

ABSTRACT
The yeast Efr3p protein is a main regulator of the Stt4p phosphatidylinositol 4-kinase at contact sites between the endoplasmic reticulum and the plasma membrane. A mutation in its fly homologue Rbo, leads to diminished light responses in the eye attributed to progressively impaired PLC signaling. Here, we find that Efr3s plays a role in maintaining responsiveness to the type-I angiotensin II (AngII) receptors. siRNA-mediated depletion of EFR3A and EFR3B impaired the sustained phase of cytosolic Ca2+ response to high concentration of AngII in HEK293 cells that express wild type but not truncated AGTR1 (AT1a receptor), missing the phosphorylation sites. Efr3 depletion had minimal effect on the recovery of plasma membrane phosphoinositides during stimulation, and AT1 receptors still underwent ligand-induced internalization. A higher level of basal receptor phosphorylation and a larger response was observed after stimulation. Moreover, Gq activation more rapidly desensitized after AngII stimulation in Efr3 downregulated cells. A similar but less pronounced effect of EFR3 depletion was observed on the desensitization of the cAMP response after stimulation with isoproterenol. These data suggest that mammalian Efr3s contribute to the control of the phosphorylation state and, hence, desensitization of AT1a receptors, and could affect responsiveness of G-protein-coupled receptors in higher eukaryotes.
KEY WORDS: EFR3, GPCR, Receptor desensitization, Angiotensin II, PI 4-kinase, Phosphoinositide
INTRODUCTION
A selected group of G-protein-coupled receptors (GPCRs) activate phospholipase C (PLC) by coupling to heterotrimeric Gq/11 proteins to induce an increase of cytosolic Ca2+ and, depending on the cell type, trigger a variety of downstream responses. PLC enzymes hydrolyze the plasma membrane lipid, phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2] to generate the two messenger molecules, Ins(1,4,5)P3 and diacylglycerol (Berridge, 1984). Owing to the limited size of the PtdIns(4,5)P2 pools at the plasma membrane, continuous signal generation requires a steady resynthesis of this lipid by PI 4-kinase and PIP 5-kinase enzymes. The PI 4-kinase implicated in this process is PI4KA (Balla et al., 2008; Balla et al., 2005), whose yeast orthologue Stt4 is also responsible for the maintenance of the PtdIns(4,5)P2 pools at the plasma membrane (Audhya and Emr, 2002).
Cells possess several protective mechanisms to prevent excessive stimulation of their GPCRs. High concentrations of GPCR ligand often lead to rapid receptor desensitization that diminishes the ability of the receptor to couple to G proteins. The most common cause of desensitization is due to the rapid phosphorylation of receptors by one of several GPCR kinases (GRKs) that lead to uncoupling from the G proteins and association with arrestins (Moore et al., 2007). Binding to arrestin triggers receptor endocytosis, thereby limiting the number of the receptors on the cell surface. However, arrestin association also lends a new signaling profile to endocytosed receptors that trigger specific cellular responses from endocytic compartments (Luttrell and Lefkowitz, 2002; Reiter et al., 2012; Wei et al., 2003). Although these molecular events have been studied and characterized in great detail, little is known about the processes that restore the sensitivity of receptors at the plasma membrane (Vasudevan et al., 2011). Early studies on β-adrenergic receptors suggested that dephosphorylation requires the receptors to be endocytosed (Yu et al., 1993) but this hypothesis has never been thoroughly investigated.
This present study was intended to investigate the possible role of the two mammalian EFR3 proteins, EFR3A and EFR3B, in the regulation of the PI4KA enzyme in mammalian cells. The yeast orthologue of these proteins, Efr3p, was found to be essential for the localization of the yeast PI4K Stt4 into discrete signaling domains and, together with another protein, Ypp1, to control Stt4 function (Baird et al., 2008). A similar role has been described in the mammalian system, where EFR3 and the TTC7 proteins were shown to keep an active pool of PI4KA at the plasma membrane (Nakatsu et al., 2012). While studying the role of EFR3 in the maintenance of the signaling phosphoinositide pools, we discovered that EFR3 proteins are important for AT1 receptors to maintain their signaling competence, an effect that was not caused directly by depletion of the phosphoinositide pool but, rather, the regulation of the phosphorylation state of the receptor. Therefore, our studies identified EFR3 as a component of the AT1 receptor signaling cascade that controls receptor re-sensitization and, thereby, could serve as a hitherto unrecognized player in determining AT1 receptor and, possibly, other GPCR responsiveness.
