

Background: Serpins are uncommon in bacteria; little is known about their function.
Results: Serpin from T. forsythia (miropin) inhibits a broad array of proteases with divergent specificities.
Conclusion: Miropin may allow T. forsythia to dwell in a highly proteolytic environment.
Significance: Miropin is the first pathogen-derived serpin with the unusual ability to efficiently inhibit different proteases at several active sites.
Keywords: Infectious Disease, Periodontal Disease, Protease Inhibitor, Serine Protease, Serpin, Tannerella forsythia
Abstract
All prokaryotic genes encoding putative serpins identified to date are found in environmental and commensal microorganisms, and only very few prokaryotic serpins have been investigated from a mechanistic standpoint. Herein, we characterized a novel serpin (miropin) from the human pathogen Tannerella forsythia, a bacterium implicated in initiation and progression of human periodontitis. In contrast to other serpins, miropin efficiently inhibited a broad range of proteases (neutrophil and pancreatic elastases, cathepsin G, subtilisin, and trypsin) with a stoichiometry of inhibition of around 3 and second-order association rate constants that ranged from 2.7 × 104 (cathepsin G) to 7.1 × 105 m−1s−1 (subtilisin). Inhibition was associated with the formation of complexes that were stable during SDS-PAGE. The unusually broad specificity of miropin for target proteases is achieved through different active sites within the reactive center loop upstream of the P1-P1′ site, which was predicted from an alignment of the primary structure of miropin with those of well studied human and prokaryotic serpins. Thus, miropin is unique among inhibitory serpins, and it has apparently evolved the ability to inhibit a multitude of proteases at the expense of a high stoichiometry of inhibition and a low association rate constant. These characteristics suggest that miropin arose as an adaptation to the highly proteolytic environment of subgingival plaque, which is exposed continually to an array of host proteases in the inflammatory exudate. In such an environment, miropin may function as an important virulence factor by protecting bacterium from the destructive activity of neutrophil serine proteases. Alternatively, it may act as a housekeeping protein that regulates the activity of endogenous T. forsythia serine proteases.
Introduction
Serpins (serine protease inhibitors) are present in all multicellular organisms analyzed to date and are abundant in plants (wheat, rice), nematodes, insects, and vertebrates, especially in mammals (1). Human serpins are the best characterized. Serpins circulating in the blood are mainly responsible for the inhibition of neutrophil-derived serine proteases and are involved in the control of inflammation, coagulation, fibrinolysis, and complement activation. On the other hand intracellular serpins exert a cytoprotective effect by inhibiting lysosomal proteases that diffuse into the cytoplasm and are also involved in host defense against infection (2). Some serpins do not inhibit proteases but instead function as storage proteins (ovalbumin), molecular chaperones (Hsp47/SERPINH1), and hormone transporting proteins (thyroxin-binding globulin, cortisol-binding globulin) as a source of a bioactive peptide (angiotensinogen) and as regulators of chromatin compaction (chicken serpin MENT). Serpins are also encoded by some viruses where they function as virulence factors by targeting proteases involved the host immune defense (1, 3).
In contrast to the wide distribution of serpins in eukaryotes, serpins in prokaryotes are sporadically distributed. Indeed, for many years serpins were considered not to be required for the functioning of unicellular organisms and thus were considered to be absent from prokaryotes. Large scale sequencing of bacterial genomes changed this misconception by revealing the presence of open reading frames (ORFs) encoding putative serpins (4, 5). The first bacterial serpin, thermopin, was expressed and characterized in 2003 (6). Advances in sequencing of prokaryotic genomes revealed that ORFs encoding putative serpins are present in 31 and 13% of the fully sequenced genomes of archaea and bacteria, respectively (7). Nevertheless, to date, only a very limited number of prokaryotic serpins have been functionally characterized. These include serpins from Clostridium thermocellum (8) and Bifidobacterium longum (9), thermopin and tengpin from the thermophilic bacteria Thermobifida fusca (6) and Thermoanaerobacter tengcongensis (10), respectively, as well as aeropin and Tk-serpin from the hyperthermophilic archaea Pyrobaculum aerophilum (11) and Thermococcus kodakaraensis (12), respectively. At present, the precise role of most bacterial serpins remains obscure with the exception of Tk-serpin from T. kodakaraensis and serpins from C. thermocellum and B. longum. It is hypothesized that Tk-serpin could protect T. kodakaraensis by inhibiting the secreted protease Tk-subtilisin (12). Similarly, it is speculated that the serpin from B. longum, a commensal gut bacterium, protects against neutrophil and pancreatic elastases present in the gastrointestinal tract (9), and the serpin from C. thermocellum protects the cellulosome from protease activity (8).
Bacterial serpins, with the exception of a few species of Bacteroidetes phylum (Prevotella buccae, Prevotella stercorea, Bacteroides ovatus, Bacteroides uniformis, and B. longum), are found in environmental microbiota (7). All these species are present in human gut microflora, and thus, these serpins may help protect bacteria against the digestive proteases of the host. In this context the presence of an ORF encoding a putative serpin (BFO_3114, GenBankTM accession number CP003191; bfor_c_1_8111 in The Bioinformatics Resource for Oral Pathogens database or TF0781 in Oralgen) in the sequenced genome of Tannerella forsythia ATCC 43037 is of particular interest (13).
T. forsythia is a Gram-negative, asaccharolytic, anaerobic rod-shaped bacteria that together with Porphyromonas gingivalis and Treponema denticola forms the so-called “red complex” (13) implicated in the development and progression of periodontitis in humans (14). It can be argued that periodontitis is the most prevalent infection-driven chronic inflammatory disease in humans, and it is estimated that 10–15% of the adult population worldwide suffers from severe forms of periodontitis (15). Progression of the disease is manifested by the loss of attachment between teeth and periodontal tissues resulting in formation of deep periodontal pockets. In severe cases the disease can lead to a loss of dentition (16). In addition, periodontitis is also associated with an increased risk of lung diseases (17), preterm low birth weight (18), endocarditis (19), cardiovascular diseases (e.g. atherosclerosis and aneurysm) (20), stroke, diabetes (21), and rheumatoid arthritis (22).
In periodontal pockets, subgingival dental plaque (a dwelling place of T. forsythia) is bathed in gingival crevicular fluid. This inflammatory exudate contains high levels of neutrophil-derived serine proteases (e.g. neutrophil elastase and cathepsin G) (23). By degrading bacterial proteins, these proteases play an important role in the innate immune system of the host (24). Therefore, we hypothesized that the serpin from T. forsythia, referred to as miropin, may contribute to the virulence of periodontal pathogens by inhibiting neutrophil serine proteases. To investigate this hypothesis, we expressed, purified, and characterized miropin. Miropin was found to be an efficient inhibitor not only of elastase and cathepsin G but also of several other serine proteases with vastly different specificities.
EXPERIMENTAL PROCEDURES
Enzymes, Inhibitors, Substrates, and Other Reagents
The following enzymes and reagents were used: human neutrophil elastase, cathepsin G, ecotin, and human α2-macroglobulin (BioCentrum, Krakow, Poland; Athens Research and Technology, Athens, GA); porcine pancreatic elastase (Promega, Fitchburg, WI); bovine pancreatic α-chymotrypsin, bovine pancreatic trypsin, thrombin from human plasma, subtilisin Carlsberg, benzoyl-Arg-pNA,2 Boc-Val-Pro-Arg-aminofluoromethylcoumarin, Boc-Gln-Ala-Arg-7-amino-4-methylcoumarin (AMC), and metoxysuccinyl-Ala-Ala-Pro-Val-pNA (Sigma); succinyl-Ala-Ala-Pro-Phe-pNA and 7-methoxycoumarin-4-acetic acid (MCA)-FVT-4-guanidine-phenylalanine (Gnf)-SW-Anb-NH2 (Anb is the amide of amino benzoic acid; Merck). Three molecular weight markers were used: PageRuler Unstained Low Range Protein Ladder (size range: 3.4–100 kDa), PageRuler Prestained Protein Ladder (size range: 20–75 kDa) from Thermo Scientific Fermentas (Vilnius, Lithuania), and Precision Plus Protein Kaleidoscope (size range: 10–250 kDa) from Bio-Rad. All other chemical reagents were obtained from BioShop Canada (Burlington, ON, Canada).
