

Abstract
Oncolytic virus therapy is being evaluated in clinical trials for human glioma. While it is widely assumed that the patient's immune response to the virus infection limits the therapy's utility, investigations into the specific cell type(s) involved in this response have been performed using non-specific pharmacological inhibitors or allogeneic models with compromised immunity. To identify the immune cells that participate in clearing an oncolytic infection in glioma, we used flow cytometry and immunohistochemistry to immunophenotype an orthotopic glioma model in immunocompetent mice after Myxoma virus (MYXV) administration. These studies revealed a large resident microglia and macrophage population in untreated tumours, and robust monocyte, T and NK cell infiltration 3 days following MYXV infection. To determine the role on the clinical utility of MYXV therapy for glioma, we used a combination of knockout mouse strains and specific immunocyte ablation techniques. Collectively, our experiments identify an important role for tumour-resident myeloid cells and overlapping roles for recruited NK and T cells in the clearance and efficacy of oncolytic MYXV from gliomas. Using a cyclophosphamide regimen to achieve lymphoablation prior and during MYXV treatment, we prevented treatment-induced peripheral immunocyte recruitment and, surprisingly, largely ablated the tumour-resident macrophage population. Virotherapy of CPA-treated animals resulted in sustained viral infection within the glioma as well as a substantial survival advantage. This study demonstrates that resistance to MYXV virotherapy in syngeneic glioma models involves a multi-faceted cellular immune response that can be overcome with CPA-mediated lymphoablation.
Keywords: Oncolytic virus, glioma, mouse models, glioma-infiltrating myeloid cells, immune response
Introduction
In North America, ∼25,000 new cases of malignant brain tumors were diagnosed in 2013.1, 2 Malignant gliomas (MGs) constitute 80% of these tumours, and have a universally fatal outcome with the most severe having an average survival of only 12-15 months despite our best treatment regimens.3 Clearly more effective treatments are needed.
Oncolytic virus (OV) therapy shows promise as an experimental therapeutic in MGs in preclinical models and has proven to be safe in early MG clinical trials.4, 5 This experimental therapy uses replication-competent viruses that exploit natural or engineered viral trophic restrictions to selectively infect and kill cancer cells. OV therapy is an appealing treatment strategy for several reasons. As a replicating virus it allows for self-potentiation, while the multiple mechanisms by which an OV can kill tumours could potentially overcome challenges that are faced by other targeted therapies. For instance, OVs have been demonstrated to direct anti-tumoural responses through direct cell killing, destroying tumour vasculature, eliciting anti-tumour cytokine responses, and provoking anti-tumour immune responses.6
The ability of OVs to elicit anti-tumoural effects will largely rely on their ability to infect and replicate within the tumour. This has been demonstrated in glioma xenograft models in immunocompromised mice, whereby tremendous efficacy is accompanied by robust viral replication.7-10 However, preclinical studies in immunocompetent glioma models have demonstrated that viral infection is dramatically reduced and rapidly cleared.11-15 We have found similar results in MG models using Myxoma virus (MYXV), a rabbit-specific member of the Poxviridae family that does not infect humans or other nonlagomorph species. In immunocompromised models of MGs, we found lengthy viral replication and durable responses, with treatment often resulting in ‘cures.’16-19 In contrast, we have demonstrated a lack of sustained viral replication within tumours in situ and no significant survival benefit in syngeneic glioma models that possess fully functional immune systems.11, 15
These results in immunocompetent preclinical models reflect those seen in early clinical trials, which demonstrated that viral clearance of oncolytic herpes viruses occurs rapidly after intratumoural injections and is accompanied by an effective cell-mediated antiviral-immune response.20, 21 Thus far, most MG patients do not respond to OV therapy, but a small proportion of long-term survivors have occurred in a recent Phase I/II safety trial treating recurrent glioblastoma with oncolytic delta24RGD Adenovirus (Frank Tufaro, Personal Communication). Hence, understanding the host factors that limit OV replication and accelerate its immune clearance would be critical to improving the efficacy of this approach in the clinic.
Several studies have looked at different immune effectors that potentially limit oncolytic infection within gliomas. For example, a robust recruitment of monocytoid cells following OV treatment of intracranial glioma models has been shown through immunohistochemical staining [oncolytic Vaccina virus (JX-594),14 herpes simplex virus (HSV),22, 23 measles virus24 and MYXV15]. Similar results have been found in clinical glioma samples after OV therapy with HSV20 and adenovirus23 administration. Targeting the recruitment of these cells with immunosuppressive compounds has been shown to inhibit the recruitment of these cells and increase virotherapy effectiveness;14, 15, 22 however, the pleiotropic nature of these reagents complicates the interpretation that immunosuppressive drugs act on specific cell types alone. Further, human MGs are heavily infiltrated with glioma-infiltrating moncoytes/macrophages (GIMs) prior to treatment,25-27 and there have been no studies to date that have attempted to separate the role of glioma resident versus treatment recruited monocytes in gliomas in vivo.
A role for NK cells in mediating OV clearance from gliomas has recently been reported. First identified as recruited in response to glioma HSV virotherapy in a syngeneic rat model,22 inhibition of these cells with various pleotropic drugs has demonstrated increased oncolytic glioma infection.22, 28 Specific interrogation of NK cells recruited by HSV glioma virotherapy suggested that viral distribution throughout the tumour is limited through the swift destruction of infected tumour cells by NK cells.29 Thus, NK cells have also been suggested to be important in mediating viral distribution and clearance in experimental models of MGs.
In this study, we immunophenotyped the glioma microenvironment before and after intratumoral (I.T.) administration of MYXV therapy in an immunocompetent orthotopic glioma model. We have previously found in this model that MYXV is cleared from these tumours between 3 and 7 days post-intratumoural infection.11 Here, we determined the contribution of various immunocytes responsible for the swift viral clearance of MYXV using a variety of transgenic animals with specific immunocyte deficiencies. To our knowledge, this is the first study to utilize transgenic animals to specifically target immunocyte populations in order to assess their involvement in mediating oncolytic virus infection, replication, immune clearance and treatment efficacy in immunocompetent MG models.
Materials and Methods
Cell Lines and Viruses
The K1492 cell line was derived from C57Bl/6J Trp53+/-/Nf1+/- mice.30 These lines were cultured in Dulbecco's modified Eagle medium (#11965, Invitrogen) containing 10% fetal bovine serum (FBS; Invitrogen) and tested for mycoplasma at regular intervals. Detailed description of these lines when orthotopically grafted into C57Bl/6 mice, as well as a characterization of their in vitro and in vivo response to MYXV treatment has previously been provided.11 Generation, propagation, and titration of the viruses (vMyx-GFP and vMyx-FLuc) have been described elsewhere.11, 15-19, 31, 32
Mouse Strains
Female wildtype (C57Bl/6J, #000664), CCR2-null (B6.129S4-Ccr2tm1Ifc/J, #004999), RAG1-null (B6.129S7-Rag1tm1Mom/J, #002216) and IL2Rγ-null (B6.129S4-Il2rgtm1Wjl/J, #003174) mice were purchased from Jackson Labs. RAG1/IL2Rγ mice were created by crossing female RAG1 mice to male IL2Rγ mice. Six to eight week-old mice were used for all experiments. The animals were housed in a vivarium maintained on a 12-hour light/dark schedule with a temperature of 22 ± 1°C and a relative humidity of 50 ± 5%. Food and water were available ad libitum.
Intracranial Surgeries
Orthotopic injections of K1492 was performed by injecting 5×104 cells in PBS in a 2 μL volume in the right striatum of mice as described previously.11, 15-17 Animals were followed until they lost ≥20% of body weight or had trouble ambulating, feeding, or grooming. Intracranial injection of vMyx-FLuc or vMyx-GFP was performed on day 14 for all lines in survival or bioluminescent studies, and on day 12 for flow cytometric studies. Viral treatments were sterotactically administered through the same burr hole created for the tumour implantation. All animal work procedures were in accordance with the Guide to the Care and Use of Experimental Animals published by the Canadian Council and all protocols were reviewed and approved by the Animal Care Committee of the University of Calgary (Protocol #AC12-0034).
Monitoring viral infection in vivo
Bioluminescence from the vMyx-FLuc-infected tumours was imaged with the Xenogen IVIS 200 system by intraperitoneal injection of D-Luciferin, Firefly, potassium salt (5 μL/g; #119222, Caliper Life Sciences) made to 30 mg/mL in PBS and filter sterilized through a 0.2 μM filter. Luminescence was analyzed by drawing a region of interest around the entire skull and measuring the total emission from that area in units of Total FLUX.
