

Background: Iodotyrosine deiodinase utilizes FMN to maintain iodide homeostasis by reductive deiodination of iodotyrosine.
Results: Crystallographic, pH, and redox studies demonstrate the role of substrate in organizing the active site for effective catalysis.
Conclusion: Stepwise single electron transfer is promoted only after coordination of a halotyrosine within iodotyrosine deiodinase.
Significance: A synergy between substrate selectivity and catalytic activity is created by the enzyme.
Keywords: Enzyme Catalysis, Enzyme Structure, Flavin Mononucleotide (FMN), Oxidation-Reduction (Redox), Thyroid, Dehalogenase, Flavoprotein, Iodotyrosine
Abstract
Reductive dehalogenation is not typical of aerobic organisms but plays a significant role in iodide homeostasis and thyroid activity. The flavoprotein iodotyrosine deiodinase (IYD) is responsible for iodide salvage by reductive deiodination of the iodotyrosine derivatives formed as byproducts of thyroid hormone biosynthesis. Heterologous expression of the human enzyme lacking its N-terminal membrane anchor has allowed for physical and biochemical studies to identify the role of substrate in controlling the active site geometry and flavin chemistry. Crystal structures of human IYD and its complex with 3-iodo-l-tyrosine illustrate the ability of the substrate to provide multiple interactions with the isoalloxazine system of FMN that are usually provided by protein side chains. Ligand binding acts to template the active site geometry and significantly stabilize the one-electron-reduced semiquinone form of FMN. The neutral form of this semiquinone is observed during reductive titration of IYD in the presence of the substrate analog 3-fluoro-l-tyrosine. In the absence of an active site ligand, only the oxidized and two-electron-reduced forms of FMN are detected. The pH dependence of IYD binding and turnover also supports the importance of direct coordination between substrate and FMN for productive catalysis.
Introduction
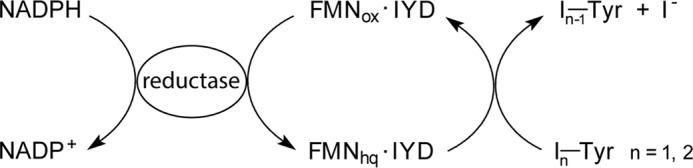
Iodide is a micronutrient necessary for thyroid hormone biosynthesis and essential for human health. Efficient use of dietary iodide depends on its accumulation in the thyroid as managed by a sodium-iodide symporter (1). Iodide is also salvaged from iodinated tyrosine by-products as promoted by iodotyrosine deiodinase (IYD)3 (2). Nature's reliance on iodine for signaling is highly unusual, and thyroid hormones represent the only example of this in higher organisms. Much of the biochemistry associated with these hormones is similarly unusual. The enzymes responsible for deiodination of the thyroid hormones represent one of two known types of reductive dehalogenases in humans. These process the iodine-containing hormones and are members of the thioredoxin structural superfamily. Their active site contains a selenocysteine residue to promote a thiol-dependent deiodination (3). The second type of reductive dehalogenase is IYD. This enzyme contains a flavin mononucleotide (FMN) rather than a selenocysteine, and its reaction appears to be driven in vivo by NADPH rather than thiols (Scheme 1). In addition, IYD is a member of the nitro-FMN reductase structural superfamily, which differs significantly from the thioredoxin superfamily. Thus, mammals maintain two quite distinct strategies for catalyzing a closely related set of unusual reactions.
SCHEME 1.
Prior studies on mouse IYD (mIYD) identified three subdomains: an N-terminal membrane anchor, an intermediate domain, and a catalytic domain (4). A soluble construct could be expressed in human and insect cells after truncating the gene to remove the membrane anchor (5, 6). More recent fusion constructs have allowed heterologous expression of IYD from many organisms in Escherichia coli (7). This same strategy has now been applied to generate sufficient quantities of human IYD (hIYD). Direct investigation of hIYD is particularly advantageous for understanding the molecular basis of genetic disease (8, 9). Previous expression of hIYD has been limited to human embryonic kidney (HEK) 293 cells (10).
The ability for NADPH to drive catalysis is lost once IYD is removed from its native membrane, and consequently reductants such as dithionite have been used for most studies in vitro (11). Likely an NADPH reductase is recruited in vivo, but it has yet to be identified. Attention has instead focused on the unusual participation of flavin in a reductive dehalogenation process that supports dechlorination and debromination as well as deiodination (12). Little precedence exists for a mechanism by which FMN may promote this reaction despite the considerable knowledge amassed on FMN from many years of investigating the broad array of flavoproteins that catalyze many diverse reactions (13, 14). Crystallographic studies of mIYD implicate a significant role for substrate in controlling enzyme activity (15). The substrates 3-iodo-l-tyrosine (I-Tyr) and 3,5-diiodo-l-tyrosine (I2-Tyr) induce closure of an active site lid that is essential for catalysis (6). In addition, these substrates simultaneously and directly coordinate to the isoalloxazine system of FMN as well. Investigations now describe the consequences of these interactions on the oxidation and reduction properties of FMN and their significance to the catalytic mechanism and substrate selectivity of IYD.
EXPERIMENTAL PROCEDURES
Materials
Biochemical reagents including xanthine oxidase were purchased from Sigma and used without purification unless specified. 3-Fluoro-l-tyrosine and Nile blue were obtained from Astatech, Inc. and Santa Cruz Biotechology, Inc., respectively.
Cloning, Expression, and Purification of a Soluble Domain of hIYD
The human iodotyrosine dehalogenase gene (GenBankTM accession number AY259176.1) was amplified by PCR with primers 5′-AAGCTTAAGCTTGGATCCGCCACCATGGCTCAAGTTCAGCCC-3′ and 5′-CTCGAGCCGAGCTAATGGTGATGGTGATGGTGTACTGTCACCATGATC-3′to generate a gene lacking codons for amino acids 1–31 and gaining a C-terminal His6 tag. The PCR product was digested with BamHI and XhoI and inserted into the pET28-SUMO vector (obtained from Dr. Christopher Lima) in which a SUMO tag is appended to the N terminus of the deiodinase. The resulting plasmid pET28-SUMO-JH1 containing the desired gene was transformed into RosettaTM 2(DE3) E. coli (Novagen). Cells were grown in LB medium with kanamycin and chloramphenicol at 37 °C to an A600 of 0.6–0.8. The medium was then cooled to 18 °C, and protein expression was induced by addition of 0.2 mm isopropyl thio-β-galactoside. After 4 h at 18 °C, cells were harvested by centrifugation and resuspended in buffer N (50 mm sodium phosphate, pH 8.0, 500 mm NaCl, and 10% glycerol). Cells were then flash frozen and stored at −80 °C until use. An alternative derivative of the hIYD gene was generated to truncate residues 1–69 by the appropriate PCR of pET28-SUMO-JH1, but this was abandoned after insoluble protein was observed to dominate expression.
To isolate the deiodinase, cells were thawed and subsequently lysed by three passages through a French press at 1000 p.s.i. Cell debris was removed by centrifugation at 40,000 × g at 4 °C for 2 h. The supernatant was fractionated with nickel ion affinity chromatography (HispurTM nickel-nitrilotriacetic acid resin, Thermo Scientific) that was equilibrated with buffer N. Successive washes used increasing concentrations of imidazole (20 mm for 5 column volumes, 60 mm for 5 column volumes, 80 mm for 2 column volumes, and 100 mm for 1 column volume) in buffer N. The SUMO fusion of hIYD was finally eluted with 350 mm imidazole in buffer N and digested with Ulp1 protease (1:200, w/w) to release hIYD from SUMO. The resulting solution was concentrated to 3 ml and passed through a gel filtration column (Sephacryl S-200 HR, GE Healthcare) equilibrated with 50 mm sodium phosphate, pH 7.4, 100 mm NaCl, 1 mm DTT, and 10% glycerol. Fractions containing hIYD were pooled, and the concentration of hIYD was determined by the A280 after correcting for the contribution from bound FMN (A280/A450 = 1.57) (7) and an extinction coefficient (ϵ280 = 37,930 m−1 cm−1) estimated by the ExPASy ProtParam tool (16). The concentration of FMN was determined by its A450 (ϵ450 = 12,500 m−1 cm−1) (17). The purified hIYD was concentrated to 10 mg/ml using Amicon® Ultra centrifugal filters prior to storage at 4 °C.
Deiodinase Activity Assay
Catalytic deiodination was measured by release of [125I]iodide from [125I]I2-Tyr as reported previously (18). Sodium dithionite was used as the reducing reagent for enzyme turnover, and I2-Tyr concentration was varied between 0 and 70 μm. Each assay was performed in triplicate, and their average was fit to Michaelis-Menten kinetics using Origin 7.0. Reported error is based on the results of least square fitting.
pH-Rate Profiles
Steady-state kinetic constants were determined from the standard deiodinase assay at pH 6.0–7.5 (100 mm potassium phosphate) and pH 7.5–8.5 (100 mm Tris-HCl) using 0.08 μm hIYD. The values of Vmax and Vmax/Km represent the average of three independent determinations. The log Vmax and log(Vmax/Km) were plotted against pH and fit to Equation 1 (19).
