

Background: CRL4CDT2 mediates replication-coupled destruction during S phase. CRL4CDT2 substrates reaccumulate by an unexplored mechanism.
Results: CDK1 activity blocks CRL4CDT2 by preventing chromatin recruitment of the substrate receptor, CDT2.
Conclusion: CDK1 activity facilitates CRL4CDT2 substrate reaccumulation upon S phase exit; several of these substrates are then required for normal mitotic progression.
Significance: We provide the first evidence that CDK1 regulates the activity of CRL4CDT2.
Keywords: Cell Cycle, Cyclin-dependent Kinase (CDK), DNA Replication, E3 Ubiquitin Ligase, Mitosis, CDT1, CDT2, p21, PCNA, SET8
Abstract
Replication-coupled destruction of a cohort of cell cycle proteins ensures efficient and precise genome duplication. Three proteins destroyed during replication via the CRL4CDT2 ubiquitin E3 ligase, CDT1, p21, and SET8 (PR-SET7), are also essential or important during mitosis, making their reaccumulation after S phase a critical cell cycle event. During early and mid-S phase and during DNA repair, proliferating cell nuclear antigen (PCNA) loading onto DNA (PCNADNA) triggers the interaction between CRL4CDT2 and its substrates, resulting in their degradation. We have discovered that, beginning in late S phase, PCNADNA is no longer sufficient to trigger CRL4CDT2-mediated degradation. A CDK1-dependent mechanism that blocks CRL4CDT2 activity by interfering with CDT2 recruitment to chromatin actively protects CRL4CDT2 substrates. We postulate that deliberate override of replication-coupled destruction allows anticipatory accumulation in late S phase. We further show that (as for CDT1) de novo SET8 reaccumulation is important for normal mitotic progression. In this manner, CDK1-dependent CRL4CDT2 inactivation contributes to efficient transition from S phase to mitosis.
Introduction
Ubiquitin-mediated degradation of cell cycle proteins is essential to ensure timely cell cycle transitions that maintain genome integrity. Conversely, cell cycle-dependent protein stabilization by preventing ubiquitination allows rapid protein accumulation at the right time to bring about robust cell cycle transitions. Replication-coupled destruction is a particularly important protein control mechanism that coordinates the degradation of a cohort of proteins with the process of DNA synthesis. Much has been learned about how replication-coupled destruction is initiated in S phase (1–5), but less is known about how it is inhibited as S phase ends.
Among the cohort of human proteins reported to be subject to replication-coupled destruction are CDT1, SET8, p12, the CDK inhibitor p21, and, most recently, thymine DNA glycosylase (6, 7). Their destruction in S phase is particularly critical to ensure precise and efficient genome duplication (1–7). CDT1 is required in G1 for rendering DNA replication origins competent for initiation in S phase (a process termed “origin licensing”) (8, 9). SET8 is the sole enzyme responsible for histone H4 lysine 20 monomethylation (H4K20me1)4 and, like CDT1, is also required for origin licensing (10). Degradation of CDT1 and SET8 at the onset of S phase restricts DNA replication to no more than once per cell cycle by preventing relicensing of replicated origins. Failure to degrade either CDT1 or SET8 results in multiple rounds of origin firing, leading to DNA rereplication and ultimately significant DNA damage and genome instability (1, 10–17). Likewise, persistence of human thymine DNA glycosylase in S phase slows proliferation (6). Degradation of p21 in early S phase stimulates CDK2 activity, which, in turn, triggers key S phase events, including DNA replication initiation and origin licensing inhibition (18, 19). During S phase, p12 levels are 35% of their G2/M and G1 levels, resulting in the presence of both Polδ3 (Polδ lacking p12) and Polδ4 (Polδ containing p12) during S phase (5). Because the two forms of Polδ have complementary biochemical properties (5, 20), CRL4CDT2-mediated degradation of p12 during S phase may be important for DNA replication fidelity.
Each of the proteins known to be subject to replication-coupled destruction follows a pattern of low abundance during S phase and then rapid reaccumulation prior to mitosis (illustrated in Fig. 1). Importantly, CDT1, p21, and SET8 also function during mitosis, making their reaccumulation critical for normal mitotic progression (21–23). Replication-coupled destruction is triggered by the ubiquitin E3 ligase, CRL4CDT2. The mechanism of CRL4CDT2 substrate recognition is unique in that the substrates must first interact with DNA-loaded PCNA (PCNADNA), and PCNA is DNA-loaded during both S phase and DNA repair (24–27). Given the robust reaccumulation of CRL4CDT2 substrates well in advance of mitosis, we sought to determine the relationship between PCNA unloading in late S phase and reaccumulation of CRL4CDT2 substrates.
FIGURE 1.

Targets of replication-coupled destruction reaccumulate prior to the end of S phase. A, in early S phase, PCNA is loaded onto DNA and is bound by CRL4CDT2 substrates (e.g. CDT1). The interaction of substrates with PCNADNA recruits the CRL4CDT2 E3 ubiquitin ligase via the CDT2 substrate receptor subunit for replication-coupled destruction. B, HCT116 cells were synchronized in early S phase by double thymidine block. Soluble proteins were extracted, and bound proteins were fixed in early S phase (during the arrest) or in late S phase 7 h after release into fresh medium. Endogenous PCNADNA and CDT1 were detected by immunostaining. C and D, as in B except that p21 or SET8 was detected by immunostaining. Scale bars, 5 μm in B–D. E, percentage of late S phase cells containing PCNADNA that also contained SET8 (n = 60), p21 (n = 100), or CDT1 (n = 122) was quantified. F, total PCNA nuclear fluorescence was quantified for the early and late S phase samples in B (n = 15). A total of 60 cells were counted over three biological replicates (≥15 cells were counted per replicate). Averages and S.D. values (error bars) are indicated in both E and F.
We have discovered that, surprisingly, CRL4CDT2 substrates accumulate prior to PCNA unloading and the completion of replication. Moreover, we demonstrate that activation of CDK1 inhibits degradation of CRL4CDT2 substrates. We show here that CDK1 activity (directly and/or indirectly) inhibits CRL4CDT2 activity itself by preventing its accumulation on chromatin, an event necessary for CRL4CDT2 activity. Activation of CDK1 as S phase completes is necessary for the normal reaccumulation of substrates, such as CDT1 and SET8, and we show that, like CDT1, failure to reaccumulate SET8 de novo prior to mitosis leads to mitotic progression defects (21). The temporal control of CRL4CDT2 activity ensures the accumulation of CRL4CDT2 substrates during mitosis, thereby preventing chromosome instability. We propose that purposeful protection from replication-coupled destruction anticipates the end of S phase and primes efficient progression through mitosis.
EXPERIMENTAL PROCEDURES
Cell Culture and Manipulations
HCT116, 293T, and HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Difco) supplemented with 10% fetal calf serum (Sigma). HCT116 and HeLa cells were synchronized in early S phase by double thymidine block (2 mm thymidine for 17 h, release into fresh medium for 9 h, 2 mm thymidine for 15 h) or in prometaphase by treatment with 2 mm thymidine for 18 h followed by release into 100 nm nocodazole for 10 h as described previously (28, 29). Kinase inhibitors were used at the following concentrations: c-Jun N-terminal kinase (JNK) inhibitor VIII at 10 μm (EMD Millipore), p38 inhibitor SB203580 at 30 μm (LC Laboratories), and the CDK1 inhibitor RO-3306 (Sigma) at 10 μm or as indicated. DNA damage was induced by a single treatment of 20 J/m2 UV. Small interfering RNA (siRNA) transfections were performed with a 400 nm concentration of each siRNA duplex using Dharmafect 1 reagent (Dharmacon) according to the manufacturer's guidelines. This high concentration of siRNA was required for efficient knockdown, given the brief siRNA treatment time. Synthetic duplexed RNA oligonucleotides were synthesized by Invitrogen: luciferase (5′-CUUACGCUGAGUACUUCGA-3′), SET8-ORF (5′-GATGCAACTAGAGAGACA-3′) (10), and SET8-UTR (5′-AAGCAUACAAGCCGAACGUU-3′) (30).
Antibodies
Antibodies were purchased from the following sources: MAPKAP kinase 2, phospho-MAPKAP kinase 2, phospho-c-JUN (Ser-63), and SET8 from Cell Signaling Technologies; hemagglutinin (HA) from Roche Applied Science; α-tubulin (DM1A) from Sigma; PCNA (PC-10), p21, cyclin A, cyclin B1, and Orc2 from Santa Cruz Biotechnology, Inc.; H4K20me1 from EMD Millipore (catalog no. 04-735); H4K20me2 from Active Motif (catalog no. 39174). CDT2 and p12 antibodies were a gift from A. Dutta. Cul4B and Cul4A antibodies were a gift from Y. Xiong. Antibodies to human CDT1 have been described previously (31). AlexaFluor 488 and Rhodamine Red-X-labeled donkey secondary antibodies for immunofluorescence microscopy were obtained from Jackson ImmunoResearch Laboratories.
