorylation of Ser910 in Response to Bombesin
In addition to tyrosine residues, FAK is phosphorylated on serine residues in response to physiological stimuli (43, 44). Ser910 is located in the N-terminal extension of FAT that binds back onto the H1/H4 FAT helices in absence of paxillin LD motifs (Fig. 2B). A previous study demonstrated that when acting on G protein-coupled receptors, bombesin stimulates FAK Ser910 phosphorylation by ERK (45). To investigate the influence of FAT hinge mutations on Ser910 phosphorylation, COS7 cells transfected with WT or mutant HA-FAK were serum deprived for 30 min and incubated with 10 nm bombesin for another 30 min. In response to bombesin, Ser910 phosphorylation was stimulated in endogenous and transfected FAK (Fig. 6A, upper panel). In T-FAK, basal phosphorylation of Ser910 was increased and the effect of bombesin was strongly enhanced as compared with WT FAK (Fig. 6, A and B). In contrast, bombesin had no effect on Ser910 in R-FAK. We verified in the same samples that bombesin also increased ERK phosphorylation and that this effect was unaltered by the presence or absence of WT or mutated FAK (Fig. 6A, bottom panels). These results demonstrate that opening of the FAT domain facilitates the phosphorylation of Ser910 by ERK in cells.
FIGURE 6.
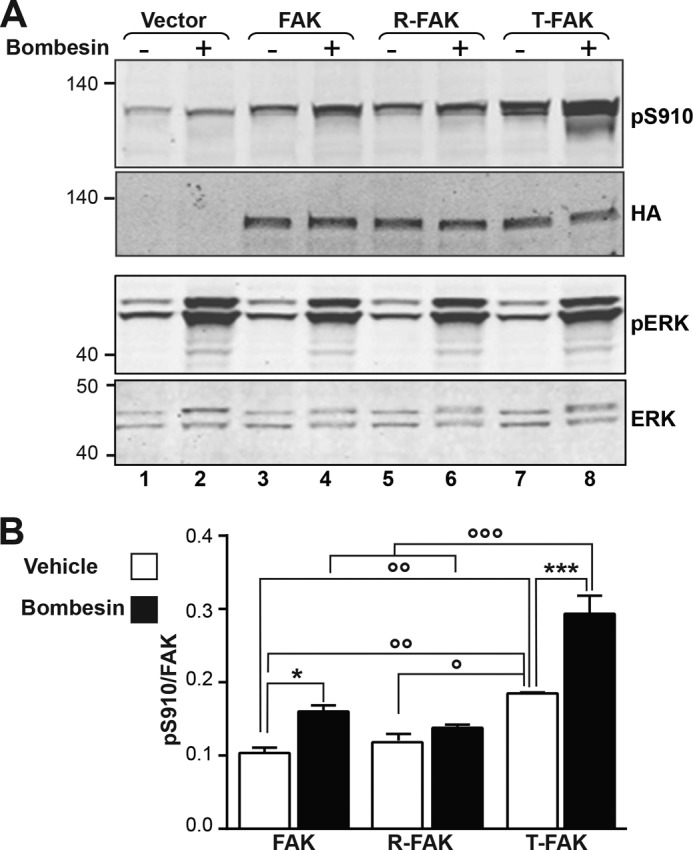
Effects of the FAT hinge mutation on Ser910 phosphorylation. A, COS7 cells transfected with HA-tagged WT FAK, R-FAK, or T-FAK were serum deprived for 30 min and then treated for another 30 min with 10 nm bombesin before cell lysis. Phosphorylation of Ser910 and levels of transfected FAK expression were analyzed by immunoblotting with phospho-specific Ser(P)910 (pS910) and HA antibodies (upper panels). In parallel, diphospho-44/42 ERK (pERK) and total ERK were monitored with the corresponding antibodies (lower panels). Molecular mass marker positions are indicated in kDa. B, quantification of three experiments for Ser(P)910 as in A, corrected for the amount of FAK (mean ± S.E.). Two-way ANOVA, interaction; F(2,8) = 5.96, p = 0.026, treatment; F(1,4) = 80.9, p = 0.0008, mutation; F(2,8) = 44.7, p < 0.0001. Sidak's multiple comparisons test, asterisks; ± bombesin, ○; comparisons between FAK constructs, 1 symbol, p < 0.05; 2 symbols, p < 0.01; 3 symbols p < 0.001.