Affinity of Halotyrosines for hIYD
Binding of halotyrosines to IYD was measured by their ability to quench the fluorescence of FMN (λex = 450 nm and λem = 527 nm) as described previously (12). Briefly, hIYD (4.5 μm) was titrated with the Tyr derivatives over a range of concentrations centered around the value necessary for quenching 50% of the initial fluorescence signal (F0). Fluorescence intensities were measured with a Hitachi F-4500 fluorescence spectrophotometer and normalized to the initial fluorescence (F0) obtained in the absence of substrate. Dissociation constants (KD) were obtained from the non-linear fit (Origin 7) of fluorescence versus ligand concentration using Equation 2 (20). Reported KD values derive from the average value of three independent determinations, and their error is based on the results of least square fitting. The pH dependence of ligand affinity was determined at pH 6.0–8.0 (100 mm potassium phosphate) and pH 8.0–9.0 (100 mm Tris-HCl). The values of log KD were plotted versus pH, fit to the sum of Equations 1 and 3, and optimized iteratively (19).
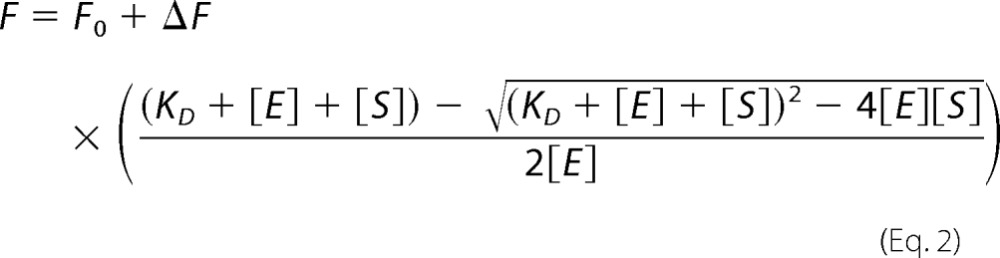 |
Protein Crystallization, Structure Determination, and Refinement
Crystallization trials utilized a Phoenix crystallization robot and sparse matrix screening WizardTM I, II, III, and IV (Emerald Biosciences), PEGSuiteTM and CryoSuiteTM (Qiagen), and IndexTM (Hampton Research). After optimization, hIYD crystals grew in ∼2 days at 20 °C by the hanging drop diffusion method with a ratio of 1 μl of hIYD (12 mg/ml; 50 mm sodium phosphate, pH 7.4, 100 mm NaCl, 1 mm DTT, and 10% glycerol) and 1 μl of precipitant (0.05 m ammonium sulfate, 50 mm BisTris, pH 6.5, and 25% pentaerythritol ethoxylate. Crystals were cryoprotected in the precipitant supplemented with 15% glycerol and 1 mm FMN prior to data collection. To generate co-crystals, hIYD was treated with 1.5 mm I-Tyr overnight and then subjected to the same hanging drop procedure using a well solution of 0.15 m sodium acetate, 85 mm Tris-HCl, pH 8.5, 25.5% (w/v) polyethylene glycol 4000, and 15% glycerol at 20 °C. Co-crystals of hIYD·I-Tyr formed within 24 h and were flash frozen in liquid nitrogen.
X-ray diffraction data were collected at Beamline 7-1 at the Stanford Synchrotron Radiation Laboratory and processed using the HKL2000 package (21). The structures of hIYD and hIYD·I-Tyr were determined by molecular replacement using AutoMR in PHENIX (22). A previous structure determined for mIYD (Protein Data Bank code 3TO0) (6) was used as the search model and yielded initial Rwork and Rfree values of 25.3 and 31.2%, respectively, from AutoBuild in PHENIX. The electron density maps showed the presence of one FMN per polypeptide as expected for IYD. Manual model building and placement of the ligand were performed with Coot (23) and refined in PHENIX (22). The electron density maps for the hIYD·I-Tyr co-crystal using hIYD as the search model showed positive densities for I-Tyr. Final refinement statistics for both structures are summarized in Table 2. All images were prepared using PyMOL (Schrödinger, LLC). Structure factors and coordinates have been deposited with the Protein Data Bank under accession codes 4TTB and 4TTC.
TABLE 2.
Data collection and refinement statistics
| Protein | hIYD | hIYD·I-Tyr |
| Ligands | FMN | FMN and I-Tyr |
| Protein Data Bank code | 4TTB | 4TTC |
| Data collection | ||
| Space group | P31 | P61 |
| No. of molecules | 2a | 6 |
| Unit cell parameters | ||
| a, b, c (Å) | 72.89, 72.89, 95.51 | 104.75, 104.75, 303.34 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 |
| Resolution range (Å)b | 50–2.45 (2.54–2.45) | 50–2.65 (2.74–2.65) |
| No. of unique reflectionsb | 20,957 (2,074) | 54,565 (5,465) |
| No. of observed reflections | 172,255 | 522,006 |
| Completeness (%)b | 100 (100) | 100 (100) |
| Redundancya | 8.2 (7.6) | 9.6 (9.6) |
| Rsym (%)b | 6.1 (61) | 8.8 (55.4) |
| I/σIa | 33.8 (2.9) | 27.1 (4.9) |
| Refinement | ||
| Resolution used in refinement (Å) | 38.09–2.45 | 44.86–2.65 |
| No. of reflections used in working set | 20,800 | 54,429 |
| No. of reflections for Rfree calculation | 1,066 | 2,722 |
| Rwork (%), Rfree (%) | 19.26, 20.26 | 17.04, 21.24 |
| No. of atoms | ||
| Protein | 3,042 (A and B) | 10,680 (A–F) |
| Heteroatoms | 62 (FMN) | 186/84 (FMN and I-Tyr) |
| Solvent | 42 | 221 |
| Mean B-factor (Å2) | ||
| Protein | 86.2 | 52.3 |
| FMN | 85.8 | 37.8 |
| I-Tyr | 38.9 | |
| Solvent | 62.7 | 42.2 |
| RMSD from ideality | ||
| Bond lengths (Å) | 0.008 | 0.008 |
| Bond angles (°) | 1.109 | 1.280 |
| Ramachandran analysis | ||
| Favored (%) | 98.1 | 97.6 |
| Allowed (%) | 100 | 100 |
| Outliers | 0 | 0 |
| Coordinate error | 0.22 | 0.29 |
a Per asymmetric unit.
b Numbers in parentheses are outer shell parameters.
Anaerobic Titration
All titrations were performed at 25 °C using xanthine/xanthine oxidase as the reducing system (24, 25). Individual samples within sealable quartz cuvettes contained 10–15 μm hIYD, 900 μm xanthine, 2 μm methyl viologen, 200 mm potassium chloride, and 100 mm potassium phosphate, pH 7.4. Molecular oxygen was removed from these solutions by continuously flushing with argon for at least 20 min prior to addition of hIYD. The reaction was initiated by addition of xanthine oxidase (40 μg/ml). Spectral changes were monitored every 2 min and compared with control samples containing no hIYD. Titrations were also repeated in the presence of F-Tyr (0.6 mm) to investigate the effect of ligand binding on the oxidation-reduction properties of the enzyme-bound FMN.
Midpoint potentials of hIYD were calculated from concurrent reduction of a reference dye in situ (25) alternatively using anthraquin-one-2,6-disulfonate (AQDS; Em = −188 mV), Nile blue (Em = −116 mV), and Safranin O (Em = −280 mV) (26). Spectral changes were recorded every 2 min for over 2 h. All potentials are reported versus a standard hydrogen electrode and represent the average of three independent determinations.
RESULTS
Expression and Purification of the Soluble Domain of hIYD
Prior sequence analysis of the IYD gene indicated that the enzyme contains three domains: an N-terminal transmembrane region, an intermediate domain, and a catalytic domain (4). Removal of the transmembrane domain from mIYD generated a soluble, stable, and active derivative of the deiodinase (5). An equivalent truncation was performed on hIYD. The gene encoding hIYD but lacking the first (1–31) amino acids was inserted into the pET28-SUMO vector and expressed in E. coli. The soluble fraction of the fusion protein (∼50%) was isolated by nickel-based affinity chromatography. The SUMO tag was then removed by the Ulp1 protease, and hIYD was separated by size exclusion chromatography in a yield of ∼10–20 mg/liter of culture. Native protein exists as a homodimer and was isolated with the expected FMN to homodimer ratio of 1.9–2.1 as determined by A280 and A450. The purity of hIYD approached 97% as estimated by SDS-PAGE, Coomassie staining, and analysis by ImageQuant TL. The desired protein migrates similarly to that of a 30-kDa marker protein as expected for a monomer of 30.5 kDa as calculated by ExPASy.