RESULTS
EFR3s are widely expressed but show distinct enrichment in various tissues
Mammalian cells contain two EFR3 proteins, EFR3A and EFR3B with several putative splice variants annotated in GenBank. To determine the expression pattern of the two EFR3 isoforms, we performed quantitative PCR (qPCR) using various mouse tissues. Fig. 1 shows that the two EFR3 isoforms are widely expressed, although they show notable differences: while EFR3A showed highest expression in the testis, EFR3B had highest expression in the brain – followed by small intestine and the eye – but showed very low expression in testis and kidney. These results did not suggest a narrow tissue-specific function for the two EFR3 proteins.
Fig. 1.
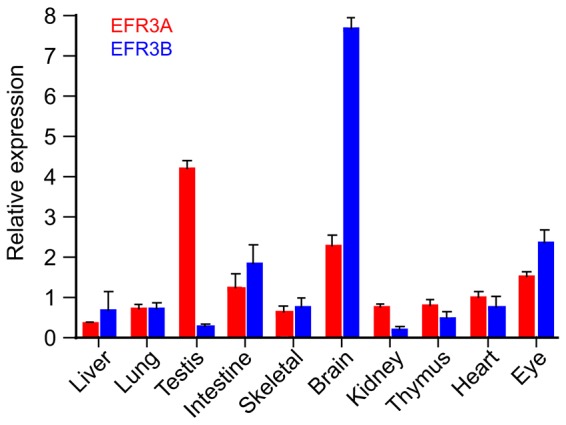
Relative expression of EFR3A and EFR3B mRNAs in various mouse tissues. Total RNA from various mouse tissues was isolated and cDNA synthesized as described in Materials and Methods. Real-time qPCR was performed using SYBR Green and primers specific for the respective mRNAs (see Materials and Methods). The signal was then normalized to that of 18S rRNA. Means+s.e.m. (n = 6) are shown from duplicate determinations of three independent mouse and tissue isolations. See the predominant expression of EFR3B in the brain and that of EFR3A in the testis.
EFR3s are localized to the plasma membrane via palmitoylation
To determine the subcellular distribution of the two EFR3 proteins, we obtained cDNAs of both of them and epitope-tagged them at their C-termini with either a hemagglutinin (HA)-tag, green fluorescent protein (GFP) or monomeric red fluorescent protein (mRFP). When these proteins were expressed in HEK293 cells or COS-7 cells, they localized to the plasma membrane although a small amount was also detected in the cytosol (Fig. 2A). Similar plasma membrane localization was found with the HA-tagged versions in fixed and immunostained cells (not shown). A shorter form of EFR3B truncated at the N-terminus (missing 148 residues) was found to be cytosolic only (Fig. 2A). The presence of cysteine residues (four in EFR3A and three in EFR3B) within this N-terminal segment – which are conserved in the EFRs of higher eukaryotes but are missing in yeast – raised the possibility that the full-length proteins are kept in the membrane by palmitoylation. Therefore, we mutated the four cysteine to serine residues in EFR3A and found this mutant protein to be located in the cytosol (Fig. 2B). By using metabolic labeling of cells that overexpress GFP-tagged EFR3A or GFP-tagged EFR3B with [3H]-palmitate, we were able to confirm that, indeed, EFR3A and EFR3B are palmitoylated proteins (Fig. 2C). Last, we determined whether the isolated N-terminus (residues 1–37 of EFR3A) is sufficient to target GFP to the plasma membrane. We found that this construct is mostly localized to the Golgi complex and that it requires the cysteine residues for the Golgi localization (Fig. 2B). This suggested that other determinants of the full-length protein are needed to localize the protein to the plasma membrane. Our results collectively suggested that, contrary to previous suggestions, EFR3 proteins are not transmembrane proteins but peripheral membrane proteins associated with the plasma membrane through palmitoylation. The same conclusion about EFR3 localization and palmitoylation was reached in a recently published study (Nakatsu et al., 2012).