FAT Hinge Mutations Alter FAK Interactions with Paxillin but Not with Talin in Cells
The FAT domain contains binding sites for the focal adhesion proteins paxillin and talin, which both participate in the localization of FAK (11, 12, 17, 46). H1 opening and concomitant structural rearrangement of FAT is expected to disrupt the H1/H4 binding site for paxillin LD motifs and compromise the site on H2/H3 (16, 32, 33). Talin does not require the structural integrity of the four-helix bundle to bind (14, 15, 17, 19). We investigated the association of mutated FAKs with endogenous paxillin and talin by co-immunoprecipitation. COS7 cells were transfected with WT or mutated VSV-FAK, alone or in combination with B-Fyn. After 48 h, cells were lysed and the phosphorylation of Tyr925 was analyzed by immunoblotting with a phospho-specific antibody (Fig. 7A, upper panel). Transfected FAK proteins were immunoprecipitated with an anti-VSV serum and precipitates were immunoblotted with FAK, paxillin, or talin antibodies (Fig. 7A, lower panels). An increase in paxillin binding was observed for R-FAK as compared with WT FAK (Fig. 7, A, lanes 2 and 3, and B), in the absence of cotransfection with B-Fyn. In contrast, less paxillin immunoreactivity was associated with T-FAK (Fig. 7, A, lane 4, and B). Interestingly, the differences in paxillin binding were amplified by B-Fyn co-expression (Fig. 7, A, lanes 5–7, and B). Fyn strongly enhanced Tyr925 phosphorylation in FAK and even more so in T-FAK, whereas R-FAK Tyr925 was less phosphorylated (Fig. 7A, upper panel, lanes 5–7). The association with paxillin was increased for R-FAK, whereas it was low for T-FAK (Fig. 7A, lanes 5–7). Thus, paxillin binding varied in the opposite direction of H1 opening, as expected from the total (H1/H4) and partial (H2/H3) loss of the LD binding sites. Importantly, neither H1-H2 hinge mutations nor cotransfection with Fyn altered the co-immunoprecipitation of talin with FAK (Fig. 7, A, lower panel, and C).
FIGURE 7.

Mutations of the FAT H1-H2 hinge alter interaction of FAK with paxillin but not talin. A, COS7 cells were transfected with VSV-tagged WT FAK, R-FAK, or T-FAK, without or with B-Fyn cotransfection, as indicated. Cells were lysed 24 h later and FAK immunoprecipitated with anti-VSV antibodies (IP VSV). Phosphorylated Tyr925 was determined in total cell lysates (pY925, upper panel). Expression of B-Fyn was detected by immunoblotting. Immune precipitates were probed with FAK, paxillin, and talin antibodies as indicated (lower panels). Mass markers positions are indicated in kDa. B, quantification of the co-immunoprecipitation of paxillin with FAK as in A expressed as a ratio of paxillin/FAK, a.u., arbitrary units. Statistical analysis, one-way ANOVA: F(5,18) = 18.09, p < 0.0001, Tukey's post hoc test; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. C, quantification of the co-immunoprecipitation of talin with FAK as in A expressed as a ratio of talin/FAK, a.u., arbitrary units. One-way ANOVA: F(5,18) = 0.35, p = 0.87. B and C, data are mean ± S.E.
FAT Hinge Mutations Alter Cell Shape and FAK Intracellular Localization
Given the effects of FAK H1-H2 hinge mutations on paxillin binding, we anticipated that they may also modify FAK targeting in cells. We examined the subcellular localization of these proteins by immunofluorescence in FAK knock-out (Ptk2−/−) MEFs (9) cotransfected with VSV-tagged WT or mutated FAK and with paxillin-GFP as the FA marker. Twenty-four hours after transfection, cells were fixed and immunostained for FAK (Fig. 8A). Cells co-transfected with FAK or T-FAK were generally less round than those transfected only with paxillin-GFP or co-transfected with R-FAK and paxillin-GFP, as shown by superimposing the cell contours (Fig. 8A, left panel). Quantification of cell circularity showed that it was lower in cells expressing FAK or T-FAK than in cells not expressing FAK (Fig. 8B). R-FAK had no effect on the circularity as compared with untransfected cells (Fig. 8B). We then examined the intracellular localization of the various forms of FAK. WT FAK was, as expected, enriched at FAs, and this enrichment was more pronounced for R-FAK (Fig. 8C). In contrast, T-FAK exhibited a diffuse cytoplasmic immunofluorescence with a decreased FA/cytoplasm ratio (Fig. 8, C and D). In summary, these experiments indicated that although T-FAK was less enriched at FAs than was WT FAK, it had an effect on cell shape, whereas R-FAK was highly enriched at FAs but had no effect on cell shape.
FIGURE 8.