Deiodination Activity
The standard [125I]iodide release assay (18) was used for initial characterization of hIYD expressed in E. coli. A Km of 31 ± 6 μm and a kcat of 12.5 ± 1 min−1 were measured for I2-Tyr. The resulting kcat/Km of 0.40 min−1 μm−1 is very close to that reported for mIYD (0.95 min−1 μm−1) also lacking its N-terminal transmembrane region (5). The native sequence of hIYD had previously been expressed in HEK293 cells to yield a microsomal deiodinase exhibiting a Km of 2.67 μm and a Vmax of 53 pmol/min-μg of membrane fraction (27).
Affinity of hIYD for 3-Halotyrosines
Binding of the various halogen-substituted tyrosines (F-Tyr, Cl-Tyr, Br-Tyr, and I-Tyr) to IYD was monitored by the decrease in fluorescence of the active site FMN upon association with these ligands (12). Binding was also evident by an increase in the visible spectrum of FMNox at 470 nm. As summarized in Table 1, hIYD associates strongly with I-Tyr, Br-Tyr, and Cl-Tyr. All share KD values close to 120 nm. Only F-Tyr has a noticeably weaker affinity for hIYD. Still, many orders of magnitude separate the KD values for all of the halotyrosines versus that for tyrosine. Similar results were observed previously with mIYD (12). This led to an assumption that binding was influenced by the acidity of the phenolic proton. Further investigation shifted to hIYD as described below.
TABLE 1.
hIYD affinity for halotyrosines
pH Dependence of I2-Tyr Deiodination and Binding with hIYD
Steady-state kinetic analysis of I2-Tyr deiodination by hIYD was repeated over a pH range of 6.0–8.5 (Fig. 1). The pH rate profiles of both log V and log(V/K) illustrate a basic limb with a slope of −1. Their respective pKa values of 7.5 ± 0.2 and 7.6 ± 0.2 as determined by Equation 1 are experimentally indistinguishable. The maximal activity at low pH is consistent with the protonation of a functional group within the active site complex. Furthermore, the similarity in the pH dependence of V and V/K indicates that their common parameter kcat dominates the profiles.
FIGURE 1.

The pH dependence for deiodination of I2-Tyr by hIYD. The pH profile of log V (A) and log(V/K) (B) were fit by non-linear least square regression using Equation 1. Data represent an average of three independent measurements in the presence of potassium phosphate (■) and Tris-HCl (○) at the indicated pH values. Error bars represent S.D.
The influence of pH on substrate binding was determined independently by measuring the KD of I2-Tyr in the absence of reductant (Fig. 2). Tightest binding was measured near a neutral pH of 7.6–7.7. Affinity diminished as the pH became alternatively more acidic and more basic. A plot of log KD versus pH suggests the importance of two ionizable groups with pKa values of 6.6 ± 0.2 and 8.7 ± 0.3 as estimated from non-linear least square fitting to the sum of Equations 1 and 3. Binding affinity decreases after protonation of an acidic group and deprotonation of a basic group. For catalysis, protonation of a group appears to enhance turnover despite the decrease of substrate affinity. Further studies will be necessary to establish the relationship between these pH profiles because binding was measured with enzyme in its oxidized form (FMNox), whereas the catalytic constants of deiodination were measured for its reduced form (FMNhq) by the necessity of using dithionite as the reductant.
FIGURE 2.
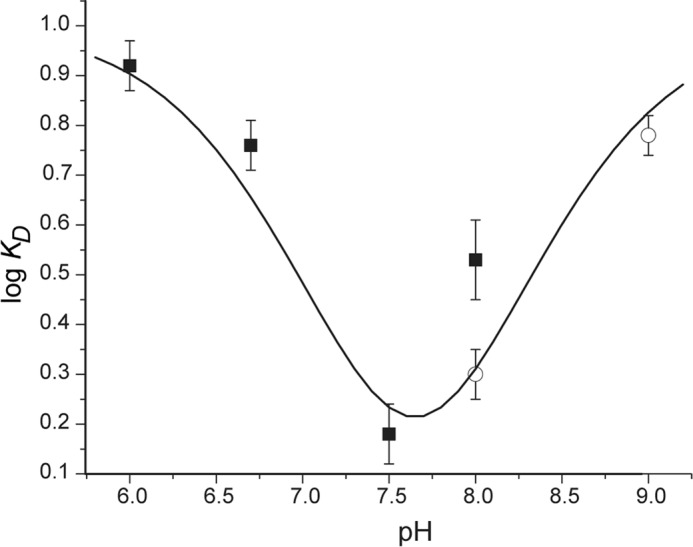
The pH dependence of substrate binding to hIYD. The affinity of I2-Tyr for hIYD (KD) is reported as the average of three independent measurements in the presence of potassium phosphate (■) and Tris-HCl (○) at the indicated pH values. The curve was generated from the calculated pKa values and pH according to the sum of Equations 1 and 3. Error bars represent S.D.
Structural Determination of hIYD
The structures of hIYD and hIYD·I-Tyr were determined by x-ray diffraction and molecular replacement based on previous data for mIYD and mIYD·I-Tyr (6, 15). hIYD crystallized with one dimer per asymmetric unit, and the hIYD·I-Tyr complex crystallized with three dimers per asymmetric unit. These and additional parameters of diffraction and refinement are summarized in Table 2. Electron density for the first 38 residues (32–70) representing part of the intermediate domain was not observed for hIYD, nor was it evident in the previously reported structures of mIYD (6, 15). Attempts to truncate this entire region generated insoluble protein almost exclusively, and no further deletions were pursued. Consequently, this intermediate domain can be expected to stabilize the homodimeric structure or at least aid in its initial folding.
The two identical subunits of hIYD form an α-β fold and domain swaps at each N and C terminus consistent with the nitro-FMN reductase superfamily. Structures determined for hIYD and its co-crystal with I-Tyr are nearly superimposable (RMSD of 0.47 Å for a 347-atom comparison) (Fig. 3A). However, electron density for residues 161–178 and 199–211 was not observed in the absence of bound I-Tyr. This is presumably due to disorder and/or dynamics of these regions. However, these regions were evident from a co-crystal containing I-Tyr. Under these conditions, a helix and loop surrounding the active site are stabilized. The helix in particular covers the active site and helps to sequester the I-Tyr·FMN complex from solvent (Fig. 4).
FIGURE 3.
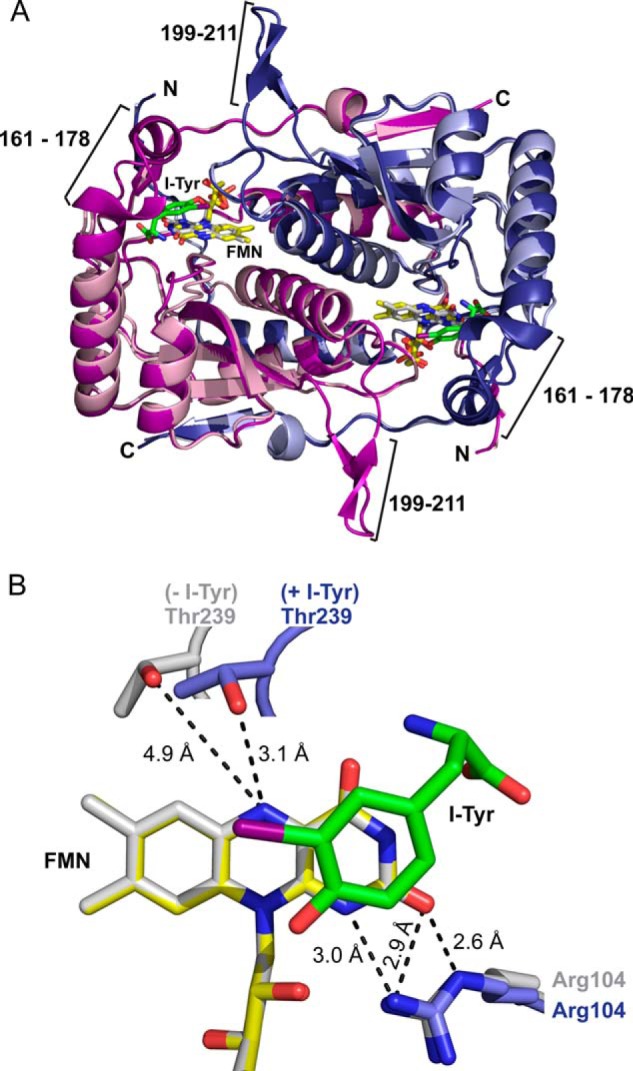
Structure of hIYD. A, the structure of hIYD·I-Tyr (deep blue and deep purple to indicate the two monomers) is overlaid from a global fit to the structure of hIYD (light blue and light pink to indicate the two monomers). The active site lid composed of a loop (residues 199–211) and a helix-turn (residues 161–178) are only detected in the hIYD·I-Tyr co-crystal. I-Tyr is illustrated with its carbon skeleton in green. FMN is indicated in yellow and gray for structures containing and lacking the substrate I-Tyr, respectively. B, binding of I-Tyr to hIYD induces a reorientation of Thr-239 to provide a hydrogen bond to the N5 of FMN. This is illustrated with the same coloring used for the full structure in A and aligned relative to FMN.