Real Time PCR
T. forsythia RNA was isolated from 5-day-old colonies on blood agar plates using innuPREP RNA Mini kit (Analytic Jena, Jena, Germany). Obtained RNA was digested with RQ1 RNase (Promega) and purified with TRI Reagent (Ambion, Carlsbad, CA). RNA (1.6 μg) was then reverse-transcribed using cDNA High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Carlsbad, CA). Real time PCR was performed on CFX96 Touch machine (Bio-Rad). Single reaction consisted of 7.5 μl of FastStart Essential DNA Green Master mix (Roche Applied Science), 1 μl of 300 nm target-specific primers for T. forsythia serpin (5′-ATGCCTTTGCCTTCGATCTG-3′, 5′-CTTCCCGTAGTGAATGGCTG-3′), 5 μl of 100× diluted cDNA, and 1.5 μl of water. The PCR reaction consisted of initial denaturation step at 95 °C for 10 min followed by 40 cycles of 10 s at 95 °C, 30 s at 56 °C, and 30 s at 72 °C.
T. forsythia Growth and Screening for Inhibitory Activity
T. forsythia ATCC 43037 was grown in Shaedler broth (Oxoid, Hampshire, UK) supplemented with l-cysteine (0.5/liter), menadione (Sigma) (1 mg/liter), hemin (Sigma) (0.5 mg/liter), and N-acetylmuramic acid (Sigma) (10 mg/liter) until an early stationary phase. Bacterial cells were pelleted by centrifugation (10 min, 5000 revolutions per minute, 4 °C) from 10 ml of culture, washed 3 times in PBS, and resuspended in 1 ml of PBS. 100 μl of suspension was withdrawn and designated as a “whole cell” sample. The remaining 900-μl cell suspension was sonicated using the Vibra-cell sonicator (Sonics and Materials, Newtown, CT), and 100 μl of homogenate was withdrawn and labeled as “cell homogenate.” Large cell debris in the remaining 800 μl of homogenate was removed by low speed centrifugation (5000 × g, 10 min, 4 °C), and the supernatant was subjected to ultracentrifugation (100,000 × g, 1 h, 4 °C). The high speed supernatant containing soluble intracellular proteins was referred to as the “cytoplasm/periplasm” fraction. The pellet containing the cell envelope components (the outer membrane, the inner membrane, and peptidoglycan) was washed in PBS, resuspended in 800 μl of PBS, and designated as a “cell envelope” fraction. Inhibitory activity of each fraction (8 μl) against neutrophil elastase was determined as described under “Screening Miropin Inhibitory Activity” below. Finally, in a separate experiment bacterial cells from 50 ml of culture were collected by centrifugation (4000 × g, 20 min, 4 °C), washed 3 times in PBS, resuspended in 2 ml of PBS, and frozen at −20 °C. Thawed cells suspension was used to determine neutrophil elastase inhibition by T. forsythia as described below under “Stoichiometry of Inhibition.”
Enzyme Titration and Measurement of Protein Concentration
Trypsin was titrated with 4-nitrophenyl 4-guanidinobenzoate (Sigma) (25). The titrated trypsin was subsequently used to titrate ecotin and α-2-macroglobulin, which were later used for active site titration of neutrophil elastase, pancreatic elastase, chymotrypsin, cathepsin G (ecotin), thrombin, and subtilisin (α2-macroglobulin). In all the calculations we assumed that protease inhibition by α-2-macroglobulin occurred with the 1:2 stoichiometry (1 native molecule of an inhibitor inhibits 2 protease molecules). A similar approximation was used for titration of proteases with ecotin, which is known to occur as a dimer presenting two independent active sites. Therefore, the concentrations of proteolytic enzymes used herein refer to the concentration of active enzymes and not to the protein concentration. The protein concentration of miropin was determined using two methods: 1) UV absorption (A) at 280 nm measured by a NanoDrop device (Thermo Fisher Scientific, Waltham, MA) using an A280 value of 0.96 for a 0.1% (1 mg/ml) solution and 2) BCA protein assay reagent (Pierce) according to the manufacturer's protocol. The mean values from both methods were considered as the final result.
Resequencing of Locus TF0781 with Surrounding Regions
A region encompassing the TF0781 gene (Oralgen) encoding the T. forsythia serpin and the surrounding 5′ and 3′ regions was amplified by PCR with Phusion DNA polymerase (Thermo Fisher Scientific) and two sets of primers (abfAPCR1 (5′-GGTAACAGGGTGACATCAAAATCAC-3′) and abfAPCR2 (5′-ACAAGGGAACGACTTTGGTGAG-3′) and exBBPCR1 (5′-CCGTAACAGCCGTAGAAATGG-3′) and exBBPCR2 (5′-GAACGAATGTGGATGTATCCTTACTG-3′), respectively. PCR products were separated on a 1% agarose gel, and fragments were excised from the gel and purified using the GeneJET Gel Extraction kit (Thermo Fisher Scientific). The purified PCR product was analyzed by DNA sequencing using the following primers: abfA PCR product abfAseq1 (5′-TCGTCGTTCAAGCCGTTGTC-3′) and abfAseq2 (5′-GGAGAGATATAGCGATGATAGGAG-3′); exBB PCR product exBBseq1 (5′-GAATAGTACTTTCACCTGTCATAC-3′) and exBBseq2 (5′-GCTCTTTTCGCGTACAATTACTTG-3′); exBBseq3 (5′-CCGGCGTCATTCTGTTTATCGG-3′).
Plasmid Construction
Genomic DNA was extracted from T. forsythia strain ATCC 43037. Three putative variants of T. forsythia serpin with different N termini were amplified by PCR, purified, and cloned into the pGEX-6P-1 expression vector (GE Healthcare) using the BamHI/EcoRI restriction sites and the following PCR primers (restriction sites are underlined): TFs62_F (5′-TCCGGATCCATGAAGTCACCGGCCGTTCC-3′) and TFs55_F (5′-GTCGGATCCGTGATTTTGATGTCACCCTGTTAC-3′); TFs46_F (5′-GTCGGATCCATGAAAACACAATGGATGTGTATCG-3′) and TFsR (5′-CGTGAATTCTTATTCTTTCACTTCCCCGATCTC-3′).
The resulting recombinant hybrid proteins include an N-terminal glutathione S-transferase (GST) tag and a PreScission protease cleavage site followed by a serpin variant. Cleavage of the GST moiety with PreScission protease leaves five residues (Gly-Pro-Leu-Gly-Ser) before the N-terminal amino acid residue; that is, Met (TFs62, TFs46) or Val (TFs55) variants of T. forsythia serpin. The correct sequences of the expression plasmids (pGEX-6P-1_Tfs62, pGEX-6P1_Tfs55 and pGEX-6P1_Tfs46) were confirmed by DNA sequencing, transformed into Escherichia coli strain Rosetta (DE3) (EMD Millipore, Billerica, MA), and expressed under the control of the T7 promoter.
Expression and Purification of Recombinant Proteins
Transformed E. coli hosts were grown in LB (Lennox) media containing ampicillin (100 μg/ml) and chloramphenicol (33 μg/ml) at 37 °C to an A600 of 0.75–1 and then incubated for 30 min at 4 °C. Expression of recombinant proteins was induced by the addition of 0.25 mm isopropyl-1-thio-β-d-galactopyranoside. After 6 h at 21 °C, cells were harvested by centrifugation (15 min, 6000 × g, 4 °C) and resuspended in PBS (15 ml/pellet from 1 liter of culture) and subsequently lysed by sonication (cycle of 30 × 0.5 s pulses at an amplitude of 70% per pellet from 1 liter of culture) using a Branson Digital 450 Sonifer (Branson Ultrasonics, Danbury, CT). Cell lysates were clarified by centrifugation (40 min, 40,000 × g, 4 °C), filtered through a 0.45-μm syringe filter (SARSTEDT, Nümbrecht, Germany), and applied to a glutathione-Sepharose 4 Fast Flow (GE Healthcare) column (bed volume 5 ml) equilibrated with PBS at 4 °C. Recombinant proteins were eluted using 50 mm Tris-HCl, pH 8.0, supplemented with fresh 10 mm reduced glutathione. Alternatively, 10 ml of PBS containing 100 μl of PreScission protease stock solution (1 units/ml) was applied to the column and incubated for 40 h at 4 °C. Protein concentration was determined by measurement of absorbance at 280 nm using a Nanodrop device. Purity of proteins was determined by SDS-PAGE. Optionally, proteins were also purified by size exclusion chromatography. The protein sample was concentrated to 2 ml using protein concentrators, 9000 molecular weight cutoff, 7 ml (Thermo Scientific Fisher), and resolved by size exclusion chromatography on HiLoad 16/600 Superdex 75 pg column (GE Healthcare) using an AKTA purifier 900 FPLC system (GE Healthcare) at a flow rate of 1 ml/min in 20 mm Tris, 150 mm NaCl, pH 8.0. The elution profile was monitored at 280 nm, and 1 ml fractions were collected. Samples containing the desired protein were pooled.
