

Background: The requirement of O-fucose monosaccharide on Notch is not fully understood.
Results: Loss of O-fucose monosaccharide on Notch caused temperature-sensitive loss of Notch signaling.
Conclusion: O-Fucose monosaccharide of Notch has a temperature-sensitive function and cooperates with O-glucose glycan in Notch signal activation.
Significance: Our findings elucidate how different forms of glycosylation on a protein influence protein functions.
Keywords: Cell Signaling, Drosophila, Endoplasmic Reticulum (ER), Glycosylation, Trafficking, Notch, O-Fucose, O-Fucosyltransferase 1, O-Glucose, Rumi
Abstract
Notch (N) is a transmembrane receptor that mediates the cell-cell interactions necessary for many cell fate decisions. N has many epidermal growth factor-like repeats that are O-fucosylated by the protein O-fucosyltransferase 1 (O-Fut1), and the O-fut1 gene is essential for N signaling. However, the role of the monosaccharide O-fucose on N is unclear, because O-Fut1 also appears to have O-fucosyltransferase activity-independent functions, including as an N-specific chaperon. Such an enzymatic activity-independent function could account for the essential role of O-fut1 in N signaling. To evaluate the role of the monosaccharide O-fucose modification in N signaling, here we generated a knock-in mutant of O-fut1 (O-fut1R245A knock-in), which expresses a mutant protein that lacks O-fucosyltransferase activity but maintains the N-specific chaperon activity. Using O-fut1R245A knock-in and other gene mutations that abolish the O-fucosylation of N, we found that the monosaccharide O-fucose modification of N has a temperature-sensitive function that is essential for N signaling. The O-fucose monosaccharide and O-glucose glycan modification, catalyzed by Rumi, function redundantly in the activation of N signaling. We also showed that the redundant function of these two modifications is responsible for the presence of N at the cell surface. Our findings elucidate how different forms of glycosylation on a protein can influence the protein's functions.
Introduction
Cell-cell signaling mediated by Notch (N)3 receptors plays crucial roles in many developmental processes in multicellular organisms (1–4). In humans, aberrant N signaling causes or is implicated in numerous diseases, including Alagille syndrome, spondylocostal dysostosis, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, multiple sclerosis, and various cancers (5–7).
N receptor family proteins have multiple EGF-like repeats in their extracellular domain (1–4), many of which can be modified with several types of O-linked glycosylation: O-fucose, O-glucose, O-xylose, and O-GlcNAc (8–12). N is O-fucosylated at serine or threonine residues in the C2XXXX(S/T)C3 consensus sequence that is present in various EGF-like repeats; for example, Drosophila has 36 EGF-like repeats, 23 of which are O-fucosylated (8, 13). This modification is mediated by protein O-fucosyltransferase 1 (O-Fut1 in Drosophila and protein O-fucosyltransferase 1 in mammals). β1,3-GlcNAc is further added to the O-fucose residue by Fringe (Fng) family proteins, which are β1,3-N-acetylglucosaminyltransferases (14, 15).
O-fut1 and Pofut1 are apparently globally required for normal N signaling in Drosophila and mammals, respectively, in that their mutant phenotypes are similar to those caused by disrupted N signaling (16–18). In contrast, mutants of genes encoding Fng family proteins reduce N signaling in only a subset of contexts that involve N signaling (15, 19–21). Therefore, the GlcNAc modification by Fng, which regulates the interaction between N and its ligands, Delta (Dl) or Serrate, is required in a tissue-specific manner for various tissue boundary formations (22).
O-Fut1 has two O-fucosyltransferase activity-independent functions (23–26). First, it is an endoplasmic reticulum (ER) resident protein and functions as a N-specific chaperon (23). Second, overexpressed O-Fut1 promotes the endocytosis of the N cell nonautonomously (25). Thus, it has been difficult to determine whether O-Fut1's O-fucosyltransferase activity or its nonenzymatic functions (or both) are required in N signaling. To address this issue, Okajima et al. (23) generated a genomic fragment, including a mutant O-fut1 locus, O-fut1R245A, which produces an O-Fut1 variant carrying an amino acid substitution in the binding site for GDP-fucose. O-Fut1R245A lacks O-fucosyltransferase activity in vitro, although it reportedly retains its N-specific chaperon activity (23). Most of the defects associated with the absence of O-fut1 are rescued by introducing the O-fut1R245A genomic locus, except for the phenotypes that are probably due to the disruption of Fng functions, which are tissue-specific and depend on the O-fucosylation of N (24). Based on these results, it was proposed that the O-fucose monosaccharide modification of N does not have a specific function in N signaling, although it is required for the Fng-dependent activation of N signaling. However, it was also reported that the ability of the O-fut1R245A genomic locus to rescue the O-fut1 null allele differs among transgenic lines, probably because the insertion site of the transgene affects the transcription efficiency of the integrated O-fut1R245A locus (23, 24).
To overcome this problem, here we generated a knock-in mutant of O-fut1R245A (O-fut1R245A knock-in) in Drosophila melanogaster. We used O-fut1R245A knock-in and other mutants affecting the O-fucosylation of Notch's EGF-like repeats to examine the specific role of the monosaccharide O-fucose in N signaling. We also studied the genetic interaction between O-fut1R245A knock-in and rumi, which encodes an O-glucosyltransferase that adds O-glucose to the EGF-like repeats of N (27–30). Based on the presented results, we propose a model showing how multiple O-glycosylation sites in EGF-like repeats might affect the function of N.
EXPERIMENTAL PROCEDURES
Drosophila Strains
Canton-S was used as the wild-type strain. The following mutant alleles were used: O-fut14R6, a null allele (17); fng13, a strong loss-of-function allele (31); GmdH78, a null allele (25); GmerSH l(2)SH1931, a P-element insertion allele (32, 33); Df(2R)BSC783, a deficiency uncovering the Gmer locus (33); and rumi44, a null allele (27). The following UAS lines were used: UAS-NFL (a gift from S. Artavanis-Tsakonas), UAS-NΔE (34), UAS-NICD (35), and UAS-Lamp-HRP (36). Decapentaplegic (dpp)-Gal4 was described previously (37). 2xUbx-FLP was used to efficiently induce somatic mosaic clones (38). To induce germ line mosaic clones, ovoD G13 (17), ovoD FRT40A, and ovoD FRT80B (39) were used.
Generation of the O-fut1R245A knock-in Fly—O-fut1R245A knock-in is a knock-in mutation generated by a homologous recombination method described previously (40, 41). Two genomic fragments covering the O-fut1 locus, referred to as the left arm and right arm, were PCR-amplified. The left arm (5005 bp) was amplified using the primers 5′-CAACCAAGCAGGGCCAATCCCA-3′ and 5′-AATTTCTTATAGTCATATAAATACAAAATA-3′, and it included the region from 4560 bp upstream of the start of the O-fut1 5′UTR to 188 bp downstream of the end of the O-fut1 3′UTR. The right arm (4996 bp) was amplified using the primers 5′-TCTTTTAGCTTTAATTCTTAAAAAGGATTT-3′ and 5′-CCGAATCGGCGACCCAGTAAAC-3′, and it included the region from 189 bp downstream of the end of the O-fut1 3′UTR to 5115-bp downstream of the end of the O-fut1 3′UTR. The left arm fragment was inserted into the AscI site of the pT7 Blue vector (Novagen), and the right arm fragment was inserted between the SphI and NotI sites of the pT7 Blue vector. The resulting constructs were pT7 Blue+left arm and pT7 Blue+right arm. To introduce a base substitution that would result in the amino acid replacement of arginine (Arg) at the 245th amino acid with alanine (Ala), an overlap extension PCR was performed using pT7 Blue+left arm and two primers, 5′-CATCTGGCCAACGGTATCGATTGGGTG-3′ and 5′-ACCGTTGGCCAGATGAATGCCCAAAAA3′. The right arm and mutated left arm were excised and cloned into an ends-out homologous recombination vector, pW25, with a selectable marker, white (40, 41). This construct was introduced into the Drosophila genome by P-element-mediated transformation (41). Using the transgenic line obtained, homologous recombination was performed as described previously (40, 41). Briefly, pW25 contains two lox sites, which make it feasible to remove the white marker by Cre-mediated recombination (41). The white marker was removed as described previously (41), and the resulting lines were maintained as O-fut1R245A knock-in. The O-fut1 locus of the O-fut1R245A knock-in line was sequenced, and the mutation was confirmed.
Generation of Somatic Mosaic Clones
Somatic clones of O-fut1R245A knock-in and fng13 were generated by mitotic recombination in wing discs isolated from the larvae of y w 2×Ubx-FLP; FRTG13 O-fut1R245A knock-in/FRTG13 Ubi-GFP and y w 2×Ubx-FLP; fng13 FRT80B/Ubi-GFP FRT80B, y w 2×Ubx-FLP; FRT82B rumi44/FRT82B Ubi-GFP, y w 2×Ubx-FLP; FRTG13 O-fut1R245A knock-in/FRTG13 Ubi-GFP; FRT82B rumi44/FRT82B rumi44, using the FLP/FRT system, as described before (42). To express UAS-Lamp-HRP in wild-type or O-fut1R245A knock-in mutants, the following males were crossed to 2×Ubx-FLP; FRTG13 tub-Gal80/CyO; tub-Gal4/TM6B females, respectively: FRTG13; UAS-Lamp-HRP/TM6B, and FRTG13 O-fut1R245A knock-in/CyO; UAS-Lamp-HRP/TM6B. Until the larval stage, cultures were maintained at the indicated temperature (18, 25, or 30 °C).
Epistasis Analysis Involving O-fut1R245A knock-in and Various N Derivatives Using the MARCM System
The MARCM system was described previously (43). The following males were crossed to 2×Ubx-FLP; FRTG13 tub-Gal80/CyO; tub-Gal4, UAS-GFP/TM6B females to obtain flies with MARCM clones: FRTG13 O-fut1R245A knock-in/CyO; UAS-NFL/TM6B, FRTG13/CyO; UAS-NFL/TM6B, FRTG13 O-fut1R245A knock-in/CyO; UAS-NΔE/TM6B, FRTG13/CyO; UAS-NΔE/TM6B, FRTG13 O-fut1R245A knock-in/CyO; UAS-NICD/TM6B, FRTG13/CyO; UAS-NICD/TM6B.
Generation of Germ Line Mosaic Clones to Remove the Maternal Contribution
To obtain embryos homozygous for O-fut1R245A knock-in that originated from an O-fut1R245A knock-in homozygous germ line, y w hs-FLP/+; FRTG13 ovoD/FRTG13 O-fut1R245A knock-in females were crossed with FRTG13 O-fut1R245A knock-in/CyO, wg-lacZ males. To obtain embryos homozygous for Gmd and lacking its maternal contribution, y w hs-FLP/+; ovoD FRT40A/GmdH78 FRT40A females were crossed with GmdH78 FRT40A/CyO, wg-lacZ males. To obtain embryos homozygous for Gmer and lacking its maternal contribution, y w hs-FLP/+; FRTG13 ovoD/FRTG13 GmerSH females were crossed with FRTG13 GmerSH/CyO, wg-lacZ males. To obtain embryos homozygous for fng and lacking its maternal contribution, y w hs-FLP/+; ovoD FRT80B/fng13 FRT80B females were crossed with fng13 FRT80B/TM3, ftz-lacZ males. To generate germ line mosaic clones, larvae were heat-shocked at 37 °C for 1 h 48–72 h after egg laying.
Western Blots
Western blotting was performed using a standard protocol (44). Wing discs of third-instar larvae were dissected and homogenized to prepare protein extracts. To detect Notch protein, 30 μg of protein extracts were resolved by electrophoresis on 4–15% Criterion TGX precast gels (Bio-Rad), and an anti-Notch intracellular domain antibody (1:5000 dilution, C17.9C6) (45) was used. As a loading control, α-tubulin was detected with an anti-α-tubulin antibody (1:2000 dilution, DM1A, Sigma) (46).
Immunostaining
Dissections and staining were performed using standard methods (44). The following antibodies were used: mouse anti-Notch intracellular domain (1:500; C17.9C6) (45); mouse anti-Notch extracellular domain (1:500, C458.2H) (47); mouse anti-Wg (1:250; 4D4) (48); mouse anti-Cut (1:250; 2B10) (49); rabbit anti-GFP (1:1,000; MBL, Nagoya, Japan); rat anti-GFP (1:1,000; Nacalai, Kyoto, Japan); rat anti-Elav (50) (1:25, 9F8A9); rabbit anti-Rab7 (1:5,000) (51); rabbit anti-Rab11 (1:10,000) (51); mouse anti-Pdi (1:500, 1D3, EnzoLife Sciences) (52); guinea pig anti-Senseless (1:500) (53); guinea pig anti-Hrs (36); guinea pig anti-O-Fut1 (1:1000) (23); and guinea pig anti-Boca (1:500) (54). Cy3-, Cy5-, and Alexa488-coupled secondary antibodies (1:500) were from Jackson ImmunoResearch and Molecular Probes. Images were acquired on an LSM5 Pascal or LSM700 microscope (Zeiss). All images were processed and assembled using ImageJ, Adobe Photoshop, and Adobe Illustrator.
