

Background: HIV-1 envelope spike protein remains a compelling but elusive target for preventing infection.
Results: Gold nanoparticle conjugates of peptide triazole Env inhibitors demonstrated impressive picomolar antiviral potencies.
Conclusion: Nanoparticle conjugates enhanced antiviral functions by multivalent attachment to virus Env spikes.
Significance: Findings reveal that multispike engagement can exploit the metastability of the virus envelope to irreversibly inactivate HIV-1.
Keywords: Drug Design, Human Immunodeficiency Virus (HIV), Nanotechnology, Peptides, Virology, Gold Nanoparticles, Microbicide, p24 Release, Virolysis
Abstract
Entry of HIV-1 into host cells remains a compelling yet elusive target for developing agents to prevent infection. A peptide triazole (PT) class of entry inhibitor has previously been shown to bind to HIV-1 gp120, suppress interactions of the Env protein at host cell receptor binding sites, inhibit cell infection, and cause envelope spike protein breakdown, including gp120 shedding and, for some variants, virus membrane lysis. We found that gold nanoparticle-conjugated forms of peptide triazoles (AuNP-PT) exhibit substantially more potent antiviral effects against HIV-1 than corresponding peptide triazoles alone. Here, we sought to reveal the mechanism of potency enhancement underlying nanoparticle conjugate function. We found that altering the physical properties of the nanoparticle conjugate, by increasing the AuNP diameter and/or the density of PT conjugated on the AuNP surface, enhanced potency of infection inhibition to impressive picomolar levels. Further, compared with unconjugated PT, AuNP-PT was less susceptible to reduction of antiviral potency when the density of PT-competent Env spikes on the virus was reduced by incorporating a peptide-resistant mutant gp120. We conclude that potency enhancement of virolytic activity and corresponding irreversible HIV-1 inactivation of PTs upon AuNP conjugation derives from multivalent contact between the nanoconjugates and metastable Env spikes on the HIV-1 virus. The findings reveal that multispike engagement can exploit the metastability built into virus the envelope to irreversibly inactivate HIV-1 and provide a conceptual platform to design nanoparticle-based antiviral agents for HIV-1 specifically and putatively for metastable enveloped viruses generally.
Introduction
There is an urgent need for new antiretroviral agents for the prevention and treatment of HIV-1 infection. Current inhibitors used in most combination antiretroviral therapies target the viral enzymes reverse transcriptase, integrase, and protease. Although the use of combination antiretroviral therapy has resulted in reducing viremia and HIV-1-related morbidity and mortality (1, 2), these drugs target stages of HIV-1 infection after cell entry and have significant challenges, such as complex regimens and treatment-resistant variants of HIV-1. Currently, there are only two Food and Drug Administration-approved antiretroviral drugs targeting the initial entry of the virus into host cells, namely T20 (enfuvirtide) and maraviroc (3–8), but these are used mainly in salvage therapy due to such limitations as injection site reaction and cost of production for the former and incomplete co-receptor breadth for the latter (7, 9–11). T20, a fusion inhibitor, targets transient conformations of the virus Env protein, gp41,2 and hence has a relatively short time window to act against the HIV-1 host cell membrane fusion process (12, 13). Maraviroc, on the other hand, targets the host cell co-receptor CCR5 and can only block R5-tropic HIV-1 virions from infecting the cell (7, 11, 14). Therefore, prolonged maraviroc treatment can result in the development of X4-tropic HIV-1 (7, 11, 14).
Host cell infection by HIV-1 is mediated by cell receptor interactions with trimeric envelope glycoprotein (Env) spikes that are exposed on the virus membrane surface (15, 16). Env is the only virally encoded protein on the virion surface (16, 17). The external gp120 component of Env is essential for cell receptor (CD4) and coreceptor (usually CCR5 or CXCR4) interactions and subsequent virus-cell fusion (17, 18). Hence, Env gp120 presents an obvious target to attack the virus directly in order to block the cascade of integrated binding and conformational change steps that lead to host cell infection. Env-specific inhibitors that could inactivate the virus before receptor encounter would hold great promise in preventing AIDS transmission and progression. Despite the potential of gp120 antagonists for prevention and intervention of infection, progress has been limited for such agents, due to such factors as the high rate of mutation of gp120 protein, leading to the threat of resistance to developing inhibitors (19–21), low potency of small molecule inhibitors (22–24), the high cost and potential toxicity of protein inhibitors (25–27), and the potential risk of infection enhancement with CD4-mimicking ligands (22, 28).
Previously, we showed that a family of entry inhibitors, peptide triazoles (29–34) derived from the peptide 12p1 (35, 36), bind specifically and with high affinity to HIV-1 Env gp120, antagonize the interactions of Env binding sites for both host cell receptors CD4 and CCR5/CXCR4, and cause gp120 shedding from the virus surface, leading to HIV-1 inactivation before host cell encounter (37, 38). The most potent compositions of peptide triazole inactivators discovered so far are multivalent forms conjugated to monodisperse gold nanoparticles. The conjugates initially derived were found to have low nanomolar inhibitory IC50 values and to act as virucidal agents, leading to the irreversible breakdown of the virus before the virus has a chance to engage the host cell (37, 38).
The functional properties of virolytic gold nanoparticle-peptide triazole (AuNP-PT) conjugates make it feasible for long term use of this type of agent to inactivate HIV-1 before host cell encounter. In the current work, we sought to investigate the mechanism of cell-free HIV-1 inactivation using the AuNP-PT multivalent nanostructures. We evaluated the impact of 1) altering the properties of the AuNP-PT constructs, by increasing size and PT coverage density of the nanoparticles, and 2) varying the HIV-1 virion functional spike density. Our investigations demonstrate the importance of multivalent contact between nanoparticle and metastable virus interaction partners. The results define Env-targeting NP designs that take advantage of HIV-1 Env metastability and cause specific and irreversible HIV-1 inactivation. Overall, the investigation demonstrates that the multivalent physical contact approach can be used as a general strategy for antagonizing HIV-1 transmission and proliferation.
EXPERIMENTAL PROCEDURES
Materials
For peptide triazole synthesis, all Fmoc-protected α- and β-amino acids, O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate, 1-hydroxybenzotriazole, Rink amide resin (4-(2′,4′-dimethoxyphenyl-Fmoc-aminomethyl)phenoxy resin) with a 0.55 mmol/g substitution, N,N-dimethylformamide, pyridine, and N,N-di-isopropylethylamine were purchased from Chem-Impex International Inc. Ethynylferrocene and Cu(I)I were purchased from Sigma-Aldrich and used without further purification. Fmoc-cis-4-azidoproline was synthesized starting with commercially available trans-hydroxyproline-OH (31). Gold(III) chloride hydrate and citric acid were obtained from Sigma-Aldrich, and bis(p-sulfonatophenyl) phenylphosphine dehydrate dipotassium salt was from Strem Chemicals. Citric acid and cysteine used in the conjugation were from Sigma-Aldrich. Modified human osteosarcoma cells (HOS.T4.R5), engineered to express CD4 receptor and CCR5 co-receptor, as well as pNL4-3 Luc+ R−E− backbone DNA, were obtained from Dr. Nathaniel Landau. The HOS.T4.R5 cells were grown in DMEM supplemented with 10% FBS, 2.5% HEPES, 1% penicillin-streptomycin, 2% l-glutamine, and 1 mg of puromycin. 293T cells were obtained from the American Type Culture Collection and grown in the same culture medium as the HOS.T4.R5 cells except without puromycin. The plasmids for HIV-1 BaL-01 gp160 and VSV-G Env DNA were obtained from the National Institutes of Health AIDS Reagent Program. Replication-competent HIV-1 BaL-01 virions were obtained from the University of Pennsylvania Center for AIDS Research. Mouse anti-p24 and rabbit anti-p24 antibodies and p24 protein were purchased from Abcam. Fully infectious HIV-1 (BaL) was a gift from Dr. Michele Kutzler and was obtained from the University of Pennsylvania Center for AIDS Research. The gp120 monomer protein was produced using an already established protocol (33), and anti-gp120 D7324 was purchased from Alto Chemicals. gp41 protein, enfurvirtide (T20), and anti-gp41 antibodies 4E10 and 2F5 were obtained from the National Institutes of Health AIDS Reagent Program. The enhanced chemiluminescence Western blot detection system was obtained from Amersham Biosciences. O-Phenylenediamine was from Sigma-Aldrich. All other materials were purchased from Fisher.
Peptide Triazole Synthesis
To conjugate peptide triazole on nanoparticles, the peptide triazole KR13 (RINNI-FtPro-WSEAMM-βA-Q-βA-C-amide, where FtPro represents ferrocenyltriazole-Pro) was synthesized to contain the 12-residue N-terminal sequence of the HNG-156 parent peptide (RINNI-FtPro-WSEAMM-amide) (39) with a C-terminal extension containing a free thiol group. KR13 itself contained the essential peptide triazole pharmacophore, Ile-triazole-Pro-Trp, and was functionally active (37). The free sulfhydryl group was incorporated to enable direct thiol conjugation to the gold surface of AuNP (38). The peptide KR13 binds to gp120 with a KD of 11.3 nm (37).
Gold Nanoparticle Synthesis
The citrate reduction method developed by Frens (40) was modified in order to synthesize size-controlled, stable, and monodisperse AuNPs. The citrate acid concentration was varied to obtain AuNP with various sizes ranging from 13 to 123 nm. The citrate reaction solution, initially at 100 °C, was cooled to room temperature, and bis(p-sulfonatophenyl) phenylphosphine dehydrate dipotassium salt (18 mg/ml for 20-nm AuNP) was added to the synthesized particles and stirred overnight at room temperature. The resulting particles were further washed with phosphate buffer at pH 7.2 and concentrated using a Millipore 100,000 kDa filter. The particle size was determined by dynamic light scattering (DLS) in the Zetasizer NS90 (Malvern Instruments), and the particle concentration was calculated using the absorbance at 450 nm and the peak absorbance of the UV spectrum (41). The AuNP particle morphology was characterized using transmission electron microscopy. Samples were prepared by adding a drop of the AuNP solution onto a lacey carbon grid film and then allowing evaporation. Transmission electron microscopy (TEM) images of AuNPs were taken with a JEM 2100 camera operated at 200 kV.
