

Background: Speculative chemical mechanism of methionine γ-lyase is formulated, kinetic and structural data concerning elementary stages of physiological reaction are mostly lacking.
Results: Pre-steady-state kinetic and structural analysis of the enzyme interaction with inhibitors was performed.
Conclusion: Results elucidate the mechanism of intermediate interconversion at initial stages of enzymatic reaction.
Significance: The data serve for understanding detailed mechanism of pyridoxal 5′-phosphate-dependent γ-elimination reaction.
Keywords: Enzyme Inhibitor, Enzyme Structure, Pre-steady-state Kinetics, Pyridoxal Phosphate, Substrate Specificity, Methionine γ-Lyase, Structure of Pyridoxal 5′-Phosphate-Cycloserine Derivative
Abstract
Methionine γ-lyase (MGL) catalyzes the γ-elimination of l-methionine and its derivatives as well as the β-elimination of l-cysteine and its analogs. These reactions yield α-keto acids and thiols. The mechanism of chemical conversion of amino acids includes numerous reaction intermediates. The detailed analysis of MGL interaction with glycine, l-alanine, l-norvaline, and l-cycloserine was performed by pre-steady-state stopped-flow kinetics. The structure of side chains of the amino acids is important both for their binding with enzyme and for the stability of the external aldimine and ketimine intermediates. X-ray structure of the MGL·l-cycloserine complex has been solved at 1.6 Å resolution. The structure models the ketimine intermediate of physiological reaction. The results elucidate the mechanisms of the intermediate interconversion at the stages of external aldimine and ketimine formation.
Introduction
Methionine γ-lyase (EC 4.4.1.11, MGL),3 a pyridoxal 5′-phosphate (PLP)-dependent enzyme, is found in many bacteria (1), including the Enterobacteriaceae family (Citrobacter freundii (2)) and in pathogenic bacteria (Bacteroides sp. (3), Aeromonas sp. (4), Clostridium sporogenes (5), Porphyromonas gingivalis (6), and Treponema denticola (7)). MGL has also been found in parasitic eukaryotes (the protozoa Entamoeba histolytica (8) and Trichomonas vaginalis (9)) and in a plant Arabidopsis thaliana (10).
The absence of the enzyme in mammals allows MGL to be considered as a drug target for the treatment of infectious diseases. In addition, MGL has been utilized to develop the therapeutic treatment of tumors by introducing recombinant proteins to deplete methionine, which is essential for the growth of cancer cells (11–13).
The biological unit of MGL is a tetramer, which can be subdivided into two so-called catalytic dimers. Every dimer contains two active sites consisting of amino acid residues from both subunits and two molecules of PLP covalently bound to Lys-210 (14).
MGL catalyzes the irreversible γ-elimination of l-methionine to give methanethiol, α-ketobutyrate, and ammonia (Reaction 1). The enzyme is also able to catalyze the β-elimination reaction of l-cysteine and the S-substituted l-cysteines (Reaction 2) and the γ- and β-replacement reactions of l-methionine and l-cysteine and their analogs (15, 16). The chemical mechanism (Scheme 1) of γ-elimination reaction was proposed by Brzovic et al. (17).
REACTION 1.
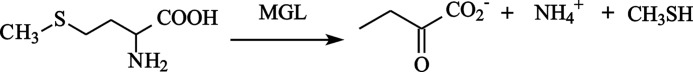
REACTION 2.
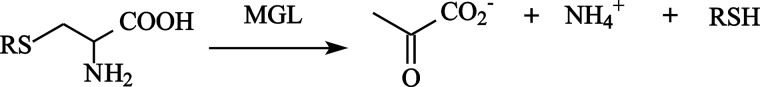
SCHEME 1.

Chemical mechanism of the γ-elimination reaction.
The initial stages of the γ-elimination occur by the exchange of the ϵ-amino group of Lys-210 in internal aldimine (I) to the α-amino group of l-methionine through the fast formation of the geminal diamine (II) and its following conversion to the external aldimine (III). In the external aldimine (III), the proton is abstracted from the α-carbon atom of substrate, and a quinonoid intermediate (IV) is formed. Subsequent protonation of the C4′ atom of the coenzyme and abstraction of a Cβ-proton of the substrate lead to the formation of ketimine (V) and enamine (VI) intermediates. The elimination of the thiol group, the sequential formation of β,γ-unsaturated ketimine (VII) and α-aminocrotonate (VIII), and hydrolysis of the Schiff base in α-aminocrotonate lead finally to the release of α-keto acid and ammonia. Intermediates of the γ-elimination reaction catalyzed by PLP-dependent enzymes possess the distinct absorption spectra (18).
Despite the spectral and structural information concerning MGL (14, 19–21), the kinetic mechanisms of β- and γ-elimination reactions catalyzed by the enzyme remain poorly understood. Therefore, the detailed analysis of the changes in the absorption spectra accompanying the binding of the amino acids allows us to elucidate the mechanisms of the interconversion of the intermediates.
In this work, we have studied the kinetic mechanisms of binding of MGL from C. freundii with competitive inhibitors glycine, l-alanine, l-norvaline, and l-cycloserine. The stopped-flow kinetic analysis of the single wavelength absorbance permitted us to attribute them separately to particular intermediates of the reaction. X-ray structure, modeling the ketimine intermediate of the γ-eliminating reaction, has been solved at 1.6 Å resolution. These data will serve for elucidation of mechanism of physiological reaction catalyzed by C. freundii MGL and can be helpful for a design of new inhibitors of MGL as potential drugs for a treatment of infection diseases.
EXPERIMENTAL PROCEDURES
Materials, Amino Acids, Enzymes
All chemicals were from Sigma.
The recombinant MGL was obtained from Escherichia coli BL21 (DE3) cells containing the pET-mgl plasmid with the inserted megL gene from the C. freundii genome. Growing the cells and purification of the enzyme were carried out as described previously (2). Protein concentrations were determined by the method of Lowry et al. (22), using bovine serum albumin as a standard. Activity of the enzyme was assayed by measuring the rate of α-ketobutyrate formation from l-methionine by the method of Friedemann and Haugen (23). One unit of enzymic activity was determined as the amount of enzyme catalyzing transformation of 1 μmol of l-methionine per min at 30 °C. The specific activity of MGL was 8.5 units/mg.
Pre-steady-state Stopped-flow Studies
Stopped-flow measurements with absorption detection were carried out using a model SX20 stopped-flow spectrometer (Applied Photophysics, UK) with a 150-watt xenon lamp and a 10-mm path length optical cell. The dead time of the instrument was 1.0 ms. All experiments were carried out at 25 °C in 0.1 m potassium phosphate buffer solution (pH 7.8), containing 0.5 mm DTT and 0.1 mm EDTA. Solutions of enzyme (12.5 μm) were mixed with various concentrations of glycine (10–500 mm), l-alanine (1.0–12.0 mm), l-cycloserine (6.35–38.1 μm), and l-norvaline (1.5–7.5 mm). Each kinetic curve was averaged over at least three independent experiments. The absorbance at 320, 420, and 500 nm was detected.
Kinetic Data Analysis
Estimation of the kinetic mechanisms as well as the number of individual reaction steps was implemented as shown in Equation 1. Each kinetic trace was fitted by sum of the exponential to define observed rate constants kobs.