Comparison of the O-Fucose and O-Glucose Sites in Various Notch Receptors
The Swiss-Prot and KEGG GENES databases were searched using the Motif search program or EGF-like repeats containing the consensus sequence for O-glucosylation between the first and second cysteines, and for O-fucosylation between the second and third cysteines (8, 13).
RESULTS
O-Fucosyltransferase Activity of O-fut1R245A knock-in Is Negligible in Vivo
In the previously reported Drosophila O-fut1R245A mutant, arginine is replaced by alanine at the 245th amino acid position of the deduced O-Fut1 protein (Fig. 1A) (23). This amino acid substitution is located in the GDP-fucose-binding motif and largely abolishes the O-fucosyltransferase activity in vitro (Fig. 1A) (23, 55, 56). We first sought to confirm that the O-fucosyltransferase activity of O-fut1R245A knock-in was also negligible in vivo. O-fut1R245A knock-in homozygotes survived until the third-instar larval stage (data not shown). In the wild-type wing discs of the third-instar larvae, N signaling is activated at the border between fng-expressing and fng-nonexpressing regions, which correspond to the dorsal and ventral (D/V) compartments, respectively. Therefore, the expression of wingless (wg), a downstream gene of N signaling, is activated along the D/V compartment boundary (D/V boundary) (Fig. 1B) (57). This activation of N signaling depends on the selective binding of N with Dl or Serrate, which is regulated by GlcNAc modification of the O-fucose on N′s EGF-like repeats by Fng (20). We found that wg expression was abolished in the wing discs isolated from O-fut1R245A knock-in homozygotes at the third instar cultured at 25 °C (Fig. 1C), probably because the fng function was disrupted in the absence of N′s O-fucosylation.
FIGURE 1.
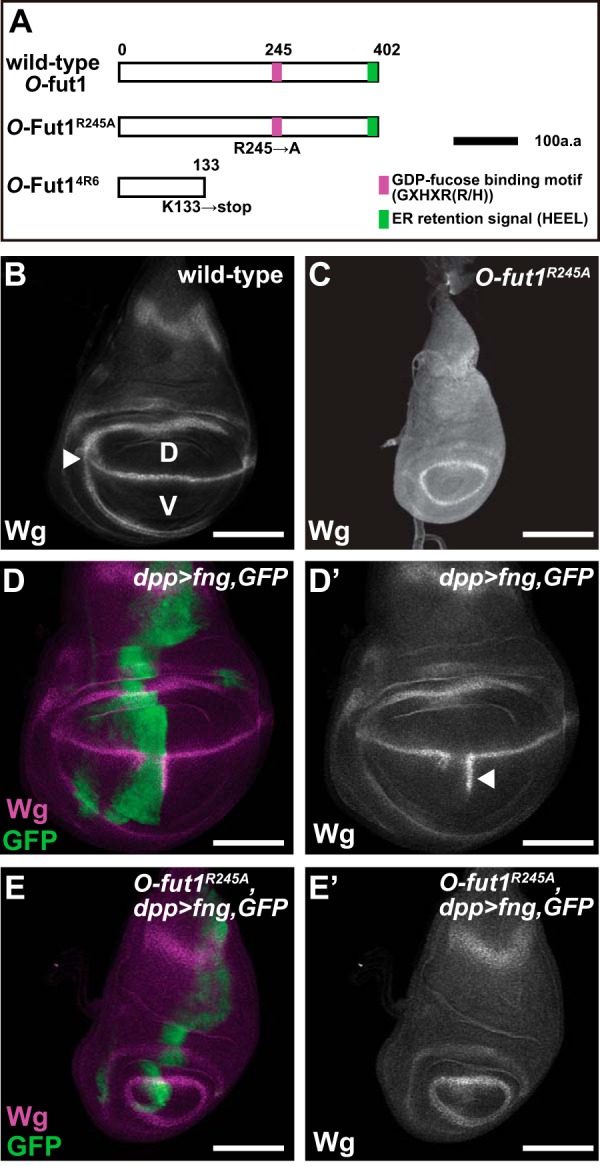
O-Fucosyltransferase activity of O-fut1R245A knock-in is negligible in vivo. A, schematic of the O-Fut1 protein and its mutant derivatives. The GDP-fucose-binding motif (GXHXR(R/H)) and ER retention signal (HEEL) are shown in magenta and green, respectively. Top, middle, and bottom bars represent wild-type, O-Fut1R245A, and O-Fut14R6, respectively. O-Fut1R245A has an amino acid substitution (Arg to Ala) at the 245th amino acid that disrupts its O-fucosyltransferase activity. O-Fut14R6 carries a nonsense mutation that introduces a premature stop codon at the 133rd amino acid. Black bar corresponds to 100 amino acids. B and C, wild-type (B) and O-fut1R245A knock-in homozygote (C) wing discs of third-instar larvae were stained with an anti-Wg antibody. The expression of wg along the D/V boundary, which depends on N signaling, is indicated by an arrowhead in B. D, dorsal compartment; V, ventral compartment. D–E′, UAS-fng was misexpressed under the control of dpp-Gal4 along the anterior-posterior boundary of wild-type (D and D′) and O-fut1R245A knock-in homozygote wing discs (E and E′). These wing discs were stained with an anti-Wg antibody (magenta in D and E and white in D′ and E′). The expression pattern of dpp-Gal4 was monitored by detecting the UAS-GFP expression using an anti-GFP antibody (green in D and E). D′ and E′ are single channel images of the magenta (anti-Wg staining) in D and E, respectively. The ectopic expression of wg is indicated by an arrowhead in D′. Scale bars, B–E′ are 100 μm.
As reported previously, misexpressed fng driven by dpp-Gal4 at 25 °C resulted in the ectopic activation of wg in the ventral compartment of the wild-type wing, where endogenous fng is not expressed (n = 99) (Fig. 1, D and D′). However, when we misexpressed fng driven by dpp-Gal4 in the wing discs of O-fut1R245A knock-in homozygotes cultured at 25 °C, no wg expression was induced in any case examined (n = 52) (Fig. 1, E and E′). We confirmed that the expression pattern of dpp-Gal4 was almost the same in the wing discs of wild-type and O-fut1R245A knock-in homozygotes at this temperature (Fig. 1, D and E). This result suggested that in the O-fut1R245A knock-in homozygotes, the O-fucose modification of N was minimal, if it occurred at all.
O-fut1 is a maternal-effect gene (17). Therefore, to observe the phenotypes associated with the O-fut1R245A knock-in mutation, we needed to remove the maternal contribution of the wild-type O-fut1 gene from the female germ line. However, these females are heterozygous for O-fut1R245A knock-in, because O-fut1R245A knock-in is a recessive lethal mutation (data not shown). Therefore, we obtained embryos homozygous for O-fut1R245A knock-in from females carrying germ line clones homozygous for O-fut1R245A knock-in, using a previously described method (17, 58). In this study, we call these embryos O-fut1R245A knock-inm/z.
O-fut1R245A knock-in Homozygotes Obtained from Females Carrying the O-fut1R245A knock-in Homozygous Germ Line Show a Temperature-sensitive Neurogenic Phenotype
In the embryonic central nervous system, the number of neuroblasts segregated from the neuroectoderm is determined by “lateral inhibition” through N signaling (59). A disruption of N signaling causes a failure of lateral inhibition, resulting in neuronal hyperplasia, known as the neurogenic phenotype (59). We studied the nervous system development in O-fut1R245A knock-inm/z embryos at 25 and 30 °C, in case O-fut1R245A had a temperature-sensitive property. Neurons were detected by anti-Elav antibody staining (Fig. 2, A–J). At 25 and 30 °C, the wild-type flies develop and reproduce normally, and their embryonic nervous system is also normal (Fig. 2, A, B, and K). In addition, the nervous system was normal in most of the O-fut1R245A knock-inm/z embryos at 25 °C (Fig. 2, C and K; 1/38 embryos showed the neurogenic phenotype). However, at 30 °C, the O-fut1R245A knock-inm/z embryos showed a highly penetrant neurogenic phenotype (Fig. 2, D and K, 15/15 embryos showed the neurogenic phenotype). This temperature-sensitive neurogenic phenotype has not been reported in previous studies using the transgenic O-fut1R245A genomic locus (23, 24).
FIGURE 2.
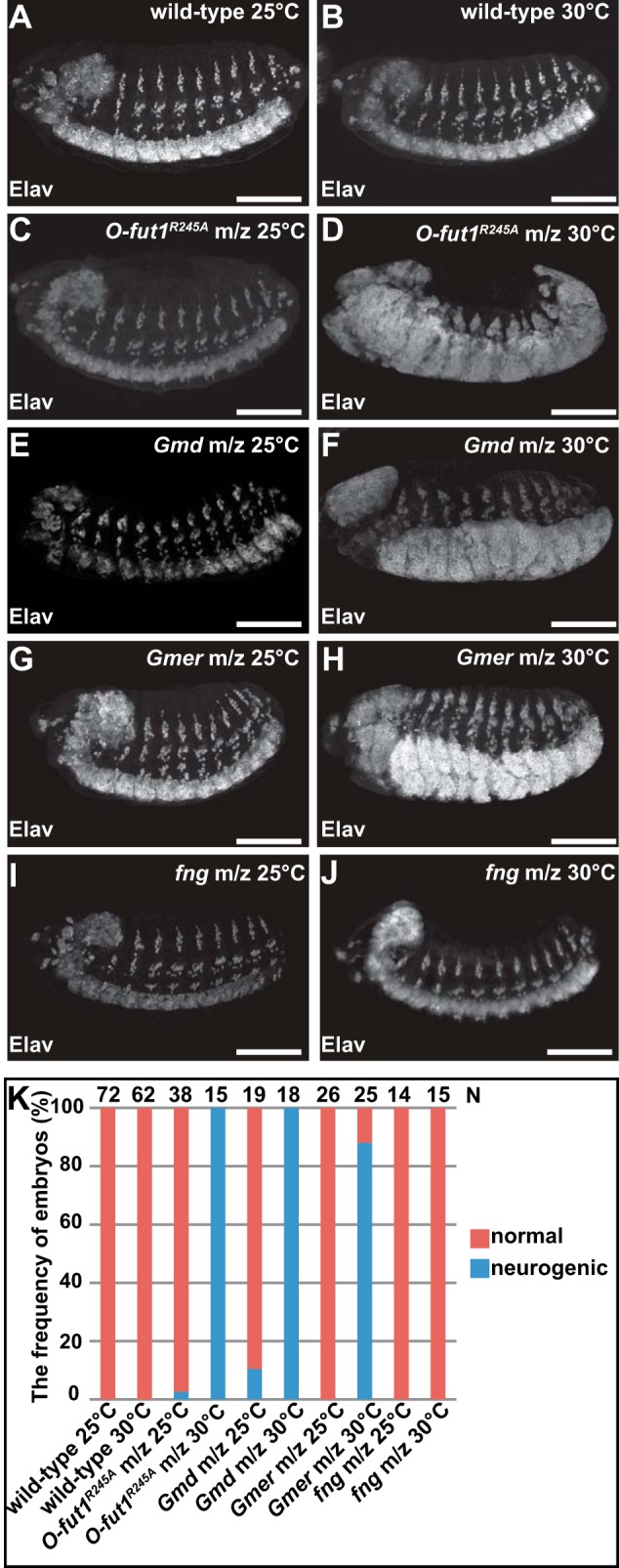
Loss of O-fucose monosaccharide modification of N causes a neurogenic phenotype at 30 °C but not at 25 °C. A–J, lateral views of embryos stained with an anti-Elav antibody (white). A and B, wild-type embryos raised at 25 °C (A) and 30 °C (B). C and D, O-fut1R245A knock-inm/z raised at 25 °C (C) and 30 °C (D). E and F, GmdH78m/z raised at 25 °C (E) and 30 °C (F). G and H, GmerSHm/z raised at 25 °C (G) and 30 °C (H). I and J, fng13m/z raised at 25 °C (I) and 30 °C (J). K, frequency of embryos (%) showing a normal central nervous system (orange) or a neurogenic phenotype (blue) at 25 and 30 °C. The number of embryos examined is shown at the top. Scale bars, 100 μm.