Gold Nanoparticle-Peptide Triazole Conjugate Production
Peptide conjugation to the AuNPs was achieved via covalent linkage between the thiol moiety (-SH) of the C-terminal cysteine residue and the gold surface of AuNP (38). A flocculation assay (42) was conducted to determine the ratio of AuNP to KR13 to use for complete peptide conjugation without peptide aggregation. The bis(p-sulfonatophenyl) phenylphosphine dehydrate dipotassium salt-stabilized AuNP-KR13 mixtures in phosphate buffer were stirred vigorously for 30 min at 25 °C. Excess peptide was removed from the nanoparticle fraction using a 10,000 Mr cut-off centrifugation filter. PT content of the AuNP-KR13 preparation was determined using amino acid analysis after acid hydrolysis (Keck Institute, Yale University). To prepare AuNP-PT particles with varying density of KR13, we used 20-nm AuNPs. These were mixed with different molar ratios of KR13 and l-cysteine during the step of peptide conjugation to AuNP. Both molecules present a single reactive thiol for covalent attachment to the bis(p-sulfonatophenyl) phenylphosphine dehydrate dipotassium salt-stabilized nanoparticle. However, AuNP-Cys has no virolytic activity (Table 2), and only the KR13 component will bind to HIV Env on the virion surface. Incorporation of KR13 was measured directly by amino acid analysis. We calculated the average areal peptide density (ρ) assuming spherical NPs,
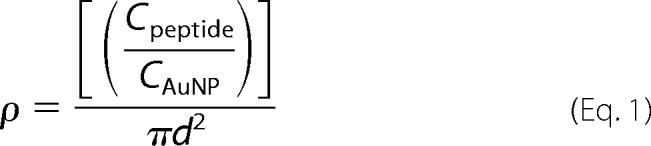 |
where πd2 is the surface area of nanoparticle and CKR13/CAuNP is the molar ratio of peptides per nanoparticle. The conjugates were validated for peptide conjugation by testing their change in zeta potential, which was measured by the Zetasizer NS90 (Malvern Instruments) (data not shown). Furthermore, the monodispersity was assessed using both DLS (Fig. 1B) and TEM (Fig. 1C).
TABLE 2.
Antiviral effects of AuNP-KR13 with varying diameter at constant peptide density per nm2 on the AuNP surface
The viral infection IC50 and p24 release EC50 were obtained from Fig. 3 using Origin Pro version 8. The average number of peptides per AuNP was obtained using amino acid analysis. n = 3.
| Inhibitor used | Viral infection inhibtion IC50 | p24 release EC50 | Coverage density (no. of peptides) | Peptide density per nm2 |
|---|---|---|---|---|
| nm | nm | |||
| KR13 | 196.6 ± 36.29 | 3250 ± 567 | 1 | 1 |
| AuNP-KR13 (20.2 nm) | 5.3 ± 1.14 | 9.7 ± 1.1 | 96 | 0.075 |
| AuNP-KR13 (42.3 nm) | 0.7 ± 0.14 | 1.2 ± 2.5 | 430 | 0.077 |
| AuNP-KR13 (80.2 nm) | 0.05 ± 0.06 | 0.2 ± 0.4 | 1387 | 0.069 |
FIGURE 1.

Characterization of the AuNP-KR13 conjugates. A, comparison of the theoretical and observed AuNP diameters as a function of citric acid concentration during AuNP synthesis. The diameters of the AuNP that were synthesized (experimental) were measured using DLS (n = 25). B, the percentage intensity of the different diameters of the AuNP-KR13 conjugates as measured on the Zetasizer using the dynamic light scattering methodology (n = 3). C, representative TEM images of the AuNP-KR13 conjugates with diameters of 13.3, 19.9, 42.8, and 92.6 nm. Error bars, S.D. (n > 3).
HIV-1 Pseudotyped Virion Production
The recombinant virus consisted of the pro-viral envelope plasmid sequence corresponding to the CCR5 targeting HIV-1BaL strain or a vesicular stomatitis virus pseudotype, and the backbone sequence corresponded to an envelope-deficient pNL4–3 luc+, env− provirus developed by N. Landau (43, 44). 4 μg of envelope DNA and 8 μg of backbone DNA were co-transfected into 293T cells using FuGene 6 as the transfection reagent (Promega). 14 h post-transfection, the medium was changed, and the virus-like particle-containing supernatants were collected at 48 h post-transfection. Soluble proteins (including Env gp120 and p24) from the viral supernatants were removed via fractionation on a 6–20% iodixanol gradient for 2 h at 210,700 × g (SW41 rotor, Beckman ultracentrifuge). The collected fractions were validated for p24 content using capture ELISA as well as gp120 content using Western blot detection. Virions purified on the 6–20% iodixanol gradient exhibited a characteristic distribution profile of p24 and gp120 content, enabling viral fractions (18–19.2% iodixanol) and soluble protein fractions (6–8% iodixanol) to be isolated. The gradient-purified virus samples, which exhibited full or greater infectivity (against HOS.T4.R5 cells (38)) compared with the unfractionated control virions, were collected, aliquoted, and stored at 80 °C until further use.
Env Spike Presentation on the Virus Surface
To make viruses with varying spike density, HEK293T cells were transfected with backbone vector, pNL4–3.Luc R-E-, and a mixture of active Env plasmid (HIV-1 BaL-WT) with an Env plasmid encoding inactive Env gp120 S375W BaL. The S375W mutation has been found previously to be fusion-competent (45, 46), but it does not bind significantly to KR13 and hence causes resistance to PT (36).3 Of note, varying density does not in itself eliminate the potential for local clustering, and indeed evidence has been obtained showing that HIV-1 Env spikes have the tendency to cluster (48, 49). Control virions included those with all BaL or all S375W Env (all active or resistant for peptide triazole binding, respectively). Protease digestion of the spike varying virion was conducted in order to eliminate non-functional envelopes. This digestion of pseudoviruses was carried out by treating the culture supernatants with a protease mixture of 1 μg of trypsin, chymotrypsin, subtilisin, and/or proteinase K (Sigma) at 37 °C as described by Crooks et al. (50). The treated virions were spun on a 6–20% iodixanol gradient as described above. Spike density was quantified using Western blot analysis of gp120, viral infection, and p24 content (ELISA) as explained above (data not shown). As expected, the S375W mutant was similar to the wild type BaL in infecting the HOS.T4.R5 cells (data not shown).
Antiviral Functions of AuNP-KR13 Conjugates
Dependence of Antiviral Effects on the Size of AuNP-KR13
To test the effects of nanoparticle size on viral inhibition and virolytic activity, we synthesized AuNPs with diameters ranging from 10 to 200 nm as described previously and functionalized them with the KR13 peptide. We utilized assays for HIV cell infectivity and for virus contents, including p24 and gp120, in order to correlate nanoparticle diameter and surface area of the AuNP-KR13s with antiviral effects. Purified virus was treated with AuNP-KR13 constructions for 30 min at 37 °C and spun on a 6–20% iodixanol gradient for 2 h at 210,700 × g (ultracentrifugation as above). The collected virus fraction and the supernatant fraction were tested for infectivity, p24 and gp120 content from the virus post-treatment. Virus treated with PBS for 30 min at 37 °C was used as negative control for background signal, and virus incubated for 5 min with 2% Triton X-100 at 98 °C was the positive control for p24 and gp120 content.
Modulating Nanoparticle Surface Density of KR13 on AuNP
KR13 coverage on 20-nm diameter AuNP was varied by mixing different molar ratios of KR13 and l-cysteine during the step of KR13 conjugation to AuNP. Both molecules present a single reactive thiol for covalent attachment to the bis(p-sulfonatophenyl) phenylphosphine dehydrate dipotassium salt-stabilized nanoparticle. However, only the KR13 was expected to bind to HIV envelope glycoproteins on the surface of the virion. AuNP and KR13 were synthesized and functionalized as described above. Cysteine/KR13 molar ratios were varied from 0:1000 to 1000:0 in increments of 50. We calculated the density of peptide coverage (ρ) as explained above. Purified virus was treated with AuNP-KR13 constructions for 30 min at 37 °C and spun on a 6–20% iodixanol gradient for 2 h at 210,700 × g (ultracentrifugation as above) (37). The collected virus fraction and the supernatant fraction were tested for infectivity and both p24 and g120 contents post-treatment.
AuNP-KR13 Effects on Replication-competent HIV-1
Replication competent HIV-1 BaL virions were obtained from the University of Pennsylvania Center for AIDS Research and purified using 6–20% iodixanol gradient, and p24 content was measured using ELISA. The inhibitor concentration that gives 50% reduction of the viral titer in tissue culture (IC50) was determined from dose-response data using nonlinear least-squares regression analysis with Origin version 8.1 (Origin Lab). The working dilution of virus was pretreated with a serial dilution of AuNP-KR13 (20 and 80 nm), starting from an effective KR13 concentration on the AuNP surface of 50 and 10 μm, respectively, for 30 min at 37 °C. HOS.T4.R5 cells, seeded at 8,000 cells/well, were incubated for 24 h, followed by the addition of preincubated inhibitor/virus mixtures. After 48 h of infection, with a growth medium wash step 24 h post-treatment, the p24 content of virions in the supernatant was measured in order to determine the IC50 value of inhibition of virus infection. For cell-free p24 release experiments, the peptide triazole and cell-free replication-competent HIV-1 BaL virus were preincubated for 30 min at 37 °C, followed by spinning for 2 h at 23,000 × g in a table top centrifuge (Eppendorf) at 4 °C. Supernatant was separated, and p24 content was determined using the p24 capture ELISA as described above. The extents of viral inhibition of, and p24 release from, fully infectious HIV-1 BaL were also evaluated for the free control peptide KR13 (data from Bastian et al. (37)).