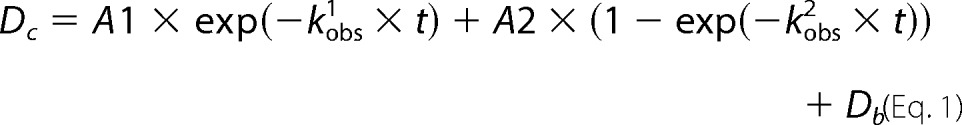 |
Dc is the absorbance intensity at any reaction time t; A1, A2 are the amplitudes; kobs1 and kobs2 are the observed rate constants; and Db is the background absorbance.
Values of the observed rate constants kobs were plotted versus amino acid concentrations to estimate the individual rate constants. As an example, the concentration dependence of kobs for the fast and slow phases at 320 nm for l-Ala are shown in supplemental Fig. S1. The fast phase fits a straight line according to the function kobs1 = k1 × [l-Ala] + k−1. The slow phase fits the function kobs2 = Kb × [l-Ala] × k2/(Kb × [l-Ala] + 1) + k−2. The dependences of kobsi on l-Ala concentration allow us to estimate all individual rate constants (supplemental Fig. S1), which were subsequently used as initial values in a global analysis.
Global nonlinear least squares fitting was performed using DynaFit4 software (BioKin Ltd.) (24) as described in Refs. 25–28. Differential equations were written for each species in the mechanisms described in Schemes 3 and 4 (see under “Results and Discussion”), and the stopped-flow absorbance traces were directly fit by expressing the absorbance intensity (Dc) at any reaction time t as the sum of the background absorbance (Db) and the absorbances of each ith intermediate Di(t) as shown in Equation 2,
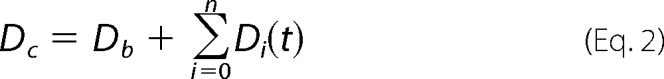 |
where Di(t) = ϵi[Ei(t)] and ϵi is the extinction coefficients at the specific wavelength for each discernible intermediate, and [Ei(t)] is the concentration of this intermediate at any given time t (i = 0 relates to the free protein; i > 0 relates to the protein-inhibitor complexes).
SCHEME 3.
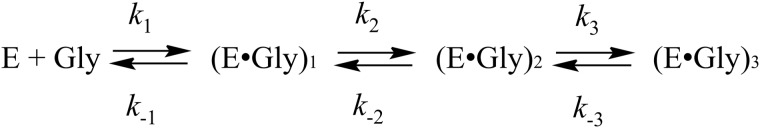
Kinetic mechanism of MGL interaction with glycine.
SCHEME 4.
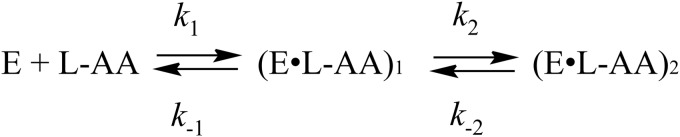
Kinetic mechanism of MGL processing of l-alanine, l-norvaline, and l-cycloserine.
In the data processing, global fits of sets of kinetic traces obtained at different concentrations of the reactants were done to derive the relevant kinetic scheme and to determine the kinetic parameters. In the fits all rate constants (including rate constants for the forward and backward reactions) as well as the specific molar extinction coefficients for all intermediate complexes were optimized.
Steady-state Kinetics
Inhibition of l-methionine γ-elimination reaction by l-cycloserine was determined in the reaction mixtures containing 100 mm potassium phosphate buffer (pH 8.0), 0.1 mm PLP, 1 mm EDTA, 5 mm DTT, and varied amounts of the substrate and the inhibitor. The rate of α-ketobutyrate accumulation was determined using dinitrophenylhydrazine (23). The reaction was initiated by addition of the enzyme (15 μg). The mixture was incubated for 15 min at 30 °C, and the reaction was stopped by addition of trichloroacetic acid to the final concentration of 12.5% (w/v). Analysis of the data was processed by Dixon (29) and Lineweaver and Burk plots (30), and the inhibition constant was determined using the EnzFitter program.
Crystallization and Data Collection
Crystals of MGL were obtained in the same conditions as described previously (31) using polyethylene glycol monomethyl ether 2000 as the precipitant. The complex of MGL with l-cycloserine was obtained by soaking holoenzyme crystals in cryoprotective mother liquid solution, containing l-cycloserine (50 mm, 7 min). The diffraction data were obtained at the BW6B DESY beamline with MAR345 MAResearch Image Plate. The detailed data collecting statistics are shown in Table 1.
TABLE 1.
Data collection and refinement statistics. Values in parentheses are for the highest resolution shell
| Space group | I222 |
| Unit cell parameters (Å) | a = 56.27, b = 122.89, c = 126.61, α = β = γ = 90 |
| Wavelength (Å) | 0.843 |
| Resolution (Å) | 15–1.60 (1.65–1.60) |
| No. of unique reflections | 55,080 |
| Mean I/σ(I) | 17.3 (3.6) |
| Completeness (%) | 94.5 (94.3) |
| Redundancy | 4.5 (3.9) |
| Rsym (%) | 4.8 (39.6) |
| Refinement | |
| Resolution range (Å) | 15–1.6 (1.64–1.60) |
| Rwork | 0.139 (0.197) |
| Rfree | 0.275 (0.293) |
| No. of protein atoms | 2993 |
| No. of water atoms | 266 |
| No. of heterogenic atoms | 57 |
| Mean temperature factor B (Å2) | 28.69 |
| Root mean square deviation from ideal values | |
| Bond lengths (Å) | 0.019 |
| Bond angles (°) | 1.985 |
| Ramachandran plot | |
| Favored region (%) | 97.11 |
| Allowed region (%) | 2.10 |
| Outlier region (%) | 0.79 |
Structure Determination and Refinement
The structure was solved by molecular replacement using the structure of C. freundii MGL (PDB code 2RFV) by rigid body procedure, implemented in CCP4 software suite (32).
The model was improved using manual rebuilding with a COOT (33) and maximum likelihood refinement using REFMAC5 (34). Flexible loops of the protein and water molecules were removed from the initial model to exclude model bias during the first round of refinement. The presence of covalently bound to the PLP cycloserine molecule is clearly visible in the experimental 2Fo − Fc and Fo − Fc electron density maps. The final model also contains a chloride ion and 266 water and 4 polyethylene glycol monomethyl ether 2000 molecules. The structure has been deposited in the Protein Data Bank with PDB code 4OMA.
RESULTS AND DISCUSSION
Reaction of Methionine γ-Lyase with Glycine
It was shown that MGL catalyzes exchange of both Cα-protons of glycine with high stereospecificity for pro-R-proton, which reacts by a factor of 1440 faster than does pro-S-proton (35). In principle, the exchange may proceed through the formation of a quinonoid intermediate or by a concerted mechanism implying fast and reversible transfer of the pro-R-proton from Cα atom to C4′ atom or a formation of a six-membered transition state incorporating a water molecule (36) (Scheme 2). To assess the formation and decay of individual intermediates, which are formed in the reaction of MGL with glycine, we carried out single wavelength stopped-flow experiments at 320, 420, and 500 nm.
SCHEME 2.

Possible intermediates in the reaction of glycine with MGL.