FIGURE 4.

The active site lid shields the substrate and FMN from solvent. A, the two identical polypeptides within the hIYD·I-Tyr structure are indicated by shades of gray. FMN and I-Tyr reside in the active site and are depicted in color. Residues 161–178 of one polypeptide are not shown to reveal the position of FMN and I-Tyr. B, the surface formed by residues 161–178 of the same polypeptide is highlighted in blue to illustrate their ability to cover the active site.
FMN binds non-covalently at the dimer interface established by a helix from each of the two identical subunits. Both subunits contribute hydrogen bonds to the ribose moiety of FMN through Ser-102 and Ser-128. Additionally, multiple Arg residues from a single subunit coordinate to the phosphate group. Arg-104 establishes the only polar interaction with the isoalloxazine moiety (N1 and O2) in the absence of substrate. The isoalloxazine is nearly planar and undergoes only a slight shift after I-Tyr association. Most side chains undergo similarly minor shifts once substrate binds. Thr-239 is an exemption. Its side chain hydroxyl group moves toward the N5 position of FMN from 4.9 to 3.1 Å and presents the potential for hydrogen bonding (Fig. 3B).
The substrate I-Tyr was previously observed to template a closure of the active site of mIYD by concurrent association with side chains from the lid and the pyrimidine region of the isoalloxazine ring (6, 15). An equivalent assembly is also evident within the co-crystal of hIYD and I-Tyr. The zwitterionic region of I-Tyr offers hydrogen bonding through its carboxylate to the N3 of FMN (3.0 Å) and through its ammonium to the O4 of FMN (2.5 Å). This same zwitterion coordinates to Glu-157, Tyr-161, and Lys-182. These three residues have become synonymous with the deiodinase and distinct from other proteins within the same superfamily (7). The aromatic ring of I-Tyr additionally stacks above the FMN with a closest approach between the FMN N10 and the phenolic oxygen of I-Tyr (3.7 Å). Additionally, the C–I bond of I-Tyr orients directly over the C4a–N5 bond of FMN that is crucial during redox cycling of many flavoproteins (13, 28–30). These substantial changes in coordination of the isoalloxazine ring suggested that substrate binding could significantly affect the oxidation-reduction properties of IYD.
Reduction of hIYD in the Absence and Presence of Halotyrosines
FMN is capable of participating in both one- and two-electron transfer processes that are typically controlled by the surrounding protein environment (13, 28–30). FMN may by reduced directly by two electrons to generate its hydroquinone form (FMNhq). Alternatively, reduction may occur via two stepwise single electron processes through an intervening semiquinone intermediate (FMNsq) (24). Each of these species has a unique absorbance signature for easy detection. The oxidized FMN bound to hIYD has an absorbance maximum at 446 nm and a shoulder near 470 nm. This absorbance decreased during reduction by xanthine and xanthine oxidase and ultimately generated a spectrum consistent with the fully reduced FMNhq (Fig. 5A). An isosbestic point at 325 nm was observed throughout the titration, indicating that no intermediates were observed during this reduction.
FIGURE 5.
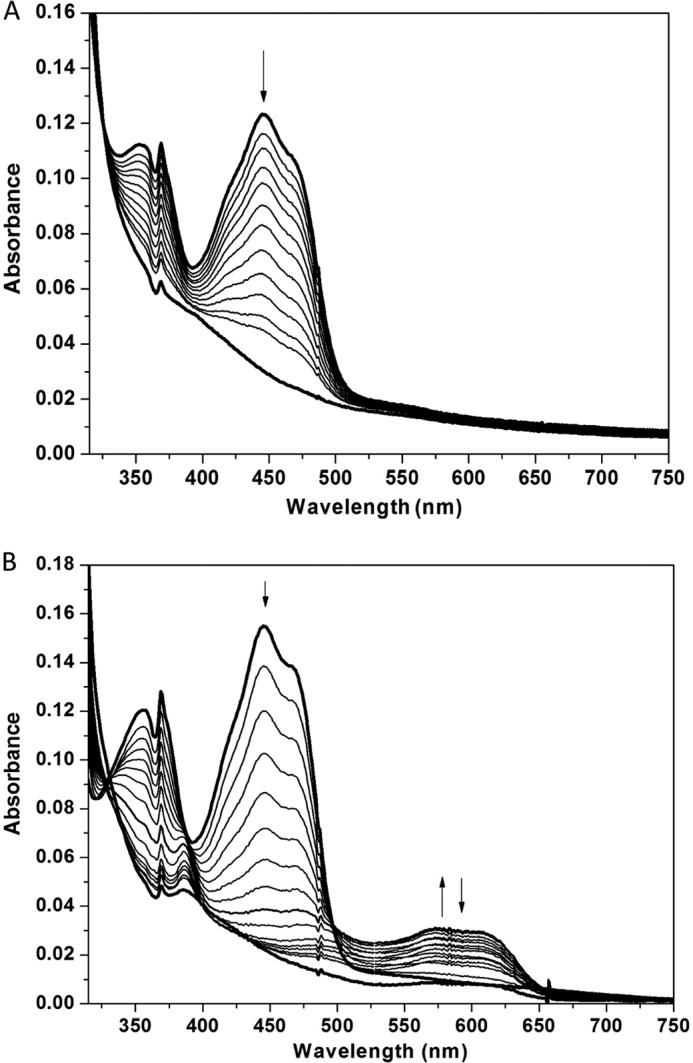
Anaerobic reduction of FMN bound to hIYD by xanthine and xanthine oxidase as monitored with absorbance spectroscopy. Sample spectra during reduction indicate that the absorbance at 446 nm decreases during reduction as indicated by the arrow in the absence (A) and presence (B) of F-Tyr. Only in the presence of F-Tyr does a spectral intermediate with a λmax of 590 nm increase and then decrease during reduction.
In contrast, the neutral FMNsq could be detected during turnover of the active halotyrosine substrates (12). This signified a dramatic change in the oxidation-reduction properties of IYD due to either catalytic turnover or perhaps just substrate binding. To focus on the effects caused by halotyrosine coordination alone, attention was directed to the effect of F-Tyr. This halotyrosine binds to the active site with reasonable affinity (Table 1) but is not subject to dehalogenation (12). Reduction of hIYD in the presence of F-Tyr generated a spectral intermediate with a λmax at 590 nm and a shoulder near 610 nm (Fig. 5B). This is characteristic of a neutral FMNsq formed after single electron reduction of FMN (24). Accumulation of the neutral semiquinone illustrates the ability of the active site structure templated by F-Tyr to stabilize and promote the one-electron chemistry of the deiodinase. Ultimately, the FMNsq is further reduced to FMHhq by the xanthine and xanthine oxidase system. Detection of the intermediate semiquinone suggests that a large separation is created between the midpoint potentials of the successive one-electron reduction steps.
Reduction Potential of FMN Bound to hIYD in the Absence and Presence of F-Tyr
Reductive titration of hIYD was repeated in the presence of suitable redox-active dyes to determine the midpoint potential of the FMN bound to IYD. AQDS was used as a reference dye because its midpoint potential is comparable with that of FMNox/FMNhq bound to hIYD in the absence of F-Tyr. The distinct spectral changes of FMN and AQDS during reduction also facilitated the measurements (Fig. 6). A plot of log(FMNox/FMNhq) versus log(AQDSox/AQDSred) produced a slope of 0.96. This value is close to the theoretical value of unity expected for the two-electron reduction of both AQDS and hIYD. From this analysis, a midpoint potential of −200 mV (pH 7.4) was determined for the FMNox/FMNhq couple. This potential is very similar to that for FMNox/FMNhq of free FMN (−205 mV, pH 7.0) in the absence of protein (31).
FIGURE 6.
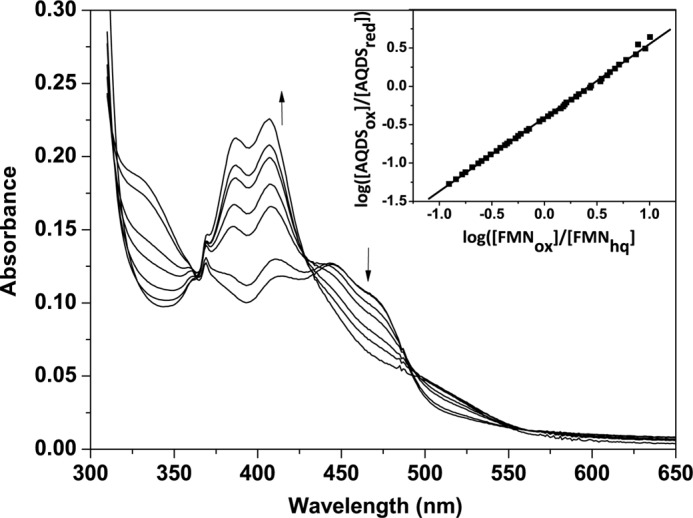
Reduction of FMN bound to hIYD and the reference dye AQDS by xanthine/xanthine oxidase. Spectral changes were monitored every 2 min over a total of 120 min. Only a selected number of spectra are illustrated for clarity, and the arrows indicate the direction of the absorbance change. The concentration of reduced AQDS was measured at 325 nm, an isosbestic point for hIYD, and conversely the concentration of reduced hIYD was measured at 355 nm, an isosbestic point of AQDS. Inset, the linear best fit of log(FMNox/FMNhq) versus log(AQDSox/AQDSred) was used to calculate the midpoint potential (24, 25).