Comparisons of Antiviral Functions of AuNP-KR13 with KR13 Alone
Exposure of gp41 in Viruses Treated with AuNP-KR13 and KR13
The HIV-1 BaL pseudotype was incubated with increasing concentration of either KR13 starting at 50 μm or AuNP-KR13 starting at 1 μm (KR13 concentration) for 30 min at 37 °C. The samples were spun at 23,000 × g (table top centrifuge) for 2 h at 4 °C, and the virion fraction for each sample was collected and fixed using a homemade fixative with 0.1% formaldehyde and 0.1% glutaraldehyde in PBS. Glycine (0.1 m) was added to stop the cross-linking reaction by the fixative. The virions then were loaded onto an ELISA plate (high protein binding) and incubated overnight on a shaker at 4 °C. The plate was blocked with 3% BSA, and ELISA was used to detect gp41 epitopes with human gp41 antibodies 2F5 and 4E10 followed by the addition of anti-human IgG HRP secondary antibody.
Effect of Fusion Inhibitor Enfurvirtide (T20) on Virolysis Induced by AuNP-KR13 and KR13
T20 was shown previously to effect the 6-helix bundle formation of the HIV-1 virion during fusion with the host cell (8, 13). Further, T20 was found to specifically inhibit KR13-induced HIV-1 cell-free virolysis (37). A corresponding experiment was conducted to test whether T20 inhibited virolysis of pseudotyped HIV-1 BaL induced by AuNP-KR13 (20.2 nm diameter of AuNP). The AuNP-KR13 was kept at an IC80 concentration of 25 nm, and serial dilutions of T20 starting at 1 μm were co-incubated with pseudotyped HIV-1 BaL for 30 min at 37 °C. Treated virion samples were fractionated on a 6–20% iodixanol gradient (above). Gradient fractions were quantified for p24 using ELISA, and relative p24 release was quantified and plotted using Origin version 8.1 (OriginLab).
Time Dependence of AuNP-KR13-induced HIV-1 Breakdown
The time dependence of gp120 shedding, p24 release, and inhibition of pseudotyped HIV-1 BaL by an IC80 concentration of 20.2 nm AuNP-KR13 (25 nm) was measured. The AuNP-KR13 was incubated with the virus for different times ranging from 1 min to 24 h at 37 °C, the samples were spun on a 6–20% iodixanol gradient, and the gradient fractions were tested for gp120 shedding, p24 release, and inhibition of viral infection. For gp120 shedding, the fractions were tested using a Western blot analysis with primary anti-gp120 antibody, D7324, and secondary antibody, anti-sheep HRP. For p24 release, the ELISA explained above was used. The Western blots were analyzed using ImageJ Western blot band quantification. For inhibition of viral infection, the luciferase reporter assay was used as explained above (37).
Antiviral Effects of AuNP-KR13 Versus KR13 on HIV-1 Pseudovirions with Modified Env Spike Presentation on the Virus Surface
Comparisons were made of the lytic deformation of viruses by KR13 and AuNP-KR13 with viruses containing a varying proportion of KR13-resistant mutant versus WT Env. In the Western blot analysis, the amount of loaded virions was normalized using both p24 content from ELISA and total protein content measured using a Bradford assay. The p24 contents of the different virions and their total protein contents were similar, within ±1.5%. Thus, virus amounts were determined by p24 content (3,000 molecules/virion (51, 52)). Antiviral assays included inhibition of infection by KR13 compared with AuNP-KR13 on HOS.T4.R5 cells and cell-free HIV-1 p24 release.
Morphological Analysis of HIV-1 after AuNP-KR13 Conjugate Treatments
TEM was conducted to visualize the morphology of the virions treated with AuNP-KR13 (20.2- and 80.2-nm diameter AuNP). Purified pseudotyped HIV-1 BaL virus and AuNP-KR13 were preincubated from 5 to 1,440 min at 37 °C. Following incubation, samples were fixed with 2% glutaraldehyde for 30 min at room temperature and then embedded in Spurr's low viscosity epoxy medium after acetone washes to dry the virions. Slices (100 nm thick) were prepared using an ultramicrotome (Leica EM UC6), loaded onto a holey carbon TEM 200 mesh grid (Electron Microscopy Science) and imaged using the JEM 2100 camera operated at 120 kV (JEOL, Tokyo, Japan). 16 images were taken per sample, and the sizes of observed particles were determined, using ImageJ software to derive average diameters of the virion particles from TEM images measured from five angles. In order to confirm the identification of collapsed virions in samples imaged, we used energy-dispersive spectroscopy and related the element ratios on untreated virions to those of the collapsed particles.
RESULTS
Monodispersity of Gold Nanoparticle-Peptide Triazole Conjugates
In this study, we investigated the mechanism underlying the antiviral functions of AuNP-KR13 conjugates against HIV-1 and, in particular, the ability of the nanoconjugates to cause specific virus lytic inactivation. To achieve this, the dependence of lysis on variations of surface characteristics, of both nanoparticle inhibitor and virion envelope protein, was measured.
The physical characteristics of gold nanoparticle-peptide triazole conjugates were validated by DLS, TEM, and UV absorbance. Particle size was obtained by DLS using the Nano S90 zetasizer (Malvern Instruments), and the particle concentration was calculated using the UV spectrum as described under “Experimental Procedures” (41). The total number of peptide molecules per AuNP was obtained by amino acid analysis. The DLS data are shown in Fig. 1, along with representative TEM images of AuNP-KR13 conjugates of various AuNP sizes. Fig. 1A also shows the comparison between the expected sizes of AuNP using varying concentrations of citric acid as well as the experimentally obtained diameters (DLS). The AuNP-KR13 conjugates showed good monodispersity, as observed by the TEM images in Fig. 1C. Further, the ζ potential values for AuNP-KR13 conjugates compared with the AuNP alone verified the successful peptide conjugation to AuNP (data not shown) (53).
Antiviral Effects of AuNP-KR13 Conjugates
Relationship between AuNP-KR13 Particle Size and Antiviral Activity
AuNP-KR13 conjugates were tested for both inhibition of HIV-1 BaL pseudotype infection of HOS.T4.R5 cells as well as cell-free p24 release, in both cases after 30-min exposure of viruses to AuNP-KR13 conjugates. Fig. 2 shows the dose responses of the effects, and IC50 values are summarized in Table 1. Strikingly, the antiviral effects were greatly enhanced with increasing size of the nanoparticle. This effect is probably due to decreasing nanoparticle curvature and corresponding increasing contact area between the AuNP-KR13 nanoparticle and the HIV-1 virion. Table 1 also shows the number of peptides per nanoparticle calculated from amino acid analysis and the KR13 area density on the AuNP surface.
FIGURE 2.
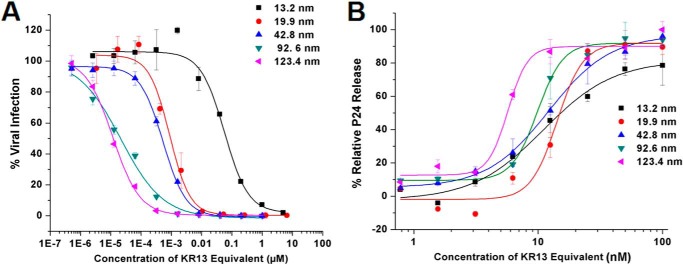
Size-dependent antiviral effects of the AuNP-KR13 conjugates on the HIV-1 BaL pseudovirions. A, inhibition of cell infection measured using a single round infection assay. The IC50 values are reported in Table 1. B, relative p24 release from the cell-free HIV-1 BaL pseudovirions with increasing concentration of the different diameter AuNP-KR13 conjugates. The EC50 values are reported in Table 1. Error bars, S.D. (n = 3).
TABLE 1.
Antiviral effects of AuNP-KR13 with varying diameter and peptide density on the AuNP surface
The viral infection IC50 and p24 release EC50 were obtained from Fig. 2 using Origin Pro version 8. The average number of peptides per AuNP was obtained using amino acid analysis. n = 3.
| Diameter | Viral infection inhibition IC50 | p24 release EC50 | Average number of peptides per AuNP | Peptide density per nm2 |
|---|---|---|---|---|
| nm | nm | nm | ||
| 13.2 | 60 ± 10 | 10.95 ± 1.66 | 34 | 0.062 |
| 19.9 | 0.9 ± 0.2 | 14.16 ± 1.52 | 73 | 0.059 |
| 42.8 | 0.18 ± 0.1 | 12.10 ± 0.83 | 139 | 0.024 |
| 92.6 | 0.02 ± 0.01 | 9.85 ± 0.61 | 220 | 0.008 |
| 123.4 | 0.01 ± 0.01 | 5.71 ± 0.60 | 652 | 0.014 |
To control for differences in peptide density on separate populations of AuNPs, we prepared AuNPs of 20.2, 42.3, and 80.2 nm diameters with nearly equal peptide densities by empirically adjusting the peptide concentration used during conjugation. The antiviral effects observed upon size variation of AuNP at (nearly) fixed KR13 area density in AuNP-KR13 conjugates are shown in Fig. 3, and the IC50 values obtained from the dose-response profiles are given in Table 2. Here, a clear correlation was observed between the effects of AuNP size on inhibition of viral infection and p24 release, with increasing size leading to increasing potency. This result suggests that the AuNP-KR13-induced p24 release from HIV-1 is related to the extent of contact of the HIV-1 virion surface with multiple KR13 monomers on the nanoparticle surface. In retrospect, the more limited increase in p24 release potency initially observed with nanoparticle size increase in initial experiments (Fig. 2) was probably due to the fact that the densities of KR13 per unit area on the AuNP surface decreased with increasing size (Table 1), whereas, in Fig. 3B, increasing size of AuNP, while maintaining a similar density of KR13 on the AuNP surface, caused a drastic increase in the potency of p24 release from HIV-1.