Screening Miropin Inhibitory Activity
All reactions were performed in 0.1 m Tris, 150 mm NaCl, 5 mm EDTA, 0.02% Tween 20, pH 7.6, with the exception of trypsin and subtilisin, which were assayed in 0.1 m Tris, 150 mm NaCl, 5 mm CaCl2, 0.02% Tween 20, pH 7.6, and thrombin, which was assayed in 0.1 m Tris, 150 mm NaCl, 5 mm CaCl2, pH 7.6. Stock solutions of substrates were prepared in DMSO and stored at −20 °C. Each protease was mixed with a 10-fold molar excess of miropin in assay buffer in a total volume of 100 μl in microtitration plates (Nunc, Roskilde, Denmark) with clear or black bottoms for chromogenic and fluorogenic substrates, respectively, and incubated for 15 min at 37 °C followed by the addition of 100 μl of substrate solution. Residual activity was monitored for 30 min at 37 °C using a SpectraMAX microplate reader (Molecular Devices, Sunnyvale, CA) for pNA substrates (absorbance measured at 410 nm) and SpectraMax Gmini XS (Molecular Devices) for Boc-Val-Pro-Arg-aminofluoromethylcoumarin (excitation = 395 nm, emission = 500 nm). The final concentration of enzyme was 1 nm for thrombin and subtilisin and 25 nm for all other enzymes. The following substrates were used: succinyl-Ala-Ala-Pro-Phe-pNA (final concentration, 500 μm) for cathepsin G, chymotrypsin, and subtilisin; methoxysuccinyl-Ala-Ala-Pro-Val-pNA (500 μm) for neutrophil and pancreatic elastases; benzoyl-Arg-pNA (500 μm) for trypsin; Boc-Val-Pro-Arg-aminofluoromethylcoumarin (40 μm) for thrombin.
Stoichiometry of Inhibition
The number of molecules of miropin needed to inhibit one molecule of target protease (stoichiometry of inhibition (SI)) was determined by incubating constant amounts of proteases with increasing concentrations of miropin and measuring the residual enzyme activity. Briefly, proteases at concentrations of 50 nm (cathepsin G, pancreatic elastase), 10 nm (neutrophil elastase, trypsin), or 1 nm (subtilisin) were mixed with increasing concentrations of miropin in a microtitration plate (total volume 100 μl) to yield molar ratios of enzyme:inhibitor ranging from 0 to 5. After 15 or 45 min of incubation (cathepsin G, pancreatic elastase) at 37 °C, 100 μl of substrate solution was added, and enzymatic hydrolysis of substrates was monitored for 30 min at 37 °C at 410 nm using a SpectraMAX microplate reader. Residual activity was plotted as a function of the molar ratio of miropin:protease. The SI was considered to be the value where the linear curve fitted to the data points crossed the x axis.
SDS-PAGE Analysis of the Interaction between Miropin and Target Proteases
For detection of covalent protease-miropin complexes, increasing concentrations of proteases (0–4 μm) were incubated with 2 μm miropin for 30 min at 37 °C in a total volume of 20 μl. The reaction was stopped by the addition of 20 μl of boiling reducing SDS-PAGE sample buffer followed by incubation for 5 min at 100 °C. Samples were resolved by 10% SDS-PAGE (acrylamide/bis-acrylamide ratio 33:1) using a Tris-HCl/Tricine buffer system (26). Gels were stained with 0.1% Coomassie Brilliant Blue R-250 in 10% acetic acid, destained in 30% methanol, 10% acetic acid, and stored in 1% acetic acid. After washing gels in distilled water, protein bands of interest were excised and subjected to mass spectroscopy. First, they were washed in water and submerged in 50% H2O, 50% acetonitrile for 15 min. After the addition of 50 μl acetonitrile, samples were incubated until the gel pieces shrunk in size. At this point the gel slices were submerged in 50 μl of 0.1 m NH4HCO3 containing trypsin, and proteins were digested overnight at 37 °C. Peptides were extracted and analyzed by NanoLC-MS/MS using an EASY-nLC II system (ThermoScientific) connected to a TripleTOF 5600 mass spectrometer (AB SCIEX; Framingham, MA). Peptides were dissolved in 5% formic acid and then injected, trapped, and desalted on a Biosphere C18 column (5 μm, 2 cm × 100 μm inner diameter; Nano Separations). Next, peptides eluted from a trap column were separated on a 15-cm analytical column (75 μm inner diameter) packed in-house in a fritted silica tip (New Objectives; Woburn, MA) with RP ReproSil-Pur C18-AQ 3 μm resin (Dr. Marisch GmbH, Ammerbuch-Entringen, Germany) and connected in-line to the mass spectrometer. Peptides were eluted at a flow rate of 250 nl/min using a 50-min gradient of 5% to 35% phase B (0.1% formic acid and 90% acetonitrile). Mass spectrometry files were converted to Mascot generic format (MGF) using the AB SCIEX MS Data Converter beta 1.1 (AB SCIEX) and “ProteinPilot MGF” parameters. The generated peak lists were searched against the Swiss-Prot database using an in-house Mascot search engine (matrix science). Search parameters used for protein identification included trypsin, two missed cleavages, propionamide (Cys) as a fixed modification, and oxidation (Met) as variable modification. Peptide tolerances were set to 10 ppm and MS/MS tolerance to 0.6 Da.
Determination of Cleavage Sites in Reactive Center Loop (RCL) of Miropin
Miropin (8 μm) and target proteases at different enzyme/inhibitor ratios (0.1875 (subtilisin), 0.25 (cathepsin G, neutrophil elastase), and 0.375 (pancreatic elastase, trypsin)) were mixed in a total volume of 20 μl and incubated for 30 min at 37 °C. The reaction was stopped by the addition of 20 μl of boiling reducing SDS-PAGE sample buffer followed by incubation for 5 min at 100 °C. Proteins were resolved by 18% SDS-PAGE (acrylamide/bis-acrylamide ratio 33:1) using a Tris-HCl/Tricine buffer system (26) and electrotransferred onto a PVDF membrane (Bio-Rad) in 10 mm CAPS, pH 11, and 10% methanol in a Semi-dry Transfer Cell (Bio-Rad). Membranes were stained with 0.1% Coomassie Brilliant Blue G-250 in 40% methanol, 1% acetic acid and destained in 50% methanol, 10% acetic acid. Peptide bands (molecular mass ∼4.5 kDa) were excised and analyzed by automated Edman degradation using a Procise 494HT amino acid sequencer (Applied Biosystems).
Kinetics of Inhibition
The kinetic parameters of inhibition of target proteases by miropin were determined by the progress curve method (27). Mixtures containing constant concentrations of substrate and increasing concentrations of miropin in a total volume of 100 μl were prepared in microtitration plates. Next, 100 μl of protease solutions were added, and the rate of substrate hydrolysis was recorded. The final concentrations of proteases (E), miropin, and substrates are listed below: cathepsin G ([E] = 1 nm; [miropin] = 0–120 nm; [7-methoxycoumarin-4-acetic acid (MCA)-FVT-Gnf-SW-Anb-NH2] = 15 μm; Anb is the amide of amino benzoic acid; Gnf is 4-guanidine-phenylalanine); neutrophil elastase ([E] = 1 nm; [Miropin] = 0–200 nm; [metoxysuccinyl-AAPV-pNA] = 250 μm); pancreatic elastase ([E] = 1 nm; [Miropin] = 0–300 nm; [metoxysuccinyl-AAPV-pNA] = 1 mm); subtilisin: ([E] = 0.05 nm; [miropin] = 0–15 nm; [Suc-AAPF-pNA] = 1 mm); trypsin: ([E] = 0.1 mm; [miropin] = 0–30 nm; [Boc-QAR-7-amino-4-methylcoumarin (AMC)] = 20 μm).