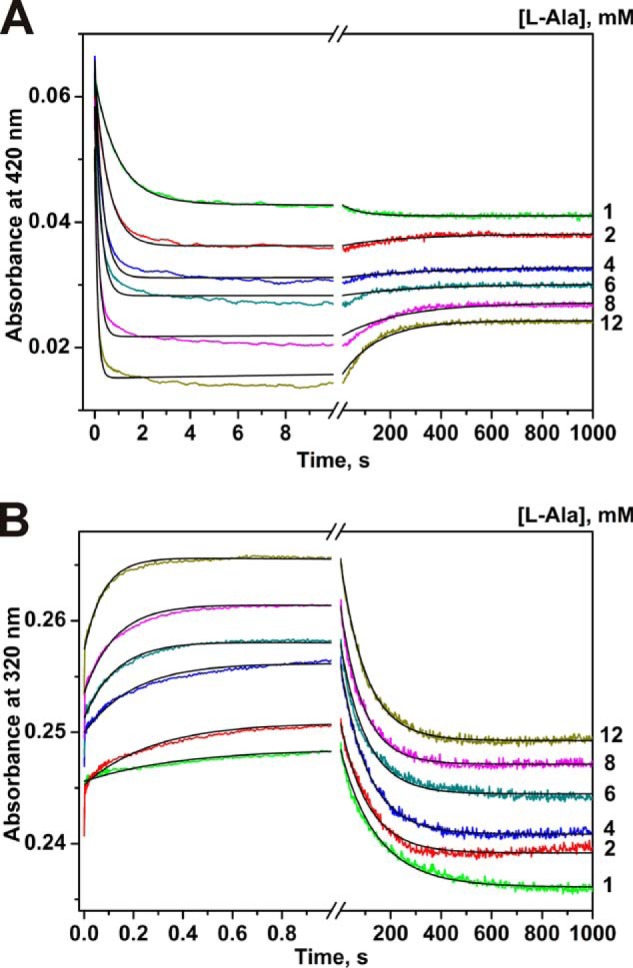
The time courses of absorbance at two single wavelengths for the interaction of MGL with glycine are presented in Fig. 1. Single wavelength kinetic curves at 420 nm indicate that the reaction of glycine with MGL consists of two phases as follows: fast (within a time interval of 20 ms) and slow (up to 100 s) increase of absorbance (Fig. 1A). However, the absorbance at 320 nm is characterized by a fast increase for about 100 ms and a slow decrease until 100 s (Fig. 1B). The absorbance at 500 nm was stable over 200 s indicating a very small level of formation of the quinonoid intermediate, which evidently is not the major intermediate.
FIGURE 1.

Interaction of MGL and glycine as monitored by changes of absorbance of the holoenzyme. Time courses were recorded at pH 7.8, 0.1 m potassium phosphate buffer solution, containing 0.5 mm DTT and 0.1 mm EDTA, with the concentration of MGL fixed at 12.5 μm, T = 25 °C. Solution of enzyme was mixed with various concentrations of glycine (10–500 mm). Each kinetic curve was averaged over at least four independent experiments. The absorbance at 420 nm (A) and 320 nm (B) was detected. Solid lines represent the fitted curves.
During the global fitting of kinetic data, a variety of kinetic models was examined to ascertain the simplest kinetic mechanism that accurately described the experimental data at 420 and 320 nm, simultaneously. Global fitting analysis showed that a reaction mechanism consisting of three steps and presented in Scheme 3 is the minimal one to appropriately describe the kinetic data. It should be noted that residuals (supplemental Fig. S2) for both 320 and 420 nm have the same amplitude of deviation from the zero line in the whole range of time that gives us confidence on the reliability of the proposed kinetic mechanism. The rate and the equilibrium constants calculated using Scheme 3 are listed in Table 2. The steady-state rate constant (kex) of the isotopic exchange of the two enantiotopic protons of glycine (35) are also presented in Table 2. Having compared the exchange rates with the other rates in Table 2, it was supposed that the complex (E·Gly)1, which absorbs at 420 nm and is formed in a millisecond time interval, probably corresponds to the external aldimine intermediate with orthogonal orientation of the “right” pro-R-proton (Hr) to the cofactor plane (Fig. 2, IIIa). Such conformation is suitable for the exchange of namely this proton. Moreover, complex (E·Gly)3, which also absorbs at 420 nm, but is formed much slower, may correspond to the external aldimine in other conformation, with an orthogonal orientation of the “wrong” pro-S-proton (Hw) (Fig. 2, IIIb). This conformation is suitable for the exchange of pro-S-proton, although exchange of the pro-R-proton is not possible. The observed rate of pro-S-proton exchange is slower than the rate of (E·Gly)3 formation, so in principle, this complex may be an intermediate in the isotopic exchange reaction.
TABLE 2.
Pre-steady-state and isotopic exchange kinetic parameters for interaction of MGL with amino acids
| Constantsa | Glycine | l-Alanine | l-Norvaline | l-Cycloserine |
|---|---|---|---|---|
| k1, m−1 s−1 | (1.0 ± 0.2) × 104 | (7.0 ± 0.1) × 102 | (4.0 ± 1.0) × 104 | (3.6 ± 1.2) × 104 |
| k-1, s−1 | 140 ± 30 | 0.6 ± 0.1 | 94.0 ± 6.0 | 1.6 ± 0.3 |
| k2, s−1 | 1.5 ± 0.1 | (6.8 ± 0.5) × 10−3 | (6.6 ± 0.4) × 10−3 | (6.5 ± 2.5) × 10−3 |
| k−2, s−1 | 33.5 ± 1.3 | (3.6 ± 0.4) × 10−3 | (6.3 ± 0.2) × 10−3 | (3.8 ± 0.4) × 10−4 |
| k3, s−1 | 0.3 ± 0.1 | |||
| k−3, s−1 | 0.08 ± 0.04 | |||
| K1, mm | 14 | 0.9 | 2.3 | 0.045 |
| K2 | 22.3 | 0.53 | 1.0 | 0.58 |
| K3 | 0.27 | |||
| Ki, mm | 49b | 3.4 | 4.7 | 0.068 ± 0.007 |
| kexα−H, s−1c | 20.2 pro-R | 2.71 | 20.6 | |
| 1.4×10−3 pro-S | ||||
| kexβ−H, s−1c | 2.63 | 11.1 |
FIGURE 2.
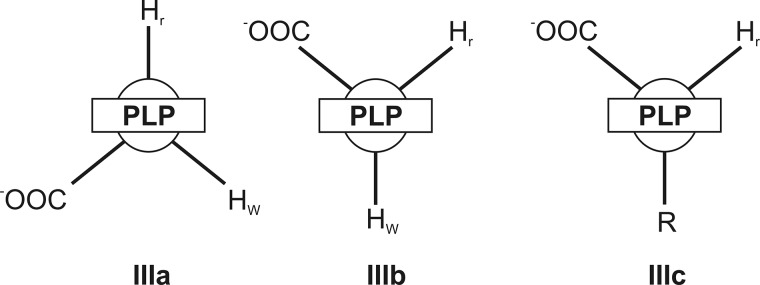
Schematic representation of possible aldimine conformers of amino acids in the active site of MGL.
