

Background: M. tuberculosis GlgB is essential for the biosynthesis of branched glucan and modulates pathogenesis and survival.
Results: Two novel small molecules demonstrated significant inhibition of M. tuberculosis GlgB enzyme activity, growth and survival.
Conclusion: Small molecules with diverse scaffolds but similar three-dimensional-structures show a similar biological effect.
Significance: Deriving scaffolds from docking and similarity search is a successful design strategy for difficult targets.
Keywords: Docking, Molecular Docking, Mycobacterium tuberculosis, Pathogenesis, Small Molecule
Abstract
GlgB (α-1,4-glucan branching enzyme) is the key enzyme involved in the biosynthesis of α-glucan, which plays a significant role in the virulence and pathogenesis of Mycobacterium tuberculosis. Because α-glucans are implicated in the survival of both replicating and non-replicating bacteria, there exists an exigent need for the identification and development of novel inhibitors for targeting enzymes, such as GlgB, involved in this pathway. We have used the existing structural information of M. tuberculosis GlgB for high throughput virtual screening and molecular docking. A diverse database of 330,000 molecules was used for identifying novel and efficacious therapeutic agents for targeting GlgB. We also used three-dimensional shape as well as two-dimensional similarity matrix methods to identify diverse molecular scaffolds that inhibit M. tuberculosis GlgB activity. Virtual hits were generated after structure and ligand-based screening followed by filters based on interaction with human GlgB and in silico pharmacokinetic parameters. These hits were experimentally evaluated and resulted in the discovery of a number of structurally diverse chemical scaffolds that target M. tuberculosis GlgB. Although a number of inhibitors demonstrated in vitro enzyme inhibition, two compounds in particular showed excellent inhibition of in vivo M. tuberculosis survival and its ability to get phagocytosed. This work shows that in silico docking and three-dimensional chemical similarity could be an important therapeutic approach for developing inhibitors to specifically target the M. tuberculosis GlgB enzyme.
Introduction
Tuberculosis (TB),6 caused by Mycobacterium tuberculosis, continues to be a debilitating disease and kills nearly 1.4 million people annually (1). Even more alarming is latent TB infection, which affects nearly 2 billion people worldwide (2). These people are potential disease carriers and are at a greater risk of reactivation of the disease. TB is continually affecting both developing and developed countries with a disturbing rise of multidrug-resistant and totally drug-resistant M. tuberculosis strains. Thus, there is an urgent need to explore new targets and develop novel therapeutics in order to achieve effective global TB control. The bacterial cell wall offers key virulence factors to M. tuberculosis. The bacterial cell wall consists of lipids, glycolipids, enzymes, proteins, etc., which are important in host-pathogen interaction as well as in bug survival under stress conditions (3). The M. tuberculosis cell wall biosynthetic pathway has been extensively studied to explore novel drug targets, and in this context, recent studies have proved that the biosynthesis pathway of α-glucan can be exploited as a novel drug target amenable for small molecule intervention. This pathway is important for the synthesis of capsular and cytosolic glucans. Capsular glucans are involved in host-pathogen interaction, thus contributing to the pathogenicity of the bacteria, whereas cytosolic glucans are important energy sources for hibernating bacteria and contribute toward M. tuberculosis latency (4). It is now well recognized that M. tuberculosis is surrounded by a capsule both in vitro and in vivo (5, 6). It has been suggested that M. tuberculosis evades both innate and acquired immune response with the help of capsular α-glucan (7). It was reported that C-type lectin DC-SIGN (dendritic cell-specific ICAM-3-grabbing non-integrin) is a novel ligand for α-glucan, which suggests that the M. tuberculosis capsule is essential for the pathogenesis and virulence of the bacterium (8). GlgB is one of the important enzymes of the pathogenic bacteria that play a crucial role in the biosynthesis of α-glucan. It was shown that the M. tuberculosis glgB (Rv1326c) gene is essential for its survival both in vitro and in vivo (9). Incidentally, the glgB gene of several other pathogenic bacteria have also been shown to be associated with their virulence (10, 11) and are expressed during pathogenesis (12, 13). It has been demonstrated that inactivation of GlgB results in the accumulation of linear α-glucan, which may feedback-inhibit polymerization of maltose 1-phosphate (a GlgE substrate), thus causing self-poisoning due to toxic build-up of this substrate (14). It has been shown that the recombinant GlgB protein is active as an enzyme and uses both amylose and starch as substrates (15). The crystal structure of full-length M. tuberculosis GlgB shows that it has four domains where the active site domain is a TIM-barrel similar to other GH13 enzymes (16). Interestingly, it was also observed that known inhibitors of Escherichia coli GlgB have no effect on the activity of M. tuberculosis GlgB, indicating that although there is sequence similarity, the structural differences between E. coli and M. tuberculosis GlgBs may explain the differential binding affinity of the two proteins (17). Recently, the crystal structure of human GlgB has been deposited in the Protein Data Bank (entry 4BZY), and comparison shows marked differences between the human and M. tuberculosis GlgB structures (18). Clearly, although these enzymes utilize similar substrates and are important for the general functioning of both human and M. tuberculosis, they seem to have evolved differently. GlgB is an essential enzyme, and selective targeting of GlgB would be an attractive target to control both “active” and “persistent” TB. This study, to the best of our knowledge, is the first report on the small molecule inhibitors that can target M. tuberculosis GlgB.
This study attempts the discovery of novel GlgB inhibitors using a structure- and ligand-based drug design strategy. We have used high throughput virtual screening (HTVS) and a shape-based ligand strategy to search compound libraries, such as Maybridge, ZINC (19), and our in-house database to discover new chemical scaffolds for targeting GlgB. These compounds were then experimentally validated by in vitro enzymatic assays, and the top hits were further validated for M. tuberculosis growth inhibition in culture and in infected human THP-1 macrophage. This study represents the first steps in the direction of feasibility of small molecules as possible therapeutics against M. tuberculosis GlgB.
EXPERIMENTAL PROCEDURES
Structure-based Virtual Screening
Protein and Ligand Preparation
For Human M. tuberculosis GlgB, homology modeling was performed using SWISS-MODEL workspace (20), which is an integrated Web-based modeling system. In order to generate a suitable homology model, libraries of experimental protein structures were searched to identify a suitable template for modeling. The crystal structure of M. tuberculosis GlgB is already known (17) (Protein Data Bank code 3KID) and was used for docking studies. Both the M. tuberculosis GlgB crystal structure and the generated human GlgB model were processed by adding missing hydrogens and assigning proper bond orders. The models were further minimized by using the optimized potentials for liquid simulations-all-atom (OPLS_AA) force field of the Schrodinger software package (21). The simulation consisted of a string of restrained and limited minimization that was designed to slowly relax the system. The Maybridge and ZINC database molecules were prepared by Glide LigPrep to account for missing hydrogens, generate ionization states and tautomers, and optimize the geometries of ligand molecules.
HTVS
To screen inhibitors against M. tuberculosis GlgB, flexible ligand-based HTVS screening was performed using Glide version 5.5 (22–26). Diverse databases containing commercially available ligands (Maybridge (MB; 14,400 molecules) and ZINC (19) (which includes an NCI (National Institutes of Health) library (316,181 molecules) and United States Food and Drug Administration-approved drugs (3,180 molecules)) were used for molecular docking. The docking score was used as a reference to identify druglike inhibitors. A receptor grid was generated to cover the active site of the enzyme by using the protocol as implemented in Glide. The van der Waals radii for the residue atoms that presumably interact with ligands were set to 1.0, whereas the partial atomic charge was not more than 0.25. After grid generation, ligands were screened through the HTVS mode and subjected to the Glide XP (extra precision) mode to perform comprehensive docking and generate significant binding poses. Ligands were considered only if they interacted with the active site residues of M. tuberculosis GlgB, but not human GlgB, in terms of hydrogen bonding. Furthermore, the ligands were assessed for their druglike properties based on Lipinski's rule of five (27) and detailed pharmacokinetic properties by using QikProp version 3.2 (Schrodinger) (28, 29).
Three-dimensional Conformer Generation
Initially, the ChemAxon software Marvin was used to draw the three-dimensional structure of the query molecule (Marvin version 6.0.2, ChemAxon). The structures were energy-minimized using a Merck molecular force field (MMFF94) (31). A three-dimensional conformational database of query molecules was generated using the software OMEGA version 2.5 (OpenEye), which utilizes model building and torsional search for efficient conformational sampling. In the first step of model building, the chemical structure was fragmented along σ bonds using the makefraglib utility, and then the structure was reconstructed by assembly of these fragments (32). These fragments were refined using a modified Merck molecular force field (MMFF94). In the second step, an ensemble of conformers was generated using torsional angle rules as described in Omega version 2.5 (33). Conformational searches were terminated when the energy cut-off exceeded 10 kcal/mol or when 100 structures were built.