The pseudo-first-order association rate constant, kobs, was determined by nonlinear regression fitting of the progress curve using Equation 1 (27):
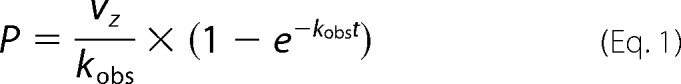 |
where P is the amount of product formation, vz is the initial velocity, and t is the reaction time. Kobs values determined for each concentration of miropin were plotted against miropin concentration. The slope of the fitted linear curve was equal to the apparent association constant k′. Taking into account that the serpin competes with a substrate for an active site of a protease, and the reaction velocity is affected by the enzyme-SI, the k′ was corrected using Equation 2 (27), which enables calculation of the second-order association rate constant (kass):
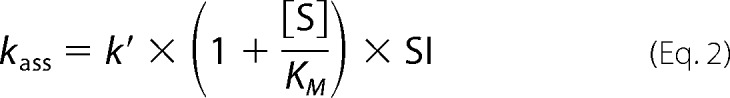 |
where Kmis the Michaelis-Menten constant determined from hyperbolic fit using GraphPad Prism macro. The calculated values of Km were as follows: 3.8 μm (cathepsin G), 114 μm (neutrophil elastase), 3.7 mm (pancreatic elastase), 465 μm (subtilisin), and 10.5 μm (trypsin).
RESULTS
Analysis of Primary Structure, Expression, Purification, and Determination of the Protease Inhibitory Activity of Miropin
Despite low levels of overall amino acid sequence identity, the tertiary structure of the inhibitory core of serpins is highly conserved and composed of three β-sheets (A-C), eight to nine α-helices, and an exposed RCL (1). These structural and sequence motifs are clearly present in miropin (Fig. 1A), in which expression by T. forsythia was confirmed by real time PCR (data not shown). Furthermore, high conservation of residues in the hinge region of the RCL predicts that this newly identified serpin should possess protease inhibitory activity. The Thr residue at the predicted P1 position together with the presence of a typical signal peptide for lipoproteins (underlined sequence in Fig. 1B) (28) suggests that miropin is the membrane-attached secretory protein and neutrophil elastase its likely target. To investigate this possibility we have preincubated elastase with increasing amounts of T. forsythia cell suspension and assay the residual activity of the enzyme, which was reduced in the concentration-dependent manner (Fig. 1C). Furthermore, comparative analysis of elastase inhibition by suspension of T. forsythia cells (WC), bacterial cell crude homogenate (CH), cell envelope (CE), and by a fraction of soluble cytoplasm/periplasm-derived proteins (C/P) revealed the highest inhibitory activity in intact bacterial cells and cell homogenate (Fig. 1C, inset). The lower level of the inhibitory activity was found in cytoplasm/periplasm and cell envelope fractions. Together these results argue that miropin is exposed to extracellular environment on the bacterial cell surface with a fraction also present in the periplasm. Such a location is supported by the predicted lipoprotein nature of miropin. Of note, no elastase inhibition was observed in any fraction of an isogenic TF0781 knock out mutant of T. forsythia.3
FIGURE 1.

T. forsythia open reading frame ID TF0781 (Oralgen) encodes an inhibitory active serpin. A, multisequence alignment of the inhibitory core of T. forsythia serpin with squamous cell carcinoma antigen 1, SCCA1 (P29508), human α1-antitrypsin (UniProt accession number P01009) and B. longum serpin (UniProt accession number Q8G7X7). The positions P1-P1′ are framed. B, possible translation initiation sites of the serpin from T. forsythia. The T. forsythia serpin locus TF0781 was resequenced together with the 5′ and 3′ flanking regions, and putative sites of initiation of translation (Tfs62, TFs55, and TFs46) according to a non-classical mechanism operating in the Bacteroidetes phylum were determined. The underline sequence in TFs46 was predicted using LipoP 1.0 Server to represent a signal peptide typical for lipoproteins. C, neutrophil elastase (NE) was preincubated with increasing amounts of T. forsythia cells suspension. The residual enzyme activity was determined and plotted against the volume of suspension (average of two independent assays). The inset shows NE inhibition by washed T. forsythia cells (WC), bacterial cells homogenate (CH), soluble cytoplasm/periplasm proteins (C/P), and cell envelope (CE) subcellular fractions standardized to the same volume of the initial culture. The mean value from two independent experiments is presented. D, three putative variants of serpin were cloned into the pGex-6P-1 vector, expressed as fusion proteins with GST in E. coli, and purified by affinity chromatography on glutathione-Sepharose. The expressed proteins were analyzed by SDS-PAGE. E, inhibitory activity of the recombinant proteins was determined using neutrophil elastase as the target protease.
To further confirm the inhibitory activity of miropin, three variants of the protein differing in the lengths of their N-terminal extensions (predicted from the three putative translation initiation sites) were expressed (Fig. 1B). The variants were obtained as fusion proteins with GST, purified on glutathione-Sepharose (Fig. 1D), and analyzed for inhibitory activity against human neutrophil elastase. This analysis firmly confirmed that miropin is an elastase inhibitor. Because the shortest variant of the protein (Tfs46) was expressed with the highest yield and greatest specific inhibitory activity (Fig. 1E), it was used to purify native miropin. GST was removed from the fusion protein by in-column digestion of glutathione-Sepharose bound GST-Tsf46 with PreScission protease (Fig. 2A), and the eluted serpin was subjected to size exclusion chromatography. The majority of miropin was found together with contaminating proteins in the void volume of the column (peak 1), whereas homogenous serpin eluted at the volume expected of a 40–50-kDa protein (peak 2) (Fig. 2, B and C). The total yield of purified miropin was 25 μg per 1 liter of E. coli culture.
FIGURE 2.

Expression and purification of recombinant miropin. A, SDS-PAGE of E. coli extracts and tag-free miropin after purification on glutathione-Sepharose and in-column digestion with the PreScission protease. Lane 1, E. coli extracts before the addition of isopropyl-1-thio-β-d-galactopyranoside; lane 2, E. coli extracts at 6 h after isopropyl-1-thio-β-d-galactopyranoside-induced protein expression; lane 3, tag-free miropin (46.1 kDa). B, profile of gel filtration chromatography of miropin on HiLoad 16/60 Superdex 75 pg column. mAU, milliabsorbance units. C, SDS-PAGE of two miropin fractions resolved in B: lane 1, miropin before gel filtration; lane 2, peak 1; lane 3, peak 2. The presence of miropin in the major band was confirmed by N-terminal sequence analysis. D, inhibitory activity against human neutrophil elastase (NE) (0.1 μg) determined in the different gel filtration fractions.
The presence of the serpin in peak 1 suggests that the protein was present as an aggregate. Nevertheless, activity assays revealed that this form of miropin retained some inhibitory activity, albeit lower than that of the monomeric form (Fig. 2D). This suggests that miropin aggregates must differ from other serpin complexes, which lack protease inhibitory activity (29).
Because monomeric miropin aggregation and/or polymerization would affect stoichiometry of inhibition, we have investigated stability of the monomer. To this end miropin stored at −20 °C and +4 °C for 6 months and 4 weeks, respectively, or incubated at 37 °C for 6 h was subjected to gel filtration chromatography and found exclusively in the monomeric form. In addition such treatment did not change the inhibitory capacity of the purified protein, clearly indicating that miropin is stable as the monomer (data not shown).