In accordance with the proposed scheme, the external aldimine is rearranged from conformation IIIa to conformation IIIb through the formation of an intermediate complex (E·Gly)2, absorbing at 320 nm. Probably, the most possible structure of complex (E·Gly)2 may be the ketimine, whose formation can be affected by abstraction of the Cα-proton of the external aldimine and protonation of C4′ atom of the cofactor. In the reaction of MGL with its natural substrate, such transformation is one of necessary catalytic stages (Scheme 1) (17). It seems possible also that the structures III, IIIa, IV, and V exist in a rapid equilibrium with each other. Alternatively, complex (E·Gly)2 may have a structure of the enolimine tautomer of the external aldimine (Scheme 2). Formation of such a structure from the cationic external aldimine (IIIa) may be possible due to a conformational change of the protein, associated with an increase of hydrophobicity of the active site (37, 38). Besides, a conformer of the external aldimine with aldimine bonds out of the plane of the cofactor, and the neutral form of the enolimine absorbs in this region. This interpretation of the absorbance changes during binding of glycine is in agreement with the occurrence of fast Cα-proton exchange. According to the crystal structure of the external aldimine of MGL with glycine (36) and the known role of active site Lys residue as abstracting pro-R-proton in external aldimines of many PLP-dependent enzymes (E·Gly)1, the conformation fits with that having an orthogonal pro-R-proton.
Reactions of Methionine γ-Lyase with l-Alanine and l-Norvaline
The kinetic traces are similar for l-alanine and l-norvaline (Figs. 3 and 4). In both cases of interaction of MGL, the latest possible intermediate is an enamine (see supplemental Scheme S1). For these amino acids, the existence of exchange of both Cα- and Cβ-protons was shown (19). Two discernable steps can be noticed on the absorbance traces. The first one is the absorbance decay at 420 nm and the increase at 320 nm (up to 1 s), and the second one is a slow increase at 420 nm and slow decrease at 320 nm (up to 1000 s). The absorbance at 500 nm during interaction MGL with l-alanine and l-norvaline is slightly changed (data not shown) indicating that the quinonoid is a temporary intermediate and is not accumulated in the reaction. The simplest mechanism that can account for the kinetic data is presented in Scheme 4, and it includes the conversion of internal aldimine, absorbing at 420 nm, into complex (E·l-AA)1, which absorbs at 320 nm, and slow conversion of this complex into the external aldimine (E·l-AA)2, which absorbs at 420 nm. The calculated rate constants of the formation of the (E·l-AA)2 complex in the reactions with l-alanine and l-norvaline are much less than the rate constants of the isotopic exchange of Cα- and Cβ-protons in the both amino acids (Table 2). Consequently, the external aldimine (E·l-AA)2 cannot be an intermediate in the exchange reactions. It seems likely that in its structure (Fig. 2, IIIc), the side chain of the bound amino acid, but not the Cα-proton, is oriented orthogonally to the PLP, and the exchange of the Cα-proton is not possible. This structure formally is analogous to that of the slowly forming external aldimine (Fig. 2, IIIb) in the reaction with glycine, responsible for the exchange of the wrong pro-S-proton. Thus, the isotopic exchange of both Cα- and Cβ-protons of l-alanine and l-norvaline should proceed through the intermediate complex (E·l-AA)1, absorbing at 320 nm. For the exchange of β-protons, the formation of ketimine is strongly required (17), and we can reasonably suppose that the complex (E·l-AA)1 corresponds to the ketimine, existing in a fast equilibrium with the enolimine tautomer of the external aldimine also absorbing at 320 nm.
FIGURE 3.
Interaction of MGL and l-alanine as monitored by changes of absorbance of the holoenzyme. Time courses were recorded at pH 7.8, 0.1 m potassium phosphate buffer solution, containing 0.5 mm DTT and 0.1 mm EDTA, with the concentration of MGL fixed at 12.5 μm, T = 25 °C. Solution of enzyme was mixed with various concentrations of l-alanine (1.0–12.0 mm). Each kinetic curve was averaged over at least four independent experiments. The absorbance at 420 nm (A) and 320 nm (B) was detected. Solid lines represent the fitted curves.
FIGURE 4.
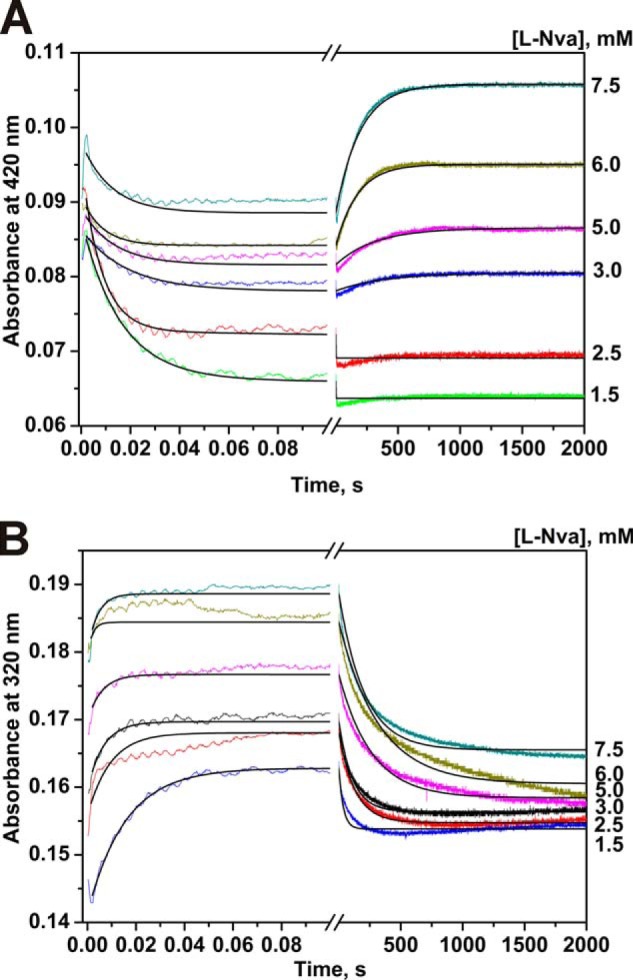
Interaction of MGL and l-norvaline as monitored by changes of absorbance of the holoenzyme. Time courses were recorded at pH 7.8, 0.1 m potassium phosphate buffer solution, containing 0.5 mm DTT and 0.1 mm EDTA, with the concentration of MGL fixed at 12.5 μm, T = 25 °C. Solution of enzyme was mixed with various concentrations of l-norvaline (1.5–7.5 mm). Each kinetic curve was averaged over at least four independent experiments. The absorbance at 420 nm (A) and 320 nm (B) was detected. Solid lines represent the fitted curves.
Inhibition of γ-Elimination Reaction by l-Cycloserine
Cycloserine is a cyclic analog of serine, and d-cycloserine is known as natural antibiotic. l- and d-enantiomers of cycloserine are inhibitors of a number of PLP-dependent enzymes belonging to different classes. The γ-eliminating activity of MGL was reduced in the course of incubation with l-cycloserine. Inactivation by l-cycloserine was completely reversed by dialysis against potassium phosphate buffer (pH 8.0), containing 0.5 mm PLP and 5 mm DTT. The Ki value determined by EnzFitter was 0.068 mm. Linearization of the data in Lineweaver and Burk plot indicated inhibition of mixed types (Fig. 5A). It was shown that application of Dixon plots in conjunction with double-reciprocal plots is useful for the identification of inhibition behavior (39). The experimental data were fitted to straight lines in the Dixon plot (Fig. 5B) thus confirming mixed type of MGL inhibition by l-cycloserine. We were not able to observe any significant isotopic exchange of cycloserine protons, at least in conditions analogous to that in which the exchange had been observed with other inhibitors.