Fig. 2.

Cellular localization and palmitoylation of the EFR3 proteins. (A) EFR3A and EFR3B proteins were tagged with GFP at their C-termini and expressed either in COS-7 cells or HEK293-AT1 cells. Cells were imaged live by confocal microscopy. Both proteins were found to primarily localize to the plasma membrane and this localization requires the N-terminus of the proteins to contain several cysteine residues. (B) Mutation of the four cysteine residues to serines eliminates localization of the full-length EFR3A (and EFR3B, not shown) proteins at the plasma membrane. The short N-terminal fragment of EFR3A, however, shows Golgi localization, suggesting that other determinants are also important for the plasma membrane localization of the protein. Without the cysteines, this short construct also loses its Golgi localization. (C) Palmitoylation of the EFR3 proteins: GFP-tagged EFR3A or EFR3B were expressed in COS-7 cells (PI4K2A-GFP was used in a separate dish as control) and cells were labeled with [3H]-palmitate for 4 hrs. Cells were lysed and the proteins immunoprecipitated using GFP-trap beads. Immunoprecipitated proteins were visualized using SDS-PAGE and the gel was incubated with EN3HANCE solution before drying and autoradiography (left). Parallel samples were run for western blotting using an anti-GFP antibody (right).
EFR3s are important for sustained Ca2+ signaling
If EFR3 proteins are important for the correct functioning of PI4KA, one expects that the phosphoinositide pools at the plasma membrane are depleted upon strong stimulation of Gq/11-coupled receptors, because the PI4KA enzyme was found to be primarily responsible for the synthesis of PtdIns4P in the plasma membrane to serve as precursor for PtdIns(4,5)P2 (Balla et al., 2008; Balla et al., 2005). Such PtdIns(4,5)P2 depletion happens when the enzyme is pharmacologically inhibited, as shown initially by using high concentrations of wortmannin or phenylarsine oxide at a concentration that primarily inhibits the PI4KA enzyme (Balla et al., 2008) or, recently, by specific PI4KA inhibitors (Bojjireddy et al., 2014). The easiest way to test for such an effect was to follow the cytosolic Ca2+ increase after stimulation because the sustained phase of this response requires continuous PtdIns(4,5)P2 resynthesis (Nakanishi et al., 1995). For this experiment, we used HEK293 cells that stably express the rat type-1A angiotensin II receptor (AT1a, also known as AGTR1) and to which we refer to hereafter as HEK-AT1. The AngII responses of these cells have been thoroughly characterized in our laboratory. We used small interfering RNA (siRNA)-mediated silencing (for 3 days) to knockdown EFR3 isoforms (either alone or in combination) and studied the Ca2+ responses of Fura-2-loaded cells following the application of AngII (100 nM). The efficiency of the EFR3 knockdown was tested using western blot analysis of either the endogenous EFR3A or transfected EFR3A and EFR3B (Fig. 3A,B). We found that, following stimulation with 100 nM AngII, EFR3A- and EFR3B-depleted cells showed a Ca2+ response with a diminished plateau phase, whereas cells treated with control siRNA maintained the sustained Ca2+ plateau that is characteristic of these cells (Fig. 3C, left panel). Notably, the Ca2+ response following stimulation with 1 nM of AngII was very similar between EFR3A and EFR3B-depleted cells (EFR3AB si) and control si cells (Fig. 3C, right panel). Knockdown of the individual EFR3s have indicated that knockdown of EFR3B had a stronger effect than that of EFR3A (not shown). Therefore, some of the later experiments were performed only by knocking down EFR3B. These data were consistent with a depletion of the PtdIns(4,5)P2 pools at the plasma membrane in the EFR3A/B-depleted cells which would be more severe at a higher level of stimulation.
Fig. 3.