Analyses of the number of functional virions was performed by crushing the tumour-bearing hemisphere in 500 μL of cold PBS. Samples were then sonicated (2 × 2sec, 10% Amplitude; Fisher Scientific Sonic Dismembrator Model 500). This slurry was then spun at 3000 × G for 15 minutes and frozen. Supernatants were freeze thawed three times and then titred on BGMK cells.
Flow Cytometry
Mice bearing 12 day K1492 glioma orthografts were treated with vMyx-GFP or PBS, or left untreated, and then sacrificed for flow cytometry 3 days following treatment. Mice were cardiac perfused with 6 mL of PBS and the tumour-bearing hemisphere placed in RPMI 1640 (GIBCO) with 2% FBS. Homogenized brains were passed through a 70 μm filter and fractionated through a Percoll (GE Healthcare) gradient. Purified immunocytes were then Fc Blocked (BD Mouse Fc Block, #553141) and then stained with CD45 [PE rat IgG2b,κ anti-mouse CD45 (BD, #553081), PE rat IgGbκ anti-mouse isotype (BD #556925)], CD11b [PerCP-Cy5.5 rat IgG2b,κ anti-mouse CD11b (BD #561114), PerCP-Cy5.5 rat IgGbκ isotype (BD #552991)], CD3ε [APC Ar Ham IgG1κ anti-mouse CD3ε (BD #533066), APC Ar Ham IgG1κ anti-mouse isotype (BD #553874)], Ly6G [PE-Cy7 rat IgG2aκ anti-mouse Ly6G isotpye (BD #560601), PE-Cy7 rat IgG2aκ (BD #552784)] and/or NK1.1 [PE-Cy7 mouse IgG2aκ anti-mouse CD161 (BD #561111), PE-Cy7 mouse IgG2aκ anti-mouse (BD #550927)]. Stained cells were then analyzed on an Attune™ Acoustic Focusing Cytometer with Blue/Red lasers using the corresponding software. All cells were initially gated on the ‘live cell’ fraction based on forward and side scatter plots, with the CD45/CD11b plots using a large initial gate to include larger macrophage and neutrophil populations. Flow experiments examining NK and T cell populations used a small initial gate on just lymphocytes to avoid off target staining in the macrophage and neutrophil populations.
Immunohistochemistry
K1492 brains were placed in 10% buffered formalin for a minimum of 48h. Brains were then cut coronally approximately 0.5 mm behind the injection site and mounted in paraffin blocks, and the first and last slides cut were stained using standard H&E by AIRG Histology Services (Calgary, Alberta). Immunohistochemistry was performed using M-T7e (McFadden Laboratory; 1:1000) and Iba1 (Wako, #019-19741; 1:4000) primary antibodies and these were detected using a biotinylated goat anti-rabbit (Vector; 1:300) and vectastain ABC elite reagent (Vector, PK-6100). Slides were mounted, counterstained with hematoxylin and viewed with a Zeiss inverted microscope (Axiovert 200M) and a Carl Zeiss camera (AxioCam MRc).
Immunocyte Ablation Studies and Inactivation Studies
Clodronate liposomes were obtained from ClodronateLiposomes.org. K1492-bearing C57Bl/6 mice were administered 200 μL of Clodronate Liposomes or control Liposomes (Vehicle) via tail vein injection at -1, 0, 1 and 2 days post-MYXV treatment.
Functional grade low-endotoxin CD161(NK1.1; PK130 antigen) or isotype control antibody was purchased from AbLab (Vancouver, BC). Antibodies were administered on 12 and 14 dpi, with vMyx-FLuc administered on 14 dpi.
Cyclophosphamide (Sigma) was diluted in sterile PBS and administered intraperitoneally at indicated doses.
Statistical Analysis
All data were processed and graphed in either MS Excel 2010 or Prism GraphPad v5.0. Statistics were also performed in these programs. All data was assumed to be normally distributed, and F-tests were used to determine if homoscedastic or hetersceodastic t- tests were needed. All t-tests were two-sided and values were considered to be statistically significant at p<0.05. In vivo luciferase monitoring produced highly variable data, thus distributions could not be considered normal, and non-parametric Mann-Whitney (Wilcoxon Rank Sum) tests were conducted. Survival curves were generated by the Kaplan-Meier method, and statistics were determined using the Log-rank Mantel-Cox test.
Results
Microglia and macrophages predominantly reside in the glioma with further peripheral recruitment of monocytoid cells following treatment
We have previously characterized the murine K1492 glioma cell line as an suitable MG model in C57Bl/6 mice to preclinically assess MYXV treatment.11 This line was derived from Trp53+/-/Nf1+/- mice that spontaneously develop high-grade gliomas that recapitulate many clinical features of the human disease.30 In vitro, K1492 is readily infected by MYXV, supports robust viral replication, and is very susceptible to viral-mediated cell death;11 however, this same cell line grafted intracranially and given intratumoral administration of MYXV was refractory to treatment, with no evidence for viral replication or treatment efficacy.
We analyzed the immunocyte composition of the K1492 tumour microenvironment with or without MYXV treatment. We focused on immunocyte populations that were present 3 days after MYXV treatment as most, but not all, of the virus is cleared from the tumour by this time.11 Using conventional neuroimmunological markers for immunocytes in the brain, we first considered CD45lowCD11b+ cells as microglia, CD45highCD11b+ as glioma-infiltrating monocytes/macrophages (GIMs), and CD45highCD11b- cells as lymphocytes.
Comparing the normal mouse brain to the untreated K1492 brain confirmed that these murine MGs, similar to human MGs, contain a significant number of immunocytes (Figure 1A). A nearly 2-fold increase in CD45lowCD11b+ microglia could be seen, making up 66.4 ± 2.1% of the total CD45+ cells found in the tumour-bearing hemisphere. These microglia appeared to have an activated phenotype, as demonstrated by the large shift in this population displaying increased levels of CD45 and CD11b.33 The remaining CD45+ cells were CD45high peripheral immunocytes (29.6 ± 1.8%), which were largely dominated by CD45highCD11b+ GIMs (80.9 ± 1.9% of CD45high), but also included CD11b- lymphocytes (20.7 ± 1.4% of CD45high).
Figure 1. Immunophenotyping of the K1492 glioma microenvironment after Myxoma virus treatment.
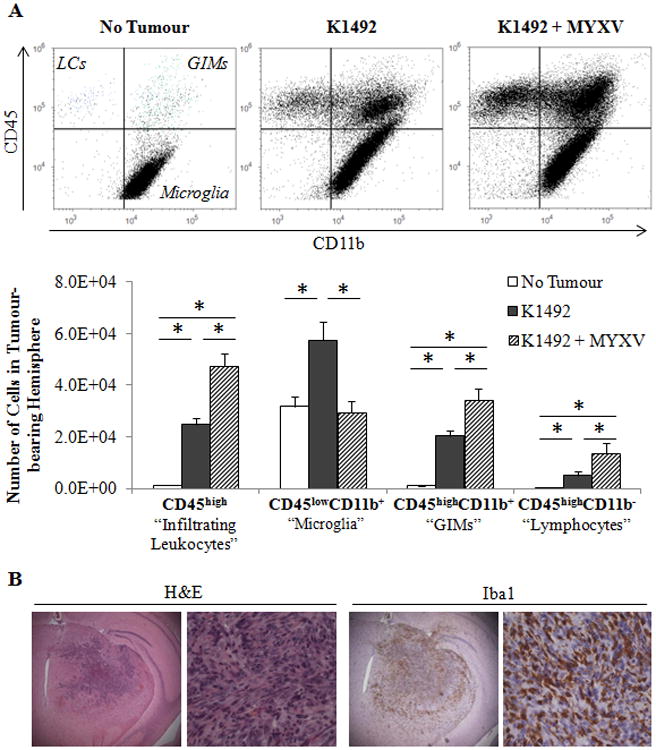
C57Bl/6 mice were implanted with K1492 cells and analyzed by flow cytometry at 15 days-post implantation, 3 days post-treatment with Myxoma virus (5×106 FFU vMyx-GFP; K1492-MYXV, n=10) or untreated (K1492, n=10). Non-tumour bearing mice were used as a control (No Tumour, n=6). Mice were assayed individually and data shown as a mean of all mice. A – Top: Representative scatter plots for CD45/CD11b gating for all experiments. LC – Lymphocytes; GIMs – Glioma infiltrating monocytes/macrophages. Bottom: Quantification of each cell type isolated from the tumour-bearing hemisphere. Error bars represent standard error, and asterisks indicate statistical difference (p<0.05). B - C57Bl/6 mice were implanted with K1492 cells and formalin-fixed paraffin sections were stained with H&E or the microglial/macrophage marker Iba1 14 days after implantation. (First row 25×; Second row 200×). Representative staining from 3 animals/group.