FIGURE 5.
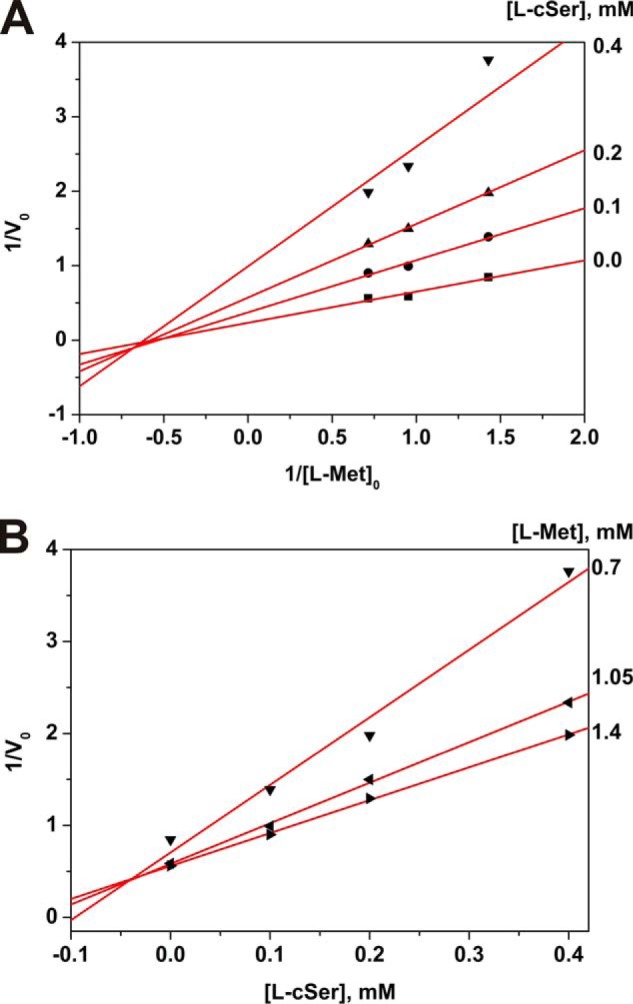
Inhibition of the γ-elimination reaction by l-cycloserine. Lineweaver and Burk (A) and Dixon (B) plots.
X-ray Structure of l-Cycloserine-PLP Derivative
Overall conformation of the polypeptide chains of the MGL holoenzyme (PDB code 2RFV) and MGL-cycloserine complex proved to be almost identical (root mean square deviation for Cα atoms of holoenzyme and the complex is 0.28 Å). There is the difference in the positions of two protein loops, N-terminal domain loop 45–60 and C-terminal domain loop 350–370, located near the active site. In the structures of the holoenzyme (PDB codes 2RFV and 1Y4I), the N-terminal loop is notably flexible. In the structure under consideration, in the structures of Michaelis complexes of MGL with a number of amino acids (20), and in the structure of external aldimine with glycine (36), it is stabilized. Most likely, the locking of the N-terminal domain loop provides optimal positioning of two cofactor-binding residues (Tyr-58 and Arg-60) and several active site hydrophobic pocket residues (Phe-49, Leu-57, and Leu-61). At the same time in spatial structures of the complexes of the enzyme with amino acids, the C-terminal segment becomes more flexible. In the presented structure, the average B-factor of residues 350–370 (around 60 Å2) increases as compared with the average B-factor of the same residues (about 45 Å2) in the holoenzyme structure (PDB code 2RFV). This flexibility may assist in a withdrawal of the products from the active site. We speculate that the N- and C-terminal segments make a “shutter” to ensure the access of a substrate to the active site, efficient catalysis, and a withdrawal of the products from the active site.
The resulting 2Fo − Fc and Fo − Fc maps indicated the presence of a cycloserine molecule covalently bound to PLP (Fig. 6). A well defined region of electron density is connected to the cofactor and is clearly separated from any protein residues, including the Nζ atom of Lys-210, which forms a covalent bond with PLP in the holoenzyme. We attempted to incorporate two tautomers of PLP-bound l-cycloserine into the electron density. One of them should possess planarity of the five-membered ring due to the presence of a double bond between the carbon and nitrogen atoms of the cycloserine; the second does not have this restrain and could be bent. Comparison of the possible fitting of the two structures revealed better matching of the planar cycloserine to the experimental electron density.
FIGURE 6.
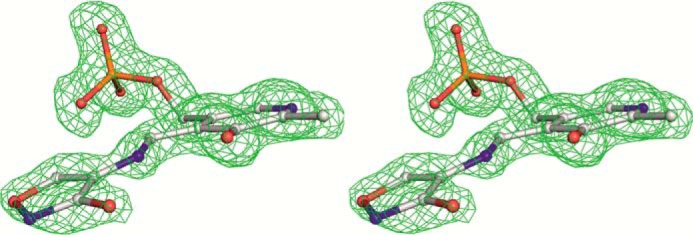
Stereo view of PLP-cycloserine derivative with the final 2Fo − Fc map contoured at the 1.5 σ level.
The C3-C4-C4A-N dihedral angle is −5.1° and is constrained by the hydrogen bond between the N and O3′ atoms. The C4-C4A-N-CA dihedral angle is 167.4°, and the C4A-N-CA-CB dihedral angle is −4.7°, so the plane of the l-cycloserine is somewhat tilted with respect to the pyridine ring (Fig. 6). The geometry of the complex is corresponding to sp3 hybridization of C4A, which is implied by the shift of the band of MGL internal aldimine at 420 to 320 nm observed in polarized absorption spectra of the holoenzyme crystal in the presence of l-cycloserine (40) and in absorption spectrum of the complex in a solution (data not shown). Accordingly, the N–CA bond of the complex should be double; the exocyclic oxygen atom connects to the cycloserine ring by a single bond and is protonated. The structure corresponds to ketimine intermediate (Scheme 5) and matches well with that described for the complex of dialkylglycine decarboxylase with l-cycloserine (41).
SCHEME 5.

Possible intermediates in the reaction of l-cycloserine with MGL.
The stereochemical position of the l-cycloserine-PLP complex in the active site is determined by firmly fixed contacts with amino acid residues of two subunits that are involved in the organization of a catalytic dimer. The endocyclic oxygen of the ring interacts with the hydrogen of the NH group of main chain Ser-339 from one side and with nearest water molecule from the other side (Fig. 7). The exocyclic oxygen makes hydrogen bonds with NH1 and NH2 atoms of Arg-374. This residue is involved in a binding of carboxylic groups in the external aldimine of MGL with glycine (36) and in Michaelis complexes of the enzyme with some substrates and inhibitors (20). The position of the bound cycloserine is additionally stabilized by stacking with side chains of Tyr-58 from the neighboring subunit and with Tyr-113 of its own protein chain.
FIGURE 7.

Stereo view of the active site. H-bonds are indicated by green dashed lines. The shown distances are in Å.