Depletion of EFR3s results in impaired cytoplasmic Ca2+ response without a notable depletion of PtdIns(4,5)P2. (A) The efficiency of an EFR3A knockdown was investigated by using two different siRNA duplexes; the levels of the endogenous EFR3A protein were measured. (B) EFR3B knockdown efficiency was tested using an expressed GFP-tagged protein because of the lack of suitable antibody to determine endogenous levels. (C) HEK293-AT1 cells cultured on 25 mm coverslips were treated with the indicated siRNAs for 3 days. Cells were loaded with Fura-2/AM and their Ca2+ responses to 100 nM (left) or 1 nM (right) AngII monitored in ratiometric imaging as described in Materials and Methods. The difference of the Ca2+ responses to 100 nM AngII was highly significant (for analyzing all time points >60 sec, P<0.0001); we either used unpaired t-test or the non-parametric Mann–Whitney test. (D,E) HEK293-AT1 cells treated with EFR3A and EFR3B siRNAs or with a control siRNA were labeled with [32P]phosphate for 3 h before stimulation with AngII (100 nM) for the indicated times. Reactions were terminated and lipids extracted and separated by TLC as detailed in Materials and Methods. Radioactivity was quantified by Phosphorimaging and expressed as the percentage of the pre-stimulatory value. Means±s.e.m. from three independent experiments are shown performed in duplicates.
EFR3-depleted cells do not show prominent defects in PtdIns4P and PtdIns(4,5)P2 resynthesis during stimulation
Next we tested the effects of EFR3 downregulation by using RNA interference (RNAi) on the PtdIns4P and PtdIns(4,5)P2 responses to stimulation with AngII using metabolic labeling of these lipids. Cells were labeled with 32P-phosphate or myo-[3H]inositol for 3 hrs or 24 hrs, respectively, before stimulation with AngII (100 nM) for the indicated times. Lipids were extracted and separated by thin layer chromatography (TLC) and quantified using PhosphorImaging (for 32P) or densitometry after exposure of plates that had been treated with EN3HANCE™ solution to X-ray film ([3H]inositol). As shown in Fig. 3D for changes in 32P-labeled PtdIns4P or PtdIns(4,5)P2 changes, we did not observe a reduction of these inositide pools in EFR3-depleted cells even after stimulation with the high concentration of AngII (100 nM). The PtdIns4P and PtdIns(4,5)P2 kinetics were very similar between the control and siRNA-treated groups (Fig. 3D,E), although the fall in the concentration of PtdIns(4,5)P2 at early time points was slightly deeper (Fig. 3D). These results suggested that the partial depletion of EFR3 (to the extent that was achieved in these experiments) had only little, if any, impact on PtdIns4P synthesis. It is worth pointing out, however, that even siRNA-mediated knockdown of PI4KA exerted only subtle effects on PtdIns(4,5)P2 resynthesis (Balla et al., 2008) or Ca2+ signaling (Korzeniowski et al., 2009) reported in our previous studies. Therefore, these results did not argue against a role of EFR3 proteins in PI4KA-mediated PtdIns4P synthesis as convincingly shown in recent studies (Nakatsu et al., 2012). However, they did raise the question whether the impaired Ca2+ response is caused by a mechanism other than the depletion of PtdIns(4,5)P2 pools under these experimental conditions.
EFR3 depletion has no major effect on the Ca2+ signal in cells that express a truncated AT1a receptor
<~?tlsb=-.015w?>There are a number of possibilities that could cause an alteration of the Ca2+ signal, including modification of Ca2+ pumps and influx pathways. However, one possible cause of a diminishing Ca2+ signal that can be easily tested is the desensitization of the receptor due to phosphorylation and internalization. Rapid receptor desensitization could also terminate PLC activation before PtdIns(4,5)P2 pools are depleted. Therefore, we generated HEK293 cells that stably express truncated (Y319stop) AT1a receptors, which lack the phosphorylation sites (HEK-AT1Δ) and, hence, is unable to interact with β-arrestins, and cannot be internalized and desensitized (Hunyady et al., 1994). We knocked down EFR3s in these cells to study their Ca2+ responses. As expected, the cells displayed a more-pronounced plateau in their cytoplasmic Ca2+ responses because of the lack of desensitization. Remarkably, this more-sustained Ca2+ elevation was only minimally affected by downregulation of EFR3 (Fig. 4A). This finding was important as it suggested that EFR3 knockdown did not have a main impact on the Ca2+-handling machinery or a massive effect on the phosphoinositide pools of the cell and, hence, the impaired Ca2+ signal observed with wild-type AT1a receptor must have been due to a process involving the receptor tail that does contain the phosphorylation sites.