Three-dimensional Shape Similarity Comparison
The three-dimensional structure of the hit molecules obtained from docking were compared with the in-house database using the Rapid Overlay of Chemical Structures (ROCS version 3.2.0.4) (34) program from OpenEye. Molecules were compared both on the basis of three-dimensional shape and chemistry of the functional groups, hydrogen bond donor, acceptor, etc. Three-dimensional similarity was quantified using the shape Tanimoto coefficient, whereas the chemistry overlap was quantified by using color score as implemented in ROCS. The implicit Mills-Dean chemistry parameters were implemented as described by Mills and Dean (35).
Docking Studies
To explore the binding mechanism of inhibitors with GlgB, compounds MB16695 and IMT007 were docked, and their binding efficiencies were evaluated in both M. tuberculosis and human GlgB. Autodock version 4.2.5 was used for docking simulations (36). The crystal structures of M. tuberculosis GlgB (Protein Data Bank code 3KID) and human GlgB (Protein Data Bank code 4BZY) were used for the docking studies (17). To prepare the receptors for docking, water molecules were removed, hydrogen atoms were added, and Kollman's charges were assigned. The docking protocol was performed with the monomeric unit of the M. tuberculosis GlgB and human GlgB because the active site residues within the receptor and not on the protein surface. The active site cavity for GlgB has been defined by seven residues (17) (Asp-341, His-346, Arg-409, Asp-411, Glu-464, His-531, and Asp-532, M. tuberculosis GlgB numbering), which are also conserved in human GlgB (Asp-286, His-291, Arg-355, Asp-357, Glu-412, His-480, and Asp-481, human GlgB numbering). In all of the docking simulations, we used induced fit docking in which the binding site residues of M. tuberculosis and human GlgB were made flexible. Each ligand was completely flexible during docking to explore torsional degrees of freedom spanned by the translational and rotational parameters. Autogrid calculates the binding energy between the protein and ligand by using atom affinity potentials that are precalculated on grid maps. The size of the docking grid was kept at 60 × 54 × 56 Å in the x, y, and z dimensions, which encompassed the active site of each receptor. The grid spacing was set to 0.375 Å. The Lamarckian genetic algorithm was used in the conformational space as defined by the grid. The Lamarckian genetic algorithm parameters were set to their default values (population size, 150; mutation rate, 0.02; crossover, 0.8). The maximum number of energy evaluations and generations was kept at 2.5 million and 27,000 respectively. The conformations of each ligand for both receptors were ranked in order of increasing energies with a root mean square deviation cut-off of 2 Å. The best pose for each docking simulation was selected by analyzing binding efficiency, energy, and the most populated cluster.
Enzyme Inhibition Assay
Chemical Sources
All chemicals were purchased from Sigma or Aldrich and used without further purification.
Cloning, Expression, and Purification of M. tuberculosis and Human GlgB
The M. tuberculosis glgB gene was PCR-amplified using gene-specific primers and cloned into the pET29a vector (with optional His tag), as reported earlier (15). The protein was expressed in E. coli BL21 (DE3) with 0.3 mm isopropyl 1-thio-β-d-galactopyranoside at 18 °C for 18 h and was purified as reported earlier. The human glgB gene was codon-optimized and synthesized from gene script. This customized human glgB was cloned into the pET28a vector. The protein was expressed in E. coli BL21 (DE3) with 0.05 mm isopropyl 1-thio-β-d-galactopyranoside at 18 °C for 18 h and was purified as reported earlier (15).
Preparation of Amylose
2 mg of amylose was freshly prepared by dissolving in 200 μl of 1 n NaOH and 400 μl of autoclaved double-distilled water, and the pH was adjusted to 7.0 by slowly adding 1 n HCl. 1 ml of final volume was made-up with 1 m sodium phosphate buffer (pH 7.0). The solution was microcentrifuged at 12,000 rpm for 10 min at 25 °C. The supernatant was collected and stored on ice for further use.
Preparation of Iodine Solution
0.26 g of I2 and 2.6 g KI were dissolved in 10 ml of autoclaved double distilled water and stored in dark as stock solution (A). To stop the enzyme reaction, freshly prepared solution containing 5 μl of the above iodine stock solution and 5 μl of 1 n HCl were mixed in water to make up the final volume to 1.3 ml (B).
Enzyme Inhibition Assay
An enzyme inhibition assay was conducted in microtiter plates with a total 100-μl reaction volume. All prospective small molecule inhibitors were dissolved in dimethyl sulfoxide (DMSO) to a final concentration of 100 mm and as far as possible used immediately or stored in −80 °C. Further dilutions were made, as desired, in DMSO (maximum concentration of DMSO in the reaction was 5%). A 100-μl reaction mix contained 50 mm phosphate buffer (pH 7.0), 50 nm purified protein (GlgB), 20 μg of substrate (amylose). For the inhibition assay, protein and inhibitors were preincubated at 25 °C in 50 mm phosphate buffer (pH 7.0) for 20 min, and then substrate was added. The reactions were further incubated for 10, 20, and 30 min before adding 100 μl of stop solution, and optical density (OD) was recorded immediately at 660 nm in an ELISA plate reader (BioTek, Powerwave XS). All assays were performed in triplicate and were repeated using freshly purified GlgB protein. Suitable positive and negative controls were maintained throughout the experiment. The percentage inhibition was calculated as follows,
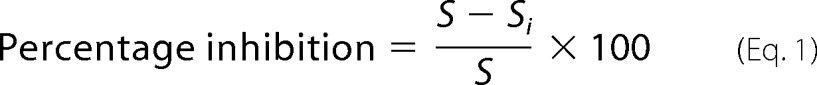 |
where S is the amount of substrate utilized by enzyme in the absence of inhibitor, and Si is the amount of substrate utilized by enzyme in the presence of inhibitor.
M. tuberculosis Growth Inhibition Assay
Bacterial Growth Conditions and Drug Preparation
M. tuberculosis virulent strain H37Rv and avirulent strain H37Ra were grown in Middlebrook 7H9 broth (Difco) along with the supplements: 0.2% (v/v) glycerol, 10% (v/v) OADC (oleic acid, albumin, dextrose, catalase; from BD Biosciences), and 0.05% Tween 80 (Sigma-Aldrich). Cultures were incubated at 37 °C at 100 rpm in a shaker incubator, and the growth was monitored by OD at 600 nm using a spectrophotometer. Log phase culture at 0.6–0.8 OD was used for experiments, and inocula were prepared by washing with 1× PBS and syringing with a 1-ml insulin syringe 10–15 times to obtain a homogeneous clump-free bacteria. Stock solutions of the candidate drugs were prepared in DMSO or deionized water. Standard frontline drugs like isoniazid (INH) and ethambutol were dissolved in deionized water, whereas rifampicin was dissolved in DMSO. Dilutions of the drugs were made in the 7H9 broth medium, added with the required supplements. pSC301 GFP vector capable of expressing in Mycobacterium sp. and E. coli was kindly provided by Dr. Yossef Av-Gay (University of British Columbia, Vancouver, Canada). GFP-H37Rv was made by electroporation and selection as described previously (37, 38).
Turbidity Assay
The M. tuberculosis growth-inhibitory property of different drugs was preliminarily tested by monitoring the turbidity of the culture tubes at different concentrations (μg/ml) of the drugs. Stock solutions of drugs in their respective solvents were subsequently diluted into a working concentration using the 7H9 broth medium. We set up the screening with a 6 log concentration series around the reported minimum inhibitory concentration (MIC) values of the standard drugs, like INH (0.25 μg/ml), rifampicin (0.25 μg/ml), and ethambutol (8 μg/ml). The first tube (sterile screw-capped glass tubes) of each row of the control and test drugs tubes was added with 0.25 × 103 μg/ml concentration (volume in each tube was 2 ml). Subsequently, from tube 1 to tube 7, a 10-fold serial dilution was made (0.2 ml was added from tube 1 to tube 2 and so on. 200 μl of log phase culture (OD of 0.6–0.8) of M. tuberculosis was added as inoculum (∼104 colony-forming units (cfu)) to tubes 1–7, and tube 8 was kept as blank and only drug was added, whereas tube 9 was a positive control for M. tuberculosis growth with no drug. The tubes were properly sealed and incubated at 37 °C in a shaker incubator and monitored regularly to check for growth and contamination in the blank or control tubes. Growth was generally observed at 8–10 days postinoculation. The anti-M. tuberculosis property of the drugs was correlated to the turbidity of the cultures, meaning that the tubes having no growth at all were scored as potential inhibitors and considered for further screening with other confirmatory techniques.