Specificity Spectrum and Stoichiometry of Inhibition of Proteases by Miropin
The chemical characteristics of the amino acid at the P1 position dictates the inhibitory spectrum of a serpin (30, 31) and is only very occasionally conferred by adjacent residues (32). From an alignment of the primary structure of miropin with those of well characterized serpins, we predicted that miropin would inhibit neutrophil elastase activity. To further characterize the inhibitory spectrum of miropin, we screened its inhibitory activity against six other serine proteases including cathepsin G, porcine pancreatic elastase, thrombin, trypsin, chymotrypsin, and the bacterial-derived protease subtilisin Carlsberg. Except for thrombin and chymotrypsin, whose activity was not affected by miropin even at a 10-fold molar excess, all other proteases were inhibited in a concentration-dependent manner. This allowed titration of the activity of five different proteases with miropin and determination of the SI. Surprisingly, all proteases, regardless of their specificity, were inhibited with a similar SI that ranged from 2.8 for trypsin to 3.4 for elastases (Fig. 3).
FIGURE 3.

Determination of the SI of serine proteases by miropin. A, cathepsin G. B, human neutrophil elastase. C, porcine pancreatic elastase. D, subtilisin Carlsberg. E, trypsin. Each protease was preincubated with increasing concentrations of miropin for 15 min, and residual enzymatic activity was determined and plotted against the serpin:enzyme molar ratio (I0/E0). Activity of enzymes in the absence of inhibitor was considered as 100%. The results presented are the mean ± S.D. from three experiments.
As inhibition of proteases by serpins is irreversible, the most important kinetic parameter characterizing inhibitory complex formation and efficiency of inhibition is the association rate constant (kass). Therefore, the second-order association rate constants for all target proteases were determined by progress curve analysis (Fig. 4). The kass values obtained varied >1 order of magnitude, from 2.7 ± 0.4 × 104 m−1s−1 for cathepsin G to 7.1 ± 0.3 × 105 m−1s−1 for subtilisin. These results show that miropin interacts with proteases with different association rate constants; miropin more effectively inhibited neutrophil elastase, trypsin, and subtilisin than cathepsin G and pancreatic elastase.
FIGURE 4.

Progress curve analysis of protease inhibition by miropin. A, cathepsin G (1 nm). B, human neutrophil elastase (1 nm). C, porcine pancreatic elastase (1 nm). D, subtilisin Carlsberg (0.05 nm). E, trypsin (0.1 nm). Proteases were added to mixtures containing a constant amount of substrate and increasing concentrations of miropin. Changes in fluorescence (RFU) or absorbance (Abs) were then recorded. The number associated with each progress curve (insets) represents the concentration of miropin (nm). The values of Kobs were plotted as a function of miropin concentration; kass was determined from the slope of the fitted linear curve to the data points and corrected for the stoichiometry factor. With the exception of cathepsin G, the mean ± S.D. from three experiments is presented.
Miropin Forms a Covalent Complex with Target Proteases
A covalent inhibitory complex with serpins is formed by an ester bond between the C-terminal carbonyl group of the P1 residue and the hydroxyl group of the active site serine residue of the target protease (33). Such a complex is resistant to denaturation. To determine if miropin interacts in the same manner, a constant amount of miropin was incubated with increasing amounts of proteases, the reaction was stopped by boiling in reducing sample buffer, and the mixture was separated by SDS-PAGE. This analysis revealed the presence of a characteristic pattern of bands. With increasing concentrations of protease, the intensity of the band corresponding to miropin (∼46 kDa) decreased with the concomitant appearance of bands with slightly lower (corresponding to RCL-cleaved serpin) and higher molecular weights (Fig. 5). In all cases mass spectroscopy analysis of bands in the range from 50 to 70 kDa revealed the presence of both the protease and miropin (Table 1), indicating that these bands represent covalent protease-inhibitor complexes. Furthermore, the lower than expected molecular mass of the complexes indicates partial degradation of the protease in the complex as was observed previously for the interaction between human serpins and target proteases (1). Interestingly, miropin were also found in 120- and >170-kDa bands that appeared after the interaction between miropin and either cathepsin G (Fig. 5A) or subtilisin (Fig. 5D), respectively. Because the molecular mass was much higher than the theoretical mass of the complex, this suggests that these bands contain multimers of miropin that cannot be dissociated by boiling in SDS-PAGE sample buffer as observed previously for other serpins (34). Such dimers/trimers or higher multimers must contain a serpin molecule covalently attached to one end of the protease. Finally, multimerization of miropin induced by cathepsin G was also observed without miropin-cathepsin G complex formation (band 1, Fig. 5A).
FIGURE 5.

Detection of stable covalent complexes of miropin with serine proteases. A–E, SDS-PAGE of miropin incubated with increasing concentrations of serine proteases at a molar ratio ranging from 0.25–2 as indicated. Little amounts of complexes of miropin with target proteases, especially in case of subtilisin (D) and trypsin (E) result from the variability in the SDS stability of the various complexes and/or different susceptibility of the covalent complexes to hydrolysis by non-inhibited proteases during SDS-PAGE sample preparation. F, effect of NaOH on the stability of the miropin-human neutrophil elastase complex. Cat G, cathepsin G; NE, neutrophil elastase; PE, pancreatic elastase.
TABLE 1.
Results from mass spectroscopy analysis of putative complexes of miropin with target proteases
The table shows the names of matched proteins, number of distinct peptide fragments identified after the tryptic digestion, and protein score. The protein score is the best total MS/MS score obtained for the indicated protein by LC-MALDI of SDS-PAGE bands.
| Band | Protein ID by MS-MS | Number of peptides matched | Sequence coverage (%) | Protein score |
|---|---|---|---|---|
| 1 | Miropin (T. forsythia serpin) | 6 | 38 | 3181 |
| 2 | Miropin (T. forsythia serpin) | 7 | 35 | 2798 |
| Cathepsin G (Homo sapiens) | 3 | 13 | 92 | |
| 3 | Miropin (T. forsythia serpin) | 6 | 18 | 694 |
| Cathepsin G (H. sapiens) | 2 | 6 | 155 | |
| 4 | Miropin (T. forsythia serpin) | 6 | 37 | 2080 |
| Cathepsin G (H. sapiens) | 5 | 21 | 1049 | |
| 5 | Miropin (T. forsythia serpin) | 6 | 36 | 2842 |
| Neutrophil Elastase (H. sapiens) | 8 | 27 | 1124 | |
| 6 | Miropin (T. forsythia serpin) | 6 | 18 | 1064 |
| Neutrophil Elastase (H. sapiens) | 4 | 14 | 429 | |
| 7 | Miropin (T. forsythia serpin) | 8 | 40 | 2847 |
| Pancreatic Elastase (Sus scrofa) | 6 | 50 | 1245 | |
| 8 | Miropin (T. forsythia serpin) | 8 | 38 | 2433 |
| Pancreatic Elastase (S. scrofa) | 6 | 74 | 2513 | |
| 9 | Miropin (T. forsythia serpin) | 7 | 34 | 2477 |
| Subtilisin Carlsberg (Bacillus licheniformis) | 4 | 31 | 1464 | |
| 10 | Miropin (T. forsythia serpin) | 7 | 30 | 1611 |
| Subtilisin Carlsberg (B. licheniformis) | 4 | 31 | 1619 | |
| 11 | Miropin (T. forsythia serpin) | 7 | 33 | 1653 |
| Subtilisin Carlsberg (B. licheniformis) | 4 | 27 | 637 | |
| 12 | Miropin (T. forsythia serpin) | 10 | 52 | 4876 |
| Trypsin (Bos taurus) | 6 | 50 | 1471 | |
| 13 | Miropin (T. forsythia serpin) | 6 | 40 | 2591 |
| Trypsin (B. taurus) | 6 | 51 | 1402 |
The ester bond responsible for the covalent complex between serpin and a protease can be hydrolyzed at alkaline pH (35). Therefore, to confirm that the same mechanism of protease inhibition is responsible for the complex between miropin and a protease, a reaction mixture containing serpin and neutrophil elastase was treated with NaOH before SDS-PAGE analysis. Indeed, after alkaline treatment, the covalent complexes were no longer present, and the free enzyme was released (Fig. 5F). Thus, miropin inhibits a broad array of target proteases through formation of ester bond-linked covalent complexes.