Fig. 4.
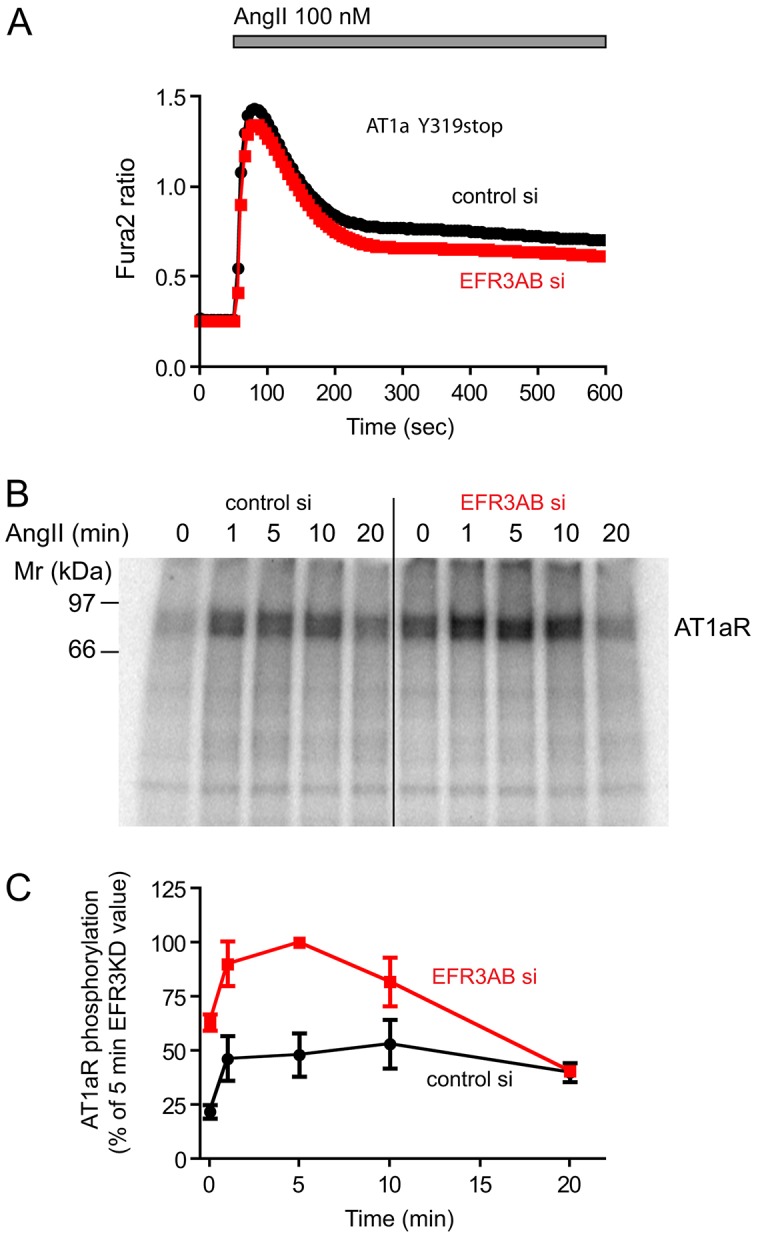
Depletion of EFR3s failed to affect the cytoplasmic Ca2+ response of cells that express a C-terminally truncated AT1a receptor and augmented phosphorylation of the wild-type AT1a receptor. (A) HEK293 cells stably transfected with an AT1a receptor truncated at its C-terminus (to remove all phosphorylation sites) were treated with siRNAs targeting EFR3A and EFR3B or with a control siRNA. Measurements of cytoplasmic Ca2+ responses to AngII were as described in the legend to Fig. 3 (B) Phosphorylation of the wild-type AT1a receptor immunoprecipitated from HEK293-AT1 cells labeled with [32P]phosphate and stimulated with AngII for the indicated times. Note the already increased phosphorylation of the receptor before and the larger response after AngII stimulation. Representative phosphorimage of three similar observations. (C) Quantification and summary of three independent experiments. All radioactivity values in each experiment were expressed as the percentage of the value measured at the 5 minute time-point in EFR3AB-depleted cells. Means±s.e.m. (n = 3).