Intracellular localization of FAT H1-H2 hinge mutants. A, FAK−/− fibroblasts were co-transfected with paxillin-GFP without (No) or with various VSV-tagged FAK constructs (WT FAK, R-FAK, or T-FAK, as indicated). FAs were visualized by GFP direct fluorescence (Pax-GFP, green) and FAK localization was determined by immunofluorescence with a FAK monoclonal antibody (4.47, anti-FAK, red). Cell contours were drawn and overlapped (left panels). Scale bars: 10 μm. B, quantification of the circularity of cell contours in A. Statistical analysis: one-way ANOVA, F(3,39) = 5.18, p = 0.004, Tukey's test versus no FAK; *, p < 0.05. C, FAK−/− cells were transfected with various FAK constructs, as indicated, and immunostained with FAK antibody. A pile of stacked confocal images is shown with color-coded FAK immunofluorescence intensity. WT and R-FAK immunoreactivity was predominantly found at FAs, whereas T-FAK immunoreactivity was very high in the cytoplasm. Scale bars: 10 μm. D, quantification of results in C. One-way ANOVA, F(2,33) = 11.73, p < 0.0001; Tukey's test, *, p < 0.05; ****, p < 0.0001. Data in B and D are mean ± S.E.
Mutations in the FAT Hinge Affect Focal Adhesion Turnover
Cell motility depends in part on the ability of FAK to facilitate FA turnover. Disassembly of FAs is correlated with the time residency of several proteins at these sites including FAK (47), paxillin (48), and talin (49). Because the rate constant for dissociation of those proteins from FAs is similar (50, 51), we analyzed FA turnover by monitoring paxillin-GFP FRAP in FAK KO MEFs co-expressing WT or mutated VSV-FAK. Although t½ was not significantly altered (Fig. 9, A and B), the mobile fraction of paxillin-GFP was increased at FAs in cells expressing WT or T-FAK, whereas R-FAK had no effect (Fig. 9C). Similarly, a point mutant of FAK in which Tyr925 was replaced by Phe (Y925F) had no effect on paxillin-GFP recovery (Fig. 9C). No change was observed in the cytoplasm away from FAs (Fig. 9D).
FIGURE 9.

FAT H1-H2 hinge mutations alter FAs turnover. A–D, FAK KO fibroblasts were co-transfected with paxillin-GFP without (−) or with various VSV-tagged FAK constructs (WT FAK, R, T, or Y925F, as indicated). FRAP was measured at FAs (A and C) and in the cytoplasm (B and D). One-way ANOVA: A, t½ at FAs, F(4,59) = 0.42, p = 0.79; B, t½ in cytoplasm, F(4,20) = 2.84, p = 0.051 FAs; C, recovery at FAs, F(4,66) = 8.17, p < 0.0001, Tukey's test, *, p < 0.05; **, p < 0.01; ****, p < 0.0001; D, recovery in cytoplasm, F(4,20) = 0.86, p = 0.50. E, FAK KO cells were co-transfected as in A and FA disassembly was analyzed by spinning disk microscopy for 60 min. The images at t0 (red) and t60 (green) were overlapped to show differences. Disassembly appears in red, newly formed FAs in green, and stable FAs in yellow. Scale bar: 10 μm. F, percentage of stable FAs determined as in C in three independent experiments with four cells per condition. One-way ANOVA, F4,56 = 8.45, p < 0.0001, Newman-Keuls post hoc test WT FAK or T-FAK versus no FAK; *, p < 0.05; ***, p < 0.001; R-FAK or Y925F versus T-FAK; ○, p < 0.05. Data in A-D, F, and G, are mean ± S.E.
To further characterize the effects of FAT mutations on the dynamic properties of FAs, FAK KO MEFs were co-transfected with VSV-FAK and paxillin-GFP and recorded for 1 h by time lapse confocal microscopy (Fig. 9E). Analysis of sequential images showed that cells lacking FAK or expressing R-FAK or Y925F-FAK displayed FAs of similar stability (Fig. 9, E and F). In contrast, expression of WT FAK decreased FA stability and this effect was more pronounced with T-FAK (Fig. 9, E and F). These results confirmed the consequences of the FAT H1-H2 hinge mutation on FA dynamics and supported the role of FAT opening in FAK function in cell motility.
DISCUSSION
Previous in vitro and modeling studies of FAT showed that FAT H1 can spontaneously dissociate from the four-helix bundle (15, 27, 32–35). This structural rearrangement allows the formation of FAT dimers in vitro through H1 swapping (27). In addition, available in vitro data suggest that partial detachment of H1 is required for Tyr925 phosphorylation by SFKs and subsequent association of Tyr(P)925 with Grb2 (27, 35). However, H1 opening in vitro is a very rare event, occurring only in 2.5% or less of the total FAT population (15). It is therefore unclear if these conformational dynamics of FAT have a biological importance. To address this question, we engineered mutations in the hinge between H1 and H2 of FAT, designed to stabilize (R) or destabilize (T) the FAT four-helix bundle, applying a strategy previously used to modulate domain-opening dynamics in p13suc1 (29–31). We reasoned that any biological effect linked to FAT dynamics would be abrogated in R-FAK and enhanced in T-FAK. Conversely, if FAT dynamics plays only a minor role in vivo, comparable with their low occurrence rate in vitro, then R-FAK would behave like WT FAK and not produce opposing effects to T-FAK.