Three days following intratumoural MYXV injection, we found a significant loss of CD45lowCD11b+ microglia, returning to levels seen in the normal mouse brain. Infection also resulted in the recruitment of additional peripheral CD45high cells, reaching levels nearly 2-fold over the untreated K1492 glioma. These CD45high cells were largely CD11b+ (GIMs; 70.3 ± 3.6%), but also contained a large proportion of CD11b- cells (lymphocytes; 30.6 ± 3.4%). Additionally, a population of cells straddling the CD11b gate in the CD45high compartment was observed. These changes were in response to viral inoculation and not a consequence of brain injury, as sterile PBS administration into K1492 tumours did not significantly alter any immunocyte populations (Suppl. Figure 1 A). Finally, as these experiments used a GFP-expressing MYXV, any infected immunocytes in which the virus could initiate replication would be visible in the FITC channel during flow measurements. We found no signal in the FITC channel suggesting that any viral infection or replication at this time point were in non-immunocyte populations.
To confirm the extent of microglial and GIM infiltration into K1492 tumours, we stained sections with the conventional myeloid marker Iba1, which stains both microglia and macrophages. We found that microglial and GIM infiltration was extensive, occurring both within and around the tumour (Figure 1 B). Microglia could be seen in a ‘gradient of activation,’ whereby ramified/unactivated microglia could be observed farther away from the tumour with the gradual transformation into the activated/amoeboid phenotype as they approached the tumour (Suppl. Figure 2 A). In response to MYXV infection, one day post-treatment (dpTx), we observed dense clusters of polymorphonuclear cells surrounding and within the infected areas (Suppl. Figure 2 and 3). By 3 dpTx, these areas of infection were largely cleared of viral protein and these polymorphonuclear cells. Areas of residual infection had intense leukocyte infiltration and the onset of necrosis. This was also apparent at 7 dpTx, where there was a complete absence of viral protein but large areas of focal necrosis (Suppl. Figure 2, black arrows).
While such extensive microglia/GIM infiltration has been known to occur in both human MGs and immunocompetent rodent models of disease,25-27 we wanted to confirm that this infiltration was not an artefact of the K1492 orthotopic model. We confirmed a similar pattern of Iba1+ cellular infiltration in the K1861 orthograft in C57 mice (Suppl. Figure 4 A). Further, we found that in spontaneous high-grade gliomas in NPcis (Trp53-/-Nf1-/-) mice, microglia and/or GIM infiltration was extensive (Suppl. Figure 4 B).
Ablation of both tumour-resident and treatment-recruited GIMs
Given that one of the largest populations of cells infiltrating the untreated and treated K1492-tumour bearing hemisphere was the CD45highCD11b+ ‘GIMs’, this was the first population assessed for their role in mediating MYXV clearance and treatment failure in our model. It has been shown under neuroinflammatory conditions that CCR2 expression is necessary for monocyte recruitment to the CNS in experimental autoimmune encephalitis,34 ischemic injury,35 and viral infection;36 but this has not been tested in a murine MGs. As expected, we found that CCR2-null mice bearing K1492 MGs had significantly fewer CD45highCD11b+ cells than glioma-bearing wildtype mice (2.8-fold decrease; Figure 2 A). This decrease was slightly less prominent after viral infection (1.4-fold), but accompanied by a large population CD45highCD11b+ Ly6G+ granulocytes (Figure 2 A, yellow). Using this additional marker, we determined that GIMs (CD45highCD11b+Ly6G-) were reduced 5.8-fold in the untreated and 2.5-fold in the MYXV-treated CCR2-null animals (Figure 2 B). Further, we found that the granulocyte population was increasingly recruited or failed to be cleared in the CCR2-null mice, resulting in a 5-fold increase in these cells (CD45highCD11b+Ly6G+) at 3 days-post MYXV treatment compared to wildtype animals.
Figure 2. CCR2-deficient mice fail to recruit GIMs to the K1492 tumour before or after Myxoma treatment.
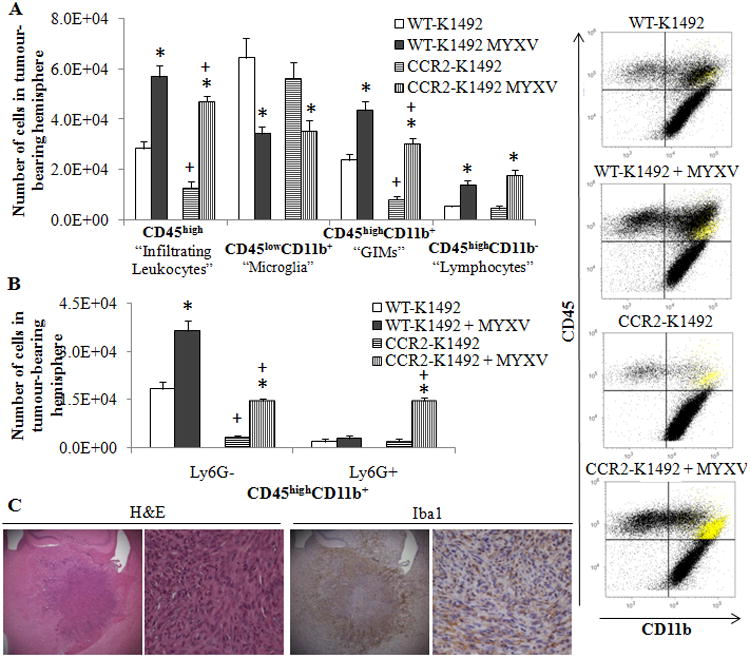
Wildtype (WT) or CCR2-deficient (CCR2) C57Bl/6 mice implanted with K1492 cells and analyzed by flow cytometry at 15 days-post implantation, 3 days post-treatment with Myxoma virus (5×106 FFU vMyx-GFP) or untreated. A - Quantified numbers of each cell type isolated from the tumour-bearing hemisphere (Left; n=6) accompanied by representative CD45/CD11b scatter plot (right). Highlighted in yellow are the Ly6G+ granulocytes. B – Addition of the Ly6G antibody to the flow cytometry analysis demonstrates granulocytic recruitment in MYXV-treated animals and refines the GIM classification of our data (n =3). Error bars represent standard error and asterisks represent significant differences within mouse strain but between treatment groups. Plus signs represent significant differences between mouse strains within treatment groups (p<0.05); there is no significant difference between WT-K1492 and CCR2-K1492 MYXV for GIMs. C - K1492-bearing CCR2-deficient mice were harvested at 14 days post-injection for formalin-fixed paraffin sections (Top row 25×; Second row 200X). Representative staining from 3 animals/group.