To confirm that the temperature-sensitive neurogenic phenotype was not due to the degradation of O-Fut1R245A at 30 °C, we compared the protein levels of O-Fut1 and its derivative in wild-type and O-fut1R245A knock-in homozygous cells, respectively. We generated genetic mosaic wing discs composed of somatic clones homozygous for O-fut14R6 or O-fut1R245A knock-in and wild-type cells, and we detected the O-Fut1 proteins with an anti-O-Fut1 antibody (Fig. 3) (24). The intensity of immunostaining was greatly reduced in the O-fut14R6 mutant cells, confirming the specificity of this antibody staining for the O-Fut1 protein, at 25 °C (Fig. 3, A–A″) (24). The level of the O-Fut1 protein derivative in the O-fut1R245A knock-in homozygous cells was slightly lower than that of the wild-type O-Fut1 protein in wild-type cells at 25 °C (Fig. 3, B–B″). However, no further reduction was observed at 30 °C (Fig. 3, B–C″), suggesting that the temperature-sensitive phenotype of O-fut1R245A knock-in was not caused by a decrease in O-Fut1 protein. Note that an anti-O-Fut1 antibody that can detect the endogenous O-Fut1 protein on Western blots was not available, so we were unable to confirm this conclusion by quantitative biochemical approaches.
FIGURE 3.
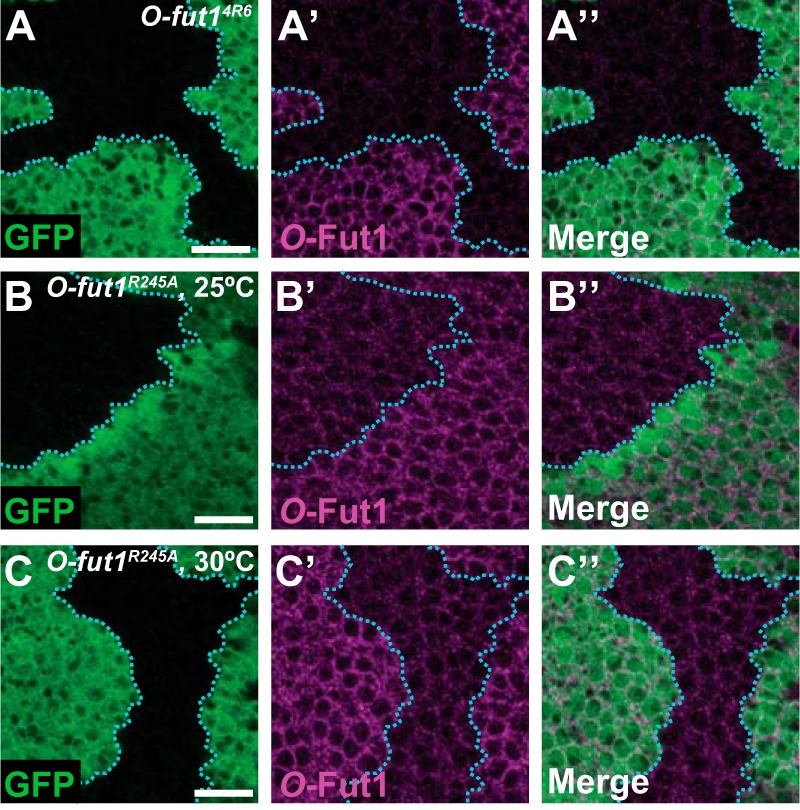
O-Fut1 protein level in O-fut1R245A knock-in homozygous cells is the same at 25 and 30 °C. A–C″, wing discs with O-fut14R6 (A–A″) and O-fut1R245A knock-in (B–C″) mosaic clones, raised at 25 °C (A–B″) or 30 °C (C–C‴), were stained with an anti-O-Fut1 antibody (magenta in A′, A″, B′, B″, and C′, C″). Mosaic clones were identified by the absence of GFP (green in A, A″, B, B″, C, and C″). A″, B″, and C″ show merged images of A and A′, B and B′, and C and C′, respectively. Boundaries of clones are indicated by blue broken lines. Scale bars, 10 μm.
A null mutation of O-fut1, O-fut14R6, is known to result in a neurogenic phenotype even at 25 °C (17). Therefore, the largely normal phenotype of the O-fut1R245A knock-inm/z embryos at this temperature may be attributable to an enzyme activity-independent function of O-Fut1.
O-Fucose Monosaccharide on the EGF-like-repeats of N Has a Temperature-sensitive Role in N Signaling, Which Is Independent of Its Further GlcNAc Modification by Fng
There are two possible explanations for the temperature-sensitive property of the O-fut1R245A knock-in mutation. First, the monosaccharide O-fucose modification of N may function in a temperature-sensitive manner. Second, the activity of the O-fut1R245A knock-in gene and/or its product may be temperature-sensitive. To distinguish between these possibilities, we examined whether mutations of other genes that abolish the O-fucosylation of N also show a temperature-sensitive neurogenic phenotype.
Fucosylation requires GDP-fucose as the donor of fucose. In Drosophila, GDP-fucose is synthesized from GDP-mannose only through the de novo synthetic pathway (25, 32, 33, 60). GDP-mannose 4,6-dehydratase (Gmd) and GDP-4 keto-6 deoxy-d-mannose epimerase/reductase (Gmer) are essential enzymes for this pathway (61). Therefore, null mutations of these genes, such as GmdH78and GmerSH, abolish GDP-fucose synthesis in Drosophila (25, 33).
We observed embryos homozygous for GmdH78 or GmerSH lacking their maternal contributions (GmdH78m/z and GmerSHm/z). We found that most of the GmdH78m/z (2/19 embryos showed the neurogenic phenotype) and GmerSHm/z (0/26 embryos showed the neurogenic phenotype) embryos had a normal nervous system at 25 °C (Fig. 2, E, G, and K). However, as found in the O-fut1R245A knock-inm/z embryos, at 30 °C the neurogenic phenotype was highly penetrant in embryos with either the GmdH78m/z (18/18 neurogenic embryos) or the GmerSHm/z (23/25 neurogenic embryos) mutation (Fig. 2, F, H, and K). In these embryos, various fucose modifications, including the fucosylation of N-glycan, should be abolished. However, the collective results obtained from the O-fut1R245A knock-inm/z, GmdH78m/z, and GmerSHm/z embryos indicated that the requirement for the monosaccharide O-fucose modification of N might be temperature-sensitive.
A contribution of fng to lateral inhibition has not been reported (22). However, to exclude the possibility that the temperature-sensitive neurogenic phenotype was caused by the lack of GlcNAc modification of O-fucose by Fng, we generated embryos homozygous for fng13 and lacking its maternal contribution (fng13m/z). The fng13m/z embryos did not show the neurogenic phenotype at either 25 °C (n = 14) or 30 °C (n = 15) in any case examined (Fig. 2, I–K). This finding indicated that the absence of GlcNAc modification by Fng is irrelevant to the temperature-sensitive neurogenic phenotype observed in the O-fut1R245A knock-inm/z, GmdH78m/z, and GmerSHm/z embryos. Together, these findings indicate that the O-fucose monosaccharide modification of N is essential for N signaling during lateral inhibition at 30 °C but not at 25 °C.
O-Fucose Monosaccharide Modification of N Is Generally Required to Activate N Signaling at 30 °C
We next examined whether the O-fucose monosaccharide modification of N is generally required for N signaling at 30 °C. For this analysis, we generated somatic mosaic clones homozygous for O-fut1R245A knock-in in the wing discs of third-instar larvae, using the FLP/FRT system (Fig. 4, B–C′). The expression of cut along the D/V boundary of wild-type wing discs is induced by N signaling (Fig. 4A). In the mosaic wing discs, cut was also ectopically expressed along the boundaries of the O-fut1R245A knock-in somatic homozygous clones (marked by the absence of GFP) located in the dorsal compartment, at 25 °C (Fig. 4, B and B′). This phenotype was similar to that of somatic mosaic clones homozygous for fng13 (Fig. 4, D–E′) at 25 and 30 °C (62). Therefore, the absence of monosaccharide O-fucose had a similar defect in cut activation at 25 °C as did the absence of GlcNAc on O-fucose. This result is consistent with the previous conclusion that O-fucose monosaccharide does not have a specific function in N signaling at 25 °C (24). However, at 30 °C, the ectopic expression of cut along the boundaries of somatic mosaic clones homozygous for O-fut1R245A knock-in was abolished (Fig. 4, C and C′), supporting the idea that the monosaccharide form of O-fucose plays an essential role in N signaling in various developmental contexts at 30 °C. We also noted that the monosaccharide O-fucose was cell-autonomously required for the activation of N signaling at 30 °C (Fig. 4, C and C′), which is consistent with the idea that the O-fucose monosaccharide attached to N is essential for N′s activity at this temperature.
FIGURE 4.
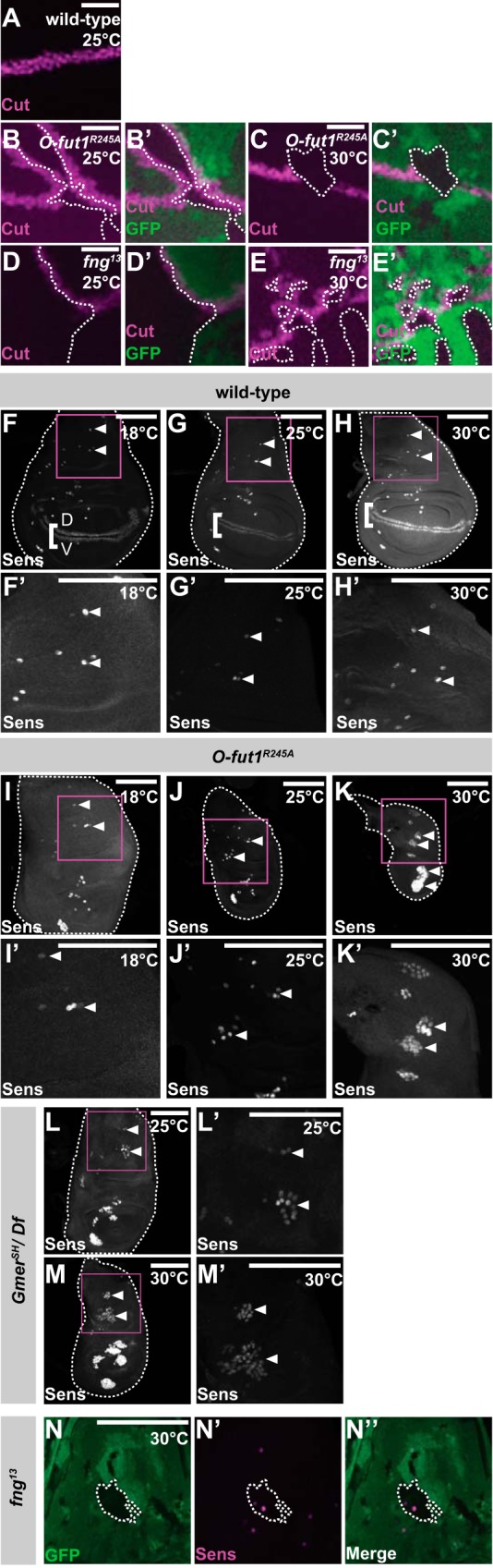
O-Fucose monosaccharide modification of N is generally required to activate N signaling at 30 °C. A–E′, anti-Cut antibody staining (magenta) of wing discs with wild-type (A), O-fut1R245A knock-in (B–C′), or fng13 (D–E′) mosaic clones. B–E′, mosaic clones were identified by the absence of GFP (green). Wing discs were isolated from larvae raised at 25 °C (A, B, B′, D, and D′) or 30 °C (C, C′, E, and E′). B′, C′, D′, and E′ show merged images of GFP and Cut staining from B to E. Boundaries of mosaic clones are indicated by broken lines. F–M′, SOPs in wing discs were detected by anti-Sens antibody staining. F–H′, wild-type wing discs were isolated from larvae raised at 18 °C (F and F′), 25 °C (G and G′), or 30 °C (H and H′). Along the D/V boundary, the expression of sens, which is induced by fng-dependent N signaling, is shown by white brackets in F–H. I–M′, SOPs in wing discs of O-fut1R245A knock-in homozygotes (I–K′) and of heterozygotes between GmerSH and a deletion uncovering the Gmer locus (GmerSH/Df) (L–M′) are shown. Wing discs were isolated from larvae raised at 18 °C (I and I′), 25 °C (J, J′, L, and L′), or 30 °C (K, K′, M, and M′). F′–M′ show magnified views of the magenta squares in F–M, respectively. Arrowheads indicate SOPs in F–M′. Wing discs are outlined by broken lines. N–N″, SOPs in wing discs with fng13 homozygote mosaic clones (detected by the absence of GFP, shown in green) were detected by anti-Sens antibody staining (magenta). Boundaries of mosaic clones are indicated by broken lines. Wing discs were isolated from larvae raised at 30 °C. N″ shows merged images of N and N′. Scale bars, 20 μm for A–E, and 100 μm for F–N.