In cells, T-FAK displayed increased Tyr925 phosphorylation but decreased paxillin binding, whereas opposite effects were observed for R-FAT. These results were in agreement with enhanced opening of the four-helix bundle in T-FAT and lost the ability to open in R-FAT. Importantly, the hinge mutations did not impair FAK expression, stability, or activation (as indicated by normal levels of phosphorylation on Tyr397 and Tyr576), showing their specific consequences on FAT functions. The T mutation also increased the ability of FAT to self-associate in vitro. Several potential mechanisms could account for FAT-FAT interactions, including H1-swapping and binding of a flexible FAT extension (residues 895–915) to FAT H1-H4 (27) (see Fig. 2). Deletion of residues 895–915 did not prevent the FAT-FAT interaction, suggesting that interactions of isolated FAT domains result from H1 swapping, although we cannot formally exclude other modes of interaction. We have recently observed that FAK can dimerize through a combination of FERM-FERM and FERM-FAT interactions (20). Interestingly, the present results showed that the T mutation increased the FAT-FAT interaction without interfering with the FAT-FERM interaction.
Despite the impaired recruitment of T-FAT to FAs, T-FAK preserved its phosphorylation on Tyr397, the autophosphorylation site (23), and on Tyr576, a residue of the activation loop phosphorylated by SFKs (41). Because autophosphorylation and SFK recruitment are promoted by FAK dimerization (20), it is possible that the increased FAT H1 opening in T-FAT favored the H1-swapped dimerization of the mutant protein and thus contributed to the persistence of normal FAK activation despite decreased FA recruitment. In cells, formation of H1-swapped WT FAK dimers is likely to be a rare event, although it could conceivably complement the FERM-FERM interaction and replace the FAT-FERM interaction under some circumstances.
In full-length FAK as in purified FAT, Tyr925 phosphorylation was increased by the T mutation and tended to be decreased by the R mutation, a contrast enhanced by Fyn co-transfection. Tyr925 is an example of a phosphorylation site that has a favorable consensus sequence but a poor conformation for phosphorylation by Src-family kinases (34). Our results indicate that the H1 opening allows the helix region surrounding Tyr925 to unfold and adopt conformations compatible with kinase interactions, in agreement with a previous report exploring the consequences of the hydrophobic core residue mutations V955A/L962A (32). Thus, the conformational dynamics of FAT appear to be a biologically important mechanism to control FAK Tyr925 phosphorylation and hence FAK interactions with the Ras-MAPK pathway (24, 25) as well as the release of FAK from FAs (26).
The T-FAT mutation also enhanced phosphorylation of Tyr861, another major substrate residue for SFKs in the FAK C-terminal region (25, 41). Tyr861 is located in a presumably flexible region of the kinase-FAT linker, more than 50 residues upstream of FAT and about 10 residues upstream of the third Pro-rich motif (PR3). Enhanced Tyr861 phosphorylation may be a direct structural consequence of altered conformational FAT dynamics and/or it may result indirectly from increased Tyr925 phosphorylation, as the latter has been reported to influence Tyr861 phosphorylation (50). FAK is also phosphorylated on several serine residues (44, 52) that regulate cell spreading and migration (53). The major such site in the C-terminal region of FAK is Ser910, a substrate of ERK (45). Phosphorylation of Ser910 in response to bombesin-induced ERK activation (45) was increased in T-FAK, suggesting that destabilization of the bundle also increased Ser910 accessibility. Ser910 is located 15 residues upstream from Tyr925, in an N-terminal extension of FAT that can bind back to the FAT surface formed between H1 and H4 (see Fig. 2B). This interaction between the N-terminal extension and FAT H1/H4 restricts the accessibility of Ser910. Opening of H1 would therefore increase exposure of Ser910. Additionally, it is possible that phosphorylation of Tyr925 hinders back-binding of the N-terminal extension and thus promotes exposure of Ser910. These observations underscore a strong interaction between the FAT domain and the upstream region of the C-terminal moiety of FAK.
Paxillin binding to full-length FAK was impaired by the T mutation and enhanced by the R mutation. These effects were expected because H1 opening and subsequent rearrangement of the FAT structure affect both paxillin binding sites (on H1/H4 and H2/H3) (14, 15). Accordingly, R-FAK accumulation at FAs was increased, whereas it was reduced for T-FAK. Interestingly, T-FAK was still found at FAs, although its binding to paxillin was dramatically decreased. This remaining enrichment may result from the residual paxillin binding as well as from paxillin-independent mechanisms (46). Indeed, consistent with previous data showing that integrity of the four-helix bundle was not required for talin binding (17, 19), the FAT hinge mutations did not alter the FAK interaction with talin.