To confirm and visualize the loss of GIMs in the K1492 tumours, we looked at Iba1 staining of K1492-tumours in wildtype or CCR2-null mice (Figure 2 C). We found that the loss of Iba1 staining was prominently from within the glioma, while the population of Iba1+ cells surrounding the glioma remained largely unchanged. This suggested that GIMs were recruited within the tumour while recruited microglia (whose activation and numbers remained unchanged by flow analysis, Figure 2 A) remained at the tumour border in the CCR-null mice. This staining pattern for glioma-associated microglia and macrophages has been previously observed in a syngeneic rat glioma model.23
We have previously shown that monitoring the bioluminescence from a luciferase reporter expressed by MYXV (vMyx-FLuc) is a reliable surrogate marker for viral titres in the tumour-bearing mouse brain.4, 11 Using this virus we found a significant increase in bioluminescent activity in K1492-beairng CCR2-null mice, resulting in 3.3-, 8.1- and 5.0-fold increase at 1, 2 and 3 dpTx, respectively (Figure 3 A). The increase in viral bioluminescence at 3 dpTx corresponded to a >10-fold increase in the amount of virus recovered from the tumour-bearing hemisphere at the same time point (Figure 3 B). The large increase in bioluminescent activity was not a result of off-target infection in the knockouts, as non-tumour CCR2-null animals had similar clearance kinetics to the wildtype, albeit there was an increase in bioluminescence at 2 dpTx (Suppl. Figure 5 A) without mouse mortality (Suppl. Figure 5 B). Further, flow cytometric analysis of the vMyx-GFP treated CCR2-null mice we did not observe any GFP+ immunocytes, suggesting that this enhanced infection was not due to the immunocyte infection. To confirm the increase in viral infection within the tumour, we stained for viral protein M-T7 in the tumour-bearing CCR2-null animals at 3 dpTx (Figure 3 C) and found large areas of infected tumour associated with large groups of polymorphonuclear cells. This was reminiscent of what we observed in 1 dpTx wildtype animals (Figure 1B), indeed suggesting that viral clearance from the tumour was delayed. These areas of infection were located adjacent to substantial areas of focal necrosis, suggesting that tumour death accompanied this increased infection. The increase in viral activity in the CCR2-null mice resulted in glioma-bearing mice living 30% longer following treatment (13 dpTx vs. 17 dpTx, p = 0.0073; Figure 3 D).
Figure 3. CCR2-deficient mice have an initial increase in viral infection but are cleared of virus with similar kinetics as the wildtype animals.
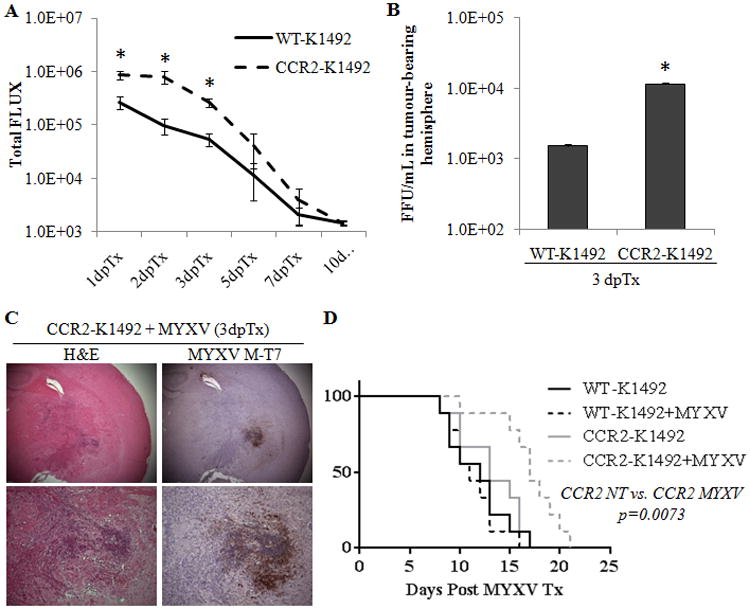
Wildtype or CCR2-/- C57Bl/6 mice implanted with K1492 cells were treated at 14 dpi with 5.0×106 PFUs of vMyxv-FLuc or left untreated. A – Measurement of viral bioluminescence. Error bars represent standard error and asterisks represent significant difference at given time point (n=9/group; p>0.05). B – Viral recovery assay from wildtype (WT) or CCR2-deficient (CCR2) mice bearing 14 day K1492 tumours and infected with 1.5×107 PFU vMyx-FLuc 3 days post-treatment. Error bars represent standard error and asterisk represents significant difference (n=3/group; p<0.05). C – Immunohistochemical staining with H&E or viral protein M-T7 of CCR2-defcient K1492-bearing animals 3 days post-infection with 5×106 FFUs of MYXV. Representative image of 3 animals. D - Kaplan-Meier survival curve of K1492-bearing wildtype (WT) or CCR2-deficeint(CCR2) C57Bl/6 mice treated with 5.0×106 PFUs of vMyxv-FLuc (MYXV). n=9/group.
Finally, as CCR2 loss could potentially inhibit recruitment of other important immunocytes to the glioma after viral infection, we analyzed the NK and T cell populations recruited after infection. We found that these cells were not inhibited in their recruitment, but tended to be enhanced, significantly in the case of NK cells (Suppl. Fig. 6). Indeed, here we identified the CD45highCD11b+ ‘straddling population’ to be NK cells (NK1.1+ in grey), suggesting that the NK cells that fall within CD11b+ gate are also causing an overrepresentation of GIMs in the wildtype and CCR2-decient animals.
Taken together, the above results show that GIMs inhibit the initial oncolytic virus infection within the glioma. However, since GIMs may either be residing within the tumor before treatment (“tumour-resident” GIMs) or recruited to the tumor after viral administration (“treatment-recruited”), and both types are reduced in CCR-null mice, we next wanted to determine which population mediated viral clearance. In order to selectively ablate treatment-recruited GIMs, we administered daily intravenous clodronate liposomes (CL) to ablate circulating monocytes.37 As CL is unable to cross the blood-brain barrier,38 treatment would not affect tumour-resident GIMs or microglia while eliminating recruited monocytoid cells. This was confirmed by flow cytometric analysis of CL and vehicle treated gliomas (Figure 4 A). Further, we found that depletion of these cells resulted in an increase in granulocytes as we had found previously in CCR2-null mice. This depletion did not result in any significant differences in terms of viral clearance as measure by bioluminescence (Figure 4 B) or viral titres (Figure 4 C), and there was no difference in animal survival (Figure 4 D). Collectively, these data suggest that it is the tumour-resident GIMs, and not the treatment-recruited GIMs, that reduce the intratumoral MYXV infection, viral clearance and efficacy in this immunocompetent MG model.
Figure 4. Clodronate liposome depletion of peripheral monocytes stops the recruitment of GIMs following Myxoma treatment but has little effect on viral clearance and treatment efficacy.
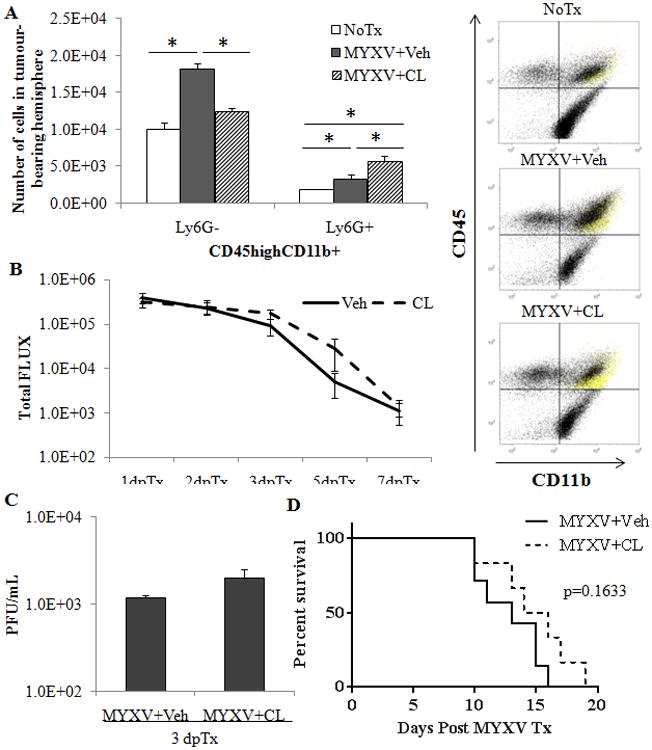
200 μL of Clodronate liposomes (CL) or Vehicle (Veh) was given via tail vein injection at -1, 0, 1 and 2 days post MYXV infection (5×106 PFU, vMyx-GFP). A – Quantified numbers of cells analyzed by flow cytometric analysis of GIM and granulocytes in CL or Veh depleted animals (n=4) accompanied by a scatter plot (right) indicating cell populations. Ly6G+ cells in yellow. B – Analysis of viral bioluminescence from vMyx-FLuc (5×106 PFU) in CL or Veh treated animals (n=7/group). C – Viral recovery assay from vehicle (Veh) or clodronate liposome (CL) treated mice bearing 14 day K1492 tumours and infected with 1.0×107 PFU vMyx-FLuc 3 days post-treatment. Error bars represent standard error and asterisk represents significant difference (n=3/group; p<0.05). D – Kaplan-Meier survival curve of CL or Veh treated animals bearing K1492 tumours and treated on day 14 with vMyx-FLuc (5×106/PFU).