The presence of F-Tyr significantly stabilized the neutral semiquinone and promoted reduction of hIYD by two single electron transfers. To characterize the first transfer, Nile blue (NB) was found to be most appropriate to monitor the FMNox/FMNsq couple of hIYD. In this case, a slope of 1.9 was obtained from plotting log(FMNox/FMNsq) versus log(NBox/NBred) (24, 25) (Fig. 7). This slope is near the theoretical value of 2.0 based on the two-electron reduction of NB and the one-electron reduction of FMNox to FMNsq. The midpoint potential for FMNox/FMNsq under these conditions is −156 mV.
FIGURE 7.
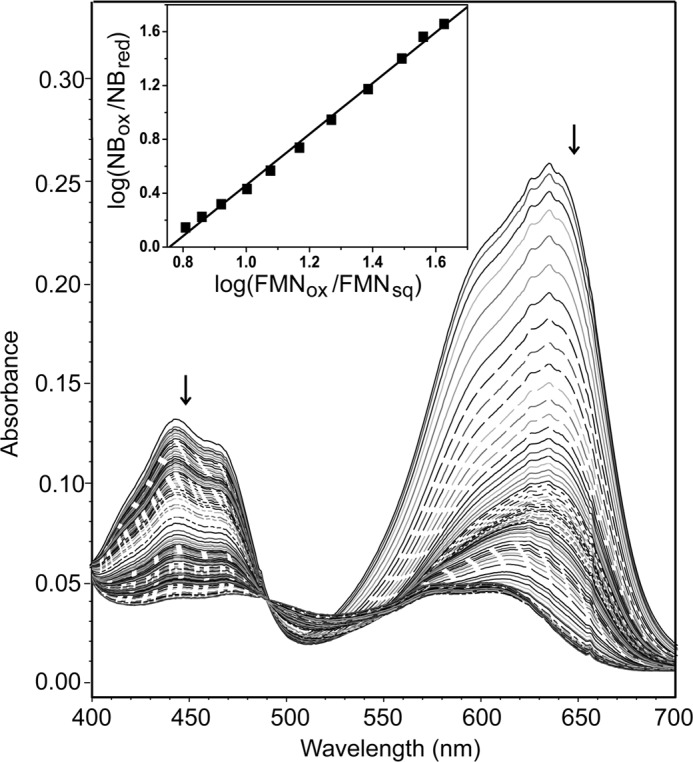
Reduction of FMNox to the neutral FMNsq bound to hIYD in the presence of F-Tyr. A selected number of spectra are illustrated for clarity, and the arrows indicate the direction of the absorbance change. Concentrations of oxidized FMNox within hIYD and the reference dye Nile blue were measured at 450 and 650 nm, respectively, during reduction by xanthine and xanthine oxidase. Inset, the linear best fit of log(FMNox/FMNsq) versus log(NBox/NBred) was used to calculate the midpoint potential (24, 25).
Safranin O (SFO) was selected as the dye to monitor the FMNsq/FMNhq couple after screening a variety of alternatives. The plot of log(FMNsq/FMNhq) versus log(SFOox/SFOred) generated a slope of 1.2 (Fig. 8). This differs from the expected slope of 2 based on the one-electron reduction of FMNsq to FMNhq and the two-electron reduction of SFO (26). Similar deviations were previously observed with an electron transfer flavoprotein and vanillyl-alcohol oxidase and ascribed to a kinetic barrier for reduction of FMNsq (32, 33). Still, a midpoint potential of −310 mV for FMNsq/FMNhq could be estimated from this titration. The difference between the midpoint potentials of FMNox/FMNsq and FMNsq/FMNhq is 154 mV, a value large enough to suggest that ∼99% of the FMN bound to IYD in the presence of F-Tyr is reduced by one electron prior to its reduction by a second electron (26, 34). These results are also consistent with the observed formation of FMNsq in the presence of either I-Tyr (12) or F-Tyr without a reference dye (Fig. 5).
FIGURE 8.
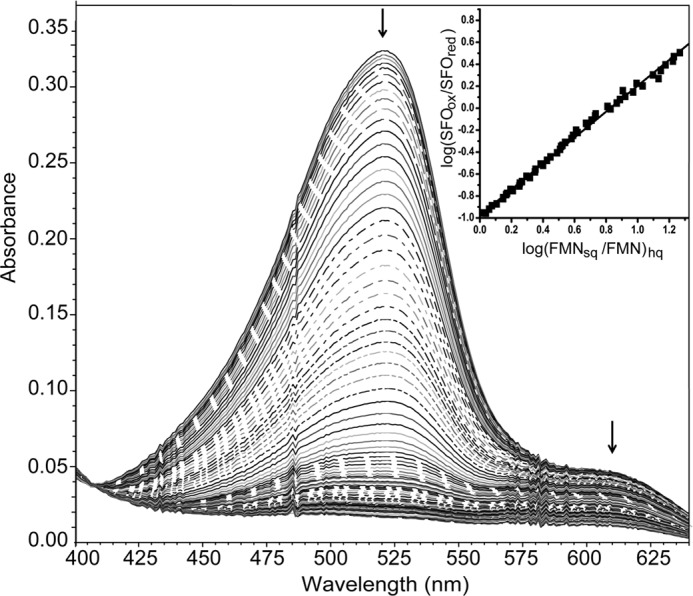
Reduction of FMNsq to FMNhq when bound to hIYD in the presence of F-Tyr. A selected number of spectra are illustrated for clarity, and the arrows indicate the direction of the absorbance change. The reduction of FMNsq and Safranine O by xanthine and xanthine oxidase was monitored at 610 and 520 nm, respectively. Inset, the linear best fit of log(FMNsq/FMNhq) versus log(SFOox/SFOred) was used to calculate the midpoint potential (24, 25).
DISCUSSION
Current studies on hIYD validate prior use of mIYD as a model for the human enzyme. Their sequences share 87% identity, and their structures are nearly superimposable with a RMSD of 0.26 Å (for a 399-atom comparison of Protein Data Bank code 4TTC versus code 3GFD, respectively). Both form active site lids consisting of a loop and helix that sequester their substrate·FMN complexes from solvent. This conformation is templated by coordination of substrate to multiple side chains of the lid and multiple sites on the isoalloxazine ring of the FMN. Additionally, the four known human mutations that compromise IYD activity and result in thyroid disease (8, 9) map to the hIYD structure as predicted earlier from the mIYD model (15). One of the most severe mutations is R101W, which effects a key interaction between the protein and the phosphate group of FMN.
The kcat/Km of 0.40 min−1 μm−1 for hIYD also falls within the range of 0.28–3.5 min−1 μm−1 that has been detected for IYD from a highly diverse series of organisms (7). Similarly, I-Tyr binds very tightly to hIYD as well as to mIYD with KD values below 1 μm (Table 1). Despite the potential for halogen bonding (35, 36), no evidence supports such an interaction between IYD and its substrates. Particularly for hIYD, the relative affinities of Cl-Tyr ≥ Br-Tyr ≥ I-Tyr are antithetical to the trend associated with halogen bonding.
The halogen substituent nonetheless appears to be critical for enzyme recognition as implied by prior investigations exploring substrate specificity (12). Neither tyrosine nor 3-methyltyrosine exhibited measurable affinity for IYD despite the common presence of the zwitterion for chelation by the active site lid and FMN. The ability of the halogen to lower the pKa of the phenolic proton was used to rationalize the previous binding data (12) and was consistent with an earlier report on the high affinity of nitro- and dinitrotyrosine for IYD (37). These results together suggested preferential binding of the phenolate form of the tyrosine derivatives and a corollary of enhanced binding at alkaline pH. The pH dependence of I2-Tyr binding now confirms an increase in affinity from pH 6.0 to 7.5 as measured by the decrease in the dissociation constant KD (Fig. 2). However, binding again weakens for I2-Tyr above pH 7.5. The two regions of the pH profile suggest an apparent deprotonation of a group with a pKa of 6.6 and protonation of a group with a pKa of 8.7 for maximum affinity. The pKa of the phenol in I2-Tyr has been reported as 6.4 and 6.5 (38, 39). These values are quite similar to the value measured from the pH dependence of binding to hIYD and consistent with a preferential association of hIYD with the phenolate form of the halotyrosines. The pH dependence of binding similarly explains the poor affinity of tyrosine because its phenol is not ionized under neutral conditions due to its corresponding pKa of 9.1 (39).