FAK is involved in the disassembly of FAs (9) and phosphorylation of Tyr925 is implicated in the exclusion of FAK from FAs and promotion of their turnover (26, 54). Despite its strongly decreased presence at FAs, T-FAK restored an elongated shape to transfected FAK−/− cells and increased global FA turnover as well as or more efficiently than WT FAK. In contrast, R-FAK was inefficient despite its higher enrichment at FAs. The phenotype of R-FAK in these assays was similar to that of Y925F-FAK, previously reported to impair FAK function (50), suggesting that decreased phosphorylation of this residue is an important aspect in R-FAK properties. Remarkably, loss of R-FAK function was very apparent in cells although the in vitro properties of R-FAK were not very different from those of the wild-type, as expected because it stabilized the closed conformation that is likely to be predominant. This contrast clearly underlines the functional importance of FAT dynamics.
Our study provides strong evidence that the structural transitions of FAT are physiologically relevant and specifically implicated in key functions of FAK (Fig. 10 summarizes the working model of the dynamics of FAT based on the current study and previous works). It also supports the proposed role of the Pro944-APP motif in the H1-H2 hinge, as the driving force behind H1 opening (27). Interestingly, this motif is conserved among vertebrate FAK, but not in other species (55) or other proteins with FAT-related domains (19). H1 dynamics may therefore be an evolutionarily recent property of FAK. Our results also reveal a functional connection between FAT and the rest of the FAK C-terminal region. An important open question is whether specific factors regulate H1 opening in physiological conditions. Partners of FAK at FAs that could promote its opening are yet to be identified. Other factors, such local pH changes, could play a role because phosphorylation and conformational dynamics of the FAT domain show some level of sensitivity to changes in pH over a physiological pH range (34). Alternatively, spontaneous FAT opening dynamics may function as a probabilistic switch mechanism, tuned to modulate a sufficiently large FAK subpopulation at sites where FAK is enriched, thus promoting Ras-MAPK signaling and FA disassembly in a time-delayed manner after FAK enrichment (56).
FIGURE 10.

Model for the role of FAT conformational dynamics in FAK function at focal adhesions. The FAT four-helix bundle exists in closed (1) and open (2) conformations in a dynamic equilibrium that is strongly shifted toward the closed state. By decreasing or increasing the propensity of the H1-H2 hinge to open, R-FAK and T-FAK mutants favor the closed and open FAT domain conformation, respectively. The scheme depicts the proposed role of these forms in FAK function at FAs. 1, the closed conformation of FAT has a strong affinity for paxillin LD motifs (two binding sites in FAT) and FAK can be strongly recruited to FAs through this interaction. However, in this configuration, Tyr925 is buried in the four-helix bundle. Ser910 and Tyr861 are also poorly accessible, possibly because of their masking by intramolecular interactions. FAT may also interact with FERM (20) (not shown in the scheme). 2, local accumulation of FAK at FAs promotes FAK dimerization through FERM-FERM interactions (20) (only the FERM domain of the second FAK molecule is drawn in dark gray). Possibly with the help of co-activators, the association between FERM and kinase domains in the dimer is loosened, which promotes autophosphorylation by intermolecular transphosphorylation of Tyr397. The Src homology 3 and 2 domains of SFK bind to a proline-rich motif 1 region and Tyr(P)397, respectively, in the FERM:kinase linker peptide. Either spontaneously (in a stochastic manner) or in response to unknown factors, H1 dissociates from the rest of the bundle. In the open conformation of FAT, the affinity of paxillin for the H1/H4 binding site on FAT is completely lost. Opening of FAT H1 is also expected to alter the stability and disposition of the other three helices, compromising paxillin binding to the H2/H3 site (32, 33); Tyr925 is exposed and can be phosphorylated by SFKs and then bind Grb2, which activates the ERK pathway. Through unfolding and/or unmasking of the linker region between FAT and the kinase domain, Ser910 and Tyr861 become exposed and accessible to ERK and SFKs, respectively. Together with the loss of paxillin affinity, phosphorylation of the above sites contributes to detachment of FAK from FAs for degradation or recycling.
Acknowledgments
We thank Dr. Dusko Ilic (Department of Cell and Tissue Biology, University of California San Francisco) for the gift of Ptk2−/− fibroblasts, Dr. Monique Arpin (Curie Institute, Paris) for providing anti-VSV antibodies and Dr. Alexandre Maucuer for the gift of His6-U2AF. We acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facilities and thank the user support team of beamline ID14-2 for assistance with data recording. J. A. Girault's team is affiliated with the ENP and the Bio-Psy laboratory of excellence.
This work was supported in part by Agence Nationale de la Recherche Grant ANR-05-2_42589, Association pour la Recherche sur le Cancer (ARC) Grant A05/3/3138, Fondation pour la Recherche Médicale, European Research Council, Inserm, UPMC, and King Abdullah University of Science and Technology (KAUST).