We next attempted to target these tumour-resident GIMs pharmacologically using minocycline tetrachloride. Minocycline is a member of the tetracycline family of antibiotics that can readily cross the blood-brain barrier, itself has anti-glioma activities, and can inhibit the activity of both microglia and monocytes/macrophages.39-41 We adopted a dosing regimen previously shown to inhibit microglia activation in the mouse brain,41 and found that administration of minocycline mimicked the viral clearance kinetics seen in the CCR2-null mice (Suppl. Figure 7A), with a significant 10-fold increase over control animals at 2 dpTx. This increase in initial infection, however, was not accompanied by a combinatory survival advantage, nor did minocycline alone have any response in this model (Suppl. Figure 7B).
The role of T and NK cell populations in mediating MYXV efficacy
We were intrigued to discover a role for tumour-resident GIMs in mediating early viral infection kinetics within syngeneic tumours. However, the enhanced viral infection did not persist for more than three days, suggesting that other recruited immunocytes may participate in its rapid clearance. Given the recent finding that NK cells mediate the premature clearance of oncolytic HSV from glioma xenografts28, 29 we next analyzed the extent of NK (CD45highCD3-NK1.1+ and NK1.1+DX5+) and T (CD45highCD3+NK1.1-) cells recruited to K1492 tumours MYXV treatment (Suppl. Figure 6). We found a resident population of both NK and T cells within the K1492 tumour prior to MYXV treatment and a significant increase (∼2.5-fold) of both cell types following MYXV administration.
To determine the importance of NK, T and B cells in MYXV clearance we next used IL2Rγ-null transgenic mice which are devoid of NK cells and have limited numbers of poorly functioning T- and B- lymphocytes.42 Immunophenotyping of these severely immunocompromised mice bearing K1492 before MYXV infection showed, as expected, that they retained the tumour-resident GIMs and microglia, but were unable to recruit any subsequent CD45high leukocytes 3 days following MYXV infection (Figure 5A). Specifically, the NK and T populations were severely depleted following MYXV infection when compared to wildtype mice. (Suppl. Figure 8)
Figure 5. IL2Rγ-deficient mice have a persistent Myxoma virus infection within the tumour that results in a significant increase in treatment efficacy.
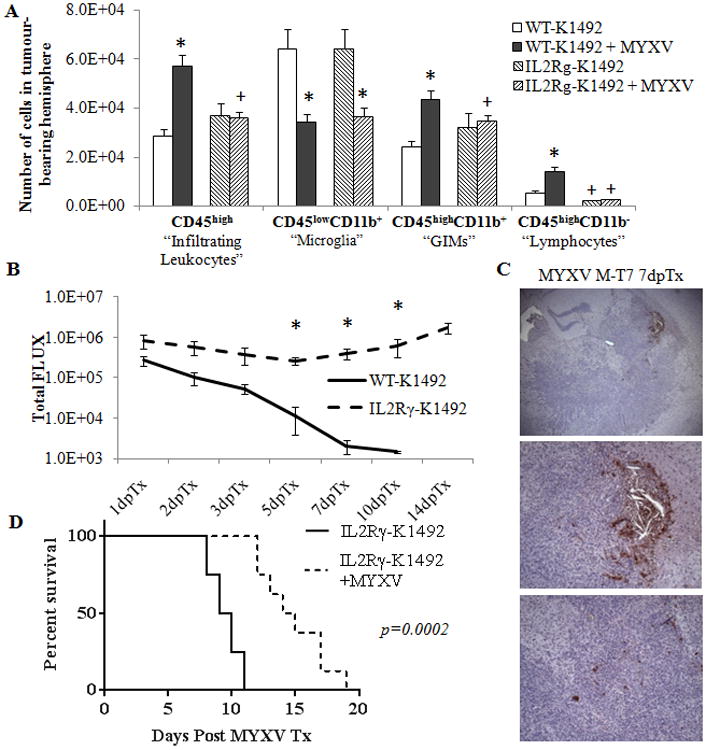
Wildtype (WT) or IL2Rγ-deficient (IL2Rγ) C57Bl/6 mice implanted with K1492 cells and analyzed by flow cytometry at 15 days-post implantation, 3 days post-treatment with Myxoma virus (5×106 FFU vMyx-GFP; MYXV) or untreated. A - Quantified numbers of each cell type isolated from the tumour-bearing hemisphere and analyzed by flow cytometry (n=6). Asterisks represent significant differences within mouse strain but between treatment groups. Plus signs represent significant differences between mouse strains within treatment groups. B – Measurement of bioluminescence emitted from WT or IL2Rγ K1492-bearing mice treated with vMyx-FLuc (5×106 PFUs; n = 9 WT, n = 8 IL2Rγ) 14 days post-implantation. C – M-7e staining of K1492-bearing IL2Rγ mice 7 days post-treatment with vMyx-FLUC (5×106 PFU). Top panel 4× objective, middle and lower panel at 10× objective. D - Kaplan-Meier survival curve of K1492-bearing IL2Rγ-mice treated vMyx-FLuc (5×106/PFU) 14 days after tumour implantation. All error bars represent standard error.
Measuring the rate of viral clearance in the K1492-bearing IL2Rγ-null animals, we found persistent bioluminescent activity that did not dissipate from the mouse brain and remained until the mice were sacrificed because of tumour burden (Figure 5 B). This signal did not significantly change from the original measurement at 1 day post-treatment, suggesting that the virus was unable to be cleared but was not effectively amplifying within the tumour. Non-tumour bearing animals also had a persistent bioluminescent signal, but it was much smaller than that of tumour-bearing animals (Suppl. Figure 9 A). We confirmed persistent infection with the tumour by staining for viral protein M-T7, and found evidence of viral infection within the tumour 7 days post-infection (Figure 5 C). The staining pattern of viral protein was interesting, with large areas of viral infection localized to the tumor margins and scattered cells expressing M-T7 within the tumor core. This persistent infection translated into significantly longer survival times, with glioma-bearing IL2Rγ-null animals living >50% longer on average after MYXV treatment (9.5 days vs 14.5 days post MYXV infection; p = 0.0002) as compared to K1492-bearing wildtype mice. Non-tumour-bearing animals did not succumb to viral treatment (Suppl. Figure 9 B).
We were intrigued by the persistent MYXV infection we observed in these IL2Rγ-null animals, and wanted to determine the potential separate roles of the T and NK cell populations. To test this we utilized K1492 implanted into T- and B- cell deficient RAG1 animals (who have intact NK function), and further treated these mice with the NK cell-depleting NK1.1 antibody. We found that a single intraperitoneal injection of this antibody depleted splenic NK cells by >80% in wildtype mice (data not shown). Interestingly, the NK1.1 treatment in wildtype animals had very little effect on viral clearance kinetics (Figure 6 A) and we did not find a survival advantage to NK cell depletion in this model (Figure 6 B). We found that treating glioma-bearing RAG1 animals with MYXV-FLUC resulted in a persistent bioluminescent that was statistically increased over wildtype beginning at 5 days post treatment (Figure 6 C); however, this sustained viral infection did not result in a survival advantage (Figure 6 D). Finally, using a combination of both B and T cell depletion (RAG1 animals) and NK cell ablation (NK1.1) we found a persistent infection that was significantly increased from day 3 through 10 over the isotype control in RAG1 animals (Figure 6 E). This elevated and persistent infection produced a significant 30% survival increase compared to the isotype treated controls (18 vs. 24 days post infection, p=0.0342; Figure 6 F). Isotype or NK1.1 antibody treatment in either wildtype or RAG1 animals without virotherapy had no change in overall survival (data not shown). These data suggest that ablation of both NK and T cells within the tumour are required to significantly enhance persistence of intratumoral viral replication and to significantly prolong survival in immunocompetent syngeneic MG animal model.
Figure 6. RAG1 animals treated with an NK cell depleting antibody have increased viral infection and have a survival advantage with Myxoma therapy.