AT1a receptors are hyperphosphorylated in EFR3-depleted cells
These findings turned our attention to receptor phosphorylation. Since our HEK-AT1 cells express receptors with a FLAG-tag at the N-terminus, we used these cells and labeled them with 32P-phosphate for 4 h followed by stimulation with AngII (100 nM) for the indicated times. Receptors were then immunoprecipitated and analyzed by western blotting and phosphorimaging. As shown in Fig. 4B, AT1a receptors isolated from EFR3-depleted cells, showed increased basal phosphorylation and a substantially larger phosphorylation response following stimulation with AngII. However, at later times (20 minutes after stimulation), the receptor phosphorylation subsided and was indistinguishable from that of the controls. This finding was consistent with impaired dephosphorylation of the AT1a receptor and could explain the faster receptor desensitization during stimulation.
AT1a receptor internalization is not impaired in EFR3-depleted cells
Previous studies have suggested that EFR3 is important for bulk endocytosis of membranes (Vijayakrishnan et al., 2009), and a genetic link was found in flies carrying mutations in dynamin and the EFR3 homologue, Rbo, suggesting that EFR3 plays a role in endocytosis (Vijayakrishnan et al., 2010). Without internalization, the receptor may not be dephosphorylated and regain its G protein signaling competence. Therefore, next we examined whether EFR3 knockdown affects endocytosis of the AT1a receptor. For this, we used several different approaches. First, we examined the translocation of GFP-tagged β-arrestin2 (GFP-β-arrestin2) in the HEK293-AT1 cell line after stimulation with AngII. In these experiments we used siRNA against EFR3B only. These experiments showed that EFR3 knockdown had no appreciable effect on the ability of the AT1 receptor to recruit GFP-β-arrestin2 to the membrane or on subsequent intracellular trafficking of the receptor–β-arrestin2 complex (Fig. 5A). Similar results were found with GFP-tagged β-arrestin1, although the association of this construct with the receptor was less pronounced both in the control and EFR3-depleted cells (not shown).
Fig. 5.

Internalization of the AT1a receptor is not inhibited by EFR3B depletion. (A) HEK293-AT1 cells treated with the indicated siRNAs were transfected with GFP-β-arrestin2 for 24 h and studied live under a confocal microscope at 30°C. Cells were treated with 100 nM AngII and images were acquired at the times indicated. (B) HEK293 cells stably expressing AT1aR-GFP were treated with the indicated siRNAs and studied live under a confocal microscope at 30°C. Cells were stimulated with 100 nM AngII and images were recorded at various time points. Note the higher fluorescence signal in the plasma membrane of control cells at 0 min compared with that of cells treated with EFR3B siRNA. These results were reproduced two times in multiple dishes (two or three) from independent knockdown experiments.
Second, we followed the distribution of the AT1 receptors in a stable cell line expressing the AT1a receptor tagged with GFP at the C-terminus (AT1aR-GFP) (Hunyady et al., 2002). In previous studies we have thoroughly characterized the trafficking of AT1aR-GFP (Hunyady et al., 2002). There was a notable decrease in the GFP signal at the plasma membrane of EFR3B-depleted cells before AngII stimulation relative to the internal signal (Fig. 5B). However, AT1aR-GFP showed similar clustering and endocytosis regardless of whether cells were treated with control or EFR3 siRNAs (Fig. 5B).