The coenzyme part of the complex is held in place by all the usual interactions observed in PLP-dependent enzymes. The phosphate is anchored by a number of polar interactions with the protein (14). The N1 atom of the pyridine ring forms an H-bond with the carboxylate of Asp-185. O3′ hydroxyl of PLP has no contacts with the protein atoms and is involved in hydrogen bonding with ketimine nitrogen atom of the complex (Fig. 7). In the structure, there are tilts of the PLP ring around the C5-C2 axis and Tyr-113 ring to the solvent in comparison with their positions in the holoenzyme (25° and 19°, respectively). Similar tilts of the cofactor (17°) and Tyr-113 (28°) rings was demonstrated in a three-dimensional structure of external aldimine of the enzyme with glycine (36). The Nϵ atom of Lys-210 occupies two slightly different positions close to the ketimine nitrogen atom (∼3.5 Å). The abstraction of a Cβ-proton from the first ketimine intermediate is the next step of γ-elimination reaction. Data on the crystal structure of C. freundii MGL external aldimine with glycine (36) allowed us to suppose that in the course of the γ-elimination reaction the 1,3-prototropic shift of Cα-proton to C4′ atom of the cofactor and the abstraction of a Cβ-proton may proceed with the participation of Lys-210. In the structure under consideration, a close positive charge of the ketimine nitrogen atom may stabilize the basic form of Lys-210, which allows the stage of Cβ-proton abstraction to proceed. In both positions, the Nζ atom of Lys-210 is close (3.75 Å, 3.15 Å) to the Cβ atom of the cycloserine ring.
In 1959, several mechanisms were proposed for interaction of cycloserine with PLP-dependent enzymes, including formation of a stable isoxazole system and an opening of the cycloserine ring with further transformations (42). Evidence for the first mechanism was obtained in 1998 for γ-aminobutyric acid aminotransferase (43) and for d-amino acid aminotransferase (44). It was proposed that an abstraction of a Cβ-proton of cycloserine from ketimine by active site lysine residue leads to stable aromatic cycloserine·PLP complex. At the resolution of our structure, we do not exclude a saturation of the N–CA bond, but the geometry of the complex is more compatible with the double N–CA bond. For mechanisms of γ-elimination and γ-replacement reactions catalyzed by PLP-dependent enzymes, it was postulated that ketimine (Scheme 1, V) is a plausible intermediate for effective labilization of the Cβ-proton to proceed (17). Its formation in the course of these reactions catalyzed by E. coli cystathionine γ-synthase was detected by stopped-flow data in the same paper. The evidence of the ketimine intermediate was obtained by X-ray data for cystathionine γ-synthase (45).
Reaction of Methionine γ-Lyase with l-Cycloserine
In a steady-state absorption spectrum of MGL complexed with l-cycloserine, the band belonging to the internal aldimine with a maximum in the region of 420 nm disappeared along with the appearance the predominant band at the region of 320–330 nm (data not shown).
The reaction of MGL with l-cycloserine leads to a single change of absorbance within 2000 s as follows: a decrease at 420 nm and an increase at 320 nm, the rates of the two processes being identical (Fig. 8). According to X-ray data, the possible intermediate of the reaction of MGL with l-cycloserine is ketimine, which has an absorbance maximum at the region of 320 nm. It can be assumed that the observed spectral changes correspond to the decomposition of external aldimine and accumulation of ketimine, respectively (Scheme 5). The external aldimine is converted to a ketimine intermediate without detectable changes at 500 nm, indicating that the intermediate quinonoid species are not accumulating in the reaction.
FIGURE 8.
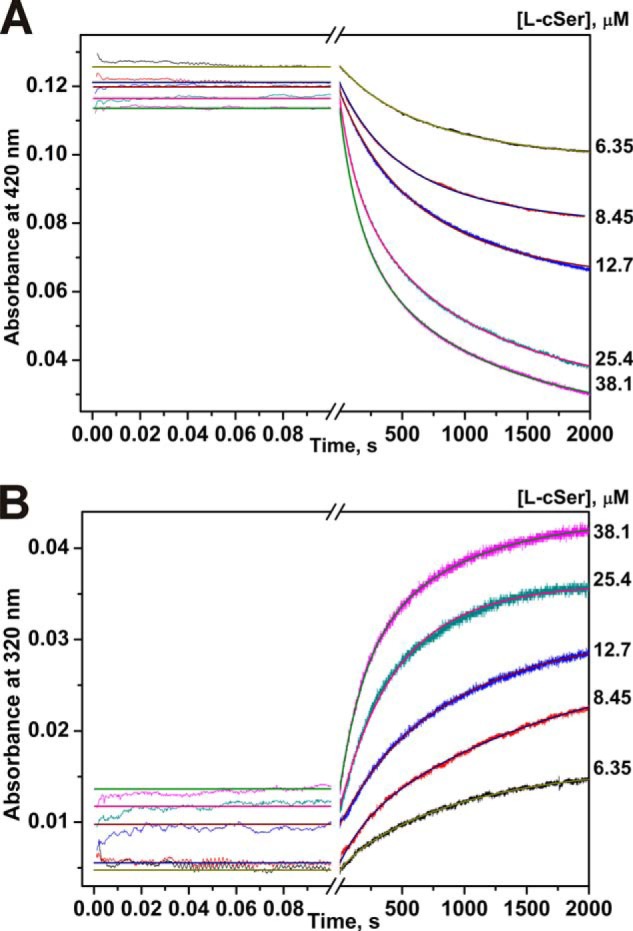
Interaction of MGL and l-cycloserine as monitored by changes of absorbance of the holoenzyme. Time courses were recorded at pH 7.8, 0.1 m potassium phosphate buffer solution, containing 0.5 mm DTT and 0.1 mm EDTA, with the concentration of MGL fixed at 12.5 μm, T = 25 °C. Solution of enzyme was mixed with various concentrations of l-cycloserine (6.35–38.1 μm). Each kinetic curve was averaged over at least three independent experiments. The absorbance at 420 nm (A) and 320 nm (B) was detected. Solid lines represent the fitted curves.
The processing of the l-cycloserine by MGL is described in kinetic Scheme 4. The values for individual kinetic constants were determined by global fitting (Table 2). (E·l-AA)1 complex more likely corresponds to external aldimine, which subsequently transforms in ketimine in the complex (E·l-AA)2 (Scheme 5). Interestingly, the forward rate constant k1 and k2 are similar to the respective constants for l-norvaline reaction. At the same time, the reverse rate constants k−1 and k−2 are 58 and 1.6 times less as compared with l-norvaline reaction, indicating that MGL complexes with l-cycloserine are more stable, which leads to a shift of the equilibrium to the ketimine intermediate.
The stability of ketimine intermediate may explain the higher inhibition of the reaction with l-methionine in comparison with glycine, l-alanine, and l-norvaline. The Ki value of inhibition of γ-elimination of l-methionine by l-cycloserine is at least 10-fold less than the Ki values for glycine, l-alanine, and l-norvaline (Table 2).