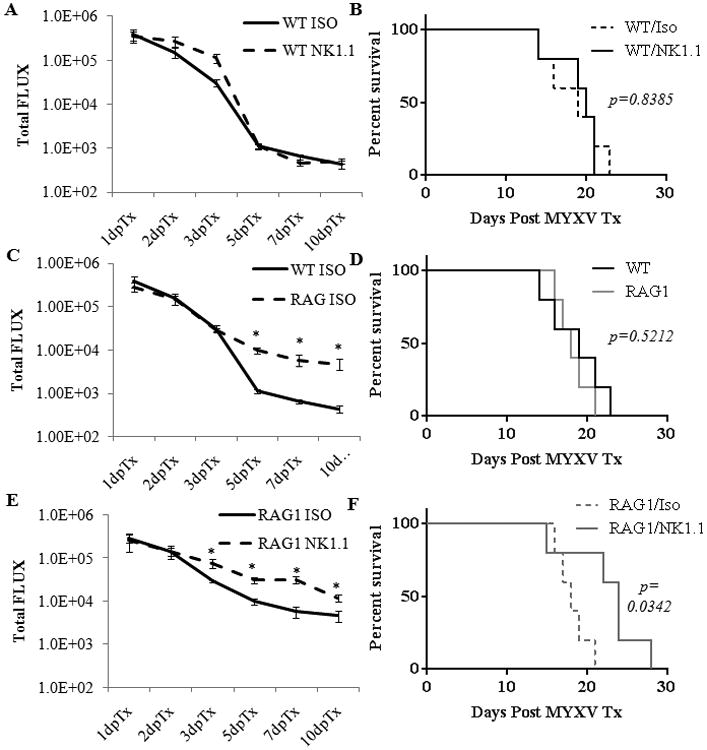
Wildtype (n=5 for each treatment) or RAG1-deficient (n=5 for each treatment) animals with 14 day K1492 tumours were treated with intraperitoneal injections of 200 μg anti-NK1.1 antibody or isotype control at -2 and 0 days post-infection with 5×106 PFU of vMyx-FLuc. Animals were monitored for bioluminescence (A, C, D) and survival via the Kaplan-Meier method (B, D, F).
Finally, we wanted to investigate whether we could mimic the immunosuppressed state of the IL2Rγ-null animals in wildtype animals using clinically relevant immunosuppressants. The alkylating agent cyclophosphamide (CPA) was the logical choice, as it is a clinically utilized anti-glioma therapeutic, has known lymphoablative properties, and has previously been shown to enhance virotherapy effectiveness.14, 22, 43 Interestingly, many studies utilizing CPA to enhance OV effectiveness in glioma syngeneic models use a single low-dose (<100mg/Kg) treatment prior to virotherapy. Initial studies in our model did not find any significant change in viral infection kinetics or treatment efficacy with single-dose CPA at 80 mg/Kg (not shown) or 150mg/Kg (Suppl. Figure 10 A). However, repeated doses of 150 mg/Kg at -2, 0 and +2 dpTx resulted in significantly sustained viral infection (Figure 7 A). This dosing schedule itself resulted in significant survival benefit (17 dpTx NoTx vs. 25 dpTx CPA; p = 0.0045); however, combination of CPA and MYXV was significantly more beneficial than either treatment alone (CPA vs. MYXV + CPA p = 0.0037). Immunohistochemistry for MYXV protein MT-7 found considerable staining within the tumours at 7 dpTx (Figure 7 C), with sizable necrotic areas surrounding the entire tumour margin.
Figure 7. Repeated low-dose cyclophosphamide treatment results in prolonged tumour infection resulting in significant combined efficacy. This dosing resulted in lymphoablation of tumour-resident and treatment-recruited immunocytes.
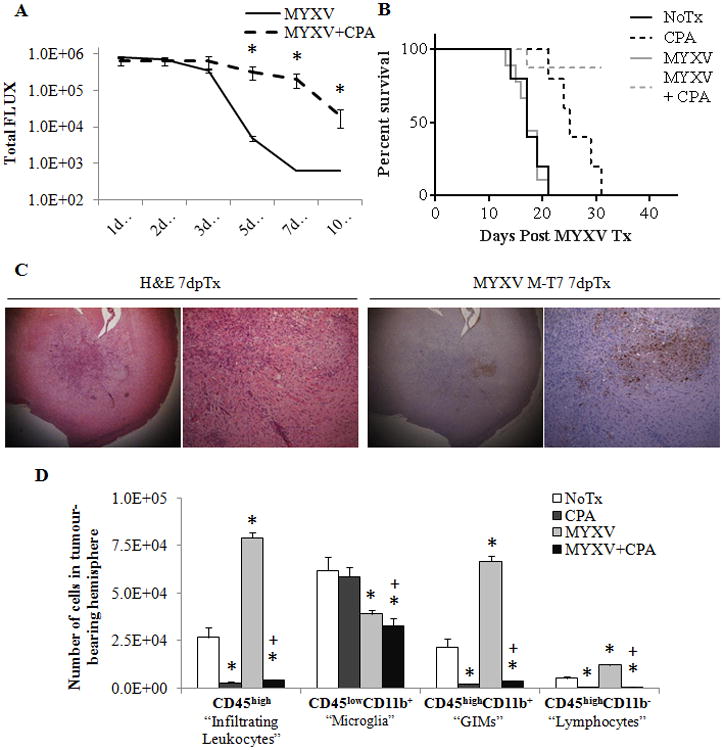
Wildtype animals bearing K1492 gliomas were treated with 150mg/Kg of cyclophosphamide two days before, the day of, and two days after (-2, 0 and +2 dpTx) intracranial administration of 5×106 PFU of vMyx-FLuc. A - Analysis of viral bioluminescence from vMyx-FLuc (MYXV n=12 1-7 dpTx, n=8 at 10 dpTx; MYXV + CPA n=11 1-7 dpTx, n=7 at 10 dpTx). B - Kaplan-Meier survival curve of non-treated (NoTx, n=5), cyclophosphamide treated (CPA, n=5), of vMyx-FLuc treated (MYXV, n=9) and cyclophosphamide and vMyx-FLuc treated (MYXV + CPA, n=8). C - Immunohistochemical staining with H&E or viral protein M-T7 in cyclophosphimide treated, K1492-bearing animals 3 days post-infection with 5×106 FFUs of MYXV. D - Quantified numbers of each cell type isolated from the tumour-bearing hemisphere and analyzed by flow cytometry (n=3). Asterisks represent significantly different no treatment (NoTx) control and plus sign significantly different from MYXV treatment. All error bars represent standard error.
We analyzed the degree of immunosuppression seen through our dosing regimen by immunophenotyping the K1492 glioma microenvironment with single and combined treatments of MYXV and CPA (Figure 7 D; Suppl. Figure 10 C). CPA treatment resulted in a complete failure to recruit any peripheral immunocytes to the tumour, suggesting strong peripheral immunosuppression. Surprisingly, however, we found that CPA treatment also resulted in the near complete abolition of tumour resident GIMs while having no effect on resident microglial populations. These results demonstrate that lymphoablation in combination with MYXV treatment is a viable therapeutic strategy in high grade gliomas.
Discussion
The human glioma microenvironment is complex, with tumour cell interactions with other glia, extracellular matrix proteins, vasculature, and infiltrating immune cells. Although all of these interactions could, and perhaps do, play some role in determining oncolytic virus therapy efficacy in patients and animal models, interrogating tumour-infiltrating immunocyte populations is a logical approach to investigate potential avenues to improve therapy.
The human glioma infiltrating immunocyte population is diverse, including an array of immunocytes. However, the most numerous infiltrating immune cells in human glioma are microglia and monocytes/macrophages,25-27 with some reports suggesting they comprise up to one-third of the tumour mass.27 The K1492 syngeneic tumours used in these studies have a similar composition, with substantial infiltration of microglia and recruited peripheral monocytes/macrophages (GIMs), making up >90% of the total immune infiltrate in the K1492 model.
We demonstrate here that glioma infiltrating monocytes are recruited to the K1492 tumours in a CCR2-dependent manner. Although monocyte recruitment to the CNS through CCR2 has been shown in other CNS pathologies,34-36 this is the first time that this has been demonstrated in a mouse model of glioma. This could potentially be an important model for testing the role of peripherally-derived GIMs in glioma treatment and biology.