Plasmids and Protein Lysate Preparation
HA-tagged mutant CDT1 and p21 were generated via PCR and cloned into pLX302 (Addgene plasmid 25896) (32) or pBABE (Addgene plasmid 51070) (33) expression vectors, respectively, as were the tagged wild type constructs. Plasmids expressing glutathione S-transferase (GST)-p21WT and GST-p21PIP fusions were generated by recombinational cloning between pENTR-p21 derivatives and pDEST15 for N-terminal GST fusion (Gateway LR clonase, Invitrogen) and expressed in BL21 cells. The PIP box was inactivated by site-directed mutagenesis to generate the p21PIP mutant (MTDFY to AAAA). Whole-cell lysates were prepared either by direct lysis of equal cell numbers in 2× Laemmli sample buffer with 10% β-mercaptoethanol or in CSK buffer (supplemented with 0.1% Triton X-100 and protease and phosphatase inhibitors) (34). Chromatin-enriched fractions were isolated as described previously (35) by the addition of micrococcal nuclease and CaCl2 to Triton-insoluble pellets to release DNA-bound material into the soluble pool and clarified by centrifugation. Alternatively, sonication was used to solubilize the chromatin-bound proteins.
Protein-Protein Interaction Assays
Recombinant GST fusion proteins GST-p21WT or GST-p21PIP (mutated PIP box) were immobilized on glutathione-Sepharose 4B beads (GE Healthcare). Whole cell extracts (from HCT116 cells) were prepared in CSK buffer (supplemented with 0.1% Triton X-100 and protease and phosphatase inhibitors), and 1 mg of cell lysate was incubated with the bound GST-p21 derivatives for 3 h at 4 °C. The bound complexes were washed three times in complete CSK buffer, resuspended in 50 μl of 2× SDS sample buffer, and boiled for 5 min to elute bound proteins.
Immunofluorescence Microscopy
Cells were rinsed briefly in 1× phosphate-buffered saline (PBS, pH 7.4) prior to fixation. Cells were typically prefixed with 4% paraformaldehyde for 60 s, permeabilized with 0.5% Triton X-100 for 5 min, and then fixed for 15 min using 4% paraformaldehyde. For all immunostaining procedures involving PCNA, the cells were first prefixed in 2% paraformaldehyde for 60 s and then permeabilized with 0.5% Triton X-100 for 5 min and fixed with 2% paraformaldehyde for 15 min followed by −20 °C methanol for 4 min. Blocking steps, antibody incubations, and washes were all performed in 1× PBS buffer plus 0.1% BSA. All antibody incubations were for 1 h at 37 °C, and washes were for 10 min at room temperature. DAPI staining (0.1 μg/ml) was performed for 10 min, and cells were mounted using either Prolong Antifade (Invitrogen/Molecular Probes) or VECTASHIELD mounting medium (Vector Laboratories, Inc.).
For indirect immunofluorescence microscopy of interphase nuclei, images were acquired using Metamorph Software and a 60×/1.4 numerical aperture (PlanApo) differential interference contrast oil immersion objective (Nikon) mounted on a Nikon TE300 inverted microscope equipped with a Yokogawa CSU10 spinning disk and a Hamamatsu Orca ER cooled CCD camera. For fluorescence intensity measurements, the average values for integrated nuclear PCNA fluorescence from control and experimental cell samples were subjected to background subtraction to obtain the specific nuclear fluorescence levels. For indirect immunofluorescence microscopy of mitotic cells, images were acquired using Metamorph Software and a 100×/1.4 numerical aperture (PlanApo) differential interference contrast oil immersion objective mounted on a Leica DMIRB inverted microscope equipped with a Photometric HQ2 cooled CCD camera. Scale bars represent 5 μm in all figures.
Flow Cytometry
Prior to flow cytometry analysis, cells were trypsinized, fixed in 70% ethanol, and treated with propidium iodide/RNase solution according to standard methods. Flow cytometry analysis was performed using a Cyan FACScan (DakoCytomation) and Summit version 4.3 software (DakoCytomation) as described previously (36).
RESULTS
CRL4CDT2 Substrate Accumulation Occurs in Late S Phase Prior to PCNA Unloading
Previous work described replication-coupled destruction of CRL4CDT2 substrates and further documented their robust accumulation at later cell cycle stages (5, 11, 19, 37) illustrated in Fig. 1A. Given that PCNA loading at replication and repair sites (PCNADNA) is a prerequisite for CRL4CDT2 targeting, the normal substrate reaccumulation as S phase ends could simply be a consequence of PCNA unloading as replication completes. Nevertheless, replication can extend close to the time of mitosis, and this late replication has been implicated in the genome instability associated with common fragile sites (38). Because PCNADNA triggers CRL4CDT2-mediated degradation, persistence of PCNADNA so late in S phase was not fully consistent with observations by us and others that CRL4CDT2 substrates reaccumulate well in advance of mitosis (5, 11, 19, 39). We therefore considered the possibility that substrates may be actively stabilized during the transition from S phase to mitosis. If so, then CRL4CDT2 substrates may in fact accumulate prior to PCNADNA unloading.
To test this idea, we interrogated the timing of CRL4CDT2 substrate accumulation in individual cells relative to the dynamics of PCNADNA unloading as cells completed S phase. We synchronized cells in early S phase, released them to proceed to late S phase, and then extracted soluble proteins prior to fixation and immunostaining for endogenous PCNADNA and the CRL4CDT2 substrates CDT1, p21, and SET8. As expected, cells in early S phase had abundant PCNADNA in a characteristic early S phase pattern (40, 41) but had very little CDT1 (Fig. 1B, top row) and similarly low levels of p21 and SET8 (Fig. 1, C and D, top rows). In late S phase (7 h postrelease), PCNA was still DNA-loaded in some cells, and CDT1, p21, and SET8 were readily detectable in those same cells (Fig. 1, B–D, bottom rows). Late S phase cells are characterized by foci of chromatin-bound PCNA at the nuclear periphery with more diffuse nuclear staining elsewhere, as seen in Fig. 1, B–D (40, 41). (Multiple mechanisms recruit these proteins to chromatin, resulting in only partial co-localization with PCNA foci.) We counted the number of cells that retained PCNADNA foci 7 h into S phase (14%, n = 150) and then scored those that also contained nuclear p21, SET8, or CDT1 using antibodies to the endogenous proteins. We found that 40–65% of PCNADNA-positive cells already had abundant CRL4CDT2 substrates (Fig. 1E; we cannot distinguish differences among the individual substrate kinetics from differences in antibody avidity). Importantly, individual late S phase cells that retained PCNADNA had nearly equivalent amounts of PCNADNA relative to their early S phase counterparts (Fig. 1F). Thus, the accumulation of substrates was not accounted for by the amount of PCNADNA declining in individual late S phase cells. The fact that cells with similar amounts of PCNADNA showed dramatically different abilities to support replication-coupled destruction indicates a qualitative difference between early and late S phase cells with regard to CRL4CDT2 activity. Moreover, the presence of CDT1, SET8, and p21 in late S phase nuclei with PCNADNA suggested an active mechanism to inhibit CRL4CDT2-mediated degradation.
CRL4CDT2 Substrates Cannot Be Targeted during Mitosis
It is worth noting that at 7 h postrelease (Fig. 1), not every cell contained both PCNADNA and CRL4CDT2 substrates, consistent with the observation that even in synchronized cells, progression through S phase is not perfectly uniform. Our time courses indicate that the onset of CRL4CDT2 substrate accumulation occurs during the final ∼10–15% of S phase based on an average 7–8-h S phase and mitotic entry at 8–9 h (e.g. see Fig. 4A). Because we could not arrest cells in very late S phase for biochemical analysis, we took advantage of the next available robust cell cycle block in prometaphase, which is 1–2 h later, to generate homogeneous cell populations. To determine if CRL4CDT2 substrates are actively protected from degradation late in the cell cycle, we deliberately stimulated synchronous replication-coupled destruction by UV-irradiating prometaphase cells. UV irradiation causes robust and simultaneous PCNA loading onto DNA during nucleotide excision repair, and thus UV treatment causes highly synchronous replication-coupled destruction of CRL4CDT2 substrates by the same mechanism that operates during S phase (reviewed in Ref. 42). (Note that CRL4CDT2-mediated degradation is stimulated by DNA synthesis per se and is not a component of the DNA damage checkpoint response (25)). We monitored the levels of CDT1 as well as three other CRL4CDT2 substrates by immunoblotting lysates from the two conditions. As expected in asynchronous cells, CDT1, SET8, p21, and p12 were degraded following UV irradiation (Fig. 2, A and B, lanes 1 and 2). In stark contrast, however, none of the four substrates were degraded in prometaphase cells (Fig. 2, A and B, lanes 3 and 4).