FIGURE 3.

Antiviral effects of AuNP-KR13 conjugates with increasing diameter of the AuNP with equal density of KR13 coverage/nm2 on the surface of the AuNP. A, inhibition of HIV-1 infection by the AuNP-KR13 conjugates measured using a single round infection assay as explained under “Experimental Procedures.” The IC50 values are reported in Table 3. B, relative p24 release from the cell-free HIV-1 BaL pseudovirions with increasing concentration of the different diameter AuNP-KR13 conjugates. The EC50 values are reported in Table 3. Error bars, S.D. (n = 3).
Impact of Density Variation of Peptide Triazoles Conjugated to AuNP on Antiviral Functions
AuNP-KR13 conjugates were prepared with varying KR13 densities on 20-nm nanoparticles and tested for both inhibition of HIV-1 BaL pseudotype cell infection with HOS.T4.R5 cells and cell-free p24 release after 30 min of exposure of virus with the conjugates. The values computed for KR13 coverage per nm2 of the AuNP surface assumed that AuNP is a perfect sphere and that individual KR13s are randomly distributed on the NP surface. As shown in Fig. 4 and Table 3, both viral infection inhibition and p24 release activities were greater with increasing area density of KR13 on the AuNP surface. The AuNP particles without KR13 had 100% cysteine coverage and did not exhibit nonspecific antiviral effects (Table 3 and Fig. 4).
FIGURE 4.

Antiviral effects of AuNP-KR13 (19.9-nm diameter) with increasing surface density coverage of KR13, with 100% being 69 peptides/AuNP, as represented in Table 3. A, inhibition of cell infection measured using a single round infection assay as explained under “Experimental Procedures.” The IC50 values are reported in Table 2. B, relative p24 release from the cell-free HIV-1 BaL pseudovirions with increasing concentration of the AuNP-KR13 conjugates with decreasing KR13 density coverage. The EC50 values are reported in Table 2. Error bars, S.D. (n = 3).
TABLE 3.
Antiviral effects of AuNP-KR13 with constant diameter (19.9 nm) and varying peptide density per nm2 on the AuNP surface
The viral infection IC50 and p24 release EC50 were obtained from Fig. 4 using Origin Pro version 8. The average number of peptides per AuNP were obtained using amino acid analysis. n = 3.
| Average no. of KR13 per AuNP | Viral infection inhibition IC50 | p24 release EC50 | Peptide density per nm2 |
|---|---|---|---|
| nm | nm | ||
| 68.6 | 0.42 | 1.64 | 0.055 |
| 28.8 | 1.1 | 6.2 | 0.023 |
| 10.9 | 8.1 | 19.5 | 0.009 |
| 0 | >50 | >50 | 0 |
AuNP-KR13 Potencies with Replication-competent HIV-1
The antiviral effects of 20- and 80-nm AuNP-KR13 particles were tested with replication-competent Bal-01 HIV-1, as shown in Fig. 5. The results demonstrate that the AuNP-KR13 is effective and has a greater potency than KR13 alone (∼300-fold greater for the 20-nm AuNP-KR13 and 1400-fold greater for the 80-nm AuNP-KR13 compared with KR13 (37)).
FIGURE 5.

Characterization of AuNP-KR13 (20 and 80 nm)-induced infection inhibition and virus breakdown of replication-competent HIV-1 BaL virus. A, inhibition of cell infection measured using a p24 ELISA of the produced virions in the presence of the inhibitor. The IC50 values of AuNP-KR13 (20 nm) and AuNP-KR13 (80 nm) to inhibit HIV-1 BaL infection were 3.16 and 0.73 nm, respectively. B, relative p24 release from the cell-free HIV-1 BaL (replication-competent) with increasing concentration of the 20- and 80-nm diameter AuNP-KR13 conjugates. The EC50 values of AuNP-KR13 (20 nm) and AuNP-KR13 (80 nm) to inhibit HIV-1 BaL infection were 8.19 and 0.49 nm, respectively. Error bars, S.D. (n = 3).
Comparison of AuNP-KR13 and KR13 Actions on Pseudoviruses
We previously observed that KR13-induced lysis of HIV-1 embodies virus breakdown characteristics reminiscent of those occurring during virus-cell fusion. These include exposure of the membrane-proximal external region (MPER) of gp41 and inhibition by fusion inhibitor T20. In the current work, we sought to evaluate these characteristics for AuNP-KR13 lysis. As shown in Fig. 6A, AuNP-KR13 treatment did not induce a concentration-dependent exposure of MPER on the residual virion, whereas treatment with KR13 did. In addition, as seen in Fig. 6B, the fusion inhibitor, T20, did not lead to the inhibition of AuNP-KR13-induced p24 release, in contrast to the inhibition seen with KR13. Fig. 6C shows that, like KR13, AuNP-KR13 induced virolysis was accompanied by gp120 shedding. However, for AuNP-KR13, gp120 shedding, p24 release and viral infection inhibition occurred simultaneously, in all cases starting within 10 min of incubation with the virus. By comparison, in KR13 virolysis, p24 release occurred with a time lag compared with the gp120 shedding and virus infection inhibition, as shown by Bastian et al. (37). Hence, from the data shown above, the AuNP-KR13-induced virolysis process appears to be controlled differently than that of KR13. In order to further compare AuNP-KR13 with KR13, we turned our attention to the surface properties of the virus.
FIGURE 6.

Comparison of the antiviral effects of AuNP-KR13 and KR13 on the HIV-1 BaL pseudotyped virus. A, MPER-specific gp41 epitope exposure on viruses treated with AuNP-KR13 and KR13, as measured with MPER-specific antibody (2F5 and 4E10) binding to HIV-1 BaL pseudotyped virion after 30 min of AuNP-KR13 (20-nm diameter) and KR13, respectively. B, T20 (fusion inhibitor) inhibition of AuNP-KR13 (50 nm)-induced and KR13 (1 μm)-induced p24 release of HIV-1 BaL pseudotyped. The IC50 value for T20 inhibition of KR13-induced p24 release was 15.9 ± 4.9 nm using Origin version 8.1 (OriginLab). No significant effect was observed on the AuNP-KR13-induced p24 release by the addition of T20. C, time-dependent effects of AuNP-KR13 (20 nm) on viral infection inhibition, p24 release, and gp120 release. The percentage of cell infection retained after peptide treatment is shown on the left y axes, and the viral protein gp120 and p24 retained in the virus fraction are shown on the right y axes. All samples were adjusted to the untreated virus as full infection and viral protein retention. Each time point of peptide treatment had a control for normalization. Error bars, S.D. (n = 3).
Impact of Env Trimer Composition on the Antiviral Potencies of Peptide-Triazole Inhibitors
The enhanced antiviral activity of AuNP-KR13 compared with KR13 suggests that multivalent interactions between the virion and nanoparticle are important determinants of AuNP-KR13 potency. Such interactions could involve multivalent attachment to single Env trimers and/or localized trimer cross-linking. To further probe the role of multivalency in inhibition, we compared the antiviral activities of AuNP-KR13 and KR13 against HIV-1 pseudotyped with both susceptible and non-susceptible BaL Env variants. The non-susceptible Env contained the gp120 S375W mutation that ablates peptide-triazole binding, by steric blockade of the PT Trp indole side chain (36),3 but has negligible impact on glycoprotein expression, processing, or fusogenic activity (data not shown). Viruses were produced from cells co-expressing WT (denoted W) and S375W (denoted M) Env at different relative levels. The resulting viral populations shared the same total Env content (W + M) but varied in the average surface density of WT Env. When co-expressed in virus producing cells, WT and S375W Env form four trimeric species (W3, W2M, WM2, and M3), the distribution among which can be approximated through binomial statistics, assuming the protomers assort randomly.
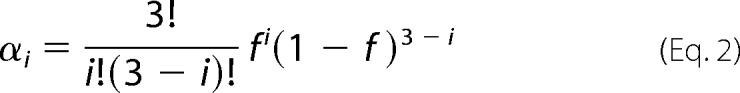 |
Here, αi is fraction of Env trimers in a viral population that contain i WT protomers (from 0 to 3), and f is the abundance of the WT protomer relative to the total protomer level. Because the mutation does not alter Env expression level, f was equated with the fraction of WT gp160 DNA (relative to total gp160 DNA) used for transfection into virus-producing cells. Values for α0, α1, α2, and α3 for each ratio of WT/S375W DNA tested are given in Fig. 7.
FIGURE 7.
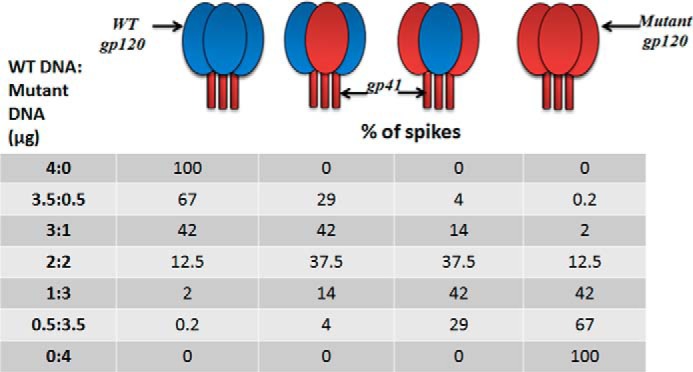
Representation of WT/mutant (S375W) monomer arrangements on the virus spike with different ratios of the WT/mutant DNA concentrations used for virus production. The percentage of spikes was calculated using the formula in Equation 2 (see “Results”). The obtained ratio, α, was multiplied by 100 to obtain the percentage of monomer.