In our study, CCR2-deficient mice, but not clodronate liposome depleted mice, had large increases in tumoural infection over the first 3 days of infection. These data suggest tumour-resident GIMs play the predominate role early in oncolytic infection. Targeting tumour-resident as of opposed to peripherally recruited monocytes to mediate oncolytic infection rates could pose a challenge clinically. Here we have introduced the idea that minocycline tetrachloride (7-dimethylamino-6-desoxytetracycline) may be able to inhibit these to enhance oncolytic infection. It is important to note that the mechanisms by which minocycline enhances the infection could be diverse, and may not in fact be due to the inhibition of microglia and/or GIMs. Minocycline has been shown to have several anti-inflammatory effects such as the inhibition of NFκB and iNOS, as well as anti-tumour activities such as inhibiting MMPs, angiogenesis, and directly killing of glioma cells.39-41 Credence to the perspective that minocycline could play an important role in glioma treatment is provided by the recently commenced RAMBO trial that has added minocycline to bevacizumab and radiation treatment for recurrent glioma (NCT01580969). Further studies investigating the combinatory effects of minocycline and OVs for glioma are warranted.
A surprising result in our study was the loss of microglia in response to MYXV infection. We considered that this could be due to a shift in microglial CD45 expression from CD45low to CD45high; however our experiments that ablated much of the infiltrating CD45highCD11b+ populations still resulted in this microglial loss. Iba1 staining after viral infection did not indicate a large loss of these microglia, albeit this stain would not distinguish dead or dying cells. It is our current opinion that these cells were lost during isolation of CNS leukocytes, perhaps as a result of changes in cellular density following viral infection or perhaps as a result of increase in fragility after the inflammatory insult. This phenomenon should be considered in future studies examining neuroinflammatory responses by flow cytometry. Further, our studies here fail to address the role that these infiltrating cells may play in mediating viral clearance from the glioma. These are technically challenging experiments, as selective ablation of microglia in vivo is very difficult to achieve. It is our hope that our future experiments investigating the mechanism by with GIMs impact OV treatment will concomitantly lead to the role these microglial cells may also be playing in altering this therapy.
We next targeted the recruited lymphoid cell types that consisted of the recruited T and NK populations upon MYXV administration using the targeted IL2Rγ-knockout animals. The persistent infection in the tumour found in the IL2Rγ-null animals suggests that these cells play a critical role in mediating viral clearance. Of note, however, is that the IL2Rγ receptor could affect the function of a number of other immunocytes. Indeed, targeting only T and B lymphocytes in RAG1-null animals with specific NK cell ablation using the NK1.1 antibody did not fully phenocopy the IL2Rγ-null viral bioluminescence kinetics; however, this could also be a function of the return of NK cells after the initial depletion.
The role of NK cells alone in mediating viral clearance in our syngeneic model does not appear to have the same impact as has been reported in the literature for immunocompromised xenograft models.28, 29 However, in RAG1 immunocomprimsed animals, NK depletion results in a large increase in viral infection in the tumour, increasing viral persistence and resulting in a significant treatment-induced survival response. This suggests that the NK and T cell populations have overlapping functions in our model. It is interesting to speculate on the nature of the T cells that are recruited so early after infection. Most studies interrogating viral-induced T cell responses in the CNS interrogate classical T cell responses, beginning seven days or later post-infection. However, the prompt recruitment seen here suggests these T cells are playing an innate-like role clearing the infection. Non-classical T cells such as natural killer T cells (NKT) and gamma-delta T cells (γδT) would be good candidates to investigate further. Both NKT and γδT cells have known innate anti-viral effector functions, are absent in RAG1-null animals, and are known to be swiftly recruited during CNS inflammation.44-46 These innate-like T cells are similar in that they have a limited, pre-determined TCR diversity that is capable of rapid effector function in response to defined foreign- or self-antigens. Further, they both contain NK cytotoxicity receptors, such as the NKG2D receptor, that upon ligation of their ligands can instigate cytotoxic responses against stressed or infected cells.47 It was recently shown that NK cell-mediated clearance in orthotopic glioma xenografts was dependent on NK cell natural cytotoxicity receptors NKp30 and NKp46 that recognized oncolytic HSV-infected glioma cells.29 It would be very interesting to investigate the roles of NKT and γδT cells in our model and to see if these cells were functioning in an NK-like fashion, perhaps dependent on virally-induced NKG2D ligand expression in MYXV-infected glioma cells.
Taken together, the results of these experiments demonstrate that resistance to Myxoma virus infection and replication in syngeneic animals is, not surprisingly, a multi-faceted mechanism. The diverse nature of the mechanisms that are limiting this therapy in our model suggests that pan-inhibitors of the immune system could be the most efficacious in enhancing OV therapeutic responses, as we and others have shown with cyclophosphamide.14-16, 22 Interestingly, in addition to peripheral lymphocyte ablation, our study here demonstrated that cyclophosmimide treatment was able to ablate GIMs in our model. This was an unexpeted observation, as the cytotoxic activity of bioactive cyclophosphamide (phosphoramide mustard) is through DNA alkylation, thus targeting actively dividing cells. We do not anticipate that these GIMs are turning over so quickly from the cirulating population, as we did not see abaltion of these GIMs with clodronate liposome-mediated monocyte depletion. It will be interesting to intertogate the loss of these GIMs with this CPA dosing regimen, and to determine whether this is a viable strategy for depleting GIMs in other models. Further, based on our transgenic animal experiments, we could predict that this is what the immune cell compostion would look like in a MYXV-treated CCR2/IL2Rγ double knockout mouse. It is curious then why we did not achieve a initial increase in viral infection following CPA treatment as observed in the CCR2-null mice; however, it is possible that the dose 2 days prior to treatment was insufficient to deplete the GIM population, and that subsequent doses were necessary. If this were the case, it could postulated that starting CPA even earlier before OV administration could result in even greater tumoural infection and combinatorial efficacy.
It is important to note that maximizing OV clinical utility exclusively through the enhancement of viral infection and virus-mediated cell death ignores the potential of this therapy to stimulate anti-tumour immune responses. Indeed, numerous OVs have been genetically modified in order to enhance anti-tumour immune responses, making OVs akin to an immunotherapy.48 We demonstrate here that for MYXV to develop an oncolytic treatment response, a threshold of infection must be reached through the inhibition of innate immune responses. Achieving this through genetic lymphoablation precludes any cellular immunotherapeutic responses (and is clinically irrelevant); however, transient lymphoablative or immune-inhibiting therapeutics may allow sufficient tumoural infection and cell death to induce a robust multi-faceted anti-tumour immune response. It is important to consider that most cancers are tremendously heterogeneous, including gliomas,49 and maximizing the viral production of tumour antigen by maximizing initial OV infection rates could help initiate a broader immunotherapeutic response. This, however, is a double-edged sword, as initial innate immune responses are what orchestrate subsequent adaptive responses. Thus, a balance must be achieved to allow sufficient viral infection and antigen generation, whilst not inhibiting the generation of appropriate adaptive responses. It is not yet clear if MYXV is capable of generating an adaptive immunotherapeutic response in gliomas, however it has been shown in other cancer models31 as well as to enhance adoptive NK immunotherapy.17 Additional investigation probing the nature of the glioma microenvironment before and after virotherapy, as well as the specific mechanisms by which the innate immune response limits MYXV infection rates, could lead to a more targeted approach to enhance viral infection while initiating anti-tumour immunity.
Supplementary Material
Acknowledgments
We would like to thank Dr. Douglas Mahoney and Dr. Jennifer Chan for critical reading of the manuscript and/or guidance throughout these experiments. Further, we would like to thank Molly Chisholm and staff in the Mouse Double Barrier Unit at the University of Calgary.
Funding: This work was funded by the Canadian Cancer Society Research Institute (CCSRI) with funds raised by the Canadian Cancer Society (PAF), a Program Project Grant from the Terry Fox Foundation (PAF), the V Foundation (PAF), and the Canadian Institutes of Health Research (CIHR) (VWY). During this work FJZ was funded by Alberta Innovates Health Solutions, Vanier Scholarship from CIHR, and the Izaak Walton Killam scholarship. BAM was funded by CIHR. GM's lab is funded by NIH grants R01 AI080607 and R01 CA138541. KMR's lab is funded by the Intramural Research Program of NIH, NCI (ZIA BC 010541).
Abbreviations used
- CL
clondronate Liposomes
- CPA
cyclophosphamide
- dpi
days post injection
- dpTx
days post treatment
- MG
malignant glioma
- IT
intratumoral
- MYXV
Myxoma virus
- HSV
Herpes simplex virus
- OV
oncolytic virus
- CL
clodronate liposomes
- Veh
vehicle
Footnotes
Authors declare no conflicts of interest.