Microwell Plate Alamar Blue Assay (MABA)
The growth inhibition or drug susceptibility of M. tuberculosis was tested against different candidate drugs using a resazurin-based oxidation-reduction dye, Alamar Blue, which is capable of delivering MICs of different M. tuberculosis isolates within a week's time. The Alamar Blue dye can indicate the cellular growth and metabolism of any bacteria based on the color conversion from blue to pink. The blue color is an oxidized form that is non-fluorescent, whereas the pink color indicates the reduced form and hence can be detected for its fluorescence. The growth of the bacilli can be monitored using a fluorimeter or spectrophotometer or a visual color change (39). For this assay, instead of a glass tube, we used microwell plates, and unlike the final volume of 2 ml in the glass tubes, we used 200 μl. The stock solutions of the drugs and the dilutions made were similar to those in the turbidity assay. The peripheral wells of a sterile flat bottomed 96-well plate were loaded with 200 μl of sterile water to avoid evaporation of the remaining experimental wells during the incubation period. A 20-μl log phase culture of M. tuberculosis (∼103 cfu) was added to all experimental wells except for drug only and blank controls (negative controls). For confirmatory experiments to determine narrow range MICs for the test drugs, serial dilutions of 1:2 were performed to make 250, 125, 62.5, 31.25, 15.62, 7.81, and 3.9 μg/ml experimental sets. For fluorescence measurement, background subtraction was made using negative controls. Microwell plates were sealed and carefully incubated at 37 °C. On day 5, we added 50 μl of 1:1 diluted Alamar Blue (in 7H9 with 10% Tween 80) to the positive control (M. tuberculosis but no drug). The plates were reincubated at 37 °C for another 24 h. On the following day, if the positive control well turned to a pink color, Alamar Blue was added to all of the wells (if not, another positive control well was added with Alamar Blue and monitored after 24 h). After 24 h of incubation, the color changes were monitored visually and were noted. Also, the fluorescence of the color changes was captured using a fluorimeter with an excitation wavelength of 560 nm and emission at 610 nm. Visually, MIC was scored as the minimum dilution at which there was no blue to pink color change, and the fluorescence value was interpreted after subtraction of the background fluorescence of the negative controls.
Macrophage Differentiation
THP-1 cells (National Centre for Cell Science, Pune, India) were maintained in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Invitrogen), 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) in a 5% CO2 incubator at 37 °C. Cells were differentiated into macrophages by PMA treatment at a concentration of 30 ng/ml for 12 h followed by a 24–48-h resting period.
M. tuberculosis Survival and Phagocytosis Assay by cfu, Flow Cytometry, and Imaging
For assays, M. tuberculosis strains were treated with MB16695 and IMT007 at a concentration of 15.6 and 125 μg/ml, respectively, for 3 days. THP-1 macrophages were infected with M. tuberculosis strains at a multiplicity of infection of 1:5 for 4–6 h. Infected macrophages were treated with drug at the concentration and time indicated. DMSO-treated M. tuberculosis culture was used as control. Any unphagocytosed or extracellular bacilli were washed with PBS, and fresh medium containing the diluted drugs was added. For the bacterial survival assay, later at day 3, the macrophages were lysed with 0.06% SDS and serially diluted. The solubilized diluted lysate was plated into 7H11 agar plates containing OADC, and plates were incubated at 37 °C. The cfu was evaluated by counting the colonies formed, and the average was considered for triplicate sets. Also, the solubilized suspension was used for the LIVE/DEAD BacLight Bacterial Viability and Counting Kit (Invitrogen) consisting of SYTO 9 and propidium iodide dye. Flow cytometry was performed to determine the percentage of live and dead bacilli. For the colocalization assay, repletion medium was supplemented with MB16695 at concentrations of 30 and 150 μg/ml and with IMT007 at concentrations of 150 μg/ml. DMSO was used as a solvent control to treat the bacteria. After 24 h of incubation, M. tuberculosis H37Rv-GFP-infected macrophage cells were treated with 100 nm LysoTracker for 30 min at 37 °C and fixation with 4% paraformaldehyde. The stained cells were washed thoroughly with 1× PBS and mounted on slides with antifade reagent. For the phagocytosis assay, infected cells were washed three times with 1× PBS after 4–6 h of infection and then fixed with 4% paraformaldehyde, followed by imaging by confocal microscopy. The cells were observed with a Nikon A1R confocal microscope. The phagocytic index was calculated according to the formula, phagocytic index = (total number of engulfed bacterial cells/total number of counted macrophages) × (number of macrophages containing engulfed bacterial cells/total number of counted macrophages) × 100 (40).
RESULTS
Virtual Screening and Chemical Similarity
Selection of Inhibitors
The refined models of M. tuberculosis and human GlgB were used for HTVS and docking as described under “Experimental Procedures,” and the protein-ligand complexes were analyzed for the active site residues involved in hydrogen bonding. The catalytic active site of M. tuberculosis GlgB contains the seven residues (17) (Asp-341, His-346, Arg-409, Asp-411, Glu-464, His-531, and Asp-532, M. tuberculosis GlgB numbering) that are also conserved in human GlgB (Asp-286, His-291, Arg-355, Asp-357, Glu-412, His-480, and Asp-481, human GlgB numbering). The ligands that interacted with at least one of the active site functional residues of M. tuberculosis, but not the human protein, were analyzed and further narrowed down to 8 (MB) and 9 (ZINC) hits, respectively. Their binding energy, chemical structure, and interacting M. tuberculosis GlgB residues are shown in Table 1. These 17 diverse molecules were subjected to further in silico ADMET properties and biological validation for inhibition of GlgB enzymatic activity (supplemental Table S1).
TABLE 1.
Glide XP results for the 17 lead molecules using Schrodinger version 9.0
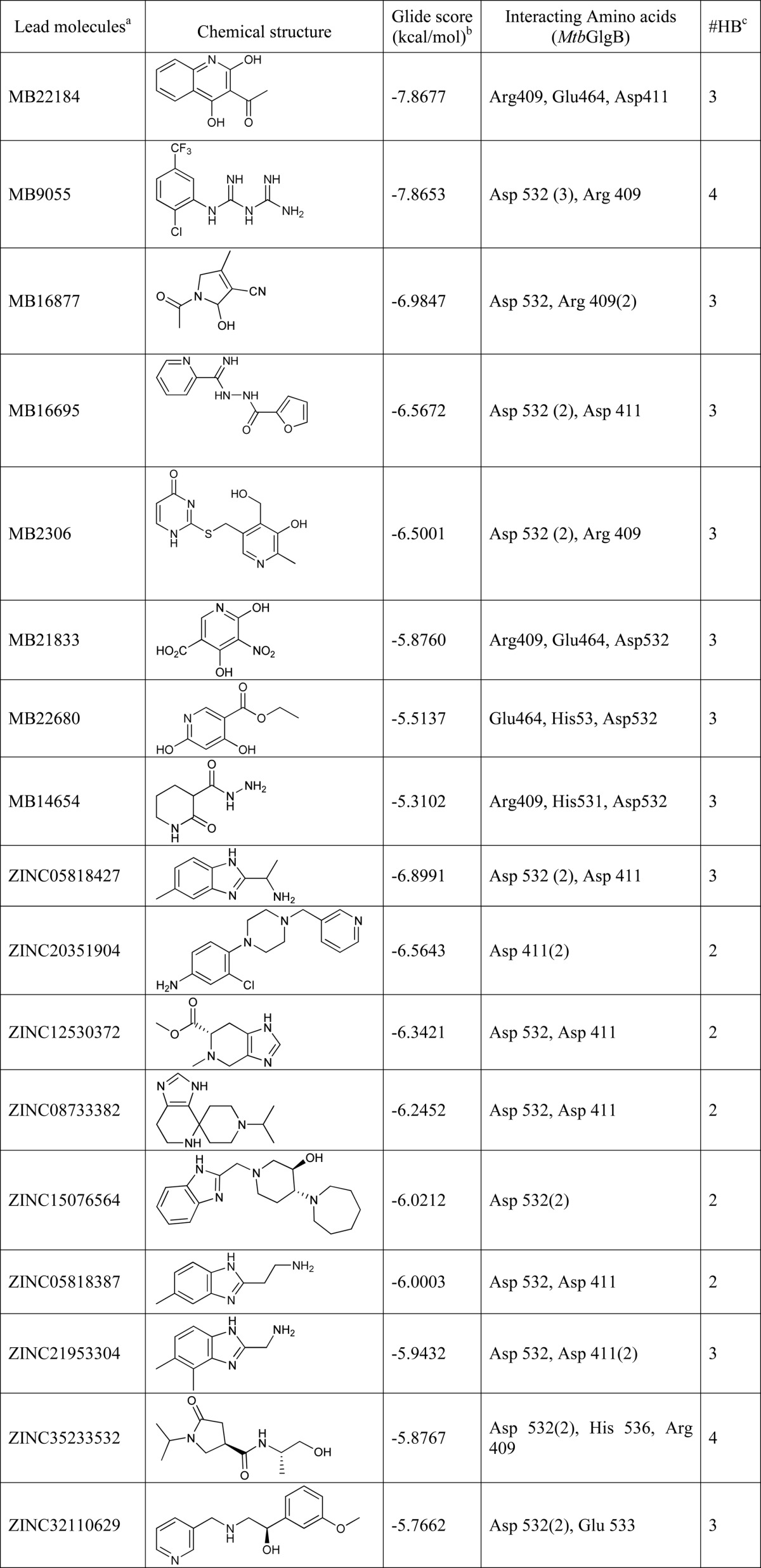
a Ligand IDs are of the Maybridge and ZINC databases.
b Glide score.
c Number of hydrogen bonds formed.