Conclusion
The comparison of values of the rate constants characterizing the processes of the amino acids binding by MGL showed that the equilibrium constant of the first step K1 = k1/k−1 is increased in the row: glycine < l-norvaline ∼ l-alanine < l-cycloserine. The rate constant of the forward reaction k1 for glycine, l-norvaline, and l-cycloserine is in the range of (1.0–4.0) × 104 m−1 s−1, being at least 10 times higher than in the case of l-alanine (Table 2). Interestingly, the rate constant of the isotopic exchange of Cα-protons for l-alanine is 10 times smaller than that of glycine and l-norvaline (19). The process characterized with the rate constant k1 should represent the complex mechanism, including not only the multistage process of the Schiff base formation between incoming amino acid and Lys-210, but possibly the enzyme reorganization during the incorporation of the amino acids into the active site. The rate constant k−1, which characterizes the decomposition of the first complex (Table 2), reflects the stability of this state. In cases of l-alanine and l-cycloserine, the magnitude of k−1 is more than 150 and 50 times lower than the same value for glycine and l-norvaline.
Formation of the “right” external aldimine enables the formation of the ketimine intermediate, which was obviously registered in the cases of l-norvaline and l-alanine and probably for glycine ([E·l-Nva]1, [E·l-Ala]1, or [E·Gly]2 complexes, respectively). The last detected complex ([E·l-norvaline]2, [E·l-Ala]2, or ([E·Gly]3) likely corresponds to the external aldimine in the wrong conformation. In the case of l-cycloserine, the formation of (E·l-cycloserine)1 complex reflects the formation of the external aldimine. The subsequent transformations (abstraction of Cα-proton and protonation of C4′ atom of PLP) lead to accumulation of stable ketimine intermediate.
The kinetic data have shown that amino acids incorporate in the active site of MGL in the proper conformation because of the conformational selectivity of the active site. The Schiff bases can be formed with an amino acid only having right orientation of the side chain, Cα-hydrogen and carboxyl group relatively to PLP plane. After the formation of the ketimine intermediate, the flat structure of the amino acid can be turned in the active site of enzyme, and thus after the reversible protonation, the amino acid would be in the wrong conformation (e.g. Fig. 2, IIIb).
In general, both X-ray and kinetic analyses of the interaction of MGL with l-cycloserine provide the direct proof of ketimine intermediate formation in the catalytic mechanism of PLP-dependent γ-elimination reaction.
This work was supported by Russian Academy of Sciences Program “Molecular & Cell Biology” (6.11), Russian Foundation for Basic Researches Grant 14-04-00349, Russian Scientific Foundation Grant 14-14-00063, President of the Russian Federation for State Support of the Leading Scientific Schools Grants SS-2064.2014.4 and SS-1205.2014.4, and Russian Ministry of Education and Science Grant SP-4012.2013.4.

This article contains supplemental Figs. S1 and S2 and Scheme S1.
The atomic coordinates and structure factors (code 4OMA) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- MGL
- methionine γ-lyase
- PLP
- pyridoxal 5′-phosphate
- PDB
- Protein Data Bank.
REFERENCES
- 1. El-Sayed A. S. (2010) Microbial l-methioninase: production, molecular characterization, and therapeutic applications. Appl. Microbiol. Biotechnol. 86, 445–467 [DOI] [PubMed] [Google Scholar]
- 2. Manukhov I. V., Mamaeva D. V., Rastorguev S. M., Faleev N. G., Morozova E. A., Demidkina T. V., Zavilgelsky G. B. (2005) A gene encoding l-methionine γ-lyase is present in Enterobacteriaceae family genomes: identification and characterization of Citrobacter freundii l-methionine γ-lyase. J. Bacteriol. 187, 3889–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ito S., Nakamura T., Eguchi Y. (1976) Purification and characterization of methioninase from Pseudomonas putida. J. Biochem. 79, 1263–1272 [DOI] [PubMed] [Google Scholar]
- 4. Nakayama T., Esaki N., Lee W. J., Tanaka I., Tanaka H., Soda K. (1984) Purification and properties of l-methionine γ-lyase from Aeromonas sp. Agric. Biol. Chem. 48, 2367–2369 [Google Scholar]
- 5. Kreis W., Hession C. (1973) Isolation and purification of l-methionine-α-deamino-γ-mercaptomethane-lyase (l-methioninase) from Clostridium sporogenes. Cancer Res. 33, 1862–1865 [PubMed] [Google Scholar]
- 6. Yoshimura M., Nakano Y., Yamashita Y., Oho T., Saito T., Koga T. (2000) Formation of methyl mercaptan from l-methionine by Porphyromonas gingivalis. Infect. Immun. 68, 6912–6916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fukamachi H., Nakano Y., Okano S., Shibata Y., Abiko Y., Yamashita Y. (2005) High production of methyl mercaptan by l-methionine-α-deamino-γ-mercaptomethane lyase from Treponema denticola. Biochem. Biophys. Res. Commun. 331, 127–131 [DOI] [PubMed] [Google Scholar]
- 8. Tokoro M., Asai T., Kobayashi S., Takeuchi T., Nozaki T. (2003) Identification and characterization of two isoenzymes of methionine γ-lyase from Entamoeba histolytica: a key enzyme of sulfur-amino acid degradation in an anaerobic parasitic protist that lacks forward and reverse trans-sulfuration pathways. J. Biol. Chem. 278, 42717–42727 [DOI] [PubMed] [Google Scholar]
- 9. Lockwood B. C., Coombs G. H. (1991) Purification and characterization of methionine γ-lyase from Trichomonas vaginalis. Biochem. J. 279, 675–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rébeillé F., Jabrin S., Bligny R., Loizeau K., Gambonnet B., Van Wilder V., Douce R., Ravanel S. (2006) Methionine catabolism in Arabidopsis cells is initiated by a γ-cleavage process and leads to S-methylcysteine and isoleucine syntheses. Proc. Natl. Acad. Sci. U.S.A. 103, 15687–15692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoffman R. M. (1997) Methioninase: a therapeutic for diseases related to altered methionine metabolism and transmethylation: cancer, heart disease, obesity, aging, and Parkinson's disease. Hum. Cell 10, 69–80 [PubMed] [Google Scholar]
- 12. Sato D., Nozaki T. (2009) Methionine γ-lyase: the unique reaction mechanism, physiological roles, and therapeutic applications against infectious diseases and cancers. IUBMB Life 61, 1019–1028 [DOI] [PubMed] [Google Scholar]
- 13. Tan Y., Xu M., Hoffman R. M. (2010) Broad selective efficacy of recombinant methioninase and polyethylene glycol-modified recombinant methioninase on cancer cells in vitro. Anticancer Res. 30, 1041–1046 [PubMed] [Google Scholar]
- 14. Nikulin A., Revtovich S., Morozova E., Nevskaya N., Nikonov S., Garber M., Demidkina T. (2008) High-resolution structure of methionine γ-lyase from Citrobacter freundii. Acta Crystallogr. D Biol. Crystallogr. 64, 211–218 [DOI] [PubMed] [Google Scholar]