References
- 1.Society. CC. [Accessed May 8th, 2013];2013 http://www.cancer.ca/en/
- 2.Society. CC. [Accessed May 8th, 2013];2013 http://www.cancer.org/
- 3.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Zemp FJ, Corredor JC, Lun X, Muruve DA, Forsyth PA. Oncolytic viruses as experimental treatments for malignant gliomas: using a scourge to treat a devil. Cytokine Growth Factor Rev. 2010;21:103–117. doi: 10.1016/j.cytogfr.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Kaufmann JK, Chiocca EA. Glioma virus therapies between bench and bedside. Neuro Oncol. 2014;16:334–351. doi: 10.1093/neuonc/not310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conrad C, Miller CR, Ji Y, et al. Delta24-hyCD adenovirus suppresses glioma growth in vivo by combining oncolysis and chemosensitization. Cancer Gene Ther. 2005;12:284–294. doi: 10.1038/sj.cgt.7700750. [DOI] [PubMed] [Google Scholar]
- 8.Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1:938–943. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- 9.Gromeier M, Lachmann S, Rosenfeld MR, Gutin PH, Wimmer E. Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc Natl Acad Sci U S A. 2000;97:6803–6808. doi: 10.1073/pnas.97.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilcox ME, Yang W, Senger D, et al. Reovirus as an oncolytic agent against experimental human malignant gliomas. J Natl Cancer Inst. 2001;93:903–912. doi: 10.1093/jnci/93.12.903. [DOI] [PubMed] [Google Scholar]
- 11.Zemp FJ, McKenzie BA, Lun X, et al. Resistance to oncolytic myxoma virus therapy in nf1(-/-)/trp53(-/-) syngeneic mouse glioma models is independent of anti-viral type-I interferon. PLoS One. 2013;8:e65801. doi: 10.1371/journal.pone.0065801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hellums EK, Markert JM, Parker JN, et al. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro Oncol. 2005;7:213–224. doi: 10.1215/S1152851705000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wakimoto H, Kesari S, Farrell CJ, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69:3472–3481. doi: 10.1158/0008-5472.CAN-08-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lun X, Chan J, Zhou H, et al. Efficacy and safety/toxicity study of recombinant vaccinia virus JX-594 in two immunocompetent animal models of glioma. Mol Ther. 2010;18:1927–1936. doi: 10.1038/mt.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lun X, Alain T, Zemp FJ, et al. Myxoma virus virotherapy for glioma in immunocompetent animal models: optimizing administration routes and synergy with rapamycin. Cancer Res. 2010;70:598–608. doi: 10.1158/0008-5472.CAN-09-1510. [DOI] [PubMed] [Google Scholar]
- 16.Zemp FJ, Lun X, McKenzie BA, et al. Treating brain tumor-initiating cells using a combination of myxoma virus and rapamycin. Neuro Oncol. 2013 doi: 10.1093/neuonc/not035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogbomo H, Zemp FJ, Lun X, et al. Myxoma virus infection promotes NK lysis of malignant gliomas in vitro and in vivo. PLoS One. 2013;8:e66825. doi: 10.1371/journal.pone.0066825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lun XQ, Zhou H, Alain T, et al. Targeting human medulloblastoma: oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res. 2007;67:8818–8827. doi: 10.1158/0008-5472.CAN-07-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lun X, Yang W, Alain T, et al. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005;65:9982–9990. doi: 10.1158/0008-5472.CAN-05-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Markert JM, Liechty PG, Wang W, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther. 2009;17:199–207. doi: 10.1038/mt.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Papanastassiou V, Rampling R, Fraser M, et al. The potential for efficacy of the modified (ICP 34.5(-)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther. 2002;9:398–406. doi: 10.1038/sj.gt.3301664. [DOI] [PubMed] [Google Scholar]
- 22.Fulci G, Breymann L, Gianni D, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12873–12878. doi: 10.1073/pnas.0605496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fulci G, Dmitrieva N, Gianni D, et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 2007;67:9398–9406. doi: 10.1158/0008-5472.CAN-07-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen C, Vongpunsawad S, Nakamura T, et al. Retargeted oncolytic measles strains entering via the EGFRvIII receptor maintain significant antitumor activity against gliomas with increased tumor specificity. Cancer Res. 2006;66:11840–11850. doi: 10.1158/0008-5472.CAN-06-1200. [DOI] [PubMed] [Google Scholar]
- 25.Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006;8:261–279. doi: 10.1215/15228517-2006-008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parney IF, Waldron JS, Parsa AT. Flow cytometry and in vitro analysis of human glioma-associated macrophages. Laboratory investigation. J Neurosurg. 2009;110:572–582. doi: 10.3171/2008.7.JNS08475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roggendorf W, Strupp S, Paulus W. Distribution and characterization of microglia/macrophages in human brain tumors. Acta Neuropathol. 1996;92:288–293. doi: 10.1007/s004010050520. [DOI] [PubMed] [Google Scholar]
- 28.Alvarez-Breckenridge CA, Yu J, Price R, et al. The histone deacetylase inhibitor valproic acid lessens NK cell action against oncolytic virus-infected glioblastoma cells by inhibition of STAT5/T-BET signaling and generation of gamma interferon. J Virol. 2012;86:4566–4577. doi: 10.1128/JVI.05545-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alvarez-Breckenridge CA, Yu J, Price R, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med. 2012;18:1827–1834. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26:109–113. doi: 10.1038/79075. [DOI] [PubMed] [Google Scholar]
- 31.Wennier ST, Liu J, Li S, Rahman MM, Mona M, McFadden G. Myxoma virus sensitizes cancer cells to gemcitabine and is an effective oncolytic virotherapeutic in models of disseminated pancreatic cancer. Mol Ther. 2012;20:759–768. doi: 10.1038/mt.2011.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smallwood SE, Rahman MM, Smith DW, McFadden G. Myxoma virus: propagation, purification, quantification, and storage. Curr Protoc Microbiol. 2010;Chapter 14 doi: 10.1002/9780471729259.mc14a01s17. Unit 14A 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 34.Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schilling M, Strecker JK, Ringelstein EB, Schabitz WR, Kiefer R. The role of CC chemokine receptor 2 on microglia activation and blood-borne cell recruitment after transient focal cerebral ischemia in mice. Brain Res. 2009;1289:79–84. doi: 10.1016/j.brainres.2009.06.054. [DOI] [PubMed] [Google Scholar]
- 36.Lim JK, Obara CJ, Rivollier A, Pletnev AG, Kelsall BL, Murphy PM. Chemokine receptor Ccr2 is critical for monocyte accumulation and survival in West Nile virus encephalitis. J Immunol. 2011;186:471–478. doi: 10.4049/jimmunol.1003003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sunderkotter C, Nikolic T, Dillon MJ, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 38.Huitinga I, van Rooijen N, de Groot CJ, Uitdehaag BM, Dijkstra CD. Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J Exp Med. 1990;172:1025–1033. doi: 10.1084/jem.172.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu WT, Huang CY, Lu IC, Gean PW. Inhibition of glioma growth by minocycline is mediated through endoplasmic reticulum stress-induced apoptosis and autophagic cell death. Neuro Oncol. 2013;15:1127–1141. doi: 10.1093/neuonc/not073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campbell JH, Burdo TH, Autissier P, et al. Minocycline inhibition of monocyte activation correlates with neuronal protection in SIV neuro AIDS. PLoS One. 2011;6:e18688. doi: 10.1371/journal.pone.0018688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao X, Shores EW, Hu-Li J, et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity. 1995;2:223–238. doi: 10.1016/1074-7613(95)90047-0. [DOI] [PubMed] [Google Scholar]
- 43.Lun XQ, Jang JH, Tang N, et al. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin Cancer Res. 2009;15:2777–2788. doi: 10.1158/1078-0432.CCR-08-2342. [DOI] [PubMed] [Google Scholar]
- 44.Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol. 2013;13:88–100. doi: 10.1038/nri3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Juno JA, Keynan Y, Fowke KR. Invariant NKT cells: regulation and function during viral infection. PLoS Pathog. 2012;8:e1002838. doi: 10.1371/journal.ppat.1002838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brait VH, Arumugam TV, Drummond GR, Sobey CG. Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J Cereb Blood Flow Metab. 2012;32:598–611. doi: 10.1038/jcbfm.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. 2013;31:413–441. doi: 10.1146/annurev-immunol-032712-095951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. 2014;2:295–300. doi: 10.1158/2326-6066.CIR-14-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sottoriva A, Spiteri I, Piccirillo SG, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. 2013;110:4009–4014. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.