Bioavailability of the Lead Molecules
The druglike properties of the lead compounds were assessed by evaluating their physicochemical properties using QikProp version 3.2 from Schrodinger. Their molecular weights, the numbers of hydrogen bond donors and acceptors, and log values were within the guidelines and acceptable range of Lipinski's rule of five for druglike molecules (Table 2). Furthermore, the pharmacokinetic parameters of the lead molecules, which include absorption, distribution, metabolism, excretion, and toxicity (ADMET) were analyzed using QikProp version 3.2.
TABLE 2.
QikProp properties of the 17 shortlisted lead molecules using Schrodinger version 9.0
| Lead moleculesa | QP log Po/wb | QP log Sc | Mrd | QP log HERGe | No. of metabf | HAg | HDh | Human oral absorptioni |
|---|---|---|---|---|---|---|---|---|
| % | ||||||||
| MB22184 | 2.042 | −2.586 | 203.1 | −4.103 | 3 | 1.7 | 1.0 | 60.2 |
| MB9055 | 1.828 | −1.647 | 226.2 | −4.384 | 2 | 3.7 | 1.0 | 59.3 |
| MB16877 | −0.937 | −0.252 | 166.1 | −1.810 | 4 | 5.2 | 1.0 | 61.7 |
| MB16695 | 0.865 | −2.194 | 230.2 | −5.264 | 2 | 6.0 | 3.0 | 80.1 |
| MB2306 | 0.615 | −2.953 | 279.3 | −5.085 | 1 | 6.9 | 3.0 | 67.5 |
| MB21833 | −0.280 | −1.064 | 200.1 | −1.366 | 2 | 3.7 | 2.0 | 37 |
| MB22680 | 1.439 | −2.113 | 183.1 | −3.818 | 4 | 2.7 | 1.0 | 77.3 |
| MB14654 | −0.766 | −0.017 | 157.1 | −1.417 | 3 | 3.5 | 2.0 | 55.99 |
| ZINC05818427 | 0.957 | −1.12 | 175.2 | −4.838 | 6 | 2.5 | 3.0 | 78.1 |
| ZINC20351904 | 2.478 | −3.196 | 302.8 | −5.978 | 1 | 5.5 | 1.5 | 80 |
| ZINC12530372 | 0.185 | −0.933 | 195.2 | −4.444 | 3 | 5.5 | 1.0 | 70 |
| ZINC08733382 | 1.154 | −1.000 | 234.3 | −5.234 | 2 | 4.5 | 2.0 | 59 |
| ZINC15076564 | 1.802 | −2.185 | 328.4 | −3.594 | 2 | 7.2 | 2.0 | 73.2 |
| ZINC05818387 | 0.756 | −0.969 | 175.2 | −4.830 | 4 | 2.5 | 3.0 | 72.3 |
| ZINC21953304 | 0.849 | −0.966 | 175.2 | −4.513 | 3 | 2.5 | 3.0 | 76.2 |
| ZINC35233532 | −0.523 | −0.383 | 228.2 | −0.858 | 2 | 7.2 | 2.0 | 66.1 |
| ZINC32110629 | 1.977 | −1.791 | 258.3 | −4.052 | 4 | 2.75 | 1.0 | 79.1 |
a Ligand IDs are of the Maybridge database and ZINC database.
b Predicted octanol/water partition co-efficient log P (acceptable range, −2.0 to 6.5).
c Predicted aqueous solubility; S in mol/liter (acceptable range, −6.5 to 0.5).
d Molecular weight (<500).
e Predicted IC50 value for blockage of HERG K+ channels (acceptable range, below −5.0).
f Number of metabolic reactions (from 1 to 8).
g Hydrogen bond acceptors (<10).
h Hydrogen bond donors (<5).
i Percentage of human oral absorption <25% is poor and >80% is high).
Three- and Two-dimensional Shape Comparison
As an additional support to our high throughput docking efforts, we also initiated three-dimensional shape-based as well as binary fingerprint screening, using the structure of the hit molecules obtained from HTVS. These hits were used as query molecules for finding additional hits from our in-house database. Fig. 1 shows some of these molecules, along with their three-dimensional shape, color, and two-dimensional similarity Tanimoto coefficients. Using this three-dimensional and two-dimensional finger print comparison, we selected a further 29 compounds for experimental validation for inhibition of GlgB enzymatic activity (see supplemental Table S2 for detailed information about these molecules).
FIGURE 1.

Schematic representation of shape similarity, functional group comparison, and two-dimensional similarity of virtual screening query molecule MB16695 with the in house database molecules.
In Vitro Evaluation of the Virtual Hits
GlgB Inhibition
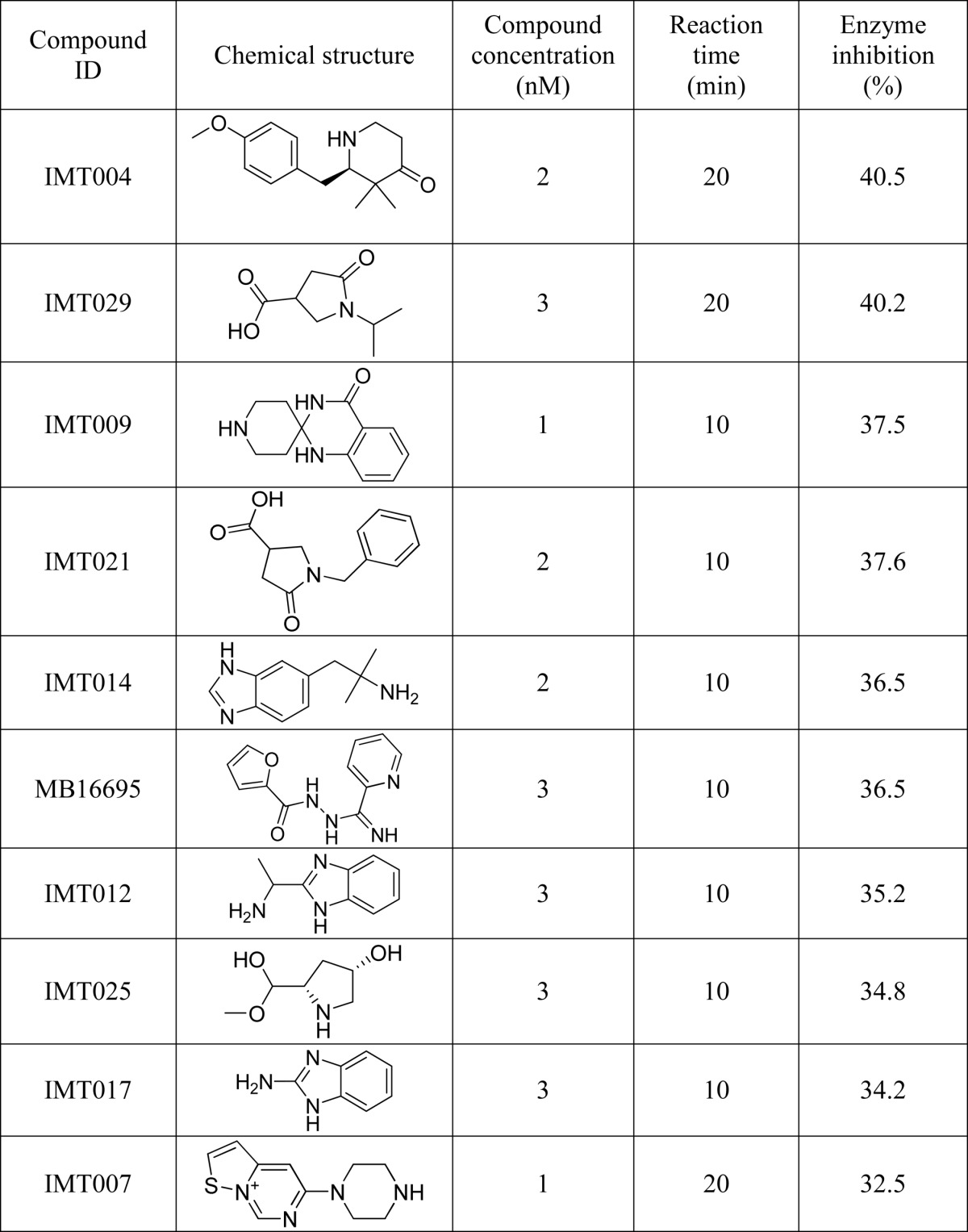
GlgB was purified using nickel-nitrilotriacetic acid resin and dialyzed against 50 mm phosphate buffer, pH 7.0. Recombinant purified GlgB is a monomer and globular in nature and has one disulfide bond, which did not seem to affect the enzyme activity. All hit molecules obtained from virtual screening (docking and chemical similarity search), except for those that were insoluble or sparingly soluble in DMSO, were tested for the inhibition of M. tuberculosis GlgB activity at different time points and different concentrations (supplemental Tables S1 and S2). At any given time, the DMSO concentration was not more than 5% in the reaction mixture. Based on the criterion of maximum GlgB inhibition in minimum time (Table 3), we selected 10 molecules to further evaluate their efficacy to inhibit in vitro M. tuberculosis growth. The maximum inhibition (enzymatic assay) was shown by compounds IMT004, IMT029, IMT009, and IMT021.