To obtain more evidence for this idea, we examined the formation of sensory organ precursors (SOPs) in the wing discs of third-instar larvae homozygous for O-fut1R245A knock-in. A single SOP is selected from each proneural cluster in the wing disc, where SOPs arise in a well defined pattern at the third instar (63). In wild-type wing discs, the SOP formation pattern, observed by anti-Senseless (Sens) staining, was essentially the same from 18 to 30 °C (Fig. 4, F–H′). In the wing discs of O-fut1R245A knock-in homozygotes, SOPs did not form along the D/V boundary, because this SOP formation depends on the expression of wg, which is induced by the fng-dependent activation of N signaling (Fig. 4, I–K) (62). At 18 and 25 °C, the number of SOPs elsewhere in the wing discs increased slightly (Fig. 4, I–J′). However, these SOPs markedly increased in these wing discs at 30 °C (Fig. 4, K and K′). These results suggest that lateral inhibition was disrupted in the wing discs of O-fut1R245A knock-in homozygotes in a temperature-sensitive manner, as found in the embryonic central nervous system.
We also examined the SOP formation in the wing discs in the absence of Gmer function. A trans-heterozygote of GmerSH and a deletion mutant uncovering the Gmer locus survived until the third-instar larval stage, when we could isolate the wing discs to study SOP formation (33). At 25 °C, the number of SOPs increased slightly in these wing discs, although they did not form along the D/V boundary due to the absence of N signaling there (Fig. 4, L and L′). Nevertheless, the SOPs, except for those located at the D/V boundary, noticeably increased at 30 °C (Fig. 4, M and M′). We also confirmed that the SOP number was not affected in somatic mosaic clones homozygous for fng13 in the wing discs of third-instar larvae, even at 30 °C (Fig. 4, N–N″). Based on these results, we concluded that the monosaccharide form of O-fucose is generally required for N signaling at 30 °C, although we could not exclude the possibility that some exceptions exist.
O-Fucose Monosaccharide Modification of N Functions Upstream of the Membrane-tethered Form of Activated N at 30 °C
To assess the function of the O-fucose monosaccharide modification of N′s EGF-like repeats, we performed a set of epistasis analyses involving O-fut1R245A knock-in and various derivatives of N. We used the MARCM system (43) to produce full-length N (NFL), a membrane-tethered form of activated N (NΔE), and the N intracellular domain (NICD) in somatic mosaic clones homozygous for O-fut1R245A knock-in at 30 °C (Fig. 5). In wild-type wing discs, the overexpression of NFL, NΔE, or NICD resulted in the ectopic induction of cut at 30 °C (Fig. 5, A, C, and E). However, the overexpression of NFL failed to induce ectopic cut expression in the somatic clones homozygous for O-fut1R245A knock-in at 30 °C (Fig. 5B). Thus, O-fut1R245A knock-in was epistatic to NFL under this condition. By contrast, the overexpression of NΔE or NICD still induced the ectopic expression of cut in somatic mosaic clones homozygous for O-fut1R245A knock-in at 30 °C (Fig. 5, D and F). Therefore, the two activated forms of N, NΔE and NICD, were epistatic to O-fut1R245A knock-in under this condition. Based on these epistasis analyses, we speculated that the monosaccharide O-fucose modification of N is required downstream of full-length N and upstream of the S3 cleavages of N.
FIGURE 5.
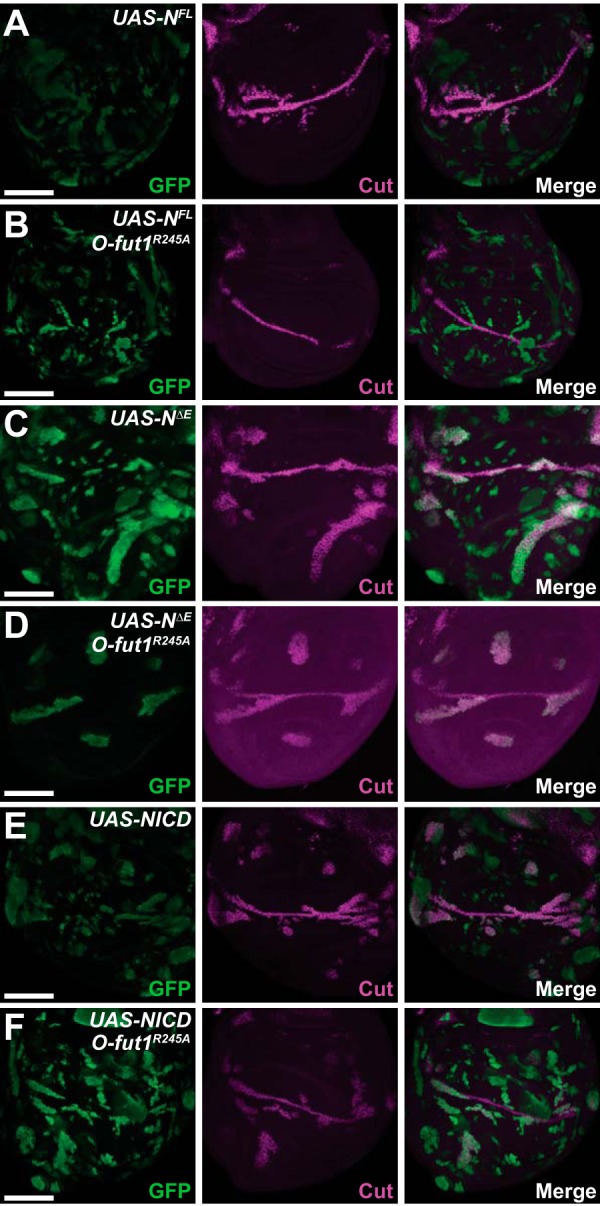
O-Fucose monosaccharide modification of N functions upstream of the membrane-tethered form of activated N. A–F, expression of cut in the wing discs of third-instar larvae was detected by anti-Cut antibody staining (magenta in A–F). A and B, mosaic clones overexpressing full-length N (NFL) (green in A and B) in wild-type (A) or O-fut1R245A knock-in homozygote mosaic clones (B). C and D, mosaic clones overexpressing a membrane-tethered form of activated N (NΔE) (green in C and D) in wild-type (C) or O-fut1R245A knock-in homozygote mosaic clones (D). E and F, mosaic clones overexpressing the intracellular domain of N (nuclear form of activated N) (NICD) (green in E and F) in wild-type (E) or O-fut1R245A knock-in homozygote mosaic clones (F). Wing discs were isolated from larvae cultured at 30 °C. Scale bars, 100 μm.
O-Fucose Monosaccharide Modification of N Is Required for the Proper Localization of N at 30 °C
To understand how the monosaccharide O-fucose modification of N affects N signaling, we compared N′s subcellular localization at 25 and at 30 °C in somatic mosaic clones homozygous for O-fut1R245A knock-in in the wing discs of third-instar larvae (Fig. 6). In epithelial cells in wild-type wing discs, most of the N protein is detected in the sub-apical region and the adherence junctions at 25 °C, although it is also detected in intracellular vesicles (26, 64).
FIGURE 6.
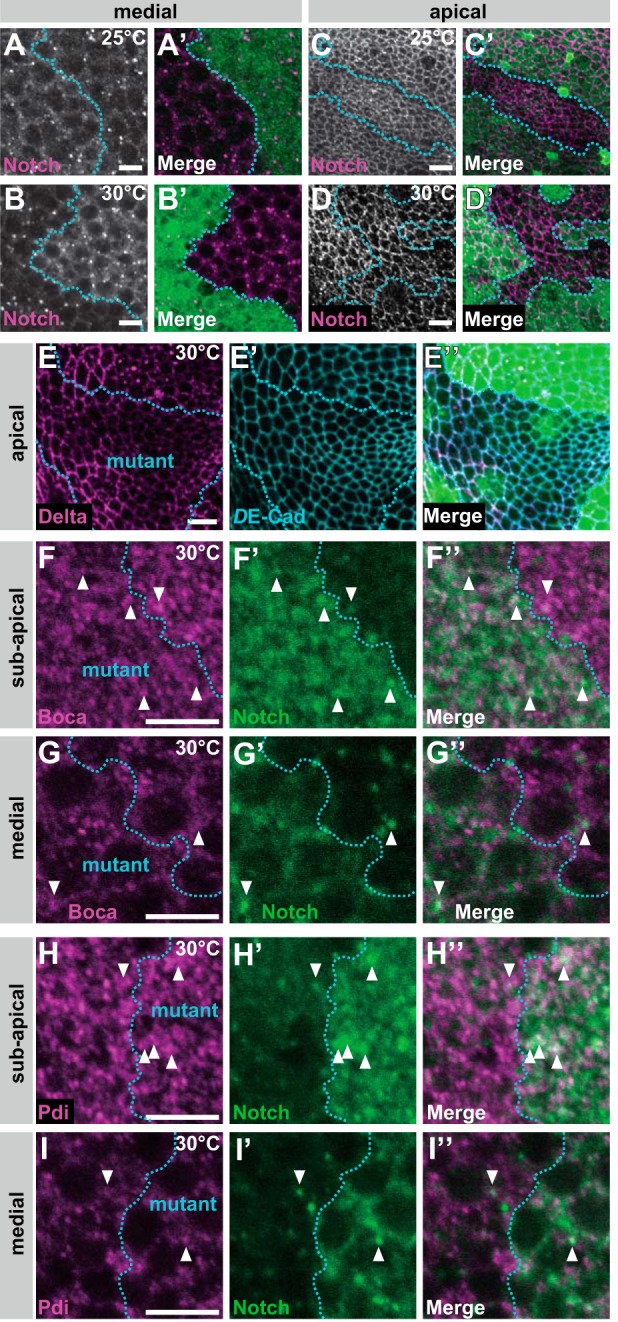
O-Fucose monosaccharides on N are required for the proper localization of N at 30 °C. A–D′, wing discs with mosaic clones homozygous for O-fut1R245A knock-in, indicated by the absence of GFP (A′, B′, C′, and D′), were isolated from larvae raised at 25 °C (A, A′, C, and C′) or 30 °C (B, B′, D, and D′). Permeabilized (A–B′; medial plane) and nonpermeabilized (C–D′; apical plane) wing discs were stained with antibodies against N′s intracellular and extracellular domains, respectively. Boundaries of clones are indicated by broken blue lines. A′, B′, C′, and D′ show merged images of N and GFP staining in A–D. E–I″, Wing discs with mosaic clones homozygous for O-fut1R245A knock-in; regions of mutants are outlined by broken blue lines. Wing discs isolated from larvae raised at 30 °C were permeabilized before antibody staining. E–E″, distributions of Delta (magenta in E and E″) and DE-Cad (blue in E′ and E″) at the apical plane are shown. E″ shows merged images of GFP, Delta (E), and DE-Cad staining (E′). F–G″, intracellular distributions of Boca (magenta in F, F″, G, and G″) and N (green in F′, F″, G′, and G″) were detected by immunostaining. F–F″ and G–G″ show optical sections of sub-apical and medial regions, respectively. F″ and G″ show merged images of F and F′ and G and G′, respectively. Arrowheads indicate vesicles with N and Boca co-localization. H–I″, intracellular distributions of Pdi (magenta in H, H″, I, and I″) and N (green in H′, H″, I′, and I″) were detected by immunostaining. H–H″ and I–I″ show optical sections of the sub-apical and medial regions, respectively. H″ and I″ show merged images of H and H′ and I and I′, respectively. Arrowheads indicate vesicles with N and Pdi co-localization. Scale bars, 5 μm.
To detect total cellular N protein, we stained permeabilized wing discs with an antibody against N′s intracellular domain (Fig. 6, A–B′), which showed N distribution at the optical plane corresponding to the middle of the epithelial cells at 25 °C (Fig. 6, A and A′) and 30 °C (Fig. 6, B and B′). At 25 °C, there were no marked abnormalities in the intracellular N distribution in somatic mosaic clones homozygous for O-fut1R245A knock-in (Fig. 6, A and A′); however, at 30 °C, N accumulated inside the cell (Fig. 6, B and B′) although other membrane proteins, such as Dl and DE-cadherin (DE-Cad), were distributed normally under the same conditions (Fig. 6, E–E″).