Inhibition of Proteases by Miropin Occurs at Multiple P1 Residues in the RCL
Proteolytic cleavage of the P1-P1′ peptide bond within the RCL is a prerequisite for covalent complex formation between a serpin and its target protease (36). Of note, non-targeted proteases can cleave the RCL without being trapped in the complex, thereby disabling the inhibitory potential of serpins (37, 38). In both cases N-terminal sequence analysis of the released peptide was performed to determine the cleavage site recognized by the protease. Toward this end, miropin was incubated with target proteases. Samples were then resolved by SDS-PAGE and transferred to PVDF membranes, and the N-terminal sequences of 4–5-kDa peptides were determined by Edman degradation (Fig. 6A). Trypsin, subtilisin, and cathepsin G all cleaved miropin at only one position within the RCL, at Lys-Thr, Glu-Met, and Lys-Thr, respectively (Fig. 6B). Interestingly, the cleavage catalyzed by the latter two proteases only marginally fitted their known specificities. Two other proteases, neutrophil and porcine pancreatic elastase, cleaved the RCL at two (Val-Thr and Val-Glu) and three non-overlapping positions (Thr-Ala, Val-Lys, and Thr-Ser), respectively (Fig. 6B). Neutrophil elastase cleavage sites at the S1 subsite were in agreement with the specificity of this protease. In contrast, pancreatic elastase cleaved after two residues (Thr and Val), which are not favored, whereas it ignored the Ala-Val peptide bond that fits perfectly with its specificity. Analysis of Edman degradation chromatograms (data not shown) showed that the released peptides were in equimolar concentrations. This suggests that cleavages at different sites of the RCL occurred at a similar rate. Of note, no other serpin-derived peptides, which would indicate miropin degradation by target proteases, were detected, ruling out a possibility that miropin was inactivated by proteolysis outside the RCL. In the context of inhibitory complex formation, it is interesting that cleavages were observed far upstream of the predicted reactive bond P1-P1′ (Thr-Ser), and only one cleavage was made at this site by pancreatic elastase.
FIGURE 6.

Identification of cleavage sites of miropin by target proteases. A, miropin was incubated with serine proteases, and the reaction was stopped by adding reducing sample buffer. Proteins were then resolved by SDS-PAGE and electrotransferred onto a PVDF membrane. The stained protein bands, labeled with frames as shown, were excised and subjected to N-terminal sequencing. B, sequences determined for each protease. Cat G, cathepsin G; NE, neutrophil elastase; PE, pancreatic elastase.
DISCUSSION
Because only a limited number of prokaryotic serpins have been thoroughly investigated at the biochemical and kinetic levels, the detailed characterization of another serpin could significantly broaden current knowledge about the inhibitory and physiological characteristics of bacterial serpins. Our findings indicate that miropin from T. forsythia is an inhibitory serpin that inhibits target proteases through formation of covalent complexes via an ester bond. The physiological efficacy of serpins as inhibitors is reflected in the kass of covalent complex formation. The kass values for the miropin range from 2.7 × 104 to 7.1 × 105 m−1s−1 and are comparable with those reported for other prokaryotic serpins: 8.4 × 104 m−1s−1 for thermopin-chymotrypsin (6), 4.7 × 104 m−1s−1 for neutrophil elastase-serpin from B. longum (9), 1.4 × 105 m−1s−1 for tengpin-elastase (10), 1.8 × 105 m−1s−1 for aeropin-chymotrypsin (11), and 7.2 × 105 for subtilisin Carlsberg-Tk-serpin (12). These rates of inhibition are similar to those of intracellular human serpins, which react with their target proteases with kass values in the range of 105-106 m−1s−1 (39, 40). Taken together, it can be assumed that the kass values determined for miropin are sufficient to inhibit target proteases in vivo.
Another parameter that describes the efficiency of serpins as inhibitors is the SI, which represents the number of serpin molecules required to inhibit one molecule of a target protease. For the majority of prokaryotic and human serpins, the SI is ∼1. In sharp contrast, miropin inhibited all target proteases tested with an SI close to three, which may suggest that miropin is a less effective inhibitor. However, it should be kept in mind that miropin efficiently inhibits a broad range of proteases with vastly different specificities.
The ability of miropin to inhibit a wide variety of proteases is apparently due to the use of different peptide bonds within the RCL. This is a very unusual feature, as all other inhibitory serpins characterized to date, with few exceptions (41–44), use a single polypeptide bond (P1-P1′) to trap a protease in a covalent complex. Of note, the RCL in miropin has the same length as the vast majority of eukaryotic and prokaryotic serpins (Figs. 1 and 7). In serpins, cleavage outside the reactive site P1-P1′ bond leads to the S (stressed) → R (relaxed) transition involving a large scale structural rearrangement that allows for accommodation of the RCL as the extra β-strand into the large β-sheet A in the center of the molecule. Cleaved serpin has no inhibitory activity. Apparently, miropin is a remarkable exception to the serpin family as both subtilisin and neutrophil elastase are inhibited at the P5-P4 (Glu-Met) and P6-P5 (Val-Glu) peptide bonds, respectively (Fig. 6B). On the other hand, both cathepsin G and trypsin are inhibited at the P2-P1 (Lys-Thr) site, and the only protease that is inhibited at the predicted P1-P1′ (Thr-Ser) site is pancreatic elastase. This enzyme is also likely to be inhibited after attacking the P3-P2 (Val-Lys) peptide bond. The role of cleavage of the P9-P8 (Val-Thr) and P8-P7 (Thr-Ala) sites in formation of the inhibitory complex seems questionable because these peptide bonds are proximal to the hinge region (Figs. 6B and 7). Unfortunately, we were unable to assess the kinetics of cleavage of individual peptide bonds to address the possibility of elastase inhibition at these sites. Nevertheless, the fact that all target proteases, regardless of the number of cleavages identified within the RCL, are inhibited with nearly the same SI suggests that all cleavage sites identified may result in both partial inhibition (30%) and partial (70%) inactivation of miropin.
FIGURE 7.

Multisequence alignment of RCL sequences of miropin, putative inhibitory serpins found in genomes of commensal bacterial species of the Bacteroidetes phylum and α-1-antitrypsin. All the sequences were downloaded from MERROPS database and derived from sequenced genomes of following bacteria: 1, T. forsythia (miropin); 2, Prevotella copri; 3, P. stercorea; 4, B. uniformis; 5, Bacteroides cellulosilyticus; 6, B. ovatus; 7, Bacteroides coprophilus; 8, Porphyromonas asaccharolytica; 9, Bacteroides finegoldii; 10, P. buccae; 11, Prevotella ruminicola; 12, Prevotella oralis; 13, Prevotella buccalis; 14, Prevotella timonensis; 15, Prevotella bergensis; 16, Prevotella sp_oral taxon_306; 17, Prevotella veroralis, 18, Prevotella sp_oral taxon_317. The sequence of α-1-antitrypsin (19) is from UniProt (accession number P01009). Amino acids at each position identical to those of miropin (1) are highlighted. The predicted active sites, P1-P1′, are framed.
The utilization of sites close to the hinge region of the RCL to form a covalent protease-serpin complex is of substantial interest in the context of the mechanism of inhibition exerted by serpins in which the inhibitor functions as a “conformational trap” (36). Briefly, RCL cleavage leads to translocation of acyl-bound protease by about 75 Å from the top to the bottom of the serpin molecule. This translocation is thermodynamically driven, with the energy derived from RCL insertion and the dramatically enhanced stability of cleaved serpin. The stability of the resulting complex is favored by the large distortion in protease structure (1). It is possible that, in the case of miropin, insertion of a much shorter stretch of the RCL into β-sheet A is sufficient to pull down the protease, at least to the side of the miropin molecule, and distort its structure so that a stable, covalent complex is formed. Such an adaptation would allow the use of different P1-P1′ sites in the RCL and greatly expand the spectrum of proteases that can be inhibited. However, this occurs at the expense of an increased stoichiometry of inhibition and relatively low kass.