- FAK
- focal adhesion kinase
- FA
- focal adhesion
- FAT
- focal adhesion targeting
- FERM
- four-point-one, ezrin, radixin, moesin
- FRAP
- fluorescence recovery after photobleaching
- H1
- first helix of the FAT domain
- H2
- second helix of the FAT domain
- MEF
- mouse embryonic fibroblasts
- R-FAT/FAK
- relaxed FAT/FAK
- SFK
- Src family kinase
- T-FAT/FAK
- tense FAT/FAK
- VSV
- vesicular stomatitis virus
- ANOVA
- analysis of variance
- ERK
- extracellular signal-regulated kinase
- SH2
- Src-homology 2
- VSV
- vesicular stomatitis virus.
REFERENCES
- 1. Hanks S. K., Calalb M. B., Harper M. C., Patel S. K. (1992) Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc. Natl. Acad. Sci. U.S.A. 89, 8487–8491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schaller M. D., Borgman C. A., Cobb B. S., Vines R. R., Reynolds A. B., Parsons J. T. (1992) pp125FAK, A structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc. Natl. Acad. Sci. U.S.A. 89, 5192–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arold S. T. (2011) How focal adhesion kinase achieves regulation by linking ligand binding, localization and action. Curr. Opin. Struct. Biol. 21, 808–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parsons J. T. (2003) Focal adhesion kinase: the first ten years. J. Cell Sci. 116, 1409–1416 [DOI] [PubMed] [Google Scholar]
- 5. Schaller M. D. (2010) Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J. Cell Sci. 123, 1007–1013 [DOI] [PubMed] [Google Scholar]
- 6. Zachary I., Rozengurt E. (1992) Focal adhesion kinase (p125FAK): a point of convergence in the action of neuropeptides, integrins, and oncogenes. Cell 71, 891–894 [DOI] [PubMed] [Google Scholar]
- 7. Mitra S. K., Hanson D. A., Schlaepfer D. D. (2005) Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68 [DOI] [PubMed] [Google Scholar]
- 8. Schaller M. D. (2001) Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim. Biophys. Acta 1540, 1–21 [DOI] [PubMed] [Google Scholar]
- 9. Ilić D., Furuta Y., Kanazawa S., Takeda N., Sobue K., Nakatsuji N., Nomura S., Fujimoto J., Okada M., Yamamoto T., Aizawa S. (1995) Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377, 539–544 [DOI] [PubMed] [Google Scholar]
- 10. Owens L. V., Xu L., Craven R. J., Dent G. A., Weiner T. M., Kornberg L., Liu E. T., Cance W. G. (1995) Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 55, 2752–2755 [PubMed] [Google Scholar]
- 11. Hildebrand J. D., Schaller M. D., Parsons J. T. (1993) Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAK, to cellular focal adhesions. J. Cell Biol. 123, 993–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tachibana K., Sato T., D'Avirro N., Morimoto C. (1995) Direct association of pp125FAK with paxillin, the focal adhesion-targeting mechanism of pp125FAK. J. Exp. Med. 182, 1089–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bertolucci C. M., Guibao C. D., Zheng J. (2005) Structural features of the focal adhesion kinase-paxillin complex give insight into the dynamics of focal adhesion assembly. Protein Sci. 14, 644–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao G., Prutzman K. C., King M. L., Scheswohl D. M., DeRose E. F., London R. E., Schaller M. D., Campbell S. L. (2004) NMR solution structure of the focal adhesion targeting domain of focal adhesion kinase in complex with a paxillin LD peptide: evidence for a two-site binding model. J. Biol. Chem. 279, 8441–8451 [DOI] [PubMed] [Google Scholar]
- 15. Hoellerer M. K., Noble M. E., Labesse G., Campbell I. D., Werner J. M., Arold S. T. (2003) Molecular recognition of paxillin LD motifs by the focal adhesion targeting domain. Structure 11, 1207–1217 [DOI] [PubMed] [Google Scholar]
- 16. Alam T., Alazmi M., Gao X., Arold S. T. (2014) How to find a leucine in a haystack? Structure, ligand recognition and regulation of leucine-aspartic acid (LD) motifs. Biochem. J. 460, 317–329 [DOI] [PubMed] [Google Scholar]
- 17. Chen H.-C., Appeddu P. A., Parsons J. T., Hildebrand J. D., Schaller M. D., Guan J.-L. (1995) Interaction of focal adhesion kinase with cytoskeletal protein talin. J. Biol. Chem. 270, 16995–16999 [DOI] [PubMed] [Google Scholar]
- 18. Lawson C., Lim S. T., Uryu S., Chen X. L., Calderwood D. A., Schlaepfer D. D. (2012) FAK promotes recruitment of talin to nascent adhesions to control cell motility. J. Cell Biol. 196, 223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hayashi I., Vuori K., Liddington R. C. (2002) The focal adhesion targeting (FAT) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nat. Struct. Biol. 9, 101–106 [DOI] [PubMed] [Google Scholar]