The phenolate form of the halotyrosines may act as a hydrogen bond acceptor by its close association with the amide backbone of Ala-130 (2.7 Å) and the 2′-hydroxy group of FMN (2.6 Å). Direct coordination between substrates and the ribose component of a flavin cofactor is not common but has precedence. This interaction serves a significant role in stabilizing the transition state of many acyl-CoA dehydrogenases (40, 41). In these examples, hydrogen bonding provided by the 2′-hydroxyl group polarizes the acyl carbonyl and stabilizes its developing charge. Nitroalkane oxidase is a member of the same acyl-CoA dehydrogenase superfamily and demonstrates an equivalent interaction between the 2′-hydroxyl of FAD and an oxygen of the nitro group (42). This same activation strategy may also be proposed for BluB, an enzyme closely related to IYD in structure but not function (7, 15). The structure of BluB reveals a similar interaction between its substrate molecular oxygen and the 2′-hydroxyl of its FMN (43). However, IYD remains unusual among these examples because the flavin side chain seems to associate with the initial electron-rich phenolate form of the substrate rather than a carbonyl group that gains charge during catalysis. There is little mechanistic logic for IYD to preferentially bind an electron-rich form of its substrate and then introduce an additional two electrons into this species. Still, the 2′-hydroxyl group of FMN could function in a manner complementary to that described for acyl-CoA dehydrogenase and BluB if the non-aromatic keto form of the I-Tyr was generated in the active site as an electrophilic acceptor of reducing equivalents (2). The only evidence currently supporting such an intermediate derives from the high affinity of IYD for non-aromatic pyridone analogs designed to mimic the keto form of the halotyrosines (44).
Another ionizable group that may affect binding is the α-ammonium group of I2-Tyr. The pKa of 8.7 estimated from the pH dependence of binding (Fig. 2) is higher than that reported for I2-Tyr (pKa of 7.8) (39) but similar to the textbook values ranging from 9.0 to 10.8 for the α-ammonium groups of all 20 common amino acids. Very few other ionizable groups are in the vicinity of the active site besides Lys-182 and Glu-157. Both of these are expected to be ionized for interaction with the α-carboxylate and the α-ammonium groups of I2-Tyr, respectively (6). The only other candidate is the N3–H of FMN. The pKa of this group is ∼10 for FMN in solution but may vary when sequestered in the IYD active site (13). The necessity for the protonated α-ammonium form of the tyrosine derivatives is easily rationalized from the structure of the hIYD·I-Tyr complex. This group provides electrostatic attraction and potential hydrogen bonding to the active site lid residue Glu-157 from a distance of 3.0 Å (6, 15) as well as the O4 of FMN (2.5 Å; Fig. 9).
FIGURE 9.
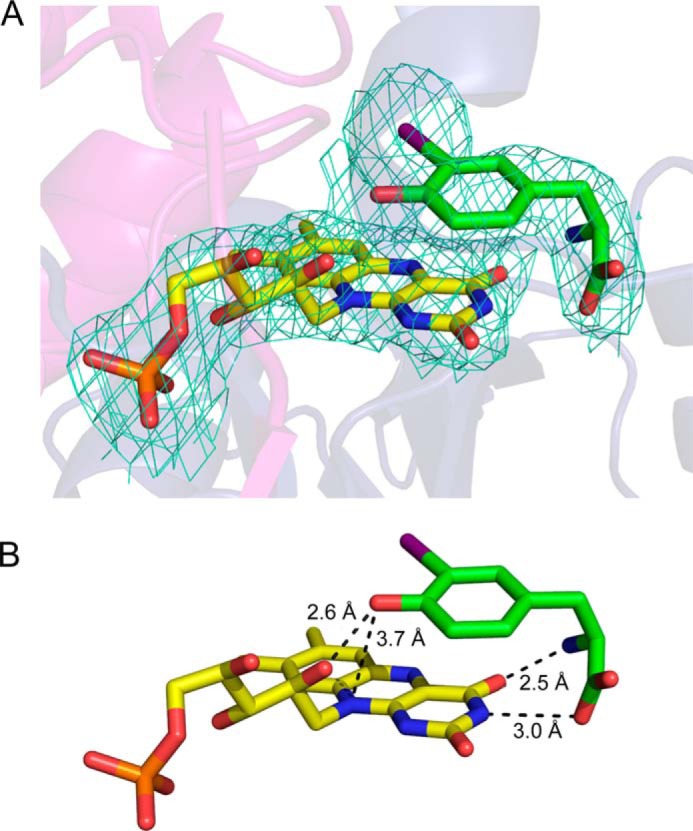
Substrate forms many contacts to FMN within the active site of hIYD. A, the simulated annealing composite omit map for FMN and I-Tyr (2Fo − Fc; contoured at 1 σ) indicates their close interaction. B, I-Tyr stacks over FMN and establishes numerous polar contacts with FMN in the active site of hIYD.
The pH dependence of both V and V/K for IYD turnover of I2-Tyr suggests that protonation of a group with a pKa of 7.5–7.6 facilitates catalysis (Fig. 1). This is intermediate between the two pKa values apparent for substrate binding. However, these data are not necessarily comparable. Binding studies involve FMNox, whereas enzyme turnover involves an initial FMNhq. Again, few candidates are immediately apparent for the ionization important in catalysis. The origin of this pH dependence will most likely be revealed once the rate-determining step of catalysis can be determined. If the α-ammonium group of I2-Tyr remains crucial for transition state stabilization, then its interaction with the O4 of FMN may help orient substrates with regard to the isoalloxazine system and modulate the oxidation-reduction properties of this system.
The midpoint potentials of flavin and its ability to promote one- and two-electron chemistry vary over the range of flavoproteins. Typically, flavin chemistry is controlled by the protein side chains surrounding the isoalloxazine ring that establish the active site polarity, hydrogen bonding, electrostatic environment, and π-stacking (13, 29). These features are all evident in IYD, but substrate rather than protein provides many of the necessary contacts (Figs. 3 and 9). The only exception is the interaction between the side chain of Arg-104 and the N1/O2 region of FMN. The guanidinium group of this Arg is poised to stabilize the increase in charge at N1 after reduction of FMN to its FMNhq form (13). A similar interaction is common to many flavoproteins and is particularly prevalent in the nitro-FMN reductase superfamily (15, 45). The protein also helps to shield the FMN·substrate complex from solvent by creating a lid over the active site (Fig. 4). However, the aromatic ring of the substrate provides direct stacking onto the isoalloxazine ring (Fig. 9). The substrate further establishes direct electrostatic and hydrogen bonding interactions with the N3 and O4 of the isoalloxazine system as described above.
Although all of these interactions may influence the redox chemistry of FMN, the region surrounding its N5 position is often considered the defining feature for many flavoproteins (29, 43, 46–48). Enzymes within a subclass of the nitro-FMN reductase superfamily that catalyze obligate two-electron reduction of their substrates typically maintain hydrogen bonding to the N5 position through a main chain amide proton (15, 45). In contrast, BluB relies on alternative hydrogen bonding from a side chain (Ser) to the N5 and represents a different subclass within this same superfamily (43). Such hydrogen bonding is critical for the function of BluB because mutation of this Ser to Gly reduces catalytic activity by ∼30-fold (43). Side chain hydrogen bonding to N5 is also evident in electron transfer flavoproteins and is generally thought to facilitate one-electron chemistry (46). This is consistent with the ability of BluB to promote an initial one-electron transfer to its substrate, molecular oxygen. IYD contains a corresponding Thr-239, but its hydroxyl group remains 4.9 Å from the N5 of FMN in the absence of the substrate I-Tyr. Once substrate binds to IYD and templates a closure of the active site lid, the hydroxyl group of Thr-239 approaches the N5 position within hydrogen bonding distance (3.1 Å) (Fig. 3B).
The significance of hydrogen bonding by Thr-239 along with the many other substrate-induced changes around FMN is best demonstrated by their collective effect on the redox chemistry of IYD. For this analysis, the substrate analog F-Tyr was used in place of a halotyrosine substrate to focus on the effect of ligand coordination to the FMN reduction without catalytic turnover. IYD has little influence on FMN in the absence of an active site ligand. Its midpoint potential is similar to that of FMN in solution, and only FMNox and FMNhq could be observed during reduction (Figs. 5A and 6). The one-electron reduced FMNsq was not evident and likely too unstable to accumulate under the experimental conditions. In contrast, a definitive switch to stepwise reduction of FMN was apparent after its association with F-Tyr. Under these conditions, FMNsq became a distinct intermediate during reduction of FMNox (Fig. 5B). The extent of semiquinone stabilization is illustrated by comparing the midpoint potential of FMNox to FMNsq when bound to hIYD (−156 mV) and free in solution (−314 mV) (49). In contrast, reduction of FMNsq to FMNhq is destabilized when bound to IYD (−310 mV) versus free in solution (−124 mV) (49). These potentials of hIYD are similar to those of flavodoxin (50), ferredoxin-NADP+ oxidoreductase (51), and nitric-oxide synthase (52) for which FMN is involved in electron transfer reactions.