To visualize N protein delivered to the epithelial cell surface at the sub-apical region, nonpermeabilized wing discs carrying somatic mosaic clones homozygous for O-fut1R245A knock-in were stained with an antibody against the extracellular domain of N (Fig. 6, C–D′) (65). The amount of cell-surface N at the sub-apical plane was the same at 25 or 30 °C in cells homozygous for O-fut1R245A knock-in under this condition (Fig. 6, C–D′). Thus, even at 30 °C, N delivery to the cell surface was not disrupted in cells homozygous for O-fut1R245A knock-in, although N accumulated within these cells (Fig. 6, B and B′). However, intracellular and cell-surface N protein did not accumulate in somatic mosaic clones homozygous for fng13 at 25 or 30 °C, suggesting that the intracellular accumulation of N could not be attributed to the GlcNAc modification of O-fucose (data not shown).
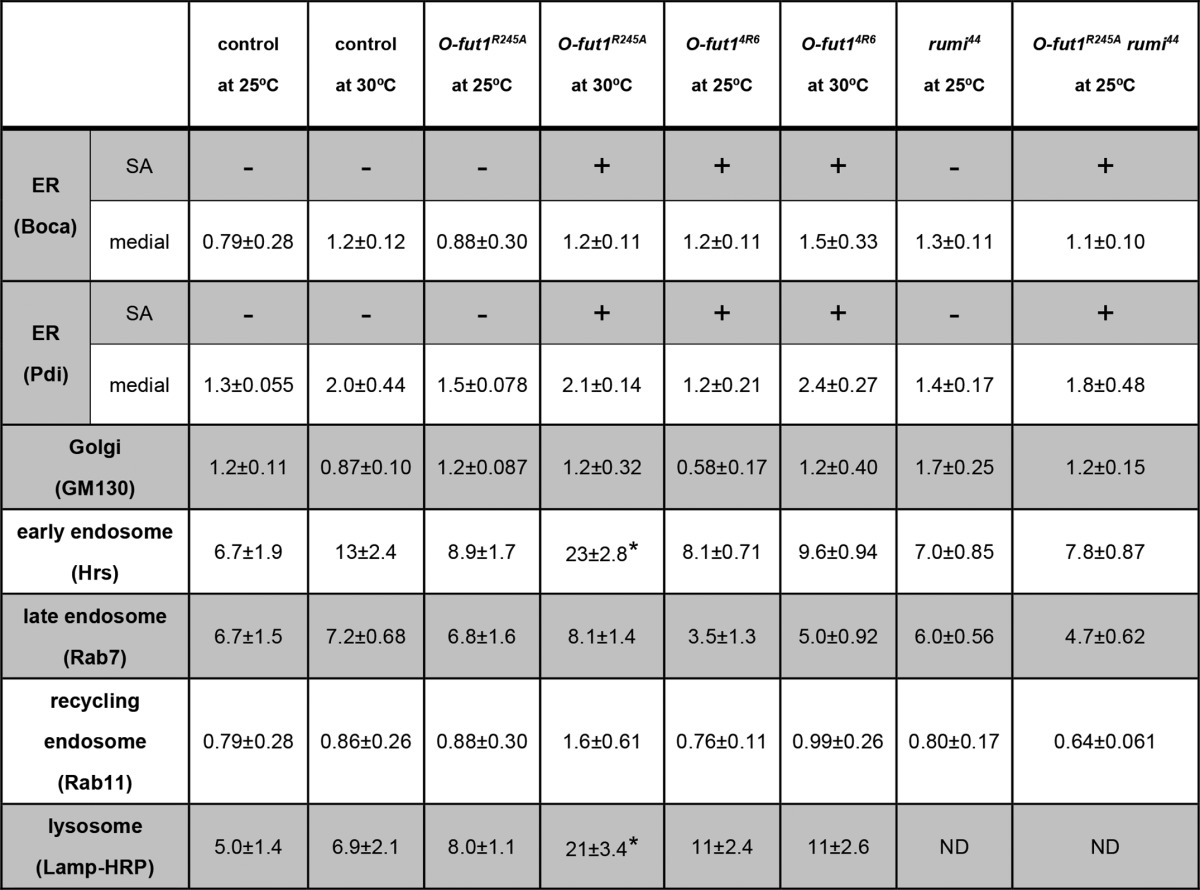
We next determined the intracellular compartments in which N accumulated in the somatic mosaic clones homozygous for O-fut1R245A knock-in at 30 °C. We identified the ER (Boca and Pdi), Golgi (GM130), early endosomes (Hrs), late endosomes (Rab7), recycling endosomes (Rab11), and lysosomes (Lamp) by immunostaining (25, 36, 51, 52, 54). In wing-disc epithelial cells, the ER was detected either as an indistinct and shapeless structure (Fig. 6, F–F″ and H–H″) or as dots (Fig. 6, G–G″ and I–I″). The former was detected mostly in the cells' apical regions (Fig. 6, F–F″ and H–H″), and the latter in more basal regions (Fig. 6, G–G″ and I–I″). These structures were recognized by antibodies against two ER-marker proteins, Boca (Fig. 6, F–G″) and Pdi (Fig. 6, H–I″). To analyze the co-localization of N and markers for various intracellular compartments quantitatively, the percentage of vesicles positive for each marker that was also positive for N is shown in Table 1. At 30 °C, the distribution of accumulated N protein overlapped partly with the indistinct and shapeless ER in somatic mosaic clones homozygous for O-fut1R245A knock-in (white arrowhead in Fig. 6, F–F″, and H–H″, and Table 1), although hardly any N protein was detected in this structure in O-fut1R245A knock-in/+ (control) cells (Fig. 6, F–F″, and H–H″, and Table 1). However, there was no increase in N-protein level in the dot-structure ER in the cells homozygous for O-fut1R245A knock-in, compared with this ER in O-fut1R245A knock-in/+ cells at 30 °C (white arrowheads in Fig. 6, G–G″, and I–I″, and Table 1). Therefore, N accumulation in the indistinct and shapeless ER structures but not the dot-shaped ER structures may coincide with the loss of N signaling activity.
TABLE 1.
Quantitative analysis of co-localization between N protein and various markers of intracellular compartments in O-fut1R245A knock-in or O-fut14R6 mutant cells, or O-fut1R245A knock-in and rumi double-mutant cells
Somatic mosaic clones homozygous or double homozygous for the indicated mutants were induced in wing discs of larvae cultured at 25 or 30 °C, as indicated. Control cells heterozygous for O-fut1R245A knock-in were in the wing discs with somatic mosaic clones of O-fut1R245A knock-in homozygotes. The percentage of vesicles positive for each marker that were also positive for N is shown. The ER was detected with two markers, Boca and Pdi, at two different optical planes, sub-apical (SA) and medial. The ER showed an indistinct and shapeless structure at the sub-apical plane. N protein distribution with Boca or Pdi is indicated by + (co-localizing) or − (not co-localizing). The MARCM system was used to express UAS-Lamp-HRP. All the N-positive vesicles and vesicles positive for each marker were counted in 32 × 32-μm square areas of representative Z-series confocal images. Three to six independent images were analyzed for each experiment. Asterisks show statistically significant results (p < 0.05), compared with control. Standard errors are shown, following ± symbol. ND means not determined.
Although N is reported to accumulate in the ER in somatic clones homozygous for O-fut14R6, a null mutant of O-fut1 (17), there are conflicting reports (23, 25). Here, we found that accumulated N partially overlapped with indistinct, shapeless ER structures, although there was no change of the amount of N in dot-shaped ER at 25 or 30 °C (Fig. 7, A–D‴, and Table 1). This may account for the previous discrepancy in the ER accumulation of N reported in O-fut14R6 mutant cells. Nonpermeabilized staining did not detect N protein at the sub-apical cell-surface region in O-fut14R6 mutant epithelial cells at 25 or 30 °C; this agreed with previously reported findings at 25 °C (data not shown) (23, 25).
FIGURE 7.
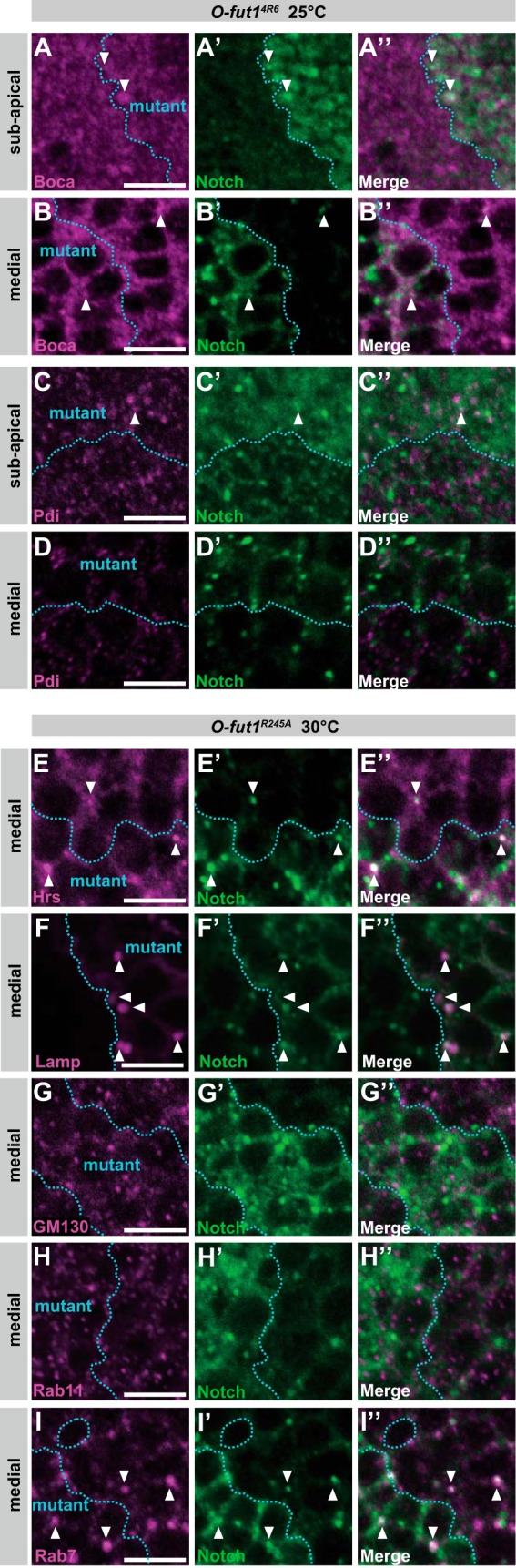
Co-localization of intracellular vesicular compartment markers with N in O-fut1R245A somatic mosaic clones. A–D″, wing discs with O-fut14R6 homozygote mosaic clones. Regions of mutants outlined by broken blue lines were double-stained with an anti-Boca (magenta in A, A″, B, and B″) or anti-Pdi (C, C″, D, and D″) antibody and an anti-N antibody (green in A′, A″, B′, B″, C′, C″, D′, and D″). Wing discs were isolated from larvae raised at 25 °C. Optical sections of sub-apical (A–A″ and C–C″) and medial regions (B–B″ and D–D″) are shown. A″–D″ show merged images from A–D and A′–D′, respectively. E–I″, wing discs with mosaic clones homozygous for O-fut1R245A knock-in; regions of mutants are outlined by broken blue lines. Wing discs isolated from larvae raised at 30 °C were permeabilized before antibody staining. E–E″, intracellular distributions of Hrs (magenta in E and E″) and N (green in E′ and E″) were detected by immunostaining at the medial plane. E″ shows merged images from E and E′. Arrowheads indicate vesicles with N and Hrs co-localization. F–F″, using the MARCM system, UAS-Lamp-HRP was overexpressed in mosaic clones homozygous for O-fut1R245A knock-in, which were detected by the presence of Lamp-HRP protein (magenta in F and F″). The intracellular distribution of N was detected by immunostaining (green in F′ and F″). F″ shows merged images from F and F′. Arrowheads indicate vesicles with N and Lamp-HRP co-localization. G–G″, intracellular distributions of GM130 (magenta in G and G″) and N (green in G′ and G″) were detected by immunostaining at the medial plane. G″ shows merged images from G and G′. H–H″, intracellular distributions of Rab11 (magenta in H and H″) and N (green in H′ and H″) were detected by immunostaining at the medial plane. H″ shows merged images from H and H′. I–I″, intracellular distributions of Rab7 (magenta in I and I″) and N (green in I′ and I″) were detected by immunostaining at the medial plane. I″ shows merged images from I and I′. Arrowheads indicate vesicles where N co-localized with rab7. Scale bars, 5 μm.