- 20. Brami-Cherrier K., Gervasi N., Arsenieva D., Walkiewicz K., Boutterin M. C., Ortega A., Leonard P. G., Seantier B., Gasmi L., Bouceba T., Kadaré G., Girault J. A., Arold S. T. (2014) FAK dimerization controls its kinase-dependent functions at focal adhesions. EMBO J. 33, 356–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Corsi J. M., Houbron C., Billuart P., Brunet I., Bouvrée K., Eichmann A., Girault J. A., Enslen H. (2009) Autophosphorylation-independent and -dependent functions of focal adhesion kinase during development. J. Biol. Chem. 284, 34769–34776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Girault J. A., Labesse G., Mornon J. P., Callebaut I. (1999) The N-termini of FAK and JAKs contain divergent band 4.1 domains. Trends Biochem. Sci. 24, 54–57 [DOI] [PubMed] [Google Scholar]
- 23. Schaller M. D., Hildebrand J. D., Shannon J. D., Fox J. W., Vines R. R., Parsons J. T. (1994) Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell. Biol. 14, 1680–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schlaepfer D. D., Hanks S. K., Hunter T., van der Geer P. (1994) Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature 372, 786–791 [DOI] [PubMed] [Google Scholar]
- 25. Schlaepfer D. D., Hunter T. (1996) Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol. Cell. Biol. 16, 5623–5633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Katz B. Z., Romer L., Miyamoto S., Volberg T., Matsumoto K., Cukierman E., Geiger B., Yamada K. M. (2003) Targeting membrane-localized focal adhesion kinase to focal adhesions: roles of tyrosine phosphorylation and SRC family kinases. J. Biol. Chem. 278, 29115–29120 [DOI] [PubMed] [Google Scholar]
- 27. Arold S. T., Hoellerer M. K., Noble M. E. (2002) The structural basis of localization and signaling by the focal adhesion targeting domain. Structure 10, 319–327 [DOI] [PubMed] [Google Scholar]
- 28. Liu G., Guibao C. D., Zheng J. (2002) Structural insight into the mechanisms of targeting and signaling of focal adhesion kinase. Mol. Cell. Biol. 22, 2751–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bergdoll M., Remy M. H., Cagnon C., Masson J. M., Dumas P. (1997) Proline-dependent oligomerization with arm exchange. Structure 5, 391–401 [DOI] [PubMed] [Google Scholar]
- 30. Bourne Y., Arvai A. S., Bernstein S. L., Watson M. H., Reed S. I., Endicott J. E., Noble M. E., Johnson L. N., Tainer J. A. (1995) Crystal structure of the cell cycle-regulatory protein suc1 reveals a β-hinge conformational switch. Proc. Natl. Acad. Sci. U.S.A. 92, 10232–10236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rousseau F., Schymkowitz J. W., Wilkinson H. R., Itzhaki L. S. (2001) Three-dimensional domain swapping in p13suc1 occurs in the unfolded state and is controlled by conserved proline residues. Proc. Natl. Acad. Sci. U.S.A. 98, 5596–5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dixon R. D., Chen Y., Ding F., Khare S. D., Prutzman K. C., Schaller M. D., Campbell S. L., Dokholyan N. V. (2004) New insights into FAK signaling and localization based on detection of a FAT domain folding intermediate. Structure 12, 2161–2171 [DOI] [PubMed] [Google Scholar]
- 33. Zhou Z., Feng H., Bai Y. (2006) Detection of a hidden folding intermediate in the focal adhesion target domain: implications for its function and folding. Proteins 65, 259–265 [DOI] [PubMed] [Google Scholar]
- 34. Cable J., Prutzman K., Gunawardena H. P., Schaller M. D., Chen X., Campbell S. L. (2012) In vitro phosphorylation of the focal adhesion targeting domain of focal adhesion kinase by Src kinase. Biochemistry 51, 2213–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Prutzman K. C., Gao G., King M. L., Iyer V. V., Mueller G. A., Schaller M. D., Campbell S. L. (2004) The focal adhesion targeting domain of focal adhesion kinase contains a hinge region that modulates tyrosine 926 phosphorylation. Structure 12, 881–891 [DOI] [PubMed] [Google Scholar]
- 36. Burgaya F., Girault J. A. (1996) Cloning of focal adhesion kinase, pp125FAK, from rat brain reveals multiple transcripts with different patterns of expression. Mol. Brain Res. 37, 63–73 [DOI] [PubMed] [Google Scholar]
- 37. Garron M. L., Arthos J., Guichou J. F., McNally J., Cicala C., Arold S. T. (2008) Structural basis for the interaction between focal adhesion kinase and CD4. J. Mol. Biol. 375, 1320–1328 [DOI] [PubMed] [Google Scholar]