The ability of substrate to lower the reduction potential of FMNhq bound to IYD suggests that substrate helps to promote its own reduction and initiation of deiodination. A related modulation of the electron donating ability of reduced flavin has been observed with certain acyl-CoA dehydrogenases. In these examples, the presence of an enoyl-CoA product affects the reduction potential of FMN to facilitate subsequent transfer of its electrons to an electron carrier (53, 54). Both processes likely satisfy physiological necessities. For IYD, deiodination of I-Tyr and I2-Tyr is required in the simultaneous presence of iodinated tyrosyl residues of thyroglobulin and the iodinated thyroid hormones. All of these would be subject to deiodination and loss of activity if the common iodophenol group were the only determinant for recognition and catalysis. Instead, the zwitterion unique to the free halotyrosines is essential for coordinating to FMN and promoting catalysis. The substrate-induced switch of FMN from two-electron to one-electron chemistry also supports the current proposal on the mechanism of IYD involving stepwise reduction of its substrate (Fig. 10) (2). A previous proposal based on a single two-electron transfer mechanism conceived in analogy to the thiol-dependent dechlorinases (20, 55) was dismissed as soon as IYD was shown to act independently of thiols and contain no Cys (nor SeCys) residues in its active site (5, 15).
FIGURE 10.
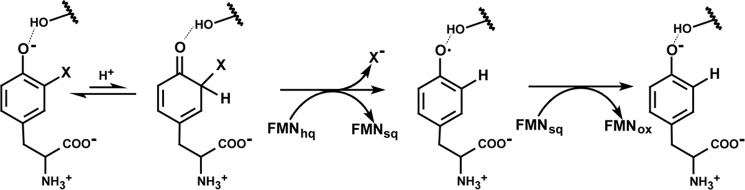
Possible stepwise reduction of I-Tyr by IYD.
Successful preparation of hIYD lacking its native membrane anchor now provides easy access to the enzyme most relevant to understanding its contribution to human health. The pH dependence of I2-Tyr binding and turnover suggests key roles for its phenolate group and perhaps its α-ammonium group in substrate recognition and catalytic efficiency. An intimate relationship among I-Tyr, FMN, and the active site lid of hIYD is illustrated by crystallographic analysis and consistent with previous observations with mIYD. However, the extent to which ligand association can modulate the redox properties of the enzyme was not appreciated before the current characterization of hIYD. This flavoprotein provides a rare example in which substrate establishes direct and multiple interactions with the isoalloxazine system. Such coordination likely guides substrate selection in vivo and provides an opportune system for future studies in vitro to measure the contributions of individual functional groups in stabilizing the active site lid and controlling the redox chemistry of FMN.
Acknowledgments
We thank Juan C. Solis-S. for initial efforts on hIYD and the Beamline 7-1 scientists at the Stanford Synchrotron Radiation Laboratory for assistance with remote data collection.
This work was supported in part by National Institutes of Health Grant RO1 DK084186 (to S. E. R.).
This work is dedicated to Professor Iwao Ojima on the occasion of his 70th birthday.
The atomic coordinates and structure factors (codes 4TTB and 4TTC) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- IYD
- iodotyrosine deiodinase
- mIYD
- mouse IYD
- hIYD
- human IYD
- AQDS
- anthraquinone-2,6-disulfonate
- Br-Tyr
- 3-bromo-l-tyrosine
- Cl-Tyr
- 3-chloro-l-tyrosine
- FMNox
- flavin mononucleotide in its oxidized form
- FMNhq
- reduced FMN hydroquinone
- FMNsq
- one-electron reduced FMN semiquinone
- F-Tyr
- 3-fluoro-l-tyrosine
- I-Tyr
- 3-iodo-l-tyrosine
- I2-Tyr
- 3,5-diiodo-l-tyrosine
- NB
- Nile blue
- SFO
- Safranin O
- SUMO
- small ubiquitin-like modifier
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- RMSD
- root mean square deviation.
REFERENCES
- 1. Nicola J. P., Carrasco N., Amzel L. M. (2014) Physiological sodium concentrations enhance the iodide affinity of the Na+/I− symporter. Nat. Commun. 5, 3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rokita S. E., Adler J. M., McTamney P. M., Watson J. A., Jr. (2010) Efficient use and recycling of the micronutrient iodide in mammals. Biochimie 92, 1227–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bianco A. C., Salvatore D., Gereben B., Berry M. J., Larsen P. R. (2002) Biochemistry, cellular and molecular biology and physiological roles of the iodothyronine selenodeiodinases. Endocr. Rev. 23, 38–89 [DOI] [PubMed] [Google Scholar]
- 4. Friedman J. E., Watson J. A., Jr., Lam D. W., Rokita S. E. (2006) Iodotyrosine deiodinase is the first mammalian member of the NADH oxidase/flavin reductase superfamily. J. Biol. Chem. 281, 2812–2819 [DOI] [PubMed] [Google Scholar]
- 5. Watson J. A., Jr., McTamney P. M., Adler J. M., Rokita S. E. (2008) The flavoprotein iodotyrosine deiodinase functions without cysteine residues. ChemBioChem 9, 504–506 [DOI] [PubMed] [Google Scholar]
- 6. Buss J. M., McTamney P. M., Rokita S. E. (2012) Expression of a soluble form of iodotyrosine deiodinase for active site characterization by engineering the native membrane protein from Mus musculus. Protein Sci. 21, 351–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Phatarphekar A., Buss J. M., Rokita S. E. (2014) Iodotyrosine deiodinase: a unique flavoprotein present in organisms of diverse phyla. Mol. BioSyst. 10, 86–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moreno J. C., Klootwijk W., van Toor H., Pinto G., D'Alessandro M., Lèger A., Goudie D., Polak M., Grüters A., Visser T. J. (2008) Mutations in the iodotyrosine deiodinase gene and hypothyroidism. N. Engl. J. Med. 358, 1811–1818 [DOI] [PubMed] [Google Scholar]
- 9. Afink G., Kulik W., Overmars H., de Randamie J., Veenboer T., van Cruchten A., Craen M., Ris-Stalpers C. (2008) Molecular characterization of iodotyrosine dehalogenase deficiency in patients with hypothyroidism. J. Clin. Endocrinol. Metab. 93, 4894–4901 [DOI] [PubMed] [Google Scholar]
- 10. Gnidehou S., Lacriox L., Sezan A., Ohayon R., Noël-Hudson M.-S., Morand S., Francon J., Courtin F., Virion A., Dupuy C. (2006) Cloning and characterization of a novel isoform of iodotyrosine dehalogenase 1 (DEHAL1) DEHAL1C from human thyroid: comparisons with DEHAL1 and DEHAL1B. Thyroid 16, 715–724 [DOI] [PubMed] [Google Scholar]
- 11. Goswami A., Rosenberg I. N. (1977) Studies on a soluble thyroid iodotyrosine deiodinase: activation by NADPH and electron carriers. Endocrinology 101, 331–341 [DOI] [PubMed] [Google Scholar]
- 12. McTamney P. M., Rokita S. E. (2009) A mammalian reductive deiodinase has broad power to dehalogenate chlorinated and brominated substrates. J. Am. Chem. Soc. 131, 14212–14213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fagan R. L., Palfey B. A. (2010) in Comprehensive Natural Products II (Begley T. P., ed) pp. 37–114, Elsevier, Oxford [Google Scholar]
- 14. Rokita S. E. (2013) in Handbook of Flavoproteins (Hille R., Miller S. M., Palfey B., eds) pp. 337–350, DeGruyter, Berlin [Google Scholar]
- 15. Thomas S. R., McTamney P. M., Adler J. M., Laronde-Leblanc N., Rokita S. E. (2009) Crystal structure of iodotyrosine deiodinase, a novel flavoprotein responsible for iodide salvage in thyroid glands. J. Biol. Chem. 284, 19659–19667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pace C. N., Vajdos F., Fee L., Grimsley G., Gray T. (1995) How to measure and predict the molar absorption coefficient of a protein. Protein. Sci. 4, 2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koziol J. (1971) Fluorometric analyses of riboflavin and its coenzymes. Methods Enzymol. 18, 235–285 [Google Scholar]
- 18. Rosenberg I. N., Goswami A. (1984) Iodotyrosine deiodinase from bovine thyroid. Methods Enzymol. 107, 488–500 [DOI] [PubMed] [Google Scholar]
- 19. Cook P. F., Cleland W. W. (2007) Enzyme Kinetics and Mechanism, pp. 325–366, Garland Science, New York [Google Scholar]