FIGURE 4.

CDK1 activity is required for substrate reaccumulation in late S phase. A, HCT116 cells were synchronized in early S phase via double thymidine block and 5 h after release were treated with RO-3306 or DMSO (control). Cells were harvested at the indicated time points and analyzed for DNA content by flow cytometry. B, HCT116 cells were treated as in A, harvested at the indicated time points, and analyzed via immunoblotting. C, HCT116 cells were treated as in A. Soluble proteins were extracted, and bound proteins were fixed in early S phase (during the arrest) or in late S phase 7 h after release into fresh medium. Endogenous PCNADNA, SET8, and CDT1 were detected by immunostaining as in Fig. 1. Late S phase cells containing PCNADNA were scored for the presence of SET8 or CDT1 as an indicator of inactive CRL4CDT2 (n ≥ 75 for each sample). Error bars, S.D.
FIGURE 2.
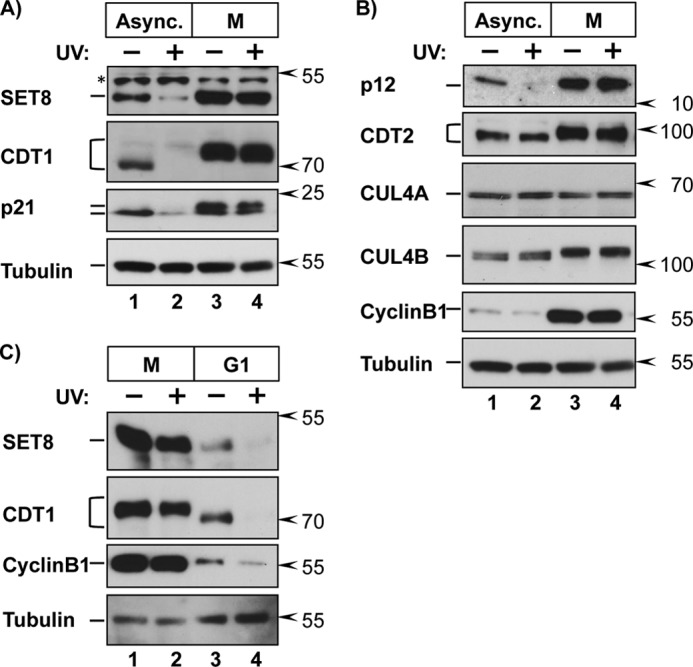
CRL4CDT2 substrates are protected in mitosis. A and B, HCT116 cells were grown asynchronously (Async.) or synchronized in prometaphase by release from a thymidine block into 100 nm nocodazole for 10 h (M). Cells were irradiated with 20 J/m2 UV to induce PCNADNA loading (DNA repair synthesis) and harvested 2 h later or left untreated as indicated. Endogenous proteins were detected in whole cell lysates by immunoblotting (asterisk in A denotes a nonspecific background band). C, HCT116 cells were synchronized in prometaphase as in A and released into fresh medium for 2.5 h to proceed to G1. Cells were UV-irradiated and harvested 90 min later, followed by immunoblotting for the indicated proteins.
CRL4CDT2 substrates are resistant to degradation in some quiescent cells because the substrate adapter subunit, CDT2, is itself degraded (43, 44). CDT2 was equally present in both asynchronous and prometaphase cells, so the substrate protection we observed could not be attributed to loss of CDT2 under these conditions (Fig. 2B, lanes 3 and 4). Importantly, releasing mitotic cells into the subsequent G1 fully restored sensitivity of SET8 and CDT1 to CRL4CDT2-mediated degradation (Fig. 2C). SET8 is also an APC/C target (23), and we observed the expected low SET8 levels in untreated G1 cells relative to mitotic cells. Taken together, we conclude that the substrates of CRL4CDT2 are protected from degradation in a cell cycle-dependent manner.
Mitotic Kinase Activity Is Required for Protection from CRL4CDT2-mediated Degradation
Our observation that CRL4CDT2 substrates are stable from late S phase through mitosis but susceptible to replication-coupled destruction during G1 suggested that a mitotic activity, such as one or more mitotic protein kinases, confers protection from CRL4CDT2-mediated degradation. To test that possibility, we employed pharmacological inhibition of candidate kinases: cyclin-dependent kinases (CDKs) and the stress-activated mitogen-activated protein kinases (MAPK) p38 and JNK, which we previously showed could stabilize CRL4CDT2 substrates (39). We briefly treated prometaphase cells with kinase inhibitors prior to UV irradiation and then tested for reversal of mitotic stability by immunoblotting (Fig. 3, A and B). In control cells as before, CRL4CDT2 substrates were stable in mitotic cells, but strikingly, treatment with the CDK1-specific inhibitor RO-3306 effectively reversed the protection (Fig. 3, A (compare lanes 4 and 6) and B (compare lanes 4 and 5)). In contrast, treatment with the p38 or JNK inhibitors had little to no effect on substrate protection (Fig. 3B, lanes 6 and 7). Note that both p38 and JNK are not only active in mitosis but also induced by UV (45, 46); the JNK and p38 inhibitors were effective for inhibiting their respective kinases at these concentrations, as measured by phosphorylation of representative substrates (Fig. 3B, bottom panels). CDK1 inhibition also caused cyclin B1 degradation (Fig. 3B, lanes 5 and 8), which is consistent with prior studies showing that inhibiting CDK1 in mitotic cells activates APCCdc20 (47, 48). (SET8 is also an APCCdc20 target, but SET8 phosphorylation during mitosis blocks APCCdc20 binding, so SET8 was not degraded in cells treated with the CDK1 inhibitor alone (23).) Importantly, only CDK1 inhibition resensitized CRL4CDT2 substrates in mitosis (Fig. 3B, compare lane 4 with lanes 5–7). Even simultaneous inhibition of p38 and JNK could not substitute for CDK1 inhibition (data not shown). Furthermore, increasing concentrations of the CDK1 inhibitor resulted in progressive loss of CRL4CDT2 substrate protection (Fig. 3C).
FIGURE 3.

CDK1 activity is required for CRL4CDT2 substrate protection. A, asynchronous or prometaphase HCT116 cells were treated with the CDK1 inhibitor RO-3306 as indicated 30 min prior to PCNADNA induction by UV and harvested 2 h later. B, top panel, asynchronous or prometaphase cells were treated with the indicated kinase inhibitors or DMSO (none) for 30 min prior to PCNADNA induction by UV irradiation. Endogenous proteins were detected by immunoblotting of whole cell lysates. Bottom panels, asynchronous cells were treated with the inhibitor JNK VIII or SB203580 (p38 inhibitor) 30 min prior to treatment with 250 mm NaCl and/or UV irradiation, as indicated. (Hyperphosphorylated c-JUN migrates significantly slower than the partially phosphorylated form). C, asynchronous or prometaphase HCT116 cells were treated with the indicated concentrations of RO-3306 for 30 min prior to UV irradiation and harvested 1 h later. D, asynchronous or prometaphase cells were untreated (−) or treated with UV (+) to induce PCNADNA and harvested 1 h after treatment. The CDK1 inhibitor RO-3306 and/or the CUL4 neddylation inhibitor MLN4924 (5 μm) were added 30 min prior to UV irradiation as indicated.
We confirmed that reacquisition of UV-induced degradation in CDK1-inhibited cells was still CUL4-dependent by co-treatment with the neddylation inhibitor, MLN4924 (Fig. 3D, compare lanes 6 and 8). Together, these data indicate that CDK1 activity is required for the protection of CRLCDT2 substrates from replication-coupled destruction.
CDK1 Activity Is Required for Substrate CRL4CDT2 Reaccumulation in Late S Phase
To understand the relationship of CDK1 activity in late S phase cells to the kinetics of CRL4CDT2 substrate reaccumulation, we treated synchronized cells in mid-S phase (5 h post-thymidine release) with the CDK1 inhibitor. We then monitored both cell cycle progression and the anticipated reaccumulation of CDT1 and SET8 as S phase completed (Fig. 4, A and B). Treatment with the CDK1 inhibitor, RO-3306, in asynchronous cells results in a G2 arrest (49), and as expected, it blocked cells released from early S synchronization, but CDK1 inhibition did not prevent S phase completion (Fig. 4A). However, CDK1 inhibition strongly and reproducibly dampened the normal late S and G2 increase in both SET8 and CDT1 protein levels (Fig. 4B, compare lanes 5–8 with lanes 10–13). The levels of cyclin A and cyclin B1 at the 7 and 8 h time points corresponding to late S and G2 phases were unperturbed by the inhibitor, but at the normal time of mitosis (at 9 h), CDK1 inhibition caused premature cyclin B1 loss, consistent with results in nocodazole-arrested cells (Figs. 3B (lane 5) and 4B (lanes 9–11)). Importantly, these effects on cyclin B1 levels were evident 2 h after the delay in CDT1 and SET8 reaccumulation.