- 15. Tanaka H., Esaki N., Soda K. (1985) A versatile bacterial enzyme: l-methionine γ-lyase. Enzyme Microbiol. Technol. 7, 530–537 [Google Scholar]
- 16. Tanaka H., Esaki N., Soda K. (1977) Properties of l-methionine γ-lyase from Pseudomonas ovalis. Biochemistry 16, 100–106 [DOI] [PubMed] [Google Scholar]
- 17. Brzović P., Holbrook E. L., Greene R. C., Dunn M. F. (1990) Reaction mechanism of Escherichia coli cystathionine γ-synthase: direct evidence for a pyridoxamine derivative of vinylglyoxylate as a key intermediate in pyridoxal phosphate dependent γ-elimination and γ-replacement reactions. Biochemistry 29, 442–451 [DOI] [PubMed] [Google Scholar]
- 18. Davis L., Metzler D. E. (1971) Pyridoxal-linked elimination and replacement reactions. Adv. Enzymol. 35, 33–74 [Google Scholar]
- 19. Morozova E. A., Bazhulina N. P., Anufrieva N. V., Mamaeva D. V., Tkachev Y. V., Streltsov S. A., Timofeev V. P., Faleev N. G., Demidkina T. V. (2010) Kinetic and spectral parameters of interaction of Citrobacter freundii methionine γ-lyase with amino acids. Biochemistry 75, 1272–1280 [DOI] [PubMed] [Google Scholar]
- 20. Revtovich S. V., Morozova E. A., Khurs E. N., Zakomirdina L. N., Nikulin A. D., Demidkina T. V., Khomutov R. M. (2011) Three-dimensional structures of noncovalent complexes of Citrobacter freundii methionine γ-lyase with substrates. Biochemistry 76, 564–570 [DOI] [PubMed] [Google Scholar]
- 21. Kudou D., Misaki S., Yamashita M., Tamura T., Takakura T., Yoshioka T., Yagi S., Hoffman R. M., Takimoto A., Esaki N., Inagaki K. (2007) Structure of the antitumour enzyme l-methionine γ-lyase from Pseudomonas putida at 1.8 A resolution. J. Biochem. 141, 535–544 [DOI] [PubMed] [Google Scholar]
- 22. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 23. Friedemann T. E., Haugen G. E. (1943) Pyruvic acid, the determination of keto acids in blood and urine. J. Biol. Chem. 147, 415–443 [Google Scholar]
- 24. Kuzmic P. (1996) Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 237, 260–273 [DOI] [PubMed] [Google Scholar]
- 25. Kuznetsov N. A., Zharkov D. O., Koval V. V., Buckle M., Fedorova O. S. (2009) Reversible chemical step and rate-limiting enzyme regeneration in the reaction catalyzed by formamidopyrimidine-DNA glycosylase. Biochemistry 48, 11335–11343 [DOI] [PubMed] [Google Scholar]
- 26. Koval V. V., Kuznetsov N. A., Ishchenko A. A., Saparbaev M. K., Fedorova O. S. (2010) Real-time studies of conformational dynamics of the repair enzyme E. coli formamidopyrimidine-DNA glycosylase and its DNA complexes during catalytic cycle. Mutat. Res. 685, 3–10 [DOI] [PubMed] [Google Scholar]
- 27. Kuznetsov N. A., Koval V. V., Zharkov D. O., Fedorova O. S. (2012) Conformational dynamics of the interaction of Escherichia coli endonuclease VIII with DNA substrates. DNA Repair 11, 884–891 [DOI] [PubMed] [Google Scholar]
- 28. Kuznetsov N. A., Vorobjev Y. N., Krasnoperov L. N., Fedorova O. S. (2012) Thermodynamics of the multi-stage DNA lesion recognition and repair by formamidopyrimidine-DNA glycosylase using pyrrolocytosine fluorescence–stopped-flow pre-steady-state kinetics. Nucleic Acids Res. 40, 7384–7392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dixon M. (1953) The determination of enzyme inhibitor constants. Biochem. J. 55, 170–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lineweaver H., Burk D. (1934) The determination of enzyme dissociation constants. J. Am. Chem. Soc. 56, 658–666 [Google Scholar]
- 31. Mamaeva D. V., Morozova E. A., Nikulin A. D., Revtovich S. V., Nikonov S. V., Garber M. B., Demidkina T. V. (2005) Structure of Citrobacter freundii l-methionine γ-lyase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61, 546–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koulikova V. V., Zakomirdina L. N., Gogoleva O. I., Tsvetikova M. A., Morozova E. A., Komissarov V. V., Tkachev Y. V., Timofeev V. P., Demidkina T. V., Faleev N. G. (2011) Stereospecificity of isotopic exchange of C-α-protons of glycine catalyzed by three PLP-dependent lyases: the unusual case of tyrosine phenol-lyase. Amino Acids 41, 1247–1256 [DOI] [PubMed] [Google Scholar]
- 36. Revtovich S. V., Faleev N. G., Morozova E. A., Anufrieva N. V., Nikulin A. D., Demidkina T. V. (2014) Crystal structure of the external aldimine of Citrobacter freundii methionine γ-lyase with glycine provides insight in mechanisms of two stages of physiological reaction and isotope exchange of α- and β-protons of competitive inhibitors. Biochimie 101, 161–167 [DOI] [PubMed] [Google Scholar]
- 37. Johnson G. F., Tu J. I., Bartlett M. L., Graves D. J. (1970) Physical-chemical studies on the pyridoxal phosphate binding site in sodium borohydride-reduced and native phosphorylase. J. Biol. Chem. 245, 5560–5568 [PubMed] [Google Scholar]
- 38. Shaltiel S., Cortijo M. (1970) The mode of binding of pyridoxal 5′-phosphate in glycogen phosphorylase. Biochem. Biophys. Res. Commun. 41, 594–600 [DOI] [PubMed] [Google Scholar]
- 39. Butterworth P. J. (1972) The use of Dixon plots to study enzyme inhibition. Biochim. Biophys. Acta 289, 251–253 [DOI] [PubMed] [Google Scholar]
- 40. Ronda L., Bazhulina N. P., Morozova E. A., Revtovich S. V., Chekhov V. O., Nikulin A. D., Demidkina T. V., Mozzarelli A. (2011) Exploring methionine γ-lyase structure-function relationship via microspectrophotometry and x-ray crystallography. Biochim. Biophys. Acta 1814, 834–842 [DOI] [PubMed] [Google Scholar]
- 41. Malashkevich V. N., Strop P., Keller J. W., Jansonius J. N., Toney M. D. (1999) Crystal structures of dialkylglycine decarboxylase inhibitor complexes. J. Mol. Biol. 294, 193–200 [DOI] [PubMed] [Google Scholar]
- 42. Kochetkov N. K., Khomutov R. M., Karpeisky M. Y., Bydovsky E. I., Severin E. S. (1959) The mechanism of antibiotic action of cycloserine. Doklady Akademii Nayk SSSR 129, 1132–1134 [Google Scholar]
- 43. Olson G. T., Fu M. M., Lau S., Rinehart K. L., Silverman R. B. (1998) An aromatization mechanism of inactivation of γ-aminobutyric acid aminotransferase for the antibiotic l-cycloserine. J. Am. Chem. Soc. 120, 2256–2267 [Google Scholar]
- 44. Peisach D., Chipman D. M., Van Ophem P. W., Manning J. M., Ringe D. (1998) d-Cycloserine inactivation of d-amino acid aminotransferase leads to a stable noncovalent protein complex with an aromatic cycloserine-PLP derivative. J. Am. Chem. Soc. 120, 2268–2274 [Google Scholar]
- 45. Steegborn C., Laber B., Messerschmidt A., Huber R., Clausen T. (2001) Crystal structures of cystathionine γ-synthase inhibitor complexes rationalize the increased affinity of a novel inhibitor. J. Mol. Biol. 311, 789–801 [DOI] [PubMed] [Google Scholar]