We also observed that N protein was detected more often in early endosomes (23 ± 2.8) and lysosomes (21 ± 3.4) in somatic mosaic clones homozygous for O-fut1R245A knock-in at 30 °C, compared with those at 25 °C or with control cells (13 ± 2.4 and 4.4 ± 0.59, respectively) (Fig. 7, E–F″ and Table 1). These differences were statistically significant (t test, p < 0.05). In contrast, there was no marked difference in N distribution in the Golgi, late endosomes, or recycling endosomes of these cells at either temperature (Fig. 7, G–I″ and Table 1). These data suggest that the O-fucose monosaccharide modification of N may be required for specific step(s) of N′s trafficking at 30 °C but not at 25 °C. In addition, the distinct defects in N′s trafficking found in O-fut1R245A knock-in mutant cells versus O-fut14R6 mutant cells suggest that the nature of the defect in these two mutants is different (Table 1). These observations further support our idea that the temperature-sensitive phenotype of O-fut1R245A knock-in is not due to a temperature-dependent loss of O-fut1 activity, associated with, for example, the breakdown of the O-Fut1R245A mutant protein (Fig. 3). To detect possible alterations in the N protein in the O-fut1R245A knock-in mutant, including changes in its stability and processing, we detected the N protein in O-fut1R245A knock-in homozygous wing discs from the third-instar larvae cultured at 25 and 30 °C, by Western blot (Fig. 8). However, we did not observe any marked differences in the amount or size of the N protein between the mutant and the wild-type wing discs (compare lanes 1 and 2 and lanes 3 and 4 in Fig. 8).
FIGURE 8.

Amount and size of N protein detected by Western blot are not altered in O-fut1R245A knock-in homozygote wing imaginal discs. N protein was detected by Western blot using an anti-N antibody (C17.9C6) (upper panel). Protein extracts were prepared from wing discs isolated from third-instar larvae. Lane 1, wild-type cultured at 25 °C; lane 2, wild-type cultured at 30 °C; lane 3, O-fut1R245A knock-in homozygote cultured at 25 °C; lane 4, O-fut1R245A knock-in homozygote cultured at 30 °C. As a loading control, α-tubulin was detected (lower panel). Positions of molecular mass markers are shown at right (kDa).
O-Fucose and O-Glucose Modifications of N Function Redundantly
The EGF-like repeats of N have consensus sequences not only for O-fucose but also for O-glucose modifications (27). O-Glucose is added by Rumi, a protein O-glucosyltransferase in Drosophila (27, 28). In the absence of rumi function, temperature-sensitive phenotypes associated with the loss of N signaling, which are similar to those of O-fut1R245A knock-in, are observed (27, 28). In somatic mosaic clones homozygous for rumi44, a null mutation, N accumulated intracellularly, which was also reminiscent of a defect in O-fut1R245A knock-in homozygous cells. Therefore, we thought that the O-fucose monosaccharide modification of N might function redundantly with the O-glucose glycan modification of N in N signaling. To test this possibility, we generated a rumi and O-fut1R245A knock-in double mutant and observed SOP formation in the wing discs of third-instar larvae. As described above, the number of SOPs increased at 30 °C in wing discs homozygous for rumi44, but it was normal at 18 and 25 °C (Fig. 9, A–C′). This phenotype is very similar to that of O-fut1R245A knock-in wing discs (Fig. 4, I–K′). However, the number of SOPs increased in the wing discs homozygous for rumi44 and O-fut1R245A knock-in even at 18 and 25 °C, suggesting that N signaling was abolished under these conditions (Fig. 9, D–E′). These results suggest that the O-fucose monosaccharide and O-glucose glycans have a redundant function during lateral inhibition that controls the number of SOPs in the wing discs.
FIGURE 9.
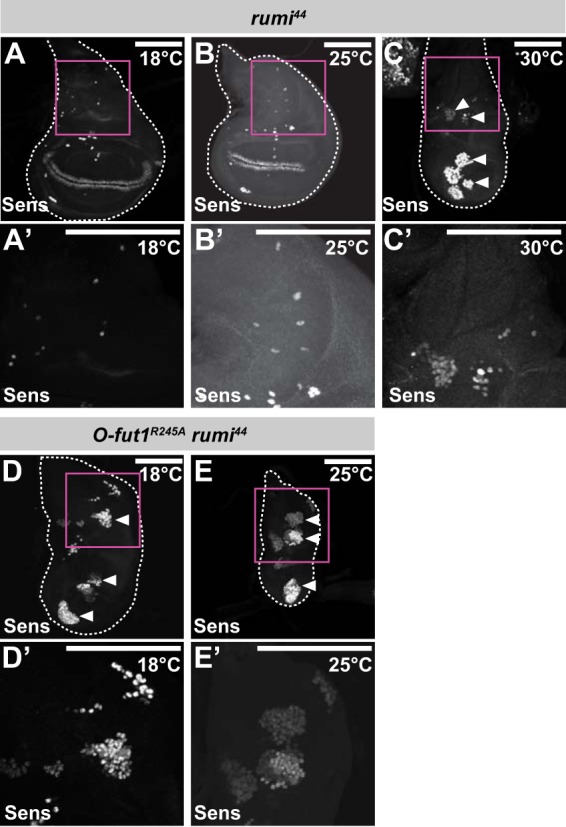
O-Fucose and O-glucose modifications of N function redundantly in the activation of N signaling. A–E′, SOPs in wing discs were detected by anti-Sens antibody staining. SOPs in rumi44 homozygote wing discs were isolated from larvae raised at 18 °C (A and A′), 25 °C (B and B′), or 30 °C (C and C′). SOPs in wing discs homozygous for O-fut1R245A knock-in and rumi44 were isolated from larvae raised at 18 °C (D and D′) or 25 °C (E and E′). Arrowheads indicate hyperplastic SOPs. Outlines of wing discs are shown by broken lines. A′–E′ show magnified views of the areas in magenta squares in A–E, respectively. Scale bars, 100 μm.
Similar redundancy in the function of the O-fucose monosaccharide and O-glucose glycans was observed in the activation of N signaling along the D/V boundary of the wing disc at the third instar. The expression of cut along the D/V boundary disappeared at 30 °C in somatic mosaic clones homozygous for rumi44 (Fig. 10, B–B″), but cut's expression was normal at 25 °C (Fig. 10, A–A″). The phenotype of the somatic mosaic clones homozygous for rumi44 at 30 °C was very similar to that of the somatic mosaic clones homozygous for O-fut1R245A knock-in at 30 °C (Fig. 4, C and C′). However, in somatic mosaic clones homozygous for rumi44 and O-fut1R245A knock-in, cut expression was abolished even at 25 °C (Fig. 10, C–C″).
FIGURE 10.
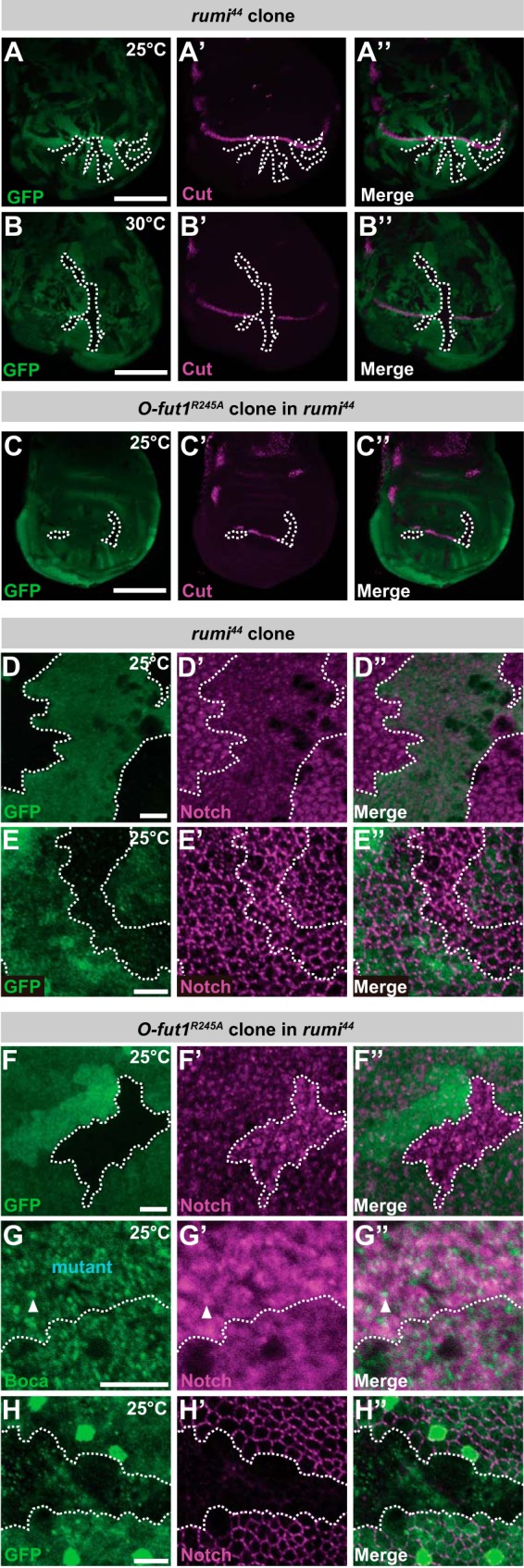
Monosaccharide O-fucose and O-glucose glycan of N function redundantly in N signaling. A–B″, anti-Cut antibody staining (magenta) of third-instar wing discs with mosaic clones homozygous for rumi44 (indicated by the absence of GFP, shown in green). The wing discs were isolated from larvae raised at 25 °C (A–A″) or 30 °C (B–B″). C–C″, anti-Cut antibody staining (magenta) of third-instar wing discs homozygous for rumi44 with mosaic clones homozygous for O-fut1R245A knock-in (indicated by the absence of GFP, shown in green). The wing discs were isolated from larvae raised at 25 °C. The boundaries of mosaic clones are indicated by broken white lines. A″, B″, and C″ show merged images of A, B, and C and A′, B′, and C′, respectively. D–H″, permeabilized (D–D″ and F–G″) or nonpermeabilized (E–E″ and H–H″) wing discs were stained with antibodies against N′s intracellular (D–D″ and F–G″) or extracellular domains (E–E″ and H–H‴). D–E″, wing discs with mosaic clones homozygous for rumi44 (indicated by the absence of GFP, shown in green). F–H″, wing discs of a rumi44 homozygote with mosaic clones homozygous for O-fut1R245A knock-in (indicated by the absence of GFP, shown in green in F, F″, H, and H″ or labeled “mutant” in G). Borders of mosaic clones are indicated by broken white lines. Scale bars, 100 μm for A–C″ and 5 μm for D–H″.
The O-fucose monosaccharide and O-glucose glycans on the EGF-like repeats of N also collaborated to increase the amount of N at the sub-apical plasma membrane. As reported previously, slight accumulations of N protein were found in cells homozygous for rumi44 alone, at 25 °C (Fig. 10, D–D″), although the amount of cell-surface N at the sub-apical region, detected by nonpermeabilized staining, was not altered in these cells (Fig. 10, E–E″) (27). However, the N-protein accumulations were more prominent in intracellular compartments in somatic mosaic clones homozygous for rumi44 and O-fut1R245A knock-in (Fig. 10, F–F″). In these double mutant cells, the accumulated N partially co-localized with ER markers (Fig. 10, G–G″, and Table 1). Curiously, nonpermeabilized staining for N revealed that the cell-surface N was diminished in these cells (Fig. 10, H–H″), but it was unaffected in cells homozygous for either O-fut1R245A knock-in (Fig. 6, C–D′) or rumi44 (Fig. 10, E–E″) (27). Therefore, O-fucose monosaccharide and O-glucose glycans have redundant roles, and in their absence N was not delivered to or stably maintained at the plasma membrane.
DISCUSSION
The functions of the monosaccharide O-fucose on the EGF-like repeats of N have been vague. In Drosophila, the monosaccharide O-fucose was proposed to serve primarily as an acceptor for Fng, with O-Fut1 being required as a specific chaperon for N (23, 24). However, in mammalian cells, the O-fucose modification was reported to be required for the optimal activation of N signaling under certain conditions (55). More recently, Fng-independent functions of fucosylation in Drosophila N signaling were reported, although these effects are relatively mild or cell type-specific (66, 67). In this study, we revealed that the O-fucose monosaccharide, independent of GlcNAc modification by Fng, is essential for N signaling at 30 °C in various developmental contexts in Drosophila. Moreover, in the absence of modifications with both O-fucose monosaccharide and O-glucose glycan, N signaling was abolished even at 25 °C, although the loss of either modification alone caused this phenotype only at 29–30 °C (27). The loss of N signaling coincides with a severe reduction of N at the sub-apical plasma membrane and an accumulation of N in a particular fraction of the ER (Figs. 10, F–G″ and 11A). These results suggest that O-fucose monosaccharide and O-glucose glycan modifications of N have redundant and collaborative roles in the activation of N signaling and the proper localization of N at the sub-apical plasma membrane (Fig. 11A). However, at this point we could not distinguish between whether N failed to reach the plasma membrane or it failed to be stably maintained on the membrane under this double mutant condition.