We once again examined individual late S phase cells (7 h postrelease) for the presence of both PCNADNA and SET8 or CDT1. In a substantial proportion of control PCNADNA-positive cells, SET8 and CDT1 had already reaccumulated, similar to Fig. 1E (Fig. 4C). When CDK1 was inhibited starting at 5 h post-S phase release, however, the percentage of double positive cells decreased nearly 2-fold (Fig. 4C). Because the effects of CDK1 inhibition on CRL4CDT2 substrate accumulation occurred well in advance of mitosis, CDK1 activity inhibits CRL4CDT2 not only in prometaphase-arrested cells but also during the transition from S phase into G2 and mitosis.
CRL4CDT2 Activity Is Itself Inhibited
Our prior work provided evidence for stabilization of CDT1 by direct phosphorylation as cells transit from S phase to M phase (39). Furthermore, our study and that of Kim et al. (50) suggested that a similar direct p21 phosphorylation mechanism could control p21 stability. To determine whether protection from CRL4CDT2-mediated degradation during late cell cycle stages is solely the consequence of direct substrate phosphorylation, we expressed epitope-tagged WT p21 (HA-p21-WT) or p21 harboring alanine substitutions at the two phosphorylation sites that were reported to stabilize p21, Thr-57 and Ser-130 (HA-p21-AA) (50). In synchronized cells, both HA-p21-WT and HA-p21-AA (as well as endogenous p21) migrated slower by SDS-PAGE compared with asynchronous cells, and this mobility shift was reversed by phosphatase treatment (Fig. 5A, lanes 5 and 10). The HA-p21-AA mutant migrated faster than WT HA-p21 in lysates of mitotic cells but was just as stable as both ectopic and endogenous WT p21 (Fig. 5A, compare lanes 4 and 8). Residual sites of p21 phosphorylation affect p21 gel mobility but have not been shown to affect p21 stability (51) (reviewed in Ref. 52). Nevertheless, mitotic phosphorylation of p21 at Thr-57 and Ser-130 is not the principal reason p21 is stable in mitotic cells.
FIGURE 5.

CDK1- dependent inhibition of CRL4CDT2 activity. A, HCT116 cells stably expressing HA-tagged WT or p21-AA (T57A and S130A mutations) were grown asynchronously or arrested in prometaphase. Cells were harvested 2 h after PCNADNA induction by UV irradiation; cell lysates were treated with λ-phosphatase (Ppase) prior to SDS-PAGE, as indicated. B, HeLa cells stably expressing HA-tagged WT or CDT1-5A (39) were grown asynchronously or arrested in prometaphase. Cells were harvested 1 h after PCNADNA induction by UV. C, HeLa cells stably expressing the illustrated PIP degron-SNAP fusion were grown asynchronously or arrested in prometaphase and treated with the CDK1 inhibitor RO-3306 or control DMSO 30 min prior to PCNADNA induction by UV irradiation; cells were harvested 1 h after UV. D, as in C except with HeLa cells stably expressing a PIP degron-RFP (mCherry) fusion.
We similarly expressed epitope-tagged versions of WT CDT1 and CDT1 harboring five alanine substitutions (CDT1-5A) at the mitotic phosphorylation sites we had previously shown to stabilize CDT1. Specifically, we had shown that CDT1–5A is not protected from CRL4CDT2 by stress MAPK activation (39). These cells were then subjected to UV irradiation during asynchronous culture or after synchronization in prometaphase. Both endogenous CDT1 and epitope-tagged WT CDT1 were protected in synchronized cells (Fig. 5B, compare lanes 6 and 8), and as expected, both WT proteins migrated slower by SDS-PAGE compared with asynchronous cells. CDT1–5A migrated slightly faster than WT CDT1 from lysates of mitotic cells, consistent with our prior analysis of this mutant. The clear residual mitotic shift of this mutant is consistent with additional phosphorylation sites in CDT1 that may or may not affect its stability (53–55). Like the p21-AA mutant, CDT1–5A was just as stable as both ectopic and endogenous CDT1 in prometaphase cells, indicating that phosphorylation at these sites previously shown to stabilize CDT1 is not solely responsible for CDT1 mitotic stability (Fig. 5B, lanes 9–12). Together, these observations indicate that protection from CRL4CDT2 probably involves more than phosphorylation of the substrates themselves. Moreover, the fact that all four of the tested CRL4CDT2 substrates were resistant to degradation suggests that CRL4CDT2 is globally suppressed during progression from late S phase into mitosis.
To probe the activity of CRL4CDT2 itself in mitosis, we designed two reporter constructs for stable expression in which the N-terminal 28 amino acids of CDT1 were fused to either red fluorescent protein (RFP) or the SNAP tag (56). The N-terminal 28 amino acids of CDT1 contain the PIP degron and was previously shown to confer CUL4-dependent degradation to a heterologous protein (57). Importantly, neither reporter construct contains canonical CDK or MAPK phosphorylation sites. In asynchronously growing cells, the SNAP reporter fusion was degraded following UV irradiation (Fig. 5C, lanes 1 and 2); however, in prometaphase cells, the reporter was stable (Fig. 5C, lanes 3 and 4). Furthermore, CDK1 inhibition resensitized the SNAP reporter to degradation (Fig. 5C, lanes 5 and 6), indicating that regulation of this reporter is similar to endogenous CRL4CDT2 substrates. We observed nearly identical CDK1-dependent stabilization of the RFP reporter (Fig. 5D). These findings indicate that the ability of CRL4CDT2 to target substrates is generally inhibited via a CDK1-dependent event independently of any additional phosphorylation-mediated mechanisms of direct substrate stabilization.
CDK1 Activity Prevents Chromatin Association of CDT2
During productive CRL4CDT2 targeting, PCNA is first loaded onto DNA for replication (S phase) or repair (e.g. post-UV). Proteins containing PIP degrons bind to PCNADNA, and the resulting complex is then recognized by the substrate receptor, CDT2 (58) (illustrated in Fig. 1A). We postulated that one or more of these steps is inhibited by CDK1. We first tested whether PCNA can be successfully loaded onto mitotic chromatin by UV-irradiating nocodazole-synchronized cells and then analyzing chromatin fractions. We detected PCNA in chromatin fractions from untreated asynchronous cells (reflecting the S phase fraction; Fig. 6A, lane 7), and PCNADNA was further induced by UV irradiation, which stimulates PCNA loading in all cell cycle phases (Fig. 6A, lane 8). As reported previously, CUL4A is also recruited to chromatin following UV irradiation (59–61), and as expected, a resident chromatin protein, ORC2, is unaffected (Fig. 6A). In otherwise untreated mitotic cells, PCNA was not DNA-associated (as expected) but was loaded following UV irradiation (Fig. 6A, lane 10). Thus, mitotic chromatin is not intrinsically resistant to PCNA loading. In addition, PCNA from extracts of mitotic and asynchronous cells showed similar binding to recombinant p21 (but not a p21 PIP box mutant) in vitro (Fig. 6B). In stark contrast to PCNA, CDT2 could not be inducibly recruited to mitotic chromatin, although it was readily recruited to chromatin in asynchronously growing cells (Fig. 6A, compare lanes 8 and 10). Finally, CDK1 inhibition restored CDT2 chromatin recruitment in mitotic cells (Fig. 6A, compare lanes 10 and 12). Thus, the CDK1-dependent inhibition of CRL4CDT2-mediated degradation operates by interfering with chromatin recruitment of the CDT2 substrate receptor. Consistent with this finding, we noted that CDT2 is lost from chromatin fractions in late S phase earlier than PCNA itself (Fig. 6C, lanes 11–13) coincident with the normal reaccumulation time points for CDT1 and SET8 (e.g. Figs. 1 (B and D) and 4B).
FIGURE 6.