TABLE 3.
Top ten hits identified for inhibiting Mtb GlgB enzymatic activity
Screening for M. tuberculosis Growth-inhibitory Activity
The discovery of various anti-TB drugs is based on the growth susceptibility to drug treatment. We have used three strategies to monitor the growth inhibition of M. tuberculosis (H37Ra and H37Rv) by 10 of our candidate drugs selected based on their enzyme activity. First, the direct measure of the growth was monitored by observing the turbidity in a sterile glass culture tubes. Second, MABA was used to check the inhibition of metabolic activity of the bacilli upon drug treatment. The reduction of the indicator dye was monitored visually for the pink color and used for reading of MICs. Third, the fluorescence intensity of the microwell plates was determined using a fluorimeter as a confirmatory read out. Unlike the sophisticated liquid broth growing techniques like the BACTEC, which require 7–14 days for screening, tetrazolium- or resazurin-based oxidation-reduction dyes are being efficiently used for simple, fast, and inexpensive high throughput drug susceptibility screening and the acquisition of MIC values against different microorganisms, including M. tuberculosis (41, 42). Preliminary standardization was done with M. tuberculosis strains H37Ra and H37Rv using the three frontline drugs (INH, rifampicin, and ethambutol) as standards. The assays were successfully established, and results with the standard drugs matched with already reported MICs. In our study, we preferred INH and rifampicin in the initial broad range screening due to their lower MICs. In the later narrow range MIC screening, we preferred ethambutol. We started our screening with a basic turbidity test to directly visualize the growth of M. tuberculosis (both H37Ra and H37Rv) upon incubation with the candidate test drugs. Drugs MB16695 and IMT007 were the only candidates that seem to have an M. tuberculosis growth-inhibitory activity because there was no growth in their tubes corresponding to 0.25 × 103 μg/ml (Table 4 and Fig. 2A) in the concentration range evaluated (0.25 × 103 to 0.25 × 10−3 μg/ml). The remaining compounds had no effect on the growth even at the higher concentration of 250 μg/ml (Table 4). A similar pattern was observed when the MABA assay was performed, confirming the M. tuberculosis growth inhibition by the compounds MB16695 and IMT007 (Table 4 and Fig. 2B). The minimum concentration at which the color remains blue was considered as the possible MIC for the compounds MB16695 and IMT007. Also, the fluorescence signal was acquired using a fluorimeter, and after background subtraction, we found a growth inhibition pattern similar to the visual reading.
TABLE 4.
Screening of the top 10 hits inhibiting GlgB enzyme activity for M. tuberculosis growth inhibition
| Drugs (0.25 × 103 to 0.25 × 10−3μg/ml) | Visual turbidity |
Visual Alamar Blue pink color change |
Fluorescence Alamar Blue |
|||
|---|---|---|---|---|---|---|
| H37Rv | H37Ra | H37Rv | H37Ra | H37Rv | H37Ra | |
| IMT004 | All | All | All | All | ||
| IMT029 | All | All | All | All | ||
| IMT009 | All | All | All | All | ||
| IMT021 | All | All | All | All | ||
| IMT014 | All | All | All | All | ||
| MB16695 | 25 μg/ml and below | 25 μg/ml and below | 25 μg/ml and below | 25 μg/ml and below | 25–250 μg/ml | 25–250 μg/ml |
| IMT012 | All | All | All | All | ||
| IMT025 | All | All | All | All | ||
| IMT017 | All | All | All | All | ||
| IMT007 | And below 25 μg/ml | 25 μg/ml and below | 25 μg/ml and below | 25 μg/ml and below | 25–250 μg/ml | 25–250 μg/ml |
| INH | 0.025 μg/ml and below | 0.025 μg/ml and below | 0.025 μg/ml and below | 0.025 μg/ml and below | 0.025–0.25 μg/ml | 0.025–0.25 μg/ml |
FIGURE 2.

Growth inhibition of M. tuberculosis H37Rv. A, preliminary turbidity screen conducted to monitor inhibition on the growth of M. tuberculosis H37Rv upon treatment with different concentrations of the 10 candidate drugs (listed in Tables 3 and 4). MB16695 and IMT007 showed no growth at all at a 250 μg/ml concentration. Standard INH also shows no growth pattern for tubes with concentrations of 0.25 μg/ml and above. The remaining drugs had no effect on the growth even at higher concentration of 250 μg/ml (arrowhead indicates the maximum concentration of the drugs with any growth observed as described in Table 4). B, a MABA assay was performed to monitor the metabolic activity of the growing bacteria. The growth of M. tuberculosis H37Rv was indicated by the reduction of the dye producing a pink color, which was visually seen, and a fluorescence reading was also acquired. The 96-well plate shows wells corresponding to 250 μg/ml MB16695 and IMT007 with no color change (remaining blue), whereas the remaining drugs had all of the wells turned into a pinkish color (arrowhead indicates the non-reduced form of the dye representing the growth inhibition).
In order to achieve a narrow range of MIC values for both MB16695 and IMT007, we carried out the same turbidity and MABA assay using the concentrations 250, 125, 62.5, 31.25, 15.62, 7.81, and 3.9 μg/ml by 1:2 serial dilutions. Both visual readings and fluorescence MABA results indicated that MB16695 and IMT007 inhibit the growth of M. tuberculosis at a concentration range of 25–31.2 and 125–250 μg/ml, respectively (Table 5 and Fig. 3).
TABLE 5.
MIC values for MB16695 and IMT007
| Drugs (0.25 × 103 to 3.9 μg/ml) | Visual Alamar Blue pink color change |
Fluorescence Alamar Blue |
||
|---|---|---|---|---|
| H37Rv | H37Ra | H37Rv | H37Ra | |
| μg/ml | μg/ml | |||
| MB16695 | 15.6 and below | 15.6 and below | 15.6–31.2 | 15.6–31.2 |
| IMT007 | 125 and below | 125 and below | 125–250 | 125–250 |
FIGURE 3.
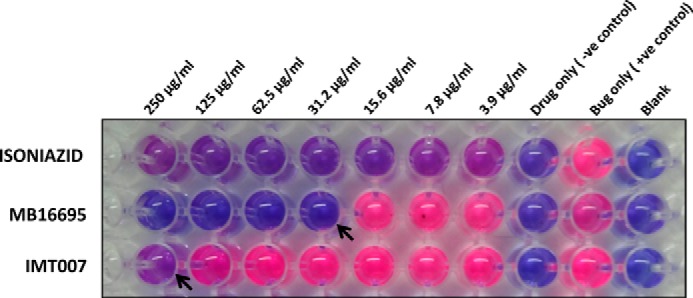
Confirmation of candidate drug's MIC value. The MIC value for MB16695 and IMT007 was evaluated by a MABA assay in the concentration range of 3.9–250 μg/ml. Visual reading and fluorescence results show MIC values of 25–31.2 and 125–250 μg/ml for MB16695 and IMT007, respectively.