FIGURE 11.
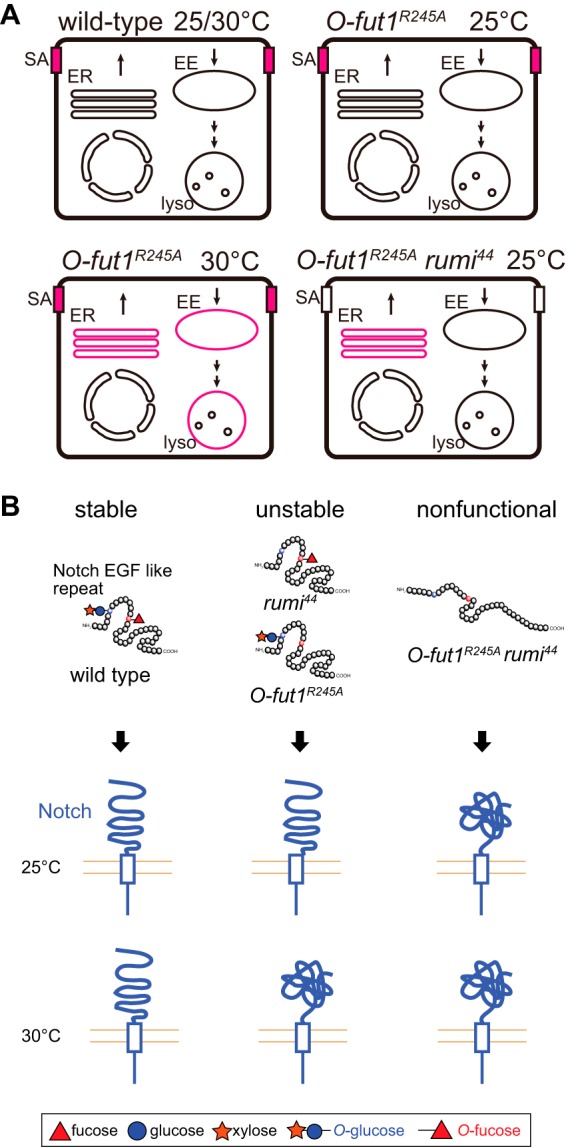
Model of O-fucose and O-glucose on N and N accumulation in various mutants. A, schematic diagram showing the intracellular compartment, in which N (magenta) is strongly accumulated in wild-type and various mutant cells. EE, early endosome; lyso, lysosome; SA, sub-apical region. B, schematic model of the redundant roles of O-fucose monosaccharide and O-glucose glycan modifications of the EGF-like repeats of N in the proper folding of the EGF-like repeats (upper), which may affect the entire extracellular structure of the full-length N (lower).
The biochemical roles of the O-fucose monosaccharide and O-glucose glycan modifications are still elusive. However, a previous NMR study revealed that the fucose moiety directly functions as a “bridge” in the formation of an antiparallel β-sheet in EGF-like repeat 12 of mouse Notch1, stabilizing its structure through an interaction between the fucose and the peptide backbone of the EGF-like repeat (68). Therefore, we speculate that structural destabilization of each EGF-like repeat may affect the folding of the full-length N (Fig. 11B). Deficient N folding may explain our observation that N accumulated in the ER of cells homozygous for O-fut1R245A knock-in at 30 °C (Fig. 11B). More severe folding defects may occur in the absence of both O-fucose monosaccharide and O-glucose glycan modifications (Fig. 11B). It was previously shown that O-Fut1 and Rumi may be involved in quality control of the N protein, because they both only modify properly folded EGF-like repeats (69–71). Therefore, it is possible that full-length N with EGF-like repeats lacking O-fucose monosaccharide and O-glucose glycan modifications may escape from its chaperon, leading to misfolding of the full-length N. However, at this point, the mechanism by which the potential destabilization of EGF-like repeats influences the global structure of full-length N is unknown. Moreover, it was previously shown that disrupting the O-fucosylation site in the 12th EGF-like repeat of mouse Notch1 does not affect the amount of cell-surface Notch1, but instead it reduces Notch1's activation, suggesting that the O-fucose modification of the 12th EGF-like repeat does not play a role in Notch1's folding (72). Therefore, it is possible that the O-fucose modification on other EGF-like repeats is required for the folding of full-length N and that the O-fucose modification of N has multiple functions in N signaling.
Considering that the O-fucose monosaccharide and O-glucose glycan modifications of the EGF-like repeats may function to increase the heat stability of full-length N, it is tempting to speculate that some sites for these modifications are specifically conserved in animals with higher body temperatures (homeothermic) but not in those with lower temperatures (poikilothermic). However, a comparison of the consensus sequences for these modifications failed to find a correlation between body temperature and the evolutionary conservation of these sites (Fig. 12).
FIGURE 12.

Predicted O-fucosylation and O-glucosylation sites in the EGF-like repeats of various N receptors. A, red bars show predicted O-fucosylation sites in the EGF-like repeats of various N proteins. B, blue bars show predicted O-glucosylation sites in the EGF-like repeats of various N proteins. The N proteins of poikilothermic and homoeothermic animals are grouped in the upper and lower boxes, respectively. Species are indicated at left.
A number of protein motifs contain multiple glycosylation sites for different types of glycan modifications (73). In this study, we found that the O-fucose monosaccharide and O-glucose glycan modifications of the EGF-like repeats of N have a redundant role, possibly in the folding of N. Thus, our results provide a case in which different types of glycosylation occurring at a protein motif may play a common and collaborative role.
Acknowledgments
We thank Hamed Jafer-Nejad, Jürgen Knoblich, David Bilder, the Bloomington Stock Center, and the Drosophila Genetic Resource Center for flies and Hugo Bellen, Kenneth D. Irvine, Richard S. Mann, Akira Nakamura, and the Developmental Studies Hybridoma Bank for antibodies.
This work was supported by grants-in-aid from the Japanese Ministry of Education, Culture, Sports and Science (to K. M. and T. A.) and grants from PRESTO, Japan Science and Technology Agency (to K. M.).
- N
- Notch
- dpp
- decapentaplegic
- DE-Cad
- DE-cadherin
- Dl
- Delta
- D/V
- dorsal and ventral
- ER
- endoplasmic reticulum
- Fng
- Fringe
- Gmer
- GDP-4 keto-6 deoxy-d-mannose epimerase/reductase
- Gmd
- GDP-mannose 4,6-dehydratase
- m/z
- maternal and zygotic
- NΔE
- a membrane-tethered form of activated Notch
- NFL
- full-length Notch
- NICD
- Notch intracellular domain
- O-Fut1
- O-fucosyltransferase 1
- Sens
- Senseless
- SOP
- sensory organ precursor
- Wg
- Wingless
- MARCM
- mosaic analysis with a repressible cell marker.
REFERENCES
- 1. Artavanis-Tsakonas S., Rand M. D., Lake R. J. (1999) Notch signaling: cell fate control and signal integration in development. Science 284, 770–776 [DOI] [PubMed] [Google Scholar]
- 2. Bray S. J. (2006) Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 7, 678–689 [DOI] [PubMed] [Google Scholar]
- 3. Kopan R., Ilagan M. X. (2009) The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fortini M. E. (2009) Notch signaling: the core pathway and its posttranslational regulation. Dev. Cell 16, 633–647 [DOI] [PubMed] [Google Scholar]
- 5. Joutel A., Corpechot C., Ducros A., Vahedi K., Chabriat H., Mouton P., Alamowitch S., Domenga V., Cécillion M., Marechal E., Maciazek J., Vayssiere C., Cruaud C., Cabanis E. A., Ruchoux M. M., Weissenbach J., Bach J. F., Bousser M. G., Tournier-Lasserve E. (1996) Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 383, 707–710 [DOI] [PubMed] [Google Scholar]
- 6. Gridley T. (2003) Notch signaling and inherited disease syndromes. Hum. Mol. Genet. 12, R9–R13 [DOI] [PubMed] [Google Scholar]
- 7. Weng A. P., Aster J. C. (2004) Multiple niches for Notch in cancer: context is everything. Curr. Opin. Genet. Dev. 14, 48–54 [DOI] [PubMed] [Google Scholar]
- 8. Moloney D. J., Shair L. H., Lu F. M., Xia J., Locke R., Matta K. L., Haltiwanger R. S. (2000) Mammalian Notch1 is modified with two unusual forms of O-linked glycosylation found on epidermal growth factor-like modules. J. Biol. Chem. 275, 9604–9611 [DOI] [PubMed] [Google Scholar]
- 9. Whitworth G. E., Zandberg W. F., Clark T., Vocadlo D. J. (2010) Mammalian Notch is modified by d-Xyl-α1–3-d-Xyl-α1–3-d-Glc-β1-O-Ser: implementation of a method to study O-glucosylation. Glycobiology 20, 287–299 [DOI] [PubMed] [Google Scholar]
- 10. Matsuura A., Ito M., Sakaidani Y., Kondo T., Murakami K., Furukawa K., Nadano D., Matsuda T., Okajima T. (2008) O-Linked N-acetylglucosamine is present on the extracellular domain of Notch receptors. J. Biol. Chem. 283, 35486–35495 [DOI] [PubMed] [Google Scholar]
- 11. Takeuchi H., Fernández-Valdivia R. C., Caswell D. S., Nita-Lazar A., Rana N. A., Garner T. P., Weldeghiorghis T. K., Macnaughtan M. A., Jafar-Nejad H., Haltiwanger R. S. (2011) Rumi functions as both a protein O-glucosyltransferase and a protein O-xylosyltransferase. Proc. Natl. Acad. Sci. U.S.A. 108, 16600–16605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rana N. A., Nita-Lazar A., Takeuchi H., Kakuda S., Luther K. B., Haltiwanger R. S. (2011) O-Glucose trisaccharide is present at high but variable stoichiometry at multiple sites on mouse Notch1. J. Biol. Chem. 286, 31623–31637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shao L., Moloney D. J., Haltiwanger R. (2003) Fringe modifies O-fucose on mouse Notch1 at epidermal growth factor-like repeats within the ligand-binding site and the Abruptex region. J. Biol. Chem. 278, 7775–7782 [DOI] [PubMed] [Google Scholar]
- 14. Brückner K., Perez L., Clausen H., Cohen S. (2000) Glycosyltransferase activity of Fringe modulates Notch-Delta interactions. Nature 406, 411–415 [DOI] [PubMed] [Google Scholar]
- 15. Moloney D. J., Panin V. M., Johnston S. H., Chen J., Shao L., Wilson R., Wang Y., Stanley P., Irvine K. D., Haltiwanger R. S., Vogt T. F. (2000) Fringe is a glycosyltransferase that modifies Notch. Nature 406, 369–375 [DOI] [PubMed] [Google Scholar]
- 16. Okajima T., Irvine K. D. (2002) Regulation of notch signaling by O-linked fucose. Cell 111, 893–904 [DOI] [PubMed] [Google Scholar]
- 17. Sasamura T., Sasaki N., Miyashita F., Nakao S., Ishikawa H. O., Ito M., Kitagawa M., Harigaya K., Spana E., Bilder D., Perrimon N., Matsuno K. (2003) Neurotic, a novel maternal neurogenic gene, encodes an O-fucosyltransferase that is essential for Notch-Delta interactions. Development 130, 4785–4795 [DOI] [PubMed] [Google Scholar]
- 18. Shi S., Stanley P. (2003) Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc. Natl. Acad. Sci. U.S.A. 100, 5234–5239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fleming R. J., Gu Y., Hukriede N. A. (1997) Serrate-mediated activation of Notch is specifically blocked by the product of the gene fringe in the dorsal compartment of the Drosophila wing imaginal disc. Development 124, 2973–2981 [DOI] [PubMed] [Google Scholar]
- 20. Panin V. M., Papayannopoulos V., Wilson R., Irvine K. D. (1997) Fringe modulates Notch-ligand interactions. Nature 387, 908–912 [DOI] [PubMed] [Google Scholar]
- 21. Hicks C., Johnston S. H., diSibio G., Collazo A., Vogt T. F., Weinmaster G. (2000) Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2. Nat. Cell Biol. 2, 515–520 [DOI] [PubMed] [Google Scholar]
- 22. Haines N., Irvine K. D. (2003) Glycosylation regulates Notch signalling. Nat. Rev. Mol. Cell Biol. 4, 786–797 [DOI] [PubMed] [Google Scholar]
- 23. Okajima T., Xu A., Lei L., Irvine K. D. (2005) Chaperone activity of protein O-fucosyltransferase 1 promotes notch receptor folding. Science 307, 1599–1603 [DOI] [PubMed] [Google Scholar]
- 24. Okajima T., Reddy B., Matsuda T., Irvine K. D. (2008) Contributions of chaperone and glycosyltransferase activities of O-fucosyltransferase 1 to Notch signaling. BMC Biol. 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sasamura T., Ishikawa H. O., Sasaki N., Higashi S., Kanai M., Nakao S., Ayukawa T., Aigaki T., Noda K., Miyoshi E., Taniguchi N., Matsuno K. (2007) The O-fucosyltransferase O-fut1 is an extracellular component that is essential for the constitutive endocytic trafficking of Notch in Drosophila. Development 134, 1347–1356 [DOI] [PubMed] [Google Scholar]
- 26. Sasaki N., Sasamura T., Ishikawa H. O., Kanai M., Ueda R., Saigo K., Matsuno K. (2007) Polarized exocytosis and transcytosis of Notch during its apical localization in Drosophila epithelial cells. Genes Cells 12, 89–103 [DOI] [PubMed] [Google Scholar]