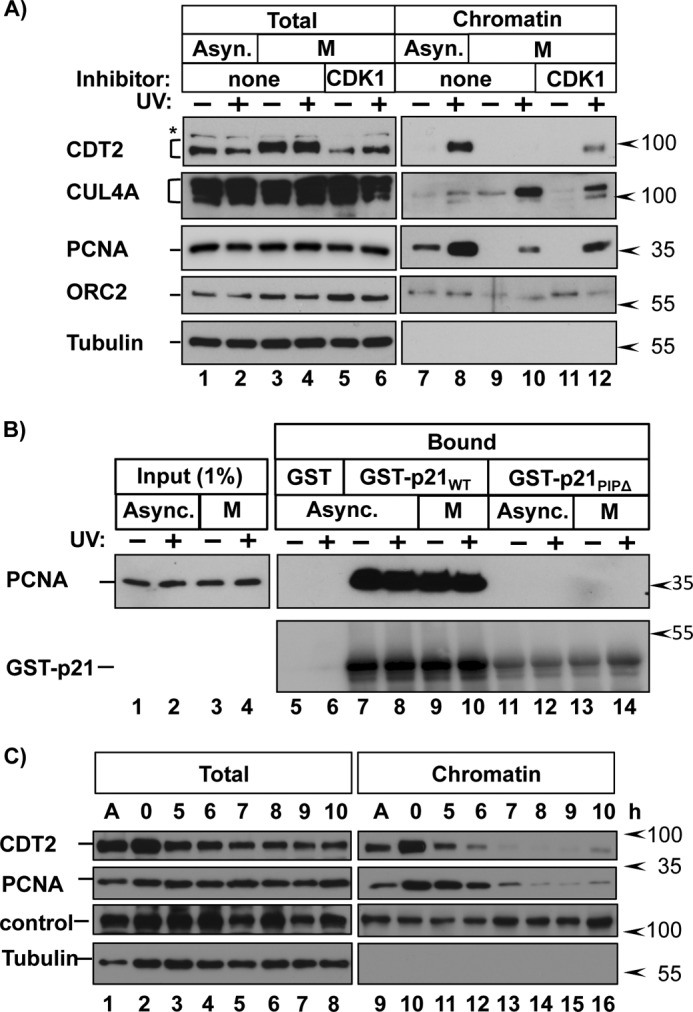
CDK1 activity prevents CDT2 chromatin recruitment. A, HCT116 cells were grown asynchronously or synchronized in prometaphase. Cells were treated with DMSO (none) or the CDK1 inhibitor RO-3306 for 30 min prior to PCNADNA induction by UV irradiation and harvested 90 min later. Endogenous proteins in whole cell extracts and chromatin-bound fractions were analyzed by immunoblotting. B, GST, GST-p21WT, or GST-p21ΔPIP was produced in E. coli and bound to glutathione beads. The protein-coated beads were then incubated with whole cell lysates from asynchronous or mitotic cells as described previously (39). Endogenous PCNA and GST-tagged p21 were detected by immunoblotting. C, HCT116 cells were synchronized in early S phase via double thymidine block and released into fresh medium. Cells were harvested at the indicated time points. Endogenous proteins in whole cell extracts and chromatin-bound fractions were analyzed by immunoblotting.
De Novo SET8 Reaccumulation Is Essential for Normal Mitotic Progression
Having demonstrated active inhibition of CRL4CDT2 targeting as cells progress from S phase to M phase, we next considered the possible biological significance of this mechanism. Replication-coupled destruction via CRL4CDT2 during S phase ensures a single round of replication through the destruction of both CDT1 and SET8; their abnormal persistence in S phase leads to repeated rounds of origin relicensing and rereplication (1, 3). On the basis of these facts alone, it should be detrimental to actively accumulate proteins like CDT1 and SET8 prior to the subsequent G1. However, CDT1 also has an essential function in mitosis that we recently documented (21), thus providing incentive for high levels of CDT1 before mitosis. In addition, sufficient levels of p21 are required in mitosis for normal mitotic progression (22). Given that all CRL4CDT2 substrates follow a similar pattern of mitotic stabilization, we postulated that CDT1 is not the only substrate needed in abundance and de novo at the end of S phase, and we turned our attention to SET8. Depleting SET8 from asynchronous cells leads to multiple defects, including not only inefficient origin licensing in G1 but also impaired chromatin condensation in mitosis. Thus far, these phenotypes have all been attributed to the loss of SET8-deposited histone H4K20me1 (3, 23, 62). However, it is not yet known whether the mitotic defects from SET8 depletion are due to the absence of SET8 specifically during G2 and mitosis or if they arise secondarily from the absence of SET8 in a prior cell cycle phase, such as G1. It is possible that SET8 activity deposits the essential methylations for chromosome condensation during G1, making SET8 dispensable during G2.
To determine whether SET8 is required specifically during transit from late S phase into mitosis, we employed a synchronization-knockdown procedure we developed during the investigation of the mitotic function of CDT1. We synchronized cells in early S phase after the G1 function of SET8 had been fully completed and released them in the presence of either control or SET8 siRNA to proceed otherwise unperturbed through S phase (Fig. 7A). SET8 siRNA effectively prevented accumulation of both SET8 and monomethylation of histone H4K20 after S phase (Fig. 7B, compare lanes 2–7 with lanes 8–13). Of note, SET8 depletion also caused a reduction in H4K20me2, indicating that establishment of histone H4K20me1 during late S phase is required to maintain H4K20me2 levels during G2 and mitosis. H4K20me1 has recently been shown to be required for proper kinetochore assembly (63). Our data clearly support this finding and further indicate that this deposition occurs during G2/M as SET8 reaccumulates (Fig. 7B).
FIGURE 7.

De novo SET8 reaccumulation after S phase is necessary for normal mitotic progression. A, illustration of the experimental approach to block SET8 reaccumulation only after S phase. B, HCT116 cells were synchronized in early S phase via double thymidine block. Cells were released into fresh medium containing control (luciferase) or SET8-ORF siRNA and harvested at the indicated time points for immunoblotting of the indicated endogenous proteins (*, a nonspecific background band). C, cells were synchronized as in A and then released into siRNA-containing medium for 10 h prior to fixation and immunofluorescence staining for tubulin and DAPI staining for DNA. Scale bar, 5 μm. D, the number of mitotic cells from C in preanaphase versus anaphase/telophase was quantified in three biological replicates (n = 300 for each replicate) using two different siRNAs as indicated. Error bars, S.E.
We examined mitotic chromosome morphology in the synchronized SET8-depleted cells by staining for DNA with DAPI and for the mitotic spindle with anti-tubulin. Control cells analyzed 10 h after release from the early S phase block contained metaphase chromosomes that were fully condensed and properly aligned at the metaphase plate (Fig. 7C, top panel). In contrast, chromosomes in cells that had normal SET8 in G1 but contained little to no SET8 only in G2 were less dense and formed a “cloud” of DNA that failed to align properly in metaphase (Fig. 7C, bottom panels). We replicated both the failure to reaccumulate normal H4K20me1 and the mitotic chromosome condensation defects using a different SET8 siRNA in synchronized cells (Fig. 7D). Furthermore, we quantified mitotic progression 10 h after thymidine release; cells with normal SET8 in G1 but blocked from reaccumulating SET8 in late S/G2 were significantly enriched in preanaphase states with a corresponding decrease in cells that had completed anaphase at 10 h (Fig. 7D). These data highlight the importance of SET8 reaccumulation and, by extension, general CRL4CDT2 inhibition prior to mitosis for proper chromosome condensation and mitotic progression. Thus, CDK1 activity in late S phase and G2 phase is required for the normally robust rebound in the levels of CRL4CDT2 substrates, and the reaccumulation (of CDT1 and SET8) is important for proper mitotic progression.
DISCUSSION
At least three substrates (CDT1, p21, and SET8) have known roles in mitosis (21–23, 64), and it is possible that other CRL4CDT2 substrates also have mitotic roles, making general substrate stabilization important for mitosis. Independent of these mitotic roles, the known substrates of replication-coupled destruction via CRL4CDT2 are proteins with clear roles in G1 or S phase. For example, the early step of origin licensing, minichromosome maintenance complex recruitment to chromatin, begins in telophase (65, 66). Thus, with respect to origin licensing, once the nuclear envelope reforms in telophase, the daughter nuclei are G1-like nuclei in a shared cytoplasm. Because telophase occurs just 2–3 h after the end of S phase in cultured somatic cells, active inhibition of replication-coupled destruction may also facilitate G1 events, a notion that has been suggested regarding CDT1 accumulation (67–69). Based on our findings, this need for early accumulation may apply to the entire cohort of CRL4CDT2 substrates.
PCNADNA is an efficient and potent trigger for replication-coupled destruction. The bulk of CDT1 can be degraded within minutes of UV irradiation (39) and occurs nearly simultaneously with S phase onset (1, 12, 70). The potency of PCNADNA then presents a challenge near the end of S phase because CRL4CDT2 substrates are needed in abundance for mitosis and the subsequent G1. (Although CDT1 can also be targeted by CRL1SKP2 (54, 57, 70), we have noted in multiple cell lines that CRL4CDT2 is the major regulator of CDT1 degradation in S phase (39).) Replication of at least 1% of the genome occurs as late as 90 min prior to the onset of mitosis (71), and this late replication requires the continued presence of PCNADNA. Instead of a long G2 phase following complete PCNADNA unloading to allow buildup of essential mitotic proteins, including those that are actively degraded at the onset of S phase, G2 can be short because that buildup begins during late S phase. We detected significant amounts of CRL4CDT2 substrates in the same cells that still contained nearly as much PCNADNA as they did at the start of S phase. Our findings indicate that CDK1 itself or a CDK1-dependent activity overrides the ability of PCNADNA to stimulate CRL4CDT2 recruitment for replication-coupled destruction. The first active CDK1 complex that appears contains cyclin A, and this form of CDK1 may be responsible for initiating CRL4CDT2 inhibition; later inhibition would be accomplished via cyclin B-CDK1. In keeping with this idea, pharmacological CDK1 inhibition blunted CRL4CDT2 substrate reaccumulation at time points that coincide with the expected time of cyclin A-CDK1 activity (Fig. 4B). Interestingly, depleting cyclin A in early S phase-arrested embryonic stem cells prevented accumulation of CDT1 8 h after release (72).