Docking and Mutagenesis Studies
Because MB16695 and IMT007 were the only compounds showing significant inhibitory effects, to determine their probable binding conformation and interaction, we docked these compounds into the active site of M. tuberculosis GlgB (Fig. 4, A and C) and subsequently checked for their propensity to bind human GlgB (Fig. 4, B and D). The crystal structures of M. tuberculosis and human GlgB were used for docking studies (17). The analysis of the MB16695-M. tuberculosis GlgB complex reveals that in the most likely binding pose, the sulfur atom of MB16695 acts as a hydrogen bond donor to Ser-466, whereas the hydrogen atoms of the ligand serve as hydrogen bond acceptors from Asp-532. Compound MB16695 occupies a groove and makes significant hydrophobic contacts with Asp-341, His-346, Arg-409, Asp-411, Ala-412, Ala-414, Ser-415, Glu-464, Glu-465, Ser-466, Met-489, His-531, and Asp-532, whereas it interacts with only five residues (Asp-286, His-291, Trp-322, Arg-355, Asp-357, and Glu-412) of human GlgB. No hydrogen bond is observed between MB16695 and human GlgB. Compound IMT007 establishes a network of interactions with amino acids Asp-341, His-346, Arg-409, Asp-411, Glu-464, Ser-466, Met-489, His-531, Asp-532, Glu-533, His-536, and Lys-538 of M. tuberculosis GlgB. The oxygen atom of IMT007 acts as hydrogen bond donor to Lys-538, whereas its hydrogen atoms make four hydrogen bonds with Asp-532, Glu-533, and Lys-538. On the other hand, IMT007 shows no significant interaction with human GlgB. On the other hand, weak binding interaction is observed in the case of human GlgB with residues Tyr-251, Asp-286, His-291, Trp-322, Arg-355, Asp-357, and Asp-481. Although the active site residues are conserved in both M. tuberculosis and human GlgB, conformational sampling and structural fluctuations may contribute toward differential binding of the ligands. It has been shown that ligand binding depends on the microenvironment of the enzyme active sites, and the conformational dynamics of the protein plays an important role in this binding event (43, 44). The ligand binding depends not only on the structural/shape complementarities of the protein but also on its chemical properties. These include the amino acids and their capability to polarize bonds and make covalent adducts etc. The binding affinity of protein is attributed to the conformational changes at the enzyme active site. Hence, orthologous proteins in two different organisms can contribute in a dissimilar way toward the binding of ligands. Further, an experimental evaluation of MB16695 and IMT007 inhibition of human GlgB enzyme activity was performed, which indeed confirmed that these compounds are not able to inhibit human GlgB enzyme activity and as such validated the above explanation (Fig. 4E). In order to ascertain that the compounds MB16695 and IMT007 inhibits M. tuberculosis GlgB activity by interacting with its active site residues, we mutated some active site residues of M. tuberculosis GlgB that were predicted to be important for binding of these compounds. Residues Asp-411, Met-489, Asp-532, and Lys-538 were mutated to alanine. All mutant M. tuberculosis GlgB proteins were purified, and an enzyme inhibition assay was performed (Fig. 4F). Mutant proteins D411A, M489A, and D532A completely lost their enzymatic activity; however, K538A retained its enzyme activity, and interestingly, compounds MB16695 and IMT007 were unable to inhibit its activity. Lys-538 is predicted to be important for hydrogen bonding with IMT007, and its mutation to alanine abrogated the binding of the compound and inhibits M. tuberculosis GlgB enzyme activity. However, the failure of MB16695 to inhibit K538A could be attributed to conformational changes. This proves that binding of both the compounds involves the predicted binding site.
FIGURE 4.

Docking comparison of MB16695 and IMT007 docked into the active site of M. tuberculosis and human GlgB. A and B, ligand IMT007 docked into M. tuberculosis and human GlgB, respectively; C and D, molecule MB16695 docked. Proteins are shown in gray (schematic), and ligands are shown in green (sticks); dashes (purple) are hydrogen bonds, and their distances are shown in Å. E, F, comparison of enzyme activity of M. tuberculosis (E) and human (F) GlgB (wild type and mutant (K538A)) in the presence or absence of MB16695 or IMT007 and at a concentration of 5 nm each. Mutant M. tuberculosis GlgB enzyme activity is normalized to wild type M. tuberculosis GlgB enzyme activity. A–D, data are representative of three independent experiments with similar results. E–F, data are represented as mean ± S.D. (error bars) of three independent experiments each performed in triplicate. *, p < 0.05.
MB16695 and IMT007 Inhibition of M. tuberculosis Growth in Infected Macrophages and Phagocytosis
To evaluate the MIC values of the candidate drugs for M. tuberculosis growth in infected macrophages, the dose-dependent antimicrobial activity of MB16695 and IMT007 in macrophage was determined. M. tuberculosis-infected macrophages were treated with both MB16695 and IMT007 at various concentration ranges, and the viability of the bacilli was enumerated in cfu (Fig. 5, A and B). The intracellular survival assay was performed with both avirulent (H37Ra) and virulent (H37Rv) M. tuberculosis. In comparison with the infected control cells (no drug treatment), MB16695-treated cells had an effect of 5–10-fold reduction in the survival of both H37Ra and H37Rv at 30 μg/ml and above, whereas IMT007-treated cells had a ∼5-fold reduction at 150 μg/ml. Similarly, the flow cytometry analysis of the percentage of dead bacilli in the case of H37Ra also clearly indicates the inhibitory effect of both the drugs MB16695 and IMT007 at a concentration of 30 and 150 μg/ml, respectively (Fig. 5C). In addition, the standard control drug, INH, had an inhibitory effect at a lower concentration of 0.2–0.3 μg/ml, as already reported. In conclusion, the in vitro or intracellular activity of the compounds (MB16695 and IMT007) against M. tuberculosis supports the MIC values.
FIGURE 5.

Intracellular antimicrobial activity of MB16695 and IMT007 against M. tuberculosis in human THP-1 macrophages. M. tuberculosis-infected macrophage cells were treated with varying concentrations of candidate drugs (MB16695 (orange), IMT007 (green), or vehicular control (red)) ranging from 15 to 150 μg/ml. H37Rv (A) and H37Ra (B) intracellular survival was determined by counting the cfu formation after the drug treatments. C, flow cytometry analysis of the percentage of dead bacilli (H37Ra) upon respective drug treatment. cfu counts are plotted as the mean ± S.D. (error bars), and flow cytometry results are plotted as the median. D, macrophages infected with drug-incubated GFP-M. tuberculosis H37Rv were treated with MB16695 (30–150 μg/ml) and IMT007 (150 μg/ml) at the indicated concentrations for 24 h and stained with LysoTracker Red for the determination of bacterial survival. Data are representative of three independent experiments with similar results (n = 3; *, p < 0.05). Each experiment was performed in triplicate.
Earlier literature on α-glucan suggests its potential role in the induction of innate immune response. Further, α-glucan has been demonstrated to be essential for the phagocytosis of fungal pathogen and induction of proinflammatory cytokines by a mechanism involving TLR2, CD14, and Myd88 (45). GlgB inhibitors affect the biosynthesis of α-glucan in M. tuberculosis, so we investigated the effect of the inhibitors on the uptake and the survival of the bacteria in the macrophage. Inhibitor-treated M. tuberculosis was used to infect the THP-1 macrophages. Confocal microscopy of LysoTracker Red-stained acidic vacuoles showed that MB16695 treatment of bacteria and cells leads to less bacterial burden inside the cells and colocalization with lysosomes in comparison with the control and a slight change in those treated with IMT007 treatment (Fig. 5D). The low number of bacteria upon MB16695 treatment is also suggestive of less phagocytosis. Because α-glucan is indicated to be important for efficient phagocytosis, we determined the bacterial burden inside the macrophages by a phagocytosis assay of shorter duration (4 h). It was observed that MB16695-treated M. tuberculosis H37Rv-GFP was much less phagocytosed as compared with IMT007-treated M. tuberculosis H37Rv-GFP and control (Fig. 6A). Quantitative determination of the number of bacteria per cell and the phagocytic property of macrophages, phagocytic index, was performed (Fig. 6, B and C).
FIGURE 6.

Effect of MB16695 and IMT007 on the phagocytosis of M. tuberculosis. A, THP-1 macrophages were infected with GFP-M. tuberculosis H37Rv preincubated in MB16995 at a concentration of 15.6 μg/ml and IMT007 at a concentration of 125 μg/ml for 4 h. Cells were fixed and imaged by a confocal microscope for the quantification of the total number of engulfed bacteria by macrophages (B) and the phagocytic index (C). Data are representative of three independent experiments with similar results (n = 3; *, p < 0.05). Error bars, S.D.