- 27. Acar M., Jafar-Nejad H., Takeuchi H., Rajan A., Ibrani D., Rana N. A., Pan H., Haltiwanger R. S., Bellen H. J. (2008) Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell 132, 247–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leonardi J., Fernandez-Valdivia R., Li Y. D., Simcox A. A., Jafar-Nejad H. (2011) Multiple O-glucosylation sites on Notch function as a buffer against temperature-dependent loss of signaling. Development 138, 3569–3578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fernandez-Valdivia R., Takeuchi H., Samarghandi A., Lopez M., Leonardi J., Haltiwanger R. S., Jafar-Nejad H. (2011) Regulation of mammalian Notch signaling and embryonic development by the protein O-glucosyltransferase Rumi. Development 138, 1925–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee T. V., Sethi M. K., Leonardi J., Rana N. A., Buettner F. F., Haltiwanger R. S., Bakker H., Jafar-Nejad H. (2013) Negative regulation of notch signaling by xylose. PLoS Genet. 9, e1003547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Irvine K. D., Wieschaus E. (1994) fringe, a Boundary-specific signaling molecule, mediates interactions between dorsal and ventral cells during Drosophila wing development. Cell 79, 595–606 [DOI] [PubMed] [Google Scholar]
- 32. Roos C., Kolmer M., Mattila P., Renkonen R. (2002) Composition of Drosophila melanogaster proteome involved in fucosylated glycan metabolism. J. Biol. Chem. 277, 3168–3175 [DOI] [PubMed] [Google Scholar]
- 33. Ayukawa T., Matsumoto K., Ishikawa H. O., Ishio A., Yamakawa T., Aoyama N., Suzuki T., Matsuno K. (2012) Rescue of Notch signaling in cells incapable of GDP-l-fucose synthesis by gap junction transfer of GDP-l-fucose in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 109, 15318–15323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fuerstenberg S., Giniger E. (1998) Multiple roles for notch in Drosophila myogenesis. Dev. Biol. 201, 66–77 [DOI] [PubMed] [Google Scholar]
- 35. Go M. J., Eastman D. S., Artavanis-Tsakonas S. (1998) Cell proliferation control by Notch signaling in Drosophila development. Development 125, 2031–2040 [DOI] [PubMed] [Google Scholar]
- 36. Lloyd T. E., Atkinson R., Wu M. N., Zhou Y., Pennetta G., Bellen H. J. (2002) Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in Drosophila. Cell 108, 261–269 [DOI] [PubMed] [Google Scholar]
- 37. Wilder E. L., Perrimon N. (1995) Dual functions of wingless in the Drosophila leg imaginal disc. Development 121, 477–488 [DOI] [PubMed] [Google Scholar]
- 38. Emery G., Hutterer A., Berdnik D., Mayer B., Wirtz-Peitz F., Gaitan M. G., Knoblich J. A. (2005) Asymmetric Rab 11 endosomes regulate delta recycling and specify cell fate in the Drosophila nervous system. Cell 122, 763–773 [DOI] [PubMed] [Google Scholar]
- 39. Windler S. L., Bilder D. (2010) Endocytic internalization routes required for delta/notch signaling. Curr. Biol. 20, 538–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rong Y. S., Golic K. G. (2000) Gene targeting by homologous recombination in Drosophila. Science 288, 2013–2018 [DOI] [PubMed] [Google Scholar]
- 41. Gong W. J., Golic K. G. (2003) Ends-out, or replacement, gene targeting in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 100, 2556–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu T., Rubin G. M. (1993) Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 117, 1223–1237 [DOI] [PubMed] [Google Scholar]
- 43. Lee T., Luo L. (2001) Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 24, 251–254 [DOI] [PubMed] [Google Scholar]
- 44. Matsuno K., Ito M., Hori K., Miyashita F., Suzuki S., Kishi N., Artavanis-Tsakonas S., Okano H. (2002) Involvement of a proline-rich motif and RING-H2 finger of Deltex in the regulation of Notch signaling. Development 129, 1049–1059 [DOI] [PubMed] [Google Scholar]
- 45. Fehon R. G., Kooh P. J., Rebay I., Regan C. L., Xu T., Muskavitch M. A., Artavanis-Tsakonas S. (1990) Molecular interactions between the protein products of the neurogenic loci Notch and Delta, two EGF-homologous genes in Drosophila. Cell 61, 523–534 [DOI] [PubMed] [Google Scholar]
- 46. Delgehyr N., Wieland U., Rangone H., Pinson X., Mao G., Dzhindzhev N. S., McLean D., Riparbelli M. G., Llamazares S., Callaini G., Gonzalez C., Glover D. M. (2012) Drosophila Mgr, a Prefoldin subunit cooperating with von Hippel Lindau to regulate tubulin stability. Proc. Natl. Acad. Sci. U.S.A. 109, 5729–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Diederich R. J., Matsuno K., Hing H., Artavanis-Tsakonas S. (1994) Cytosolic interaction between deltex and Notch ankyrin repeats implicates deltex in the Notch signaling pathway. Development 120, 473–481 [DOI] [PubMed] [Google Scholar]
- 48. Brook W. J., Cohen S. M. (1996) Antagonistic interactions between wingless and decapentaplegic responsible for dorsal-ventral pattern in the Drosophila Leg. Science 273, 1373–1377 [DOI] [PubMed] [Google Scholar]
- 49. Blochlinger K., Bodmer R., Jan L. Y., Jan Y. N. (1990) Patterns of expression of cut, a protein required for external sensory organ development in wild-type and cut mutant Drosophila embryos. Genes Dev. 4, 1322–1331 [DOI] [PubMed] [Google Scholar]
- 50. O'Neill E. M., Rebay I., Tjian R., Rubin G. M. (1994) The activities of two Ets-related transcription factors required for Drosophila eye development are modulated by the Ras/MAPK pathway. Cell 78, 137–147 [DOI] [PubMed] [Google Scholar]
- 51. Tanaka T., Nakamura A. (2008) The endocytic pathway acts downstream of Oskar in Drosophila germ plasm assembly. Development 135, 1107–1117 [DOI] [PubMed] [Google Scholar]
- 52. Yamakawa T., Yamada K., Sasamura T., Nakazawa N., Kanai M., Suzuki E., Fortini M. E., Matsuno K. (2012) Deficient Notch signaling associated with neurogenic pecanex is compensated for by the unfolded protein response in Drosophila. Development 139, 558–567 [DOI] [PubMed] [Google Scholar]
- 53. Nolo R., Abbott L. A., Bellen H. J. (2000) Senseless, a Zn finger transcription factor, is necessary and sufficient for sensory organ development in Drosophila. Cell 102, 349–362 [DOI] [PubMed] [Google Scholar]
- 54. Culi J., Mann R. S. (2003) Boca, an endoplasmic reticulum protein required for wingless signaling and trafficking of LDL receptor family members in Drosophila. Cell 112, 343–354 [DOI] [PubMed] [Google Scholar]
- 55. Stahl M., Uemura K., Ge C., Shi S., Tashima Y., Stanley P. (2008) Roles of Pofut1 and O-fucose in mammalian Notch signaling. J. Biol. Chem. 283, 13638–13651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lira-Navarrete E., Valero-González J., Villanueva R., Martínez-Júlvez M., Tejero T., Merino P., Panjikar S., Hurtado-Guerrero R. (2011) Structural insights into the mechanism of protein O-fucosylation. PLoS One 6, e25365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rulifson E. J., Blair S. S. (1995) Notch regulates wingless expression and is not required for reception of the paracrine wingless signal during wing margin neurogenesis in Drosophila. Development 121, 2813–2824 [DOI] [PubMed] [Google Scholar]
- 58. Chou T. B., Perrimon N. (1996) The autosomal FLP-DFS technique for generating germ line mosaics in Drosophila melanogaster. Genetics 144, 1673–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Simpson P. (1990) Lateral inhibition and the development of the sensory bristles of the adult peripheral nervous system of Drosophila. Development 109, 509–519 [DOI] [PubMed] [Google Scholar]
- 60. Ishikawa H. O., Higashi S., Ayukawa T., Sasamura T., Kitagawa M., Harigaya K., Aoki K., Ishida N., Sanai Y., Matsuno K. (2005) Notch deficiency implicated in the pathogenesis of congenital disorder of glycosylation IIc. Proc. Natl. Acad. Sci. U.S.A. 102, 18532–18537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tonetti M., Sturla L., Bisso A., Zanardi D., Benatti U., De Flora A. (1998) The metabolism of 6-deoxyhexoses in bacterial and animal cells. Biochimie 80, 923–931 [DOI] [PubMed] [Google Scholar]
- 62. Rauskolb C., Correia T., Irvine K. D. (1999) Fringe-dependent separation of dorsal and ventral cells in the Drosophila wing. Nature 401, 476–480 [DOI] [PubMed] [Google Scholar]
- 63. Pitsouli C., Delidakis C. (2005) The interplay between DSL proteins and ubiquitin ligases in Notch signaling. Development 132, 4041–4050 [DOI] [PubMed] [Google Scholar]
- 64. Fehon R. G., Johansen K., Rebay I., Artavanis-Tsakonas S. (1991) Complex cellular and subcellular regulation of notch expression during embryonic and imaginal development of Drosophila: implications for notch function. J. Cell Biol. 113, 657–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang W., Struhl G. (2004) Drosophila Epsin mediates a select endocytic pathway that DSL ligands must enter to activate Notch. Development 131, 5367–5380 [DOI] [PubMed] [Google Scholar]
- 66. Glavic A., López-Varea A., de Celis J. F. (2011) The balance between GMD and OFUT1 regulates Notch signaling pathway activity by modulating Notch stability. Biol. Res. 44, 25–34 [DOI] [PubMed] [Google Scholar]
- 67. Perdigoto C. N., Schweisguth F., Bardin A. J. (2011) Distinct levels of Notch activity for commitment and terminal differentiation of stem cells in the adult fly intestine. Development 138, 4585–4595 [DOI] [PubMed] [Google Scholar]
- 68. Hiruma-Shimizu K., Hosoguchi K., Liu Y., Fujitani N., Ohta T., Hinou H., Matsushita T., Shimizu H., Feizi T., Nishimura S. (2010) Chemical synthesis, folding, and structural insights into O-fucosylated epidermal growth factor-like repeat 12 of mouse Notch-1 receptor. J. Am. Chem. Soc. 132, 14857–14865 [DOI] [PubMed] [Google Scholar]
- 69. Wang Y., Spellman M. W. (1998) Purification and characterization of a GDP-fucose:polypeptide fucosyltransferase from Chinese hamster ovary cells. J. Biol. Chem. 273, 8112–8118 [DOI] [PubMed] [Google Scholar]
- 70. Luo Y., Haltiwanger R. S. (2005) O-Fucosylation of Notch occurs in the endoplasmic reticulum. J. Biol. Chem. 280, 11289–11294 [DOI] [PubMed] [Google Scholar]
- 71. Takeuchi H., Kantharia J., Sethi M. K., Bakker H., Haltiwanger R. S. (2012) Site-specific O-glucosylation of the epidermal growth factor-like (EGF) repeats of notch: efficiency of glycosylation is affected by proper folding and amino acid sequence of individual EGF repeats. J. Biol. Chem. 287, 33934–33944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ge C., Stanley P. (2008) The O-fucose glycan in the ligand-binding domain of Notch1 regulates embryogenesis and T cell development. Proc. Natl. Acad. Sci. U.S.A. 105, 1539–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Moremen K. W., Tiemeyer M., Nairn A. V. (2012) Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462 [DOI] [PMC free article] [PubMed] [Google Scholar]