- 38. Dunty J. M., Gabarra-Niecko V., King M. L., Ceccarelli D. F., Eck M. J., Schaller M. D. (2004) FERM domain interaction promotes FAK signaling. Mol. Cell. Biol. 24, 5353–5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kadaré G., Toutant M., Formstecher E., Corvol J. C., Carnaud M., Boutterin M. C., Girault J. A. (2003) PIAS1-mediated sumoylation of focal adhesion kinase activates its autophosphorylation. J. Biol. Chem. 278, 47434–47440 [DOI] [PubMed] [Google Scholar]
- 40. Ishibe S., Joly D., Liu Z. X., Cantley L. G. (2004) Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol. Cell 16, 257–267 [DOI] [PubMed] [Google Scholar]
- 41. Calalb M. B., Polte T. R., Hanks S. K. (1995) Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol. Cell. Biol. 15, 954–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lim Y., Han I., Jeon J., Park H., Bahk Y. Y., Oh E. S. (2004) Phosphorylation of focal adhesion kinase at tyrosine 861 is crucial for Ras transformation of fibroblasts. J. Biol. Chem. 279, 29060–29065 [DOI] [PubMed] [Google Scholar]
- 43. Hunger-Glaser I., Fan R. S., Perez-Salazar E., Rozengurt E. (2004) PDGF and FGF induce focal adhesion kinase (FAK) phosphorylation at Ser-910: dissociation from Tyr-397 phosphorylation and requirement for ERK activation. J. Cell Physiol. 200, 213–222 [DOI] [PubMed] [Google Scholar]
- 44. Ma A., Richardson A., Schaefer E. M., Parsons J. T. (2001) Serine phosphorylation of focal adhesion kinase in interphase and mitosis: a possible role in modulating binding to p130Cas. Mol. Biol. Cell 12, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hunger-Glaser I., Salazar E. P., Sinnett-Smith J., Rozengurt E. (2003) Bombesin, lysophosphatidic acid, and epidermal growth factor rapidly stimulate focal adhesion kinase phosphorylation at Ser-910: requirement for ERK activation. J. Biol. Chem. 278, 22631–22643 [DOI] [PubMed] [Google Scholar]
- 46. Cooley M. A., Broome J. M., Ohngemach C., Romer L. H., Schaller M. D. (2000) Paxillin binding is not the sole determinant of focal adhesion localization or dominant-negative activity of focal adhesion kinase/focal adhesion kinase-related nonkinase. Mol. Biol. Cell 11, 3247–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hamadi A., Bouali M., Dontenwill M., Stoeckel H., Takeda K., Rondé P. (2005) Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J. Cell Sci. 118, 4415–4425 [DOI] [PubMed] [Google Scholar]
- 48. Schober M., Raghavan S., Nikolova M., Polak L., Pasolli H. A., Beggs H. E., Reichardt L. F., Fuchs E. (2007) Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. J. Cell Biol. 176, 667–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Millon-Frémillon A., Bouvard D., Grichine A., Manet-Dupé S., Block M. R., Albiges-Rizo C. (2008) Cell adaptive response to extracellular matrix density is controlled by ICAP-1-dependent β1-integrin affinity. J. Cell Biol. 180, 427–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Deramaudt T. B., Dujardin D., Hamadi A., Noulet F., Kolli K., De Mey J., Takeda K., Rondé P. (2011) FAK phosphorylation at Tyr-925 regulates cross-talk between focal adhesion turnover and cell protrusion. Mol. Biol. Cell 22, 964–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Webb D. J., Donais K., Whitmore L. A., Thomas S. M., Turner C. E., Parsons J. T., Horwitz A. F. (2004) FAK-Src signalling through paxillin, ERK, and MLCK regulates adhesion disassembly. Nat. Cell. Biol. 6, 154–161 [DOI] [PubMed] [Google Scholar]
- 52. Grigera P. R., Jeffery E. D., Martin K. H., Shabanowitz J., Hunt D. F., Parsons J. T. (2005) FAK phosphorylation sites mapped by mass spectrometry. J. Cell Sci. 118, 4931–4935 [DOI] [PubMed] [Google Scholar]
- 53. Jiang X., Sinnett-Smith J., Rozengurt E. (2007) Differential FAK phosphorylation at Ser-910, Ser-843 and Tyr-397 induced by angiotensin II, LPA and EGF in intestinal epithelial cells. Cell Signal. 19, 1000–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hamadi A., Deramaudt T. B., Takeda K., Ronde P. (2010) Hyperphosphorylated FAK delocalizes from focal adhesions to membrane ruffles. J. Oncol. 2010, pii: 932803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Corsi J. M., Rouer E., Girault J. A., Enslen H. (2006) Organization and post-transcriptional processing of focal adhesion kinase gene. BMC Genomics 7, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ladbury J. E., Arold S. T. (2012) Noise in cellular signaling pathways: causes and effects. Trends Biochem. Sci. 37, 173–178 [DOI] [PMC free article] [PubMed] [Google Scholar]