Dose-response curves of infection inhibition and viral lysis for the different viral populations are shown in Fig. 8, A–D, and the IC50 values are given in Table 4. As expected, both AuNP-KR13 and KR13 were most potent against the 100% WT Env HIV-1 population (4:0) and demonstrated no antiviral activity against the 100% S375W Env HIV-1 population (0:4). For inhibition of viral infection, there was a similar loss of both AuNP-KR13 and KR13 potency as the fraction of WT Env decreased. However, even at the lowest WT Env fraction tested (f = 0.125 for the 0.5:3.5 population), infection was almost fully blocked at high inhibitor concentrations. In this viral population, the amount of Env trimers with two or three WT protomers (W3 and W2M) is small (∼4%), and viruses lacking such Env trimers (i.e. containing only WM2 and M3 trimers) should form a nontrivial fraction of the population. The fact that all viruses of the 0.5:3.5 WT DNA/mutant DNA population appear sensitive to AuNP-KR13 and KR13 suggests that these inhibitors can effectively block the function of Env trimers containing only one susceptible gp120.
FIGURE 8.

Impact of WT/mutant gp120 variation on antiviral functions of AuNP-KR13 (infection inhibition (A) and p24 release (C)) and KR13 (infection inhibition (B) and p24 release (D)). Assays used HIV-1 BaL pseudovirions with varying concentrations of incorporated S375W mutant spikes. Inhibition of HIV-1 infection was measured using a single round infection assay as explained under “Experimental Procedures.” The cell-free KR13 and AuNP-KR13 (20 nm)-induced p24 release from these virions was measured using the p24 ELISA of the released protein, as explained under “Experimental Procedures.” The EC50 values are reported in Table 4. E, linear relationship between the natural log of the IC50 values of the virus infection inhibition (Table 4) by AuNP-KR13 and KR13 and the fraction of the WT DNA in the HIV-1 virions with linear fits that have R values of >0.97. F, data analysis showing that only the natural log of the EC50 values of the p24 release, induced by AuNP-KR13 (Table 4), and the fraction of WT DNA in the HIV-1 virion have a linear relationship, with R value = 0.973, whereas the KR13-induced p24 release does not follow this pattern. Error bars, S.D. (n = 3).
TABLE 4.
Antiviral effects of AuNP-KR13 (19.9 nm) and KR13 with HIV-1 pseudovirions expressed with varying WT/mutant (S375W) gp160 DNA ratio
The viral infection IC50 and p24 release EC50 were obtained from Fig. 8 using Origin Pro version 8. The relative amounts of DNA of WT versus S375W gp160 used for transfection to produce virions with different ratios of WT and S375W gp120 are listed. n = 3. NA, not available.
| AuNP-KR13 |
KR13 |
||||
|---|---|---|---|---|---|
| μg of DNA BAL-01 WT/S375W | IC50 of viral infection inhibition | EC50 of p24 release | μg of DNA BAL-01 WT/S375W | IC50 of viral infection inhibition | EC50 of p24 release |
| nm | μm | nm | μm | ||
| 0:4 | NA | NA | 0:4 | NA | NA |
| 0.5:3.5 | 2930 ± 316 | 215.1 ± 89.6 | 0.5:3.5 | 22,800 ± 3191 | NA |
| 1:3 | 469.6 ± 67 | 159.9 ± 39.5 | 1:3 | 1412.8 ± 213.3 | NA |
| 2:2 | 151.4 ± 60 | 22.5 ± 1.6 | 2:2 | 614.1 ± 89.4 | 4.26 ± 0.32 |
| 3:1 | 40.6 ± 4.3 | 10.2 ± 0.8 | 3:1 | 243.4 ± 32.1 | 6.03 ± 0.54 |
| 3.5:0.5 | 16.5 ± 2.2 | 1.8 ± 0.3 | 3.5:0.5 | 119.2 ± 45.6 | 6.74 ± 1.31 |
| 4:0 | 10.4 ± 2.6 | 1.3 ± 0.1 | 4:0 | 53.2 ± 12.8 | 3.10 ± 2.5 |
The logarithm of the IC50 values for infection inhibition varied linearly with the fraction of WT protomer in the viral populations (Fig. 8E). This linearity suggests that inhibitory potency directly depends on the energy of inhibitor binding. Suppose the energy of dissociation for a single KR13 is ΔG, then the energy of n such events is nΔG. The average value of this occupancy number n can be reasonably assumed to vary linearly with the average number of the available sites on HIV-1, which, in turn, is proportional to f. In this simplistic model, effective inhibitory concentrations can be treated as de facto equilibrium binding constants, leading to the observed proportionality.
The linear relationships for AuNP-KR13 and KR13 share the same slope, as expected because single inhibitor-binding energy ΔG is the same for both species. The result suggests that the two inhibitors utilize the same mechanism to block viral infection and that the enhanced inhibitory activity of AuNP-KR13 is a consequence of its ability to concentrate additional KR13 near the viral surface after the first peptide triazole is bound.
Although similar dependence on f was observed for viral lysis induced by AuNP-KR13, the lytic activity of KR13 was markedly different (Fig. 8, B and D). These KR13 titrations were largely independent of the WT protomer fraction until f dropped to 0.25. The KR13 titrations for f = 0.25 (1:3) and f = 0.125 (0.5:3.5) were significantly shallower than those for f > 0.25 and for all of the viral lysis titrations of AuNP-KR13. The divergence in lytic activity between AuNP-KR13 and KR13 is reflected in the different slopes of the ln(EC50) versus f plots (Fig. 8F). The AuNP-KR13 data had the same slope as the plots for infection inhibition (Fig. 8E), suggesting a similar dependence of lytic activity on KR13 binding energy. By contrast, the KR13 data for f > 0.25 had zero slope, implying a fundamental difference in how AuNP-KR13 and KR13 encounter the virus to cause lysis.
Morphological Analysis of AuNP-KR13 Conjugate Effects on HIV-1
We sought to image the virus-nanoparticle interface and consequent virus lysis using transmission electron microscopy. Viruses were treated with the AuNP-KR13 (20 and 80 nm) conjugates for 10 min and then fixed and loaded on a TEM grid. Uranyl acetate was used as a contrast agent to stain the lipids in order to visualize the virion. Representative images obtained are shown in Fig. 9. In the images for both the 20- and 80-nm AuNP-KR13, a limited number of AuNP-KR13 particles (an average of 1–3) were found associated with individual virions, suggesting that a small number of NPs is sufficient to induce virus lysis. The 80-nm AuNP-KR13 appeared to cause a more drastic disruption of the virion compared with the 20-nm AuNP-KR13. As shown by the representative images in Fig. 9, we observed that AuNP-KR13 led to a drastic disruption and collapse of the virus particle, with the residual virion partially engulfing the nanoparticle conjugate. In contrast, KR13 was shown previously (37) to lead to a more subtle virion collapse and at the same time greater virion shrinkage, as observed using TEM.
FIGURE 9.

Representative TEM images for HIV-1 BaL pseudotyped virus treated with PBS (top), 20-nm AuNP (middle), or 80-nm AuNP (bottom). After treatment for 10 min at 37 °C, samples were fixed as explained under “Experimental Procedures.” Collapsed virions of the type shown were validated to be virus-derived, as opposed to impurities, by detecting distinctive element signatures using the energy-dispersive spectroscopy system attached to the TEM instrument. The samples were stained with 1% uranyl acetate, loaded onto a TEM copper grid, and imaged using the JEM 2100 camera operated at 120 kV (JEOL).
DISCUSSION
The current study has demonstrated the ability to dramatically enhance the potency of anti-HIV-1 function of PT inhibitors by gold nanoparticle display and at the same time has derived a conceptual model of nanoparticle-virus encounter that can hijack the intrinsic metastability of the virus spike protein complex (15, 54–56). Previously, we demonstrated that conjugating PT thiols on 20-nm AuNP improved their antiviral functions (38). Here, we found that altering the AuNP properties of core size and PT density coverage led to dramatic enhancement in potency of both viral infection inhibition and cell-free virolysis (Figs. 2–4). More importantly, potency enhancement was also observed in replication-competent HIV-1 (Fig. 5). The potent antiviral functions of nanoparticle-PT conjugates, in particular irreversible inactivation of viruses by specific interaction with the virus surface protein gp120, makes this a worthy type of inhibition to understand mechanistically.
Considering that both KR13 and AuNP-KR13 exhibit a similar pattern of antiviral activities although with vastly different potencies, we asked whether there are fundamental mechanistic differences for these two inhibitor compositions. Observations made in this study (Fig. 6) demonstrated important differences. The MPER of gp41 on the HIV-1 Env is known to be exposed during the HIV-1/host cell fusion process but to be unexposed in the ground (unliganded) state of the virus (57). For KR13, we previously observed both concentration-dependent and time-dependent exposure of the MPER epitope on the HIV-1 virion when viruses were treated with KR13 (37). However, in the case of AuNP-KR13, we did not observe MPER epitope exposure (Fig. 6A). In addition, the data in Fig. 6B show that T20, a fusion inhibitor that targets 6-helix bundle formation of gp41 during HIV-1 fusion with the host cell, inhibits KR13-induced p24 release (as reported previously (37)) but does not inhibit such release by AuNP-KR13 (Fig. 6B). Further, the data of Fig. 6C show that gp120 shedding, infection inhibition, and virolysis tracked by p24 release of the HIV-1 BaL pseudotype all had a similar time dependence for the AuNP-KR13 exposure, in contrast to the unequal kinetics for KR13 (p24 release lagged compared with gp120 shedding and infection inhibition (37)). We note that the difference in kinetics of virus breakdown could have an impact on the extent of T20 sensitivity detected with AuNP-KR13 treatment. However, the lack of exposure of 4E10 and 2F5 epitopes with AuNP-KR13, versus exposure with KR13, argues that kinetics alone cannot explain the differences seen between the modes of action of these two types of virus inactivators. Collectively, the characteristics of MPER exposure, T20 sensitivity, and time dependence of antiviral effects observed for KR13 all argue that KR13-induced lysis bears a relationship to the physiological events of virus-cell fusion. In contrast, the lack of these phenotypes with AuNP-KR13 suggests that the latter responds to different forces. Several other differences between the mechanism of action of KR13 and AuNP-KR13 have emerged during ongoing investigations of KR13-induced lysis and its relationship to HIV-1 biological functions.4 A gp120 antibody, 2G12, that binds to the V3 loop and inhibits KR13-induced virolysis, has been observed4 not to inhibit AuNP-KR13-induced virolysis. In addition, DTNB (dithionitrobenzene), a thiol-blocking agent that inhibits KR13-induced virolysis, did not affect AuNP-KR13-mediated virolysis.5 These data clearly reinforce the need to consider mechanisms specific to gold nanoparticles in order to explain the virolytic activity of AuNP-KR13.