DISCUSSION
Structure and ligand-based virtual screening approaches have been used previously to design inhibitors for important targets, such as FabI and M. tuberculosis malate synthase (46, 47). It has been shown that virtual screening involving the fusion of structure- and ligand-based methods is an effective tool to achieve a higher hit rate and provide wider structural coverage of chemical space (30), and a combination of these methodologies was used in this study. A ligand was accepted if it bound to at least one conserved active site residue because it is likely that it would interfere with the binding or catalysis of substrate and inactivate the protein function. Out of the 333,761 database molecules used for docking, 400 hits from the MB database and 1,020 hits from the ZINC database were initially retained with the HTVS mode. The Glide XP cut-off score was used to identify low or no affinity ligands, which were then discarded. These initial hits were further narrowed down to 75 (MB) and 128 (ZINC), based on their pharmacokinetics properties. This list was further narrowed down to 17 (8 from MB and 9 from ZINC database) based on the interaction of ligands with at least one of the active site residues of M. tuberculosis and not the human sequence. The partition coefficient (QP log Po/w) and water solubility (QP log S), which are critical for the estimation of absorption and distribution of drugs within the body, ranged from ∼−0.9 to ∼2.23 and from ∼−0.05 to ∼−3.65, respectively, whereas the bioavailability and toxicity were ∼−0.1 to ∼0.3. Overall, the percentage human oral absorption of the compounds ranged from ∼57 to ∼72%. These pharmacokinetic parameters lie within the acceptable range defined for human use, thus indicating their potential as druglike molecules. These compounds have diverse structures and functional groups; for example, compound IMT004 is methoxybenzyl piperidinone derivative, compound MB16695 is furancarbohydrazide derivative, and compound IMT007 is isothiazole derivative. Molecular docking predicts that all of these compound could interact with active site residue Asp-532 of M. tuberculosis GlgB, whereas compounds MB16695 and IMT007 show additional interaction with the residue Arg-409. Both the turbidity and MABA results suggested that among the broad range concentrations of the drugs used, MB16695 and IMT007 exhibited an MIC value in the range of 25–250 μg/ml. The sensitivity, specificity, and validity of both of the assays were based on the activity of the positive and negative controls and the standard used. Thus, our screen successfully identified two important compounds, MB16695 and IMT007, with different MIC on M. tuberculosis (H37Ra and H37Rv strains) growth. Compound IMT007 shows three-dimensional shape similarity to MB16695 and also exhibited significant inhibition of M. tuberculosis GlgB enzymatic activity and in vitro growth. Thus, these compounds represent new scaffolds for targeting M. tuberculosis GlgB. These results clearly show that docking- and ligand-based screening as a combined effort will be useful in discovering new molecular scaffolds for targeting important bacterial cell wall enzymes as these two compounds demonstrated better antimycobacterial activity, indicating that these particular molecular scaffolds are better for further derivatization and could potentially be developed as highly selective inhibitors for M. tuberculosis GlgB using the structure-activity relationship through medicinal chemistry applications. The results also indicate that M. tuberculosis GlgB can be targeted at different sites. Although, due to the lack of ligand-bound crystal structure, it is not possible to predict which molecule targets a particular amino acid residue, we believe that these compounds interact differently because they show different binding capacity and also have different chemical scaffolds. Mutagenesis studies confirm the role of Lys-538 in binding to IMT007 and MB16695. Whereas the former could be attributed to hydrogen bonding, the latter seemingly is an indirect effect. Therefore, it may be possible to develop each one independently as a lead molecule for further inhibition studies toward M. tuberculosis GlgB. It will also be possible to use the structural information of ligand-protein interaction obtained from molecular docking to develop further derivatives of MB16695 and IMT007, leading to rational drug design of selective M. tuberculosis GlgB inhibitors.
In this work, we have used a unique strategy of high throughput docking and the use of diverse ligand databases to explore a novel chemical space for inhibiting M. tuberculosis GlgB. Using three-dimensional shape matching, we were able to expand the chemical diversity for finding new series of chemical scaffolds for inhibiting M. tuberculosis GlgB. The lead molecules were further validated for M. tuberculosis GlgB enzyme- and growth-inhibitory activity. On an initial screen, compounds MB16695 and IMT007 have significant growth-inhibitory effects and can further be optimized by scaffold analysis using medicinal chemistry applications. MB16695 and IMT007 inhibit the enzyme activity of branching enzyme GlgB and alter the biosynthesis of α-glucan, a vital cell wall and storage polysaccharide. It has been shown in the literature that inactivation of GlgB results in toxicity due to accumulation of maltose 1-phosphate (14). We evaluated that the observed MIC values for the individual drugs were effective in infected macrophages as well. We also show by confocal imaging that compound MB16695 results in less uptake of bacteria due to defective α-glucan synthesis and slow clearance from the macrophage cells. Compound IMT007 also affects the uptake of bacteria but to a lesser extent. Reduction in the phagocytic index of MB16695-treated cells indicate marked the inhibitory effect of the compound.
This work proves the utility of both structure- and ligand-based virtual screening approaches to identify hit compounds for difficult targets, such as GlgB, for which no prior small molecule inhibitors are reported. We are currently exploring the future studies involving the structure-activity relationship of the scaffolds through computational and medicinal chemistry efforts, screening of other diverse libraries, testing of potent lead molecules for additional pathogenic bacteria, and in vivo pharmacokinetic studies.
Acknowledgment
We thank the Council of Scientific and Industrial Research Institute of Microbial Technology for providing facilities. R.P. thanks OpenEye for free academic license.
This work was supported by the Council of Scientific and Industrial Research (CSIR) 12th Plan project Infectious Disease (BSC0210), Genesis (BSC0121), and OLP projects (to P. G., R. P., and P. A.).

This article contains supplemental Tables S1 and S2.
- TB
- tuberculosis
- HTVS
- high throughput virtual screening
- MABA
- microwell plate Alamar Blue assay
- MB
- Maybridge
- INH
- isoniazid
- MIC
- minimum inhibitory concentration.