It is clear that the ultimate target(s) of CDK1 (either direct or indirect) is neither PCNADNA nor PCNADNA-substrate interactions but rather the subsequent chromatin recruitment of CDT2 (illustrated in Fig. 8). This inhibition is distinct from the recently reported SCF-dependent CDT2 degradation and underscores the importance of regulating the CDT2 substrate receptor for cell cycle control (43, 44). In fact, phosphorylation of CDT2 during S phase promotes interaction with 14-3-3 proteins that protect CDT2 from this SCF-mediated degradation (73). We and others have noted extensive mitotic phosphorylation of CDT2 in mitotic cells, and human CDT2 bears 19 conserved Ser/Thr-Pro sites, but it is currently unknown if phosphorylation at these sites affects CDT2 substrate interactions. We have found, however, that CDT2 binding to CRL4 is unaffected in mitosis (data not shown). Given the number of potential mitotic proline-directed kinases, the number of potential CDT2 phosphorylation sites, and observations that other members of the CRL4CDT2 complex (CUL4A/B and Roc1) are phosphorylated in mitosis (74, 75), further studies are required to define the mechanism of CRL4CDT2 inhibition. Because the details of how CDT2 contacts the substrate-PCNADNA remain unknown, we cannot yet explain how a CDK1-dependent event blocks this interaction. Importantly, however, CDT2 chromatin recruitment could conceivably be achieved by any PIP degron-PCNA complex in sufficient abundance; we infer that not only is the interaction between CDT2 and the substrates tested here inhibited, but the interaction between CDT2 and any as yet undiscovered substrates is also inhibited. For example, two recent reports identified thymine DNA glycosylase as a target of CRL4CDT2 in Xenopus and human cells (6, 7). The behavior of our two reporter substrates and accumulated findings herein suggest that thymine DNA glycosylase is also protected via CDK1.
FIGURE 8.
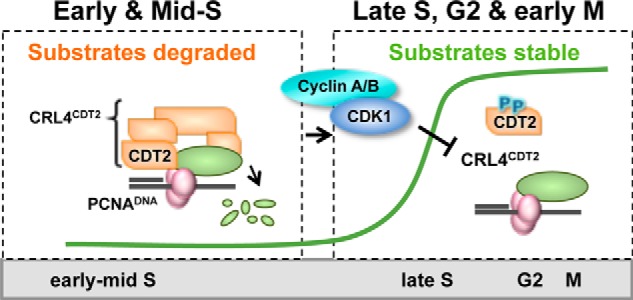
Model. CRL4CDT2 substrates are efficiently degraded in early S phase as a result of PCNA loading onto DNA (PCNADNA). Beginning in late S phase, active CDK1 blocks substrate-mediated chromatin recruitment of the CDT2 receptor, allowing reaccumulation of the cohort of CRL4CDT2 substrates that were subject to replication-coupled destruction. Many of these substrates are required for normal mitotic progression.
Substrates of replication-coupled destruction accumulate beginning in late S phase and are abundant in mitosis. In contrast, experimental manipulations that stabilize the substrates CDT1 or SET8 throughout S phase cause severe genome damage from origin relicensing and refiring (1, 10–12, 14, 37, 76). If the continued presence of either CDT1 or SET8 during S phase is toxic, how then do G2 cells tolerate the high levels of both CDT1 and SET8 prior to mitosis? In other words, why do the high levels of CDT1 and SET8 not trigger rereplication in late S/G2/M? Replication-coupled destruction is only one of many mechanisms that prevent rereplication, but interestingly, many of the other mechanisms are not fully in place until late in S phase. For example, the CDT1 inhibitor geminin is induced in S phase, but its levels are substantially lower in early S phase than in late S phase (77, 78). Other mechanisms, such as the degradation of the largest subunit of the origin recognition complex, Orc1, and the nuclear export of the CDC6 licensing protein are also most evident in middle to late S phase (reviewed in Ref. 24). Further, cyclin A-CDK2 (middle to late S phase) is a better inhibitor of origin licensing than cyclin E-CDK2 (early to middle S phase) (79). In addition, we recently provided evidence that the mitotic phosphorylation of CDT1 not only stabilizes CDT1 but also inhibits its licensing activity (39). Of note, these additional mechanisms impacting CRL4CDT2 substrate targeting probably fine tune the kinetics of individual substrate degradation and reaccumulation. Therefore, in early S phase, while both geminin and cyclin A are low, we postulate that replication-coupled destruction of CDT1 and SET8 is most critical, but by late S phase, it is “safe” for CDT1 and SET8 to accumulate to high levels because the many other mechanisms that block origin relicensing are by then fully established.
The anticipatory accumulation of proteins and activities prior to the cell cycle phase in which they are needed is well established. For example, a series of mid-G1 events, including CDC6 accumulation, results in replication origin licensing during late G1 that is in turn essential for S phase execution (80). Likewise, cyclin B1 accumulation in G2 occurs in advance of M phase. Moreover, the concept of a checkpoint that prevents a cell cycle transition until the preceding one is complete is also well known. We have explored here a variation on these themes in which an essential regulatory mechanism (replication-coupled destruction) is actively inhibited immediately before and during the next cell cycle phase (prior to and during M phase). In contrast to a checkpoint that blocks the transition to the next phase, this mechanism blocks regulation of the current phase so that the next phase can be launched efficiently. Reaccumulation of CRL4CDT2 targets to their maximal levels in anticipation of their requirement primes cells for subsequent cell cycle events. As such, our discovery of CDK1-mediated inhibition of CRL4CDT2 adds a new dimension to a general cell cycle theme: proactive preparation for cell cycle phase launch.
Acknowledgments
We thank M. Emanuele, Y. Xiong, M. Lee, C. Vaziri, and A. Dutta for the generous gifts of antibodies and reagents. We thank T. Salmon for use of microscopy instruments. We also thank Y. Wang, K. Brantley, and J. Hall for technical assistance and members of the Cook, Strahl, and Emanuele laboratories for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM102413A (to J. G. C.), K99CA178177 (to D. V.), and F31CA165891 (to K. C.). This work was also supported by a University of North Carolina (UNC) Chapel Hill dissertation completion fellowship (to L. F. R.). The UNC Flow Cytometry Core Facility is supported in part by National Institutes of Health, NCI, Center Core Support Grant P30CA016086 to the UNC Lineberger Comprehensive Cancer Center.
- H4K20me1 and H4K20me2
- histone H4 lysine 20 monomethylation and dimethylation, respectively
- PCNA
- proliferating cell nuclear antigen
- PCNADNA
- DNA-loaded PCNA
- CDK
- cyclin-dependent kinase
- PIP
- PCNA interaction protein.