The responses of both p24 release and infection inhibition potency enhancements to nanoparticle and virus surface properties argue that multivalent contact between virus and nanoparticle surfaces is a major driving force for the enhanced antiviral potencies of AuNP-KR13. Three lines of evidence obtained in this work support the conclusion of multivalency. 1) Increasing the size of the AuNP particle leads to dramatic potency enhancement (Fig. 2 and Table 1). Because size increase causes a decrease of nanoparticle curvature, the area of contact of virus with nanoparticle would be expected to increase, and more points of contact would ensue between the peptide triazole NP surface and multiple spikes on the virus surface. 2) Increased density of KR13 on the AuNP surface strongly enhances antiviral potency (Fig. 3 and Tables 2 and 3). Increased area peptide density is expected to increase the extent of multipoint attachment between peptide triazoles and Env spikes on the nanoparticle and virus surfaces, respectively. 3) Decreasing the amount of wild-type (fully active) spikes on the HIV-1 virion surface by replacing the gp120 components with the KR13-insensitive mutant gp120, S375W, suppressed KR13-induced p24 release more drastically than AuNP-KR13 activity (Fig. 8, C and D). The functional data (Fig. 8) can be correlated with the statistically predicted relative abundance of heterogeneous and homogenous spikes expected for different mutant/WT mixtures (Fig. 7). We observed that, whereas KR13 induced p24 release required a statistical average of ≥1 fully active spike (containing 3 active WT gp120 monomers in individual spikes per virion), AuNP-KR13 induced up to 50% p24 release even with an average of close to zero fully active spikes (the case of 0.5:3.5 WT/mutant). The contrasting effects of KR13- versus AuNP-KR13-induced p24 release (Fig. 8F) suggest that AuNP-KR13 can cause lysis by engaging multiple spikes with an average of only 1 active gp120/spike, whereas KR13 probably requires a greater number of active gp120 monomers per spike trimer. It is worth noting that the ability of both KR13 and AuNP-KR13 to fully inhibit virus at spike populations containing a significant (although not complete) proportion of S375W mutant shows that mixed trimers can still exhibit PT sensitivity to the extent that they contain WT gp120 protomers. The difference in HIV-1 virion encounter by KR13 and AuNP-KR13 is shown schematically in Fig. 10. The physical nature of encounter in the model is based on the previously known dimensions of the virus Env (17, 55) and assumes that KR13 components on the AuNP are isotropic and equidistant from each other.
FIGURE 10.
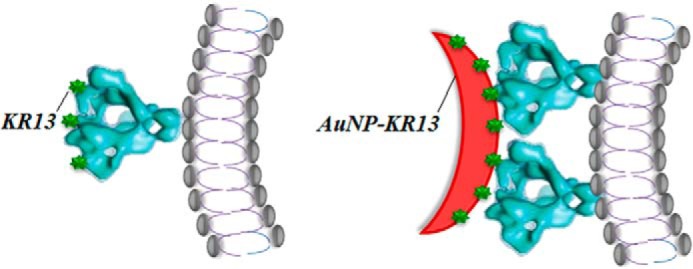
Model comparing the lysis-inducing encounter of KR13 and AuNP-KR13 with HIV-1 virus. The diameters of the HIV-1 virus as well as the AuNP-KR13 conjugates are taken from the measured values by DLS and TEM, respectively. The width and height of a spike were taken from the cryo-TEM modeling of Mao et al. (55).
One may ask why multivalent contact should cause such a profound effect leading to irreversible inactivation. We speculate that multivalent contact as envisioned in Fig. 10 causes an elevated stress on the HIV-1 membrane of the metastable HIV-1 virion, leading to irreversible collapse of the virion particle. As envisioned in the model in Fig. 10, KR13 action requires an Env spike with full or close to full native Env protomer content to exert a lytic force on the virus. In contrast, AuNP-KR13 exerts a lytic force by cross-linking Env spikes and hence can carry out this function as long as multiple Env spikes each contain at least one active protomer. That AuNP-KR13 causes significant loss in virus integrity was visualized by TEM imaging (Fig. 9). Here, the physical impact on the virion is much greater for AuNP-KR13 as compared with KR13 alone (37). Further investigation is needed to better understand the nature of the virus membrane transformation caused by AuNP-KR13 and how this type of effect compares with membrane transformations triggered by other types of virus-lysing agents (59–61).
The work presented here reinforces the potential usefulness of inhibitor display on nanoparticles to improve potency for disease prevention and intervention. Although nanocarriers have been used as vehicles for drug delivery, their use for forming therapeutic agents has been more limited (62–65). Jiang et al. have shown that gold nanoparticles ranging from 2 to 100 nm in diameter conjugated to multiple trastuzumab antibodies enable targeting and cross-linking of HER-2 (human epidermal growth factor receptor-2) in human SK-BR-3 breast cancer cells, by causing reduced HER-2 receptor expression and inducing receptor-mediated endocytosis (66). Jiang et al. (66) also showed that the anti-cancer potencies were altered by variations of the diameter of the gold nanoparticles, with the larger particles having more protein on their surface and increased avidity, whereas the smaller particles exhibited greater receptor mediated endocytosis. In the area of HIV-1, nanomaterial design for HIV-1 infection inhibition has been limited (67). Gold nanoparticle display was found to increase the antiviral potency of the HIV-1 inhibitor SDC-1721, a derivative of TAK-779 that targets the cellular coreceptor, CCR5 (68, 69). However, such studies have used very small (2-nm) AuNP that could be taken up by cells nonspecifically (70). Heterogeneity of the HIV-1 also has limited antibody and drug targeting (71). The current work argues that gold nanoparticle display could help overcome the heterogeneity of Env spikes by enabling multivalent attachment of neighboring partially defective spikes. The current work provides a generalizable approach for nanoparticle drug design not only for HIV-1 but also for other infectious enveloped viruses, such as influenza and hepatitis B virus.
Several types of inhibitors that bind to the HIV-1 envelope protein gp120 have now been found to be lytic inactivators of HIV-1. These include DAVEI (dual acting virus entry inhibitor containing cyanovirin-N fused to MPER) (61), the dodecameric CD4 construct (72), KR13 and other PT thiols (37, 38),4 and PT thiols on AuNPs. There are differences in the modes of action of these inactivators at the molecular level. DAVEI causes lysis but no observed gp120 shedding, and the requirement for the MPER domain for DAVEI lysis argues for unique interactions of this sequence with gp41 and/or membrane of the virus envelope (61). KR13 and AuNP-KR13 both cause gp120 shedding and membrane lysis but differ mechanistically as enumerated above. Nonetheless, despite these differences, the common feature of cell-independent virus lysis hints that there is probably a common mechanistic underpinning for all of these inhibitors. We posit that this lies in the intrinsic metastability that has been argued to be a property of HIV-1 Env (15, 54–56) and of the HIV-1 virion itself (47, 74). Metastability is a feature common to all viruses because these must protect their genetic contents and at the same time be able to open up to transfer their contents during cell entry to propagate more virus (47, 58, 75). For HIV-1, we propose that virus metastability appears to be a general property that can be hijacked for specific antiviral activities, as observed previously (37, 38, 61, 72). At the same time, the actions of lytic inactivators can inform about fundamental properties of virus metastability, including the membrane transformations that underlie both virolysis by gp120-inhibitor interactions and virus-cell fusion by gp120-cell receptor interactions.
Acknowledgments
We thank the Drexel University core facilities for access to the transmission electron microscope. We thank Beth Haggarty (University of Pennsylvania) from Dr. James Hoxie's laboratory, for help with fully infectious virus handling and access to a BSL-2 Plus facility for infectious virus assays.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1GM11029. This work was also supported by a W. W. Smith Foundation Award, National Science Foundation Grant CBET 0853680, and a Schlumberger Foundation Faculty of the Future Award (to A. R. B.).
R. Aneja, A. A. Rashad, H. Li, R. V. Kalyana Sundaram, C. Duffy, L. D. Bailey, and I. Chaiken, manuscript in preparation.
L. D. Bailey, H. Li, C. Duffy, R. V. Kalyana Sundaram, R. Aneja, A. Rosemary Bastian, A. P. Holmes, K. Kamanna, A. A. Rashad, and I. Chaiken, manuscript in preparation.
L. D. Bailey and I. Chaiken, unpublished results.
- gp41
- glycoprotein 41 (viral envelope protein)
- gp120
- glycoprotein 120 (viral envelope protein)
- AuNP
- gold nanoparticle
- PT
- peptide triazole
- AuNP-PT
- gold nanoparticle-peptide triazole conjugate
- HOS.T4.R5
- human osteosarcoma cell line with stable expression of CD4 and CCR5
- p24
- viral capsid protein
- MPER
- membrane-proximal external region
- DLS
- dynamic light scattering
- TEM
- transmission electron microscope
- 293T
- human embryonic kidney cell line
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl.