REFERENCES
- 1. Mitchison D. A. (2005) The diagnosis and therapy of tuberculosis during the past 100 years. Am. J. Respir. Crit. Care Med. 171, 699–706 [DOI] [PubMed] [Google Scholar]
- 2. Wirth T., Hildebrand F., Allix-Béguec C., Wölbeling F., Kubica T., Kremer K., van Soolingen D., Rüsch-Gerdes S., Locht C., Brisse S., Meyer A., Supply P., Niemann S. (2008) Origin, spread and demography of the Mycobacterium tuberculosis complex. PLoS Pathog. 4, e1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Silhavy T. J., Kahne D., Walker S. (2010) The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2, a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kalscheuer R., Jacobs W. R., Jr. (2010) The significance of GlgE as a new target for tuberculosis. Drug News Perspect. 23, 619–624 [DOI] [PubMed] [Google Scholar]
- 5. Daffé M., Draper P. (1998) The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb. Physiol. 39, 131–203 [DOI] [PubMed] [Google Scholar]
- 6. Schwebach J. R., Casadevall A., Schneerson R., Dai Z., Wang X., Robbins J. B., Glatman-Freedman A. (2001) Expression of a Mycobacterium tuberculosis arabinomannan antigen in vitro and in vivo. Infect. Immun. 69, 5671–5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gagliardi M. C., Lemassu A., Teloni R., Mariotti S., Sargentini V., Pardini M., Daffé M., Nisini R. (2007) Cell wall-associated α-glucan is instrumental for Mycobacterium tuberculosis to block CD1 molecule expression and disable the function of dendritic cell derived from infected monocyte. Cell Microbiol. 9, 2081–2092 [DOI] [PubMed] [Google Scholar]
- 8. Geurtsen J., Chedammi S., Mesters J., Cot M., Driessen N. N., Sambou T., Kakutani R., Ummels R., Maaskant J., Takata H., Baba O., Terashima T., Bovin N., Vandenbroucke-Grauls C. M., Nigou J., Puzo G., Lemassu A., Daffé M., Appelmelk B. J. (2009) Identification of mycobacterial α-glucan as a novel ligand for DC-SIGN: involvement of mycobacterial capsular polysaccharides in host immune modulation. J. Immunol. 183, 5221–5231 [DOI] [PubMed] [Google Scholar]
- 9. Sambou T., Dinadayala P., Stadthagen G., Barilone N., Bordat Y., Constant P., Levillain F., Neyrolles O., Gicquel B., Lemassu A., Daffé M., Jackson M. (2008) Capsular glucan and intracellular glycogen of Mycobacterium tuberculosis: biosynthesis and impact on the persistence in mice. Mol. Microbiol. 70, 762–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Spatafora G., Rohrer K., Barnard D., Michalek S. (1995) A Streptococcus mutans mutant that synthesizes elevated levels of intracellular polysaccharide is hypercariogenic in vivo. Infect. Immun. 63, 2556–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nguyen B. D., Valdivia R. H. (2012) Virulence determinants in the obligate intracellular pathogen Chlamydia trachomatis revealed by forward genetic approaches. Proc. Natl. Acad. Sci. U.S.A. 109, 1263–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lombardo M. J., Michalski J., Martinez-Wilson H., Morin C., Hilton T., Osorio C. G., Nataro J. P., Tacket C. O., Camilli A., Kaper J. B. (2007) An in vivo expression technology screen for Vibrio cholerae genes expressed in human volunteers. Proc. Natl. Acad. Sci. U.S.A. 104, 18229–18234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jones S. A., Jorgensen M., Chowdhury F. Z., Rodgers R., Hartline J., Leatham M. P., Struve C., Krogfelt K. A., Cohen P. S., Conway T. (2008) Glycogen and maltose utilization by Escherichia coli O157:H7 in the mouse intestine. Infect. Immun. 76, 2531–2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kalscheuer R., Syson K., Veeraraghavan U., Weinrick B., Biermann K. E., Liu Z., Sacchettini J. C., Besra G., Bornemann S., Jacobs W. R., Jr. (2010) Self-poisoning of Mycobacterium tuberculosis by targeting GlgE in an α-glucan pathway. Nat. Chem. Biol. 6, 376–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garg S. K., Alam M. S., Kishan K. V., Agrawal P. (2007) Expression and characterization of α-(1,4)-glucan branching enzyme Rv1326c of Mycobacterium tuberculosis H37Rv. Protein Expr. Purif. 51, 198–208 [DOI] [PubMed] [Google Scholar]
- 16. Garg S., Alam M. S., Bajpai R., Kishan K. R., Agrawal P. (2009) Redox biology of Mycobacterium tuberculosis H37Rv: protein-protein interaction between GlgB and WhiB1 involves exchange of thiol-disulfide. BMC Biochem. 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pal K., Kumar S., Sharma S., Garg S. K., Alam M. S., Xu H. E., Agrawal P., Swaminathan K. (2010) Crystal structure of full-length Mycobacterium tuberculosis H37Rv glycogen branching enzyme: insights of N-terminal β-sandwich in substrate specificity and enzymatic activity. J. Biol. Chem. 285, 20897–20903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Agrawal P., Gupta P., Swaminathan K., Parkesh R. (2014) α-Glucan pathway as a novel M. tuberculosis drug target: structural insights and cues for polypharmcological targeting of GlgB and GlgE. Curr. Med. Chem. 21, 4074–4084 [DOI] [PubMed] [Google Scholar]
- 19. Irwin J. J., Sterling T., Mysinger M. M., Bolstad E. S., Coleman R. G. (2012) ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 52, 1757–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 21. Jorgensen W., Tirado-Rives J. (1988) The OPLS (optimized potentials for liquid simulations) potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110, 1657–1666 [DOI] [PubMed] [Google Scholar]
- 22. Jorgensen W. L., Maxwell D. S., Tirado-Rives J. (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 [Google Scholar]
- 23. Shivakumar D., Williams J., Wu Y., Damm W., Shelley J., Sherman W. (2010) Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 6, 1509–1519 [DOI] [PubMed] [Google Scholar]
- 24. Friesner R. A., Murphy R. B., Repasky M. P., Frye L. L., Greenwood J. R., Halgren T. A., Sanschagrin P. C., Mainz D. T. (2006) Extra precision Glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196 [DOI] [PubMed] [Google Scholar]
- 25. Halgren T. A., Murphy R. B., Friesner R. A., Beard H. S., Frye L. L., Pollard W. T., Banks J. L. (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759 [DOI] [PubMed] [Google Scholar]
- 26. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., Shenkin P. S. (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 [DOI] [PubMed] [Google Scholar]
- 27. Lipinski C. A., Lombardo F., Dominy B. W., Feeney P. J. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46, 3–26 [DOI] [PubMed] [Google Scholar]
- 28. Duffy E. M., Jorgensen W. L. (2000) Prediction of properties from simulations: free energies of solvation in hexadecane, octanol, and water. J. Am. Chem. Soc. 122, 2878–2888 [Google Scholar]
- 29. Jorgensen W. L., Duffy E. M. (2000) Prediction of drug solubility from Monte Carlo simulations. Bioorg. Med. Chem. Lett. 10, 1155–1158 [DOI] [PubMed] [Google Scholar]
- 30. Krüger D. M., Evers A. (2010) Comparison of structure- and ligand-based virtual screening protocols considering hit list complementarity and enrichment factors. ChemMedChem 5, 148–158 [DOI] [PubMed] [Google Scholar]
- 31. Halgren T. A. (1996) Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 17, 490–519 [Google Scholar]
- 32. Halgren T. A. (1996) Merck molecular force field. V. Extension of MMFF94 using experimental data, additional computational data, and empirical rules. J. Comput. Chem. 17, 616–641 [Google Scholar]
- 33. Hawkins P. C., Skillman A. G., Warren G. L., Ellingson B. A., Stahl M. T. (2010) Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 50, 572–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haigh J. A., Pickup B. T., Grant J. A., Nicholls A. (2005) Small molecule shape-fingerprints. J. Chem. Inf. Model. 45, 673–684 [DOI] [PubMed] [Google Scholar]
- 35. Mills J. E., Dean P. M. (1996) Three-dimensional hydrogen-bond geometry and probability information from a crystal survey. J. Comput. Aided Mol. Des. 10, 607–622 [DOI] [PubMed] [Google Scholar]
- 36. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cowley S. C., Av-Gay Y. (2001) Monitoring promoter activity and protein localization in Mycobacterium spp. using green fluorescent protein. Gene 264, 225–231 [DOI] [PubMed] [Google Scholar]
- 38. Chandra V., Mahajan S., Saini A., Dkhar H. K., Nanduri R., Raj E. B., Kumar A., Gupta P. (2013) Human IL10 gene repression by Rev-erbα ameliorates Mycobacterium tuberculosis clearance. J. Biol. Chem. 288, 10692–10702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martin A., Camacho M., Portaels F., Palomino J. C. (2003) Resazurin microtiter assay plate testing of Mycobacterium tuberculosis susceptibilities to second-line drugs: rapid, simple, and inexpensive method. Antimicrob. Agents Chemother. 47, 3616–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sano H., Hsu D. K., Apgar J. R., Yu L., Sharma B. B., Kuwabara I., Izui S., Liu F. T. (2003) Critical role of galectin-3 in phagocytosis by macrophages. J. Clin. Invest. 112, 389–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Collins L., Franzblau S. G. (1997) Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 41, 1004–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Franzblau S. G., Witzig R. S., McLaughlin J. C., Torres P., Madico G., Hernandez A., Degnan M. T., Cook M. B., Quenzer V. K., Ferguson R. M., Gilman R. H. (1998) Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin. Microbiol. 36, 362–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hammes G. G. (2002) Multiple conformational changes in enzyme catalysis. Biochemistry 41, 8221–8228 [DOI] [PubMed] [Google Scholar]
- 44. Petsko G. A., Ringe D. (2004) Protein Structure and Function, pp. 50–82, New Science Press, London [Google Scholar]
- 45. Bittencourt V. C., Figueiredo R. T., da Silva R. B., Mourão-Sá D. S., Fernandez P. L., Sassaki G. L., Mulloy B., Bozza M. T., Barreto-Bergter E. (2006) An α-glucan of Pseudallescheria boydii is involved in fungal phagocytosis and Toll-like receptor activation. J. Biol. Chem. 281, 22614–22623 [DOI] [PubMed] [Google Scholar]
- 46. Hevener K. E., Mehboob S., Su P. C., Truong K., Boci T., Deng J., Ghassemi M., Cook J. L., Johnson M. E. (2012) Discovery of a novel and potent class of F. tularensis enoyl-reductase (FabI) inhibitors by molecular shape and electrostatic matching. J. Med. Chem. 55, 268–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krieger I. V., Freundlich J. S., Gawandi V. B., Roberts J. P., Gawandi V. B., Sun Q., Owen J. L., Fraile M. T., Huss S. I., Lavandera J. L., Ioerger T. R., Sacchettini J. C. (2012) Structure-guided discovery of phenyl-diketo acids as potent inhibitors of M. tuberculosis malate synthase. Chem. Biol. 19, 1556–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]