REFERENCES
- 1. Arias E. E., Walter J. C. (2005) Replication-dependent destruction of Cdt1 limits DNA replication to a single round per cell cycle in Xenopus egg extracts. Genes Dev. 19, 114–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kim Y., Starostina N. G., Kipreos E. T. (2008) The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 22, 2507–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tardat M., Murr R., Herceg Z., Sardet C., Julien E. (2007) PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J. Cell Biol. 179, 1413–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Terai K., Shibata E., Abbas T., Dutta A. (2013) Degradation of p12 subunit by CRL4Cdt2 E3 ligase inhibits fork progression after DNA damage. J. Biol. Chem. 288, 30509–30514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang S., Zhao H., Darzynkiewicz Z., Zhou P., Zhang Z., Lee E. Y. C., Lee M. Y. W. T. (2013) A novel function of CRL4(Cdt2): regulation of the subunit structure of DNA polymerase δ in response to DNA damage and during the S phase. J. Biol. Chem. 288, 29550–29561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shibata E., Dar A., Dutta A. (2014) CRL4Cdt2 E3 ubiquitin ligase and PCNA cooperate to degrade thymine DNA glycosylase in S-phase. J. Biol. Chem. 289, 23056–23064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Slenn T. J., Morris B., Havens C. G., Freeman R. M., Jr., Takahashi T. S., Walter J. C. (2014) Thymine DNA glycosylase is a CRL4Cdt2 substrate. J. Biol. Chem. 289, 23043–23055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maiorano D., Moreau J., Méchali M. (2000) XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis. Nature 404, 622–625 [DOI] [PubMed] [Google Scholar]
- 9. Nishitani H., Lygerou Z., Nishimoto T., Nurse P. (2000) The Cdt1 protein is required to license DNA for replication in fission yeast. Nature 404, 625–628 [DOI] [PubMed] [Google Scholar]
- 10. Tardat M., Brustel J., Kirsh O., Lefevbre C., Callanan M., Sardet C., Julien E. (2010) The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat. Cell Biol. 12, 1086–1093 [DOI] [PubMed] [Google Scholar]
- 11. Abbas T., Shibata E., Park J., Jha S., Karnani N., Dutta A. (2010) CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol. Cell 40, 9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arias E. E., Walter J. C. (2006) PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat. Cell Biol. 8, 84–90 [DOI] [PubMed] [Google Scholar]
- 13. Kerns S. L., Torke S. J., Benjamin J. M., McGarry T. J. (2007) Geminin prevents rereplication during Xenopus development. J. Biol. Chem. 282, 5514–5521 [DOI] [PubMed] [Google Scholar]
- 14. Li A., Blow J. J. (2005) Cdt1 downregulation by proteolysis and geminin inhibition prevents DNA re-replication in Xenopus. EMBO J. 24, 395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takeda D. Y., Parvin J. D., Dutta A. (2005) Degradation of Cdt1 during S phase is Skp2-independent and is required for efficient progression of mammalian cells through S phase. J. Biol. Chem. 280, 23416–23423 [DOI] [PubMed] [Google Scholar]
- 16. Vaziri C., Saxena S., Jeon Y., Lee C., Murata K., Machida Y., Wagle N., Hwang D. S., Dutta A. (2003) A p53-dependent checkpoint pathway prevents rereplication. Mol. Cell 11, 997–1008 [DOI] [PubMed] [Google Scholar]
- 17. Yoshida K., Takisawa H., Kubota Y. (2005) Intrinsic nuclear import activity of geminin is essential to prevent re-initiation of DNA replication in Xenopus eggs. Genes Cells 10, 63–73 [DOI] [PubMed] [Google Scholar]
- 18. Abbas T., Sivaprasad U., Terai K., Amador V., Pagano M., Dutta A. (2008) PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 22, 2496–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nishitani H., Shiomi Y., Iida H., Michishita M., Takami T., Tsurimoto T. (2008) CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J. Biol. Chem. 283, 29045–29052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meng X., Zhou Y., Lee E. Y. C., Lee M. Y. W. T., Frick D. N. (2010) The p12 subunit of human polymerase delta modulates the rate and fidelity of DNA synthesis. Biochemistry 49, 3545–3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Varma D., Chandrasekaran S., Sundin L. J. R., Reidy K. T., Wan X., Chasse D. A. D., Nevis K. R., DeLuca J. G., Salmon E. D., Cook J. G. (2012) Recruitment of the human Cdt1 replication licensing protein by the loop domain of Hec1 is required for stable kinetochore-microtubule attachment. Nat. Cell Biol. 14, 593–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kreis N.-N., Sanhaji M., Rieger M. A., Louwen F., Yuan J. (2013) p21Waf1/Cip1 deficiency causes multiple mitotic defects in tumor cells. Oncogene 10.1038/onc.2013.518 [DOI] [PubMed] [Google Scholar]
- 23. Wu S., Wang W., Kong X., Congdon L. M., Yokomori K., Kirschner M. W., Rice J. C. (2010) Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev. 24, 2531–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arias E. E., Walter J. C. (2007) Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev. 21, 497–518 [DOI] [PubMed] [Google Scholar]
- 25. Higa L. A. A., Mihaylov I. S., Banks D. P., Zheng J., Zhang H. (2003) Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat. Cell Biol. 5, 1008–1015 [DOI] [PubMed] [Google Scholar]
- 26. Machida Y. J., Hamlin J. L., Dutta A. (2005) Right place, right time, and only once: replication initiation in metazoans. Cell 123, 13–24 [DOI] [PubMed] [Google Scholar]
- 27. Teer J. K., Machida Y. J., Labit H., Novac O., Hyrien O., Marheineke K., Zannis-Hadjopoulos M., Dutta A. (2006) Proliferating human cells hypomorphic for origin recognition complex 2 and pre-replicative complex formation have a defect in p53 activation and Cdk2 kinase activation. J. Biol. Chem. 281, 6253–6260 [DOI] [PubMed] [Google Scholar]
- 28. Whitfield M. L., Sherlock G., Saldanha A. J., Murray J. I., Ball C. A., Alexander K. E., Matese J. C., Perou C. M., Hurt M. M., Brown P. O., Botstein D. (2002) Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell 13, 1977–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grant G. D., Brooks L., 3rd, Zhang X., Mahoney J. M., Martyanov V., Wood T. A., Sherlock G., Cheng C., Whitfield M. L. (2013) Identification of cell cycle-regulated genes periodically expressed in U2OS cells and their regulation by FOXM1 and E2F transcription factors. Mol. Biol. Cell 24, 3634–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pesavento J. J., Yang H., Kelleher N. L., Mizzen C. A. (2008) Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol. Cell Biol. 28, 468–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cook J. G., Chasse D. A., Nevins J. R. (2004) The regulated association of Cdt1 with minichromosome maintenance proteins and Cdc6 in mammalian cells. J. Biol. Chem. 279, 9625–9633 [DOI] [PubMed] [Google Scholar]
- 32. Yang X., Boehm J. S., Yang X., Salehi-Ashtiani K., Hao T., Shen Y., Lubonja R., Thomas S. R., Alkan O., Bhimdi T., Green T. M., Johannessen C. M., Silver S. J., Nguyen C., Murray R. R., Hieronymus H., Balcha D., Fan C., Lin C., Ghamsari L., Vidal M., Hahn W. C., Hill D. E., Root D. E. (2011) A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 8, 659–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Greulich H., Kaplan B., Mertins P., Chen T.-H., Tanaka K. E., Yun C.-H., Zhang X., Lee S.-H., Cho J., Ambrogio L., Liao R., Imielinski M., Banerji S., Berger A. H., Lawrence M. S., Zhang J., Pho N. H., Walker S. R., Winckler W., Getz G., Frank D., Hahn W. C., Eck M. J., Mani D. R., Jaffe J. D., Carr S. A., Wong K.-K., Meyerson M. (2012) Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc. Natl. Acad. Sci. U.S.A. 109, 14476–14481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Todorov I. T., Attaran A., Kearsey S. E. (1995) BM28, a human member of the MCM2-3-5 family, is displaced from chromatin during DNA replication. J. Cell Biol. 129, 1433–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cook J. G., Park C. H., Burke T. W., Leone G., DeGregori J., Engel A., Nevins J. R. (2002) Analysis of Cdc6 function in the assembly of mammalian prereplication complexes. Proc. Natl. Acad. Sci. U.S.A. 99, 1347–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hall J. R., Lee H. O., Bunker B. D., Dorn E. S., Rogers G. J., Duronio R. J., Cook J. G. (2008) Cdt1 and Cdc6 are destabilized by rereplication-induced DNA damage. J. Biol. Chem. 283, 25356–25363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jin J., Arias E. E., Chen J., Harper J. W., Walter J. C. (2006) A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 23, 709–721 [DOI] [PubMed] [Google Scholar]
- 38. Debatisse M., Le Tallec B., Letessier A., Dutrillaux B., Brison O. (2012) Common fragile sites: mechanisms of instability revisited. Trends Genet. 28, 22–32 [DOI] [PubMed] [Google Scholar]