The ability to inhibit many proteases with different substrate specificities using a single serpin may be especially advantageous for bacteria living in crowded microbiomes, where they are exposed to proteases produced by other bacteria and released by the host. In this context, it is worthy to note that although serpins are rare among bacteria, they are relatively abundant in commensal species of Bacteroides and Prevotella (phylum Bacteroidetes), which are part of diversified bacterial flora in animals found in both the gastrointestinal tract and dental surfaces (dental plaque). Alignment of the primary sequences of putative serpins from many species revealed that there is high homology in 51 highly conserved residues (data not shown) and that nearly all contain an alanine (or similarly small R group) repeat motif in the so-called hinge region, at position P17-P9 (Fig. 7). This motif is characteristic of all inhibitory serpins. Accordingly, Bacteroidetes serpins are expected to contain a serpin fold and act as protease inhibitors, which may protect bacteria against proteases such as trypsin, pancreatic elastase, chymotrypsin, and neutrophil elastase. It remains to be determined if Bacteroidetes serpins can inhibit a number of proteases using different sites in the RCL.
Because T. forsythia also belongs to the Bacteroidetes phylum, it is not surprising that miropin shares a high degree of identity with a putative inhibitory serpin from B. uniformis (43%) and P. buccae (37%) but very little similarity to serpins produced by other prokaryotes, including that from the intestinal bacterium B. longum (24%). In this context it is interesting to note that miropin is fairly closely related to human intracellular serpin SCCA1 (36% identity). This suggests that Bacteroidetes serpins and other bacterial serpins do not share the same ancestor. In this regard it is tempting to speculate that their unique properties were acquired through a very rare event involving xenogenous horizontal gene transfer from humans or other animals during their long, intimate coexistence. Such a possibility has been proposed for serpins (5, 45), and one such example has already been demonstrated for the T. forsythia metalloprotease karilysin (46).
In contrast to the prokaryotic serpins identified to date, miropin is the first one to be derived from a microbial pathogen. Therefore, the contribution of miropin to virulence merits consideration as inhibition of neutrophil serine proteases by miropin may attenuate the bactericidal activity of neutrophils and facilitate survival of T. forsythia. This hypothesis is strongly supported by the fact that miropin is predicted to be a secretory lipoprotein, which is likely associated with the outer membrane. A similar function has been described for the canonical, non-covalent protease inhibitor ecotin located in the periplasm of E. coli (47). Alternatively, the main function of miropin could be related to control of the activity of the bacteria's own proteases. This hypothesis is supported by the presence of a significant number of ORFs encoding putative chymotrypsin-like (family S1) and subtilisin-like (family S8) proteases in the T. forsythia genome. In this regard the ability of miropin to inhibit a broad spectrum of proteases from different families and with divergent specificity would be essential.
In conclusion, miropin is a unique serpin that is most closely related to human SCCA1. At the expense of high SI and relatively low kass, miropin has acquired the ability to inhibit efficiently an unusually broad range of proteases by using different sites in the RCL. This evolution was probably driven by adaptation to the biological niche of subgingival bacterial biofilm on the dental surface, which is rich in serine proteases originating from other bacteria and the host. In such an environment, miropin may function as an essential housekeeping protein and/or an important virulence factor. Elucidation of the pathophysiological role of miropin is the subject of ongoing investigations.
This work was supported, in whole or in part, by National Institutes of Health Grants DE 09761 and DE 022597). This work was also supported by Grants 2013/08/T/NZ1/00315 (a doctoral scholarship) and 2011/03/N/NZ1/00586 from the National Science Center (NCN, Krakow, Poland; to M. K.) and by grants from the NCN (2012/04/A/NZ1/00051), European Commission (FP7-HEALTH-2010-261460 “Gums and Joints,” FP7-PEOPLE-2011-ITN-290246 “RAPID,” and FP7-HEALTH-F3–2012-306029 “TRIGGER”), and Polish Ministry of Science and Higher Education (project 137/7.PR-EU/2011/2; to J. P.). The Faculty of Biochemistry, Biophysics, and Biotechnology of the Jagiellonian University is a beneficiary of structural funds from the European Union (POIG.02.01.00-12-064/08).
M. Ksiazek, D. Mizgalska, J. J. Enghild, C. Scavenius, I. B. Thogersen, and J. Potempa, manuscript in preparation.
- pNA
- p-nitroanilide
- SI
- stoichiometry of inhibition
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- RCL
- reactive center loop
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- Boc
- t-butoxycarbonyl.
REFERENCES
- 1. Gettins P. G. (2002) Serpin structure, mechanism, and function. Chem. Rev. 102, 4751–4804 [DOI] [PubMed] [Google Scholar]
- 2. Kantyka T., Plaza K., Koziel J., Florczyk D., Stennicke H. R., Thogersen I. B., Enghild J. J., Silverman G. A., Pak S. C., Potempa J. (2011) Inhibition of Staphylococcus aureus cysteine proteases by human serpin potentially limits staphylococcal virulence. Biol. Chem. 392, 483–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Silverman G. A., Whisstock J. C., Bottomley S. P., Huntington J. A., Kaiserman D., Luke C. J., Pak S. C., Reichhart J. M., Bird P. I. (2010) Serpins flex their muscle: I. Putting the clamps on proteolysis in diverse biological systems. J. Biol. Chem. 285, 24299–24305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Irving J. A., Steenbakkers P. J., Lesk A. M., Op den Camp H. J., Pike R. N., Whisstock J. C. (2002) Serpins in prokaryotes. Mol. Biol. Evol. 19, 1881–1890 [DOI] [PubMed] [Google Scholar]
- 5. Roberts T. H., Hejgaard J., Saunders N. F., Cavicchioli R., Curmi P. M. (2004) Serpins in unicellular Eukarya, Archaea, and Bacteria: sequence analysis and evolution. J. Mol. Evol. 59, 437–447 [DOI] [PubMed] [Google Scholar]
- 6. Irving J. A., Cabrita L. D., Rossjohn J., Pike R. N., Bottomley S. P., Whisstock J. C. (2003) The 1.5 Å crystal structure of a prokaryote serpin: controlling conformational change in a heated environment. Structure 11, 387–397 [DOI] [PubMed] [Google Scholar]
- 7. Kantyka T., Rawlings N. D., Potempa J. (2010) Prokaryote-derived protein inhibitors of peptidases: A sketchy occurrence and mostly unknown function. Biochimie 92, 1644–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kang S., Barak Y., Lamed R., Bayer E. A., Morrison M. (2006) The functional repertoire of prokaryote cellulosomes includes the serpin superfamily of serine proteinase inhibitors. Mol. Microbiol. 60, 1344–1354 [DOI] [PubMed] [Google Scholar]
- 9. Ivanov D., Emonet C., Foata F., Affolter M., Delley M., Fisseha M., Blum-Sperisen S., Kochhar S., Arigoni F. (2006) A serpin from the gut bacterium Bifidobacterium longum inhibits eukaryotic elastase-like serine proteases. J. Biol. Chem. 281, 17246–17252 [DOI] [PubMed] [Google Scholar]
- 10. Zhang Q., Buckle A. M., Law R. H., Pearce M. C., Cabrita L. D., Lloyd G. J., Irving J. A., Smith A. I., Ruzyla K., Rossjohn J., Bottomley S. P., Whisstock J. C. (2007) The N terminus of the serpin, tengpin, functions to trap the metastable native state. EMBO Rep. 8, 658–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cabrita L. D., Irving J. A., Pearce M. C., Whisstock J. C., Bottomley S. P. (2007) Aeropin from the extremophile Pyrobaculum aerophilum bypasses the serpin misfolding trap. J. Biol. Chem. 282, 26802–26809 [DOI] [PubMed] [Google Scholar]