- 20. Warner J. R., Copley S. D. (2007) Pre-steady-state kinetic studies of the reductive dehalogenation catalyzed by tetrachlorohydroquinone dehalogenase. Biochemistry 46, 13211–13222 [DOI] [PubMed] [Google Scholar]
- 21. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 22. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 24. Massey V. (1991) in Flavins Flavoproteins, Proceedings of the 10th International Symposium, Como, Italy, July 15–20, 1990 (Curti B., Ronchi S., Zanetti G., eds) pp. 59–66, DeGruyter, Berlin [Google Scholar]
- 25. van den Heuvel R. H., Fraaije M. W., van Berkel W. J. (2002) Redox properties of vanillyl-alcohol oxidase. Methods Enzymol. 353, 177–186 [DOI] [PubMed] [Google Scholar]
- 26. Clark W. M. (1960) Oxidation-Reduction Potentials of Organic Systems, Williams & Wilkins, Baltimore [Google Scholar]
- 27. Gnidehou S., Caillou B., Talbot M., Ohayon R., Kaniewski J., Noël-Hudson M.-S., Morand S., Agnangji D., Sezan A., Courtin F., Virion A., Dupuy C. (2004) Iodotyrosine dehalogenase 1 (DEHAL1) is a transmembrane protein involved in the recycling of iodide close to the thyroglobulin iodination site. FASEB J. 18, 1574–1576 [DOI] [PubMed] [Google Scholar]
- 28. Massey V. (2000) The chemical and biological versatility of riboflavin. Biochem. Soc. Trans. 28, 283–296 [PubMed] [Google Scholar]
- 29. Fraaije M. W., Mattevi A. (2000) Flavoenzymes: diverse catalysts with recurrent features. Trends Biochem. Sci. 25, 126–132 [DOI] [PubMed] [Google Scholar]
- 30. Mansoorabadi S. O., Thibodeaux C. J., Liu H.-W. (2007) The diverse roles of flavin coenzymes—nature's most versatile thespians. J. Org. Chem. 72, 6329–6342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Draper R. D., Ingraham L. L. (1968) A potentiometric study of the flavin semiquinone equilibrium. Arch. Biochem. Biophys. 125, 802–8808 [DOI] [PubMed] [Google Scholar]
- 32. Byron C. M., Stankovich M. T., Husain M., Davidson V. L. (1989) Unusual redox properties of electron-transfer flavoprotein from Methylophilus methylotrophus. Biochemistry 28, 8582–8587 [DOI] [PubMed] [Google Scholar]
- 33. van den Heuvel R. H., Fraaije M. W., Mattevi A., van Berkel W. J. (2000) Asp-170 is crucial for the redox properties of vanillyl-alcohol oxidase. J. Biol. Chem. 275, 14799–14808 [DOI] [PubMed] [Google Scholar]
- 34. Hamill M. J., Jost M., Wong C., Bene N. C., Drennan C. L., Elliott S. J. (2012) Electrochemical characterization of Escherichia coli adaptive response protein AidB. Int. J. Mol. Sci. 13, 16899–16915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Auffinger P., Hays F. A., Westhof E., Ho P. S. (2004) Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. U.S.A. 101, 16789–16794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Metrangolo P., Meyer F., Pilati T., Resnati G., Terraneo G. (2008) Halogen bonding in supramolecular chemistry. Angew. Chem. Int. Ed. Engl. 47, 6114–6127 [DOI] [PubMed] [Google Scholar]
- 37. Green W. L. (1968) Inhibition of thyroidal iodotyrosine deiodination by tyrosine analogues. Endocrinology 83, 336–347 [DOI] [PubMed] [Google Scholar]
- 38. Gemmill C. L. (1955) The apparent ionization constants of the phenolic hydroxyl group of thyroxine and related compounds. Arch. Biochem. Biophys. 54, 359–367 [DOI] [PubMed] [Google Scholar]
- 39. Greenstein J. P., Winitz M. (1961) Chemistry of the Amino Acids, pp. 486–487, Wiley, New York [Google Scholar]
- 40. Thorpe C., Kim J.-J. (1995) Structure and mechanism of action of the acyl-CoA dehydrogenases. FASEB J. 9, 718–725 [DOI] [PubMed] [Google Scholar]
- 41. Engst S., Vock P., Wang M., Kim J.-J., Ghisla S. (1999) Mechanism of activation of acyl-CoA substrates by medium chain acyl-CoA dehydrogenase: interaction of the thioester carbonyl with the flavin adenine dinucleotide ribityl side chain. Biochemistry 38, 257–267 [DOI] [PubMed] [Google Scholar]
- 42. Héroux A., Bozinovski D. M., Valley M. P., Fitzpatrick P. F., Orville A. M. (2009) Crystal structure of intermediates in the nitroalkane oxidase reaction. Biochemistry 48, 3407–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Taga M. E., Larsen N. A., Howard-Jones A. R., Walsh C. T., Walker G. C. (2007) BluB cannibalizes flavin to form the lower ligand of vitamin B12. Nature 446, 449–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kunishima M., Friedman J. E., Rokita S. E. (1999) Transition-state stabilization by a mammalian reductive dehalogenase. J. Am. Chem. Soc. 121, 4722–4723 [Google Scholar]
- 45. Roldán M. D., Pérez-Reinado E., Castillo F., Moreno-Vivián C. (2008) Reduction of polynitroaromatic compounds: the bacterial nitroreductases. FEMS Microbiol. Rev. 32, 474–500 [DOI] [PubMed] [Google Scholar]
- 46. Yang K.-Y., Swenson R. P. (2007) Modulation of the redox properties of the flavin cofactor through hydrogen-bonding interactions with the N5 atom: role of αser254 in the electron-transfer flavoprotein from the methylotrophic bacterium W3A1. Biochemistry 46, 2289–2297 [DOI] [PubMed] [Google Scholar]
- 47. Pitsawong W., Sucharitakul J., Prongjit M., Tan T. C., Spadiut O., Haltrich D., Divne C., Chaiyen P. (2010) A conserved active-site threonine is important for both sugar and flavin oxidations of pyranose 2-oxidase. J. Biol. Chem. 285, 9697–9705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McDonald C. A., Fagan R. L., Collard F., Monnier V. M., Palfey B. A. (2011) Oxygen reactivity in flavoenzymes: context matters. J. Am. Chem. Soc. 133, 16809–16811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Anderson R. F. (1983) Energetics of the one-electron reduction steps of riboflavin, FMN and FAD to their fully reduced forms. Biochim. Biophys. Acta 722, 158–162 [DOI] [PubMed] [Google Scholar]
- 50. Zhou Z., Swenson R. P. (1996) The cumulative electrostatic effect of aromatic stacking interactions and the negative electrostatic environment of the flavin mononucleotide binding site is a major determinant of the reduction potential for the flavodoxin from Desulfovibrio vulgaris [Hildenborough]. Biochemistry 35, 15980–15988 [DOI] [PubMed] [Google Scholar]
- 51. Bowsher C. G., Eyres L. M., Gummadova J. O., Hothi P., McLean K. J., Munro A. W., Scrutton N. S., Hanke G. T., Sakakibara Y., Hase T. (2011) Identification of N-terminal regions of wheat leaf ferredoxin NADP+ oxidoreductase important for interactions with ferredoxin. Biochemistry 50, 1778–1787 [DOI] [PubMed] [Google Scholar]
- 52. Panda S. P., Gao Y. T., Roman L. J., Martásek P., Salerno J. C., Masters B. S. (2006) The role of a conserved serine residue within hydrogen bonding distance of FAD in redox properties and the modulation of catalysis by Ca2+/calmodulin of constitutive nitric-oxide synthases. J. Biol. Chem. 281, 34246–34257 [DOI] [PubMed] [Google Scholar]
- 53. Becker D. F., Fuchs J. A., Stankovich M. T. (1994) Product binding modulates the thermodynamic properties of a Megasphaera elsdenii short-chain Acyl-CoA dehydrogenase active-site mutant. Biochemistry 33, 7082–7087 [DOI] [PubMed] [Google Scholar]
- 54. Gorelick R. J., Schopfer L. M., Ballou D. P., Massey V., Thorpe C. (1985) Interflavin oxidation-reduction reactions between pig kidney general acyl-CoA dehydrogenase and electron-transferring flavoprotein. Biochemistry 24, 6830–6839 [DOI] [PubMed] [Google Scholar]
- 55. Velazquez F., Peak-Chew S. Y., Fernández I. S., Neumann C. S., Kay R. R. (2011) Identification of a eukaryotic reductive dechlorinase and characterization of its mechanism of action on its natural substrate. Chem. Biol. 18, 1252–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stradins J., Hansanli B. (1993) Anodic voltammetry of phenol and benzenethiol derivatives. 1. Influence of pH on electrooxidation potentials of substitutes phenols and evaluation of pKa from anodic voltammetry data. J. Electroanal. Chem. 353, 57–69 [Google Scholar]