- 39. Chandrasekaran S., Tan T. X., Hall J. R., Cook J. G. (2011) Stress-stimulated mitogen-activated protein kinases control the stability and activity of the Cdt1 DNA replication licensing factor. Mol. Cell Biol. 31, 4405–4416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ersoy I., Bunyak F., Chagin V., Cardoso M. C., Palaniappan K. (2009) Segmentation and classification of cell cycle phases in fluorescence imaging. Med. Image Comput. Comput. Assist. Interv. 12, 617–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leonhardt H., Rahn H. P., Weinzierl P., Sporbert A., Cremer T., Zink D., Cardoso M. C. (2000) Dynamics of DNA replication factories in living cells. J. Cell Biol. 149, 271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Abbas T., Dutta A. (2011) CRL4Cdt2: master coordinator of cell cycle progression and genome stability. Cell Cycle 10, 241–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Abbas T., Mueller A. C., Shibata E., Keaton M., Rossi M., Dutta A. (2013) CRL1-FBXO11 promotes Cdt2 ubiquitylation and degradation and regulates Pr-Set7/Set8-mediated cellular migration. Mol. Cell 49, 1147–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rossi M., Duan S., Jeong Y.-T., Horn M., Saraf A., Florens L., Washburn M. P., Antebi A., Pagano M. (2013) Regulation of the CRL4(Cdt2) ubiquitin ligase and cell-cycle exit by the SCF(Fbxo11) ubiquitin ligase. Mol. Cell 49, 1159–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yujiri T., Fanger G. R., Garrington T. P., Schlesinger T. K., Gibson S., Johnson G. L. (1999) MEK kinase 1 (MEKK1) transduces c-Jun NH2-terminal kinase activation in response to changes in the microtubule cytoskeleton. J. Biol. Chem. 274, 12605–12610 [DOI] [PubMed] [Google Scholar]
- 46. Takenaka K., Moriguchi T., Nishida E. (1998) Activation of the protein kinase p38 in the spindle assembly checkpoint and mitotic arrest. Science 280, 599–602 [DOI] [PubMed] [Google Scholar]
- 47. Pesin J. A., Orr-Weaver T. L. (2008) Regulation of APC/C activators in mitosis and meiosis. Annu. Rev. Cell Dev. Biol. 24, 475–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu H. (2007) Cdc20: a WD40 activator for a cell cycle degradation machine. Mol. Cell 27, 3–16 [DOI] [PubMed] [Google Scholar]
- 49. Vassilev L. T. (2006) Cell cycle synchronization at the G2/M phase border by reversible inhibition of CDK1. Cell Cycle 5, 2555–2556 [DOI] [PubMed] [Google Scholar]
- 50. Kim G.-Y., Mercer S. E., Ewton D. Z., Yan Z., Jin K., Friedman E. (2002) The stress-activated protein kinases p38 α and JNK1 stabilize p21Cip1 by phosphorylation. J. Biol. Chem. 277, 29792–29802 [DOI] [PubMed] [Google Scholar]
- 51. Hodeify R., Tarcsafalvi A., Megyesi J., Safirstein R. L., Price P. M. (2011) Cdk2-dependent phosphorylation of p21 regulates the role of Cdk2 in cisplatin cytotoxicity. Am. J. Physiol. Renal Physiol. 300, F1171–F1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Child E. S., Mann D. J. (2006) The intricacies of p21 phosphorylation: protein/protein interactions, subcellular localization and stability. Cell Cycle 5, 1313–1319 [DOI] [PubMed] [Google Scholar]
- 53. Miotto B., Struhl K. (2011) JNK1 phosphorylation of Cdt1 inhibits recruitment of HBO1 histone acetylase and blocks replication licensing in response to stress. Mol. Cell 44, 62–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu E., Li X., Yan F., Zhao Q., Wu X. (2004) Cyclin-dependent kinases phosphorylate human Cdt1 and induce its degradation. J. Biol. Chem. 279, 17283–17288 [DOI] [PubMed] [Google Scholar]
- 55. Sugimoto N., Tatsumi Y., Tsurumi T., Matsukage A., Kiyono T., Nishitani H., Fujita M. (2004) Cdt1 phosphorylation by cyclin A-dependent kinases negatively regulates its function without affecting geminin binding. J. Biol. Chem. 279, 19691–19697 [DOI] [PubMed] [Google Scholar]
- 56. Keppler A., Gendreizig S., Gronemeyer T., Pick H., Vogel H., Johnsson K. (2003) A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 21, 86–89 [DOI] [PubMed] [Google Scholar]
- 57. Senga T., Sivaprasad U., Zhu W., Park J. H., Arias E. E., Walter J. C., Dutta A. (2006) PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J. Biol. Chem. 281, 6246–6252 [DOI] [PubMed] [Google Scholar]
- 58. Havens C. G., Shobnam N., Guarino E., Centore R. C., Zou L., Kearsey S. E., Walter J. C. (2012) Direct role for proliferating cell nuclear antigen in substrate recognition by the E3 ubiquitin ligase CRL4Cdt2. J. Biol. Chem. 287, 11410–11421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Groisman R., Polanowska J., Kuraoka I., Sawada J., Saijo M., Drapkin R., Kisselev A. F., Tanaka K., Nakatani Y. (2003) The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 113, 357–367 [DOI] [PubMed] [Google Scholar]
- 60. Guerrero-Santoro J., Kapetanaki M. G., Hsieh C. L., Gorbachinsky I., Levine A. S., Rapić-Otrin V. (2008) The Cullin 4B-based UV-damaged DNA-binding protein ligase binds to UV-damaged chromatin and ubiquitinates histone H2A. Cancer Res. 68, 5014–5022 [DOI] [PubMed] [Google Scholar]
- 61. Kapetanaki M. G., Guerrero-Santoro J., Bisi D. C., Hsieh C. L., Rapić-Otrin V., Levine A. S. (2006) The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc. Natl. Acad. Sci. U.S.A. 103, 2588–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Oda H., Okamoto I., Murphy N., Chu J., Price S. M., Shen M. M., Torres-Padilla M. E., Heard E., Reinberg D. (2009) Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol. Cell Biol. 29, 2278–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hori T., Shang W.-H., Toyoda A., Misu S., Monma N., Ikeo K., Molina O., Vargiu G., Fujiyama A., Kimura H., Earnshaw W. C., Fukagawa T. (2014) Histone H4 Lys 20 monomethylation of the CENP-A nucleosome is essential for kinetochore assembly. Dev. Cell 29, 740–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Centore R. C., Havens C. G., Manning A. L., Li J.-M., Flynn R. L., Tse A., Jin J., Dyson N. J., Walter J. C., Zou L. (2010) CRL4Cdt2-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol. Cell 40, 22–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nishitani H., Lygerou Z. (2002) Control of DNA replication licensing in a cell cycle. Genes Cells 7, 523–534 [DOI] [PubMed] [Google Scholar]
- 66. Dimitrova D. S., Prokhorova T. A., Blow J. J., Todorov I. T., Gilbert D. M. (2002) Mammalian nuclei become licensed for DNA replication during late telophase. J. Cell Sci. 115, 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ballabeni A., Zamponi R., Moore J. K., Helin K., Kirschner M. W. (2013) Geminin deploys multiple mechanisms to regulate Cdt1 before cell division thus ensuring the proper execution of DNA replication. Proc. Natl. Acad. Sci. U.S.A. 110, E2848–E2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ballabeni A., Melixetian M., Zamponi R., Masiero L., Marinoni F., Helin K. (2004) Human geminin promotes pre-RC formation and DNA replication by stabilizing CDT1 in mitosis. EMBO J. 23, 3122–3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tsunematsu T., Takihara Y., Ishimaru N., Pagano M., Takata T., Kudo Y. (2013) Aurora-A controls pre-replicative complex assembly and DNA replication by stabilizing geminin in mitosis. Nat. Commun. 4, 1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nishitani H., Sugimoto N., Roukos V., Nakanishi Y., Saijo M., Obuse C., Tsurimoto T., Nakayama K. I., Nakayama K., Fujita M., Lygerou Z., Nishimoto T. (2006) Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J. 25, 1126–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Widrow R. J., Hansen R. S., Kawame H., Gartler S. M., Laird C. D. (1998) Very late DNA replication in the human cell cycle. Proc. Natl. Acad. Sci. U.S.A. 95, 11246–11250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ballabeni A., Park I.-H., Zhao R., Wang W., Lerou P. H., Daley G. Q., Kirschner M. W. (2011) Cell cycle adaptations of embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 108, 19252–19257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dar A., Wu D., Lee N., Shibata E., Dutta A. (2014) 14-3-3 proteins play a role in the cell cycle by shielding Cdt2 from ubiquitin-mediated degradation. Mol. Cell. Biol. 34, 4049–4061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. [DOI] [PubMed] [Google Scholar]
- 75. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dorn E. S., Chastain P. D., 2nd, Hall J. R., Cook J. G. (2009) Analysis of re-replication from deregulated origin licensing by DNA fiber spreading. Nucleic Acids Res. 37, 60–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. McGarry T. J., Kirschner M. W. (1998) Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 93, 1043–1053 [DOI] [PubMed] [Google Scholar]
- 78. Wohlschlegel J. A., Dwyer B. T., Dhar S. K., Cvetic C., Walter J. C., Dutta A. (2000) Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science 290, 2309–2312 [DOI] [PubMed] [Google Scholar]
- 79. Wheeler L. W., Lents N. H., Baldassare J. J. (2008) Cyclin A-CDK activity during G1 phase impairs MCM chromatin loading and inhibits DNA synthesis in mammalian cells. Cell Cycle 7, 2179–2188 [DOI] [PubMed] [Google Scholar]
- 80. Mailand N., Diffley J. F. X. (2005) CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 122, 915–926 [DOI] [PubMed] [Google Scholar]