REFERENCES
- 1. Doyle T., Geretti A. M. (2012) Low-level viraemia on HAART: significance and management. Curr. Opin. Infect. Dis. 25, 17–25 [DOI] [PubMed] [Google Scholar]
- 2. Joint United Nations Programme on HIV/AIDS (2013) Global Report: UNAIDS Report on the Global AIDS Epidemic 2013, p 198, United Nations, Geneva [Google Scholar]
- 3. Kromdijk W., Huitema A. D., Mulder J. W. (2010) Treatment of HIV infection with the CCR5 antagonist maraviroc. Expert Opin. Pharmacother. 11, 1215–1223 [DOI] [PubMed] [Google Scholar]
- 4. Flores A., Quesada E. (2013) Entry inhibitors directed towards glycoprotein gp120: an overview on a promising target for HIV-1 therapy. Curr. Med. Chem. 20, 751–771 [PubMed] [Google Scholar]
- 5. Palmisano L., Vella S. (2011) A brief history of antiretroviral therapy of HIV infection: success and challenges, Ann. Ist. Super. Sanita 47, 44–48 [DOI] [PubMed] [Google Scholar]
- 6. Granich R., Crowley S., Vitoria M., Lo Y. R., Souteyrand Y., Dye C., Gilks C., Guerma T., De Cock K. M., Williams B. (2010) Highly active antiretroviral treatment for the prevention of HIV transmission, J. Int. AIDS Soc. 13, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haqqani A. A., Tilton J. C. (2013) Entry inhibitors and their use in the treatment of HIV-1 infection. Antiviral Res. 98, 158–170 [DOI] [PubMed] [Google Scholar]
- 8. Qiu S., Yi H., Hu J., Cao Z., Wu Y., Li W. (2012) The binding mode of fusion inhibitor T20 onto HIV-1 gp41 and relevant T20-resistant mechanisms explored by computational study. Curr. HIV Res. 10, 182–194 [DOI] [PubMed] [Google Scholar]
- 9. Henrich T. J., Kuritzkes D. R. (2013) HIV-1 entry inhibitors: recent development and clinical use. Curr. Opin. Virol. 3, 51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wallace B. J., Tan K. B., Pett S. L., Cooper D. A., Kossard S., Whitfeld M. J. (2011) Enfuvirtide injection site reactions: a clinical and histopathological appraisal. Australas. J. Dermatol. 52, 19–26 [DOI] [PubMed] [Google Scholar]
- 11. Perry C. M. (2010) Maraviroc: a review of its use in the management of CCR5-tropic HIV-1 infection. Drugs 70, 1189–1213 [DOI] [PubMed] [Google Scholar]
- 12. Pan C., Liu S., Jiang S. (2010) HIV-1 gp41 fusion intermediate: a target for HIV therapeutics. J. Formos. Med. Assoc. 109, 94–105 [DOI] [PubMed] [Google Scholar]
- 13. Champagne K., Shishido A., Root M. J. (2009) Interactions of HIV-1 inhibitory peptide T20 with the gp41 N-HR coiled coil. J. Biol. Chem. 284, 3619–3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Emmelkamp J. M., Rockstroh J. K. (2007) CCR5 antagonists: comparison of efficacy, side effects, pharmacokinetics and interactions: review of the literature. Eur. J. Med. Res. 12, 409–417 [PubMed] [Google Scholar]
- 15. Chan D. C., Kim P. S. (1998) HIV entry and its inhibition. Cell 93, 681–684 [DOI] [PubMed] [Google Scholar]
- 16. Wyatt R., Kwong P. D., Desjardins E., Sweet R. W., Robinson J., Hendrickson W. A., Sodroski J. G. (1998) The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393, 705–711 [DOI] [PubMed] [Google Scholar]
- 17. Kwong P. D., Wyatt R., Robinson J., Sweet R. W., Sodroski J., Hendrickson W. A. (1998) Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393, 648–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wyatt R., Sodroski J. (1998) The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280, 1884–1888 [DOI] [PubMed] [Google Scholar]
- 19. Hermann F. G., Egerer L., Brauer F., Gerum C., Schwalbe H., Dietrich U., von Laer D. (2009) Mutations in gp120 contribute to the resistance of human immunodeficiency virus type 1 to membrane-anchored C-peptide maC46. J. Virol. 83, 4844–4853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park E. J., Vujcic L. K., Anand R., Theodore T. S., Quinnan G. V., Jr. (1998) Mutations in both gp120 and gp41 are responsible for the broad neutralization resistance of variant human immunodeficiency virus type 1 MN to antibodies directed at V3 and non-V3 epitopes. J. Virol. 72, 7099–7107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson V. A., Calvez V., Gunthard H. F., Paredes R., Pillay D., Shafer R. W., Wensing A. M., Richman D. D. (2013) Update of the drug resistance mutations in HIV-1: March 2013. Top. Antivir. Med. 21, 6–14 [PMC free article] [PubMed] [Google Scholar]
- 22. Madani N., Princiotto A. M., Schön A., LaLonde J., Feng Y., Freire E., Park J., Courter J. R., Jones D. M., Robinson J., Liao H. X., Moody M. A., Permar S., Haynes B., Smith A. B., 3rd, Wyatt R., Sodroski J. (2014) CD4-mimetic small molecules sensitize human immunodeficiency virus (HIV-1) to vaccine-elicited antibodies. J. Virol. 88, 6542–6555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nettles R. E., Schürmann D., Zhu L., Stonier M., Huang S. P., Chang I., Chien C., Krystal M., Wind-Rotolo M., Ray N., Hanna G. J., Bertz R., Grasela D. (2012) Pharmacodynamics, safety, and pharmacokinetics of BMS-663068, an oral HIV-1 attachment inhibitor in HIV-1-infected subjects. J. Infect. Dis. 206, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 24. Zhao Q., Ma L., Jiang S., Lu H., Liu S., He Y., Strick N., Neamati N., Debnath A. K. (2005) Identification of N-phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 339, 213–225 [DOI] [PubMed] [Google Scholar]
- 25. Hughes A., Barber T., Nelson M. (2008) New treatment options for HIV salvage patients: an overview of second generation PIs, NNRTIs, integrase inhibitors and CCR5 antagonists. J. Infect. 57, 1–10 [DOI] [PubMed] [Google Scholar]
- 26. Nichols W. G., Steel H. M., Bonny T., Adkison K., Curtis L., Millard J., Kabeya K., Clumeck N. (2008) Hepatotoxicity observed in clinical trials of aplaviroc (GW873140). Antimicrob. Agents Chemother. 52, 858–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nichols W. K., Mehta R., Skordos K., Macé K., Pfeifer A. M., Carr B. A., Minko T., Burchiel S. W., Yost G. S. (2003) 3-Methylindole-induced toxicity to human bronchial epithelial cell lines, Toxicol. Sci. 71, 229–236 [DOI] [PubMed] [Google Scholar]
- 28. Madani N., Schön A., Princiotto A. M., Lalonde J. M., Courter J. R., Soeta T., Ng D., Wang L., Brower E. T., Xiang S. H., Kwon Y. D., Huang C. C., Wyatt R., Kwong P. D., Freire E., Smith A. B., 3rd, Sodroski J. (2008) Small-molecule CD4 mimics interact with a highly conserved pocket on HIV-1 gp120. Structure 16, 1689–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cocklin S., Gopi H., Querido B., Nimmagadda M., Kuriakose S., Cicala C., Ajith S., Baxter S., Arthos J., Martín-García J., Chaiken I. M. (2007) Broad-spectrum anti-human immunodeficiency virus (HIV) potential of a peptide HIV type 1 entry inhibitor. J. Virol. 81, 3645–3648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emileh A., Tuzer F., Yeh H., Umashankara M., Moreira D. R., Lalonde J. M., Bewley C. A., Abrams C. F., Chaiken I. M. (2013) A model of peptide triazole entry inhibitor binding to HIV-1 gp120 and the mechanism of bridging sheet disruption. Biochemistry 52, 2245–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gopi H. N., Tirupula K. C., Baxter S., Ajith S., Chaiken I. M. (2006) Click chemistry on azidoproline: high-affinity dual antagonist for HIV-1 envelope glycoprotein gp120. ChemMedChem 1, 54–57 [DOI] [PubMed] [Google Scholar]
- 32. McFadden K., Fletcher P., Rossi F., Kantharaju, Umashankara M., Pirrone V., Rajagopal S., Gopi H., Krebs F. C., Martin-Garcia J., Shattock R. J., Chaiken I. (2012) Antiviral breadth and combination potential of peptide triazole HIV-1 entry inhibitors, Antimicrob. Agents Chemother. 56, 1073–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tuzer F., Madani N., Kamanna K., Zentner I., LaLonde J., Holmes A., Upton E., Rajagopal S., McFadden K., Contarino M., Sodroski J., Chaiken I. (2013) HIV-1 Env gp120 structural determinants for peptide triazole dual receptor site antagonism. Proteins 81, 271–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Umashankara M., McFadden K., Zentner I., Schön A., Rajagopal S., Tuzer F., Kuriakose S. A., Contarino M., Lalonde J., Freire E., Chaiken I. (2010) The active core in a triazole peptide dual-site antagonist of HIV-1 gp120. ChemMedChem 5, 1871–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ferrer M., Harrison S. C. (1999) Peptide ligands to human immunodeficiency virus type 1 gp120 identified from phage display libraries. J. Virol. 73, 5795–5802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Biorn A. C., Cocklin S., Madani N., Si Z., Ivanovic T., Samanen J., Van Ryk D. I., Pantophlet R., Burton D. R., Freire E., Sodroski J., Chaiken I. M. (2004) Mode of action for linear peptide inhibitors of HIV-1 gp120 interactions. Biochemistry 43, 1928–1938 [DOI] [PubMed] [Google Scholar]