- 12. Tanaka S., Koga Y., Takano K., Kanaya S. (2011) Inhibition of chymotrypsin- and subtilisin-like serine proteases with Tk-serpin from hyperthermophilic archaeon Thermococcus kodakaraensis. Biochim. Biophys. Acta 1814, 299–307 [DOI] [PubMed] [Google Scholar]
- 13. Socransky S. S., Haffajee A. D., Cugini M. A., Smith C., Kent R. L., Jr. (1998) Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25, 134–144 [DOI] [PubMed] [Google Scholar]
- 14. Darveau R. P. (2010) Periodontitis: a polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 8, 481–490 [DOI] [PubMed] [Google Scholar]
- 15. Fox C. H. (1992) New considerations in the prevalence of periodontal disease. Curr. Opin. Dent. 2, 5–11 [PubMed] [Google Scholar]
- 16. Flemmig T. F. (1999) Periodontitis. Ann. Periodontol. 4, 32–38 [DOI] [PubMed] [Google Scholar]
- 17. Garcia R. I., Nunn M. E., Vokonas P. S. (2001) Epidemiologic associations between periodontal disease and chronic obstructive pulmonary disease. Ann. Periodontol. 6, 71–77 [DOI] [PubMed] [Google Scholar]
- 18. Offenbacher S., Katz V., Fertik G., Collins J., Boyd D., Maynor G., McKaig R., Beck J. (1996) Periodontal infection as a possible risk factor for preterm low birth weight. J. Periodontol. 67, 1103–1113 [DOI] [PubMed] [Google Scholar]
- 19. Wang C. Y., Wang H. C., Li J. M., Wang J. Y., Yang K. C., Ho Y. K., Lin P. Y., Lee L. N., Yu C. J., Yang P. C., Hsueh P. R. (2010) Invasive infections of Aggregatibacter (Actinobacillus) actinomycetemcomitans. J. Microbiol. Immunol. Infect. 43, 491–497 [DOI] [PubMed] [Google Scholar]
- 20. Behle J. H., Papapanou P. N. (2006) Periodontal infections and atherosclerotic vascular disease: an update. Int. Dent. J. 56, 256–262 [DOI] [PubMed] [Google Scholar]
- 21. Pihlstrom B. L., Michalowicz B. S., Johnson N. W. (2005) Periodontal diseases. Lancet 366, 1809–1820 [DOI] [PubMed] [Google Scholar]
- 22. Wegner N., Wait R., Sroka A., Eick S., Nguyen K. A., Lundberg K., Kinloch A., Culshaw S., Potempa J., Venables P. J. (2010) Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and a-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 62, 2662–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mailhot J. M., Potempa J., Stein S. H., Travis J., Sterrett J. D., Hanes P. J., Russell C. M. (1998) A relationship between proteinase activity and clinical parameters in the treatment of periodontal disease. J. Clin. Periodontol. 25, 578–584 [DOI] [PubMed] [Google Scholar]
- 24. Belaaouaj A. (2002) Neutrophil elastase-mediated killing of bacteria: lessons from targeted mutagenesis. Microbes Infect. 4, 1259–1264 [DOI] [PubMed] [Google Scholar]
- 25. Chase T., Jr., Shaw E. (1967) p-Nitrophenyl-p′-guanidinobenzoate HCl: a new active site titrant for trypsin. Biochem. Biophys. Res. Commun. 29, 508–514 [DOI] [PubMed] [Google Scholar]
- 26. Schägger H., von Jagow G. (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 27. Morrison J. F., Walsh C. T. (1988) The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 61, 201–301 [DOI] [PubMed] [Google Scholar]
- 28. Juncker A. S., Willenbrock H., Von Heijne G., Brunak S., Nielsen H., Krogh A. (2003) Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 12, 1652–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mast A. E., Stadanlick J. E., Lockett J. M., Dietzen D. J. (1999) Solvent/detergent-treated plasma has decreased antitrypsin activity and absent antiplasmin activity. Blood 94, 3922–3927 [PubMed] [Google Scholar]
- 30. Owen M. C., Brennan S. O., Lewis J. H., Carrell R. W. (1983) Mutation of antitrypsin to antithrombin. α1-Antitrypsin Pittsburgh (358 Met leads to Arg), a fatal bleeding disorder. N. Engl. J. Med. 309, 694–698 [DOI] [PubMed] [Google Scholar]
- 31. Kaiserman D., Bird P. I. (2010) Control of granzymes by serpins. Cell Death Differ. 17, 586–595 [DOI] [PubMed] [Google Scholar]
- 32. Djie M. Z., Le Bonniec B. F., Hopkins P. C., Hipler K., Stone S. R. (1996) Role of the P2 residue in determining the specificity of serpins. Biochemistry 35, 11461–11469 [DOI] [PubMed] [Google Scholar]
- 33. Huntington J. A., Read R. J., Carrell R. W. (2000) Structure of a serpin-protease complex shows inhibition by deformation. Nature 407, 923–926 [DOI] [PubMed] [Google Scholar]
- 34. Stanley P., Serpell L. C., Stein P. E. (2006) Polymerization of human angiotensinogen: insights into its structural mechanism and functional significance. Biochem. J. 400, 169–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peter J., Unverzagt C., Hoesel W. (2000) Analysis of free prostate-specific antigen (PSA) after chemical release from the complex with α1-antichymotrypsin (PSA-ACT). Clin. Chem. 46, 474–482 [PubMed] [Google Scholar]
- 36. Huntington J. A., Carrell R. W. (2001) The serpins: nature's molecular mousetraps. Sci. Prog. 84, 125–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carrell R. W., Owen M. C. (1985) Plakalbumin, α1-antitrypsin, antithrombin, and the mechanism of inflammatory thrombosis. Nature 317, 730–732 [DOI] [PubMed] [Google Scholar]
- 38. Potempa J., Fedak D., Dubin A., Mast A., Travis J. (1991) Proteolytic inactivation of α-1-anti-chymotrypsin. Sites of cleavage and generation of chemotactic activity. J. Biol. Chem. 266, 21482–21487 [PubMed] [Google Scholar]
- 39. Schick C., Pemberton P. A., Shi G. P., Kamachi Y., Cataltepe S., Bartuski A. J., Gornstein E. R., Brömme D., Chapman H. A., Silverman G. A. (1998) Cross-class inhibition of the cysteine proteinases cathepsins K, L, and S by the serpin squamous cell carcinoma antigen 1: a kinetic analysis. Biochemistry 37, 5258–5266 [DOI] [PubMed] [Google Scholar]
- 40. Dahlen J. R., Jean F., Thomas G., Foster D. C., Kisiel W. (1998) Inhibition of soluble recombinant furin by human proteinase inhibitor 8. J. Biol. Chem. 273, 1851–1854 [DOI] [PubMed] [Google Scholar]
- 41. Potempa J., Shieh B. H., Travis J. (1988) α-2-antiplasmin: a serpin with two separate but overlapping reactive sites. Science 241, 699–700 [DOI] [PubMed] [Google Scholar]
- 42. Dahl S. W., Rasmussen S. K., Hejgaard J. (1996) Heterologous expression of three plant serpins with distinct inhibitory specificities. J. Biol. Chem. 271, 25083–25088 [DOI] [PubMed] [Google Scholar]
- 43. Riewald M., Schleef R. R. (1996) Human cytoplasmic antiproteinase neutralizes rapidly and efficiently chymotrypsin and trypsin-like proteases utilizing distinct reactive site residues. J. Biol. Chem. 271, 14526–14532 [DOI] [PubMed] [Google Scholar]
- 44. Ostergaard H., Rasmussen S. K., Roberts T. H., Hejgaard J. (2000) Inhibitory serpins from wheat grain with reactive centers resembling glutamine-rich repeats of prolamin storage proteins. Cloning and characterization of five major molecular forms. J. Biol. Chem. 275, 33272–33279 [DOI] [PubMed] [Google Scholar]
- 45. Irving J. A., Pike R. N., Lesk A. M., Whisstock J. C. (2000) Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res. 10, 1845–1864 [DOI] [PubMed] [Google Scholar]
- 46. Cerdà-Costa N., Guevara T., Karim A. Y., Ksiazek M., Nguyen K. A., Arolas J. L., Potempa J., Gomis-Rüth F. X. (2011) The structure of the catalytic domain of Tannerella forsythia karilysin reveals it is a bacterial xenologue of animal matrix metalloproteinases. Mol. Microbiol. 79, 119–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Eggers C. T., Murray I. A., Delmar V. A., Day A. G., Craik C. S. (2004) The periplasmic serine protease inhibitor ecotin protects bacteria against neutrophil elastase. Biochem. J. 379, 107–118 [DOI] [PMC free article] [PubMed] [Google Scholar]