- 37. Bastian A. R., Contarino M., Bailey L. D., Aneja R., Moreira D. R., Freedman K., McFadden K., Duffy C., Emileh A., Leslie G., Jacobson J. M., Hoxie J. A., Chaiken I. (2013) Interactions of peptide triazole thiols with Env gp120 induce irreversible breakdown and inactivation of HIV-1 virions. Retrovirology 10, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bastian A. R., Kantharaju, McFadden K., Duffy C., Rajagopal S., Contarino M. R., Papazoglou E., Chaiken I. (2011) Cell-free HIV-1 virucidal action by modified peptide triazole inhibitors of Env gp120, ChemMedChem 6, 1335–1339, 1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gopi H., Cocklin S., Pirrone V., McFadden K., Tuzer F., Zentner I., Ajith S., Baxter S., Jawanda N., Krebs F. C., Chaiken I. M. (2009) Introducing metallocene into a triazole peptide conjugate reduces its off-rate and enhances its affinity and antiviral potency for HIV-1 gp120. J. Mol. Recognit. 22, 169–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frens G. (1973) Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nature 10.1038/physci241020a0 [DOI] [Google Scholar]
- 41. Haiss W., Thanh N. T., Aveyard J., Fernig D. G. (2007) Determination of size and concentration of gold nanoparticles from UV-vis spectra. Anal. Chem. 79, 4215–4221 [DOI] [PubMed] [Google Scholar]
- 42. Xie H., Tkachenko A. G., Glomm W. R., Ryan J. A., Brennaman M. K., Papanikolas J. M., Franzen S., Feldheim D. L. (2003) Critical flocculation concentrations, binding isotherms, and ligand exchange properties of peptide-modified gold nanoparticles studied by UV-visible, fluorescence, and time-correlated single photon counting spectroscopies. Anal. Chem. 75, 5797–5805 [DOI] [PubMed] [Google Scholar]
- 43. He J., Choe S., Walker R., Di Marzio P., Morgan D. O., Landau N. R. (1995) Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 69, 6705–6711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Connor R. I., Chen B. K., Choe S., Landau N. R. (1995) Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206, 935–944 [DOI] [PubMed] [Google Scholar]
- 45. Xiang S. H., Kwong P. D., Gupta R., Rizzuto C. D., Casper D. J., Wyatt R., Wang L., Hendrickson W. A., Doyle M. L., Sodroski J. (2002) Mutagenic stabilization and/or disruption of a CD4-bound state reveals distinct conformations of the human immunodeficiency virus type 1 gp120 envelope glycoprotein. J. Virol. 76, 9888–9899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dey B., Pancera M., Svehla K., Shu Y., Xiang S. H., Vainshtein J., Li Y., Sodroski J., Kwong P. D., Mascola J. R., Wyatt R. (2007) Characterization of human immunodeficiency virus type 1 monomeric and trimeric gp120 glycoproteins stabilized in the CD4-bound state: antigenicity, biophysics, and immunogenicity. J. Virol. 81, 5579–5593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Flint S. J., Enquist L. W., Racaniello V. R. (2008) Principles of Virology, pp. 83–85, ASM Press, Washington, D. C. [Google Scholar]
- 48. Chojnacki J., Staudt T., Glass B., Bingen P., Engelhardt J., Anders M., Schneider J., Müller B., Hell S. W., Kräusslich H. G. (2012) Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science 338, 524–528 [DOI] [PubMed] [Google Scholar]
- 49. Muranyi W., Malkusch S., Müller B., Heilemann M., Kräusslich H. G. (2013) Super-resolution microscopy reveals specific recruitment of HIV-1 envelope proteins to viral assembly sites dependent on the envelope C-terminal tail. PLoS Pathog. 9, e1003198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Crooks E. T., Tong T., Osawa K., Binley J. M. (2011) Enzyme digests eliminate nonfunctional Env from HIV-1 particle surfaces, leaving native Env trimers intact and viral infectivity unaffected. J. Virol. 85, 5825–5839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Summers M. F., Henderson L. E., Chance M. R., Bess J. W., Jr., South T. L., Blake P. R., Sagi I., Perez-Alvarado G., Sowder R. C., 3rd, Hare D. R. (1992) Nucleocapsid zinc fingers detected in retroviruses: EXAFS studies of intact viruses and the solution-state structure of the nucleocapsid protein from HIV-1. Protein Sci. 1, 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vogt V. M., Simon M. N. (1999) Mass determination of Rous sarcoma virus virions by scanning transmission electron microscopy. J. Virol. 73, 7050–7055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim T., Lee K., Gong M. S., Joo S. W. (2005) Control of gold nanoparticle aggregates by manipulation of interparticle interaction. Langmuir 21, 9524–9528 [DOI] [PubMed] [Google Scholar]
- 54. Mao Y., Wang L., Gu C., Herschhorn A., Désormeaux A., Finzi A., Xiang S. H., Sodroski J. G. (2013) Molecular architecture of the uncleaved HIV-1 envelope glycoprotein trimer. Proc. Natl. Acad. Sci. U.S.A. 110, 12438–12443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mao Y., Wang L., Gu C., Herschhorn A., Xiang S. H., Haim H., Yang X., Sodroski J. (2012) Subunit organization of the membrane-bound HIV-1 envelope glycoprotein trimer. Nat. Struct. Mol. Biol. 19, 893–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Myszka D. G., Sweet R. W., Hensley P., Brigham-Burke M., Kwong P. D., Hendrickson W. A., Wyatt R., Sodroski J., Doyle M. L. (2000) Energetics of the HIV gp120-CD4 binding reaction. Proc. Natl. Acad. Sci. U.S.A. 97, 9026–9031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dimitrov A. S., Jacobs A., Finnegan C. M., Stiegler G., Katinger H., Blumenthal R. (2007) Exposure of the membrane-proximal external region of HIV-1 gp41 in the course of HIV-1 envelope glycoprotein-mediated fusion. Biochemistry 46, 1398–1401 [DOI] [PubMed] [Google Scholar]
- 58. White J. M., Delos S. E., Brecher M., Schornberg K. (2008) Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43, 189–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Xu Y., Zhang C., Jia L., Wen C., Liu H., Wang Y., Sun Y., Huang L., Zhou Y., Song H. (2009) A novel approach to inhibit HIV-1 infection and enhance lysis of HIV by a targeted activator of complement. Virol. J. 6, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nakamura M., Terada M., Sasaki H., Kamada M., Ohno T. (2000) Virolysis and in vitro neutralization of HIV-1 by humanized monoclonal antibody hNM-01. Hybridoma 19, 427–434 [DOI] [PubMed] [Google Scholar]
- 61. Contarino M., Bastian A. R., Kalyana Sundaram R. V., McFadden K., Duffy C., Gangupomu V., Baker M., Abrams C., Chaiken I. (2013) Chimeric Cyanovirin-MPER recombinantly engineered proteins cause cell-free virolysis of HIV-1. Antimicrob. Agents Chemother. 57, 4743–4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Farokhzad O. C., Langer R. (2009) Impact of nanotechnology on drug delivery. ACS Nano 3, 16–20 [DOI] [PubMed] [Google Scholar]
- 63. Zhang L., Gu F. X., Chan J. M., Wang A. Z., Langer R. S., Farokhzad O. C. (2008) Nanoparticles in medicine: therapeutic applications and developments, Clin. Pharmacol. Ther. 83, 761–769 [DOI] [PubMed] [Google Scholar]
- 64. Hamidi M., Azadi A., Rafiei P., Ashrafi H. (2013) A pharmacokinetic overview of nanotechnology-based drug delivery systems: an ADME-oriented approach. Crit. Rev. Ther. Drug Carrier Syst. 30, 435–467 [DOI] [PubMed] [Google Scholar]
- 65. Heidel J. D., Davis M. E. (2011) Clinical developments in nanotechnology for cancer therapy. Pharm. Res. 28, 187–199 [DOI] [PubMed] [Google Scholar]
- 66. Jiang W., Kim B. Y., Rutka J. T., Chan W. C. (2008) Nanoparticle-mediated cellular response is size-dependent. Nat. Nanotechnol. 3, 145–150 [DOI] [PubMed] [Google Scholar]
- 67. Mamo T., Moseman E. A., Kolishetti N., Salvador-Morales C., Shi J., Kuritzkes D. R., Langer R., von Andrian U., Farokhzad O. C. (2010) Emerging nanotechnology approaches for HIV/AIDS treatment and prevention. Nanomedicine 5, 269–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bowman M. C., Ballard T. E., Ackerson C. J., Feldheim D. L., Margolis D. M., Melander C. (2008) Inhibition of HIV fusion with multivalent gold nanoparticles. J. Am. Chem. Soc. 130, 6896–6897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mahmoud K. A., Luong J. H. (2008) Impedance method for detecting HIV-1 protease and screening for its inhibitors using ferrocene-peptide conjugate/Au nanoparticle/single-walled carbon nanotube modified electrode. Anal. Chem. 80, 7056–7062 [DOI] [PubMed] [Google Scholar]
- 70. Chithrani B. D., Ghazani A. A., Chan W. C. (2006) Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 6, 662–668 [DOI] [PubMed] [Google Scholar]
- 71. Poignard P., Moulard M., Golez E., Vivona V., Franti M., Venturini S., Wang M., Parren P. W., Burton D. R. (2003) Heterogeneity of envelope molecules expressed on primary human immunodeficiency virus type 1 particles as probed by the binding of neutralizing and nonneutralizing antibodies. J. Virol. 77, 353–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bennett A., Liu J., Van Ryk D., Bliss D., Arthos J., Henderson R. M., Subramaniam S. (2007) Cryoelectron tomographic analysis of an HIV-neutralizing protein and its complex with native viral gp120. J. Biol. Chem. 282, 27754–27759 [DOI] [PubMed] [Google Scholar]
- 73. Deleted in proof.
- 74. Kol N., Shi Y., Tsvitov M., Barlam D., Shneck R. Z., Kay M. S., Rousso I. (2007) A stiffness switch in human immunodeficiency virus. Biophys. J. 92, 1777–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wallin M., Ekström M., Garoff H. (2005) The fusion-controlling disulfide bond isomerase in retrovirus Env is triggered by protein destabilization. J. Virol. 79, 1678–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]