Third, we performed a more-quantitative assessment of receptor distribution by using flow cytometry. We determined the total number of receptors and that of cell-surface receptors by using HEK293 cells expressing AT1a receptors that were HA-tagged at the N-terminus and GFP-tagged at the C-terminus (HA-AT1aR-GFP). Cells were treated with siRNAi targeting EFR3A and EFR3B or with control siRNA prior to trypsinization, pelleting and fixation in 4% PFA. Cells were next immunostained using a mouse antibody against the extracellular HA epitope under non-permeabilizing conditions. Secondary antibody staining was performed using PerCP-conjugated goat anti-mouse antibody, and the total receptor population (GFP; Fig. 6A, left panel) and the surface receptor population (Ab signal; Fig. 6A, right panel) were analyzed using flow cytometry. These studies showed no decrease in the amount of cell-surface receptors in EFR3-depleted cells and a slight but statistically not significant increase in the amount of total receptors (Fig. 6A,B). To examine the extent of receptor internalization, we used HEK293-AT1 cells. Cells were treated with EFR3 A/B or control siRNA duplexes. Two days after the second knockdown treatment, cells were incubated with 1 µM Alexa-Fluor-488-conjugated AngII for 5 min at 37°C to allow receptor internalization. At that point the cell-surface-bound ligand was washed away with an acid wash and the cells were subjected to live-cell flow cytometry. These experiments showed no significant difference in the amount of acid-resistant signal between control and EFR3-depleted cells (Fig. 6CD).
Fig. 6.

Effects of EFR3 depletion on AT1aR levels and AngII uptake. (A,B) HEK19 cells stably expressing HA-AT1aR-GFP were treated with control siRNA (control RNAi) or with EFR3A/B-targeting siRNA (EFR3 A/B RNAi). Cells were then fixed and immunostained against the HA epitope under non-permeabilizing conditions and analyzed using flow cytometry, recording the GFP signal of the total AT1aR population (left panel) and the cell-surface HA epitope signal (right panel). In B, control knockdown levels (control RNAi; black) were compared to those in EFR3 A/B knockdown cells (EFR3 A/B RNAi; gray) (n = 10,000 cells). Shown are grand averages ± range from two experiments. (C,D) HEK293-AT1 cells treated with control siRNA or siRNA directed against EFR3 A/B, were incubated with Alexa-Fluor-488-conjugated AngII for 5 min prior to acid wash and detection of internalized AngII by flow cytometry. EFR3 A/B depletion (right panel) did not alter the rate of AngII internalization when compared to the control knockdown (left panel) (n = 10,000 cells). Red, signals (C) and bars (D) indicate acid resistant fluorescent AngII uptake; black signals (C) and bars (D) indicate total cell-associated (surface + internalized) AngII fluorescence.. AU, arbitrary units.
AT1a receptors uncouple from Gq proteins more rapidly in cells depleted in EFR3
These results did not indicate a major problem with AT1 receptor endocytosis. In fact, if anything, the receptor internalized more rapidly. However, hyperphosphorylation of the receptor could mean more internalization as well as a more rapid uncoupling of the receptor from its partner G protein(s). To test this latter possibility, we next examined the kinetics of Gq activation in HEK293 cells transfected with the AT1a receptor, together with a plasmid that drives the stoichiometric expression of the three Gq subunits: an mTurquoise (mTq)-tagged Gαq, a YFP-tagged Gγ2 and an untagged Gβ subunit (Goedhart et al., 2011). This reporter system is capable of monitoring Gq activation directly based on a decreasing Förster resonance energy transfer (FRET) between the two FP-tagged subunits once they dissociate (or change orientation) following activation by the ligand-bound receptor (Fig. 7A). As shown in Fig. 7B, stimulation of AT1 receptors induced rapid activation of Gq, which was reflected in the rapid decrease in the FRET ratios. This activation was followed by a rapid dose-dependent deactivation, which became more prominent at high AngII concentrations (>30 nM). Depletion of EFR3B (or EFR3A and EFR3B) caused no change in the activation kinetics, but the receptors uncoupled from the Gq protein more quickly than in control siRNA-treated cells (Fig. 7C). In cells expressing the truncated AT1 receptors, the activation of Gq was more robust and without deactivation and it was not affected by depletion of the EFR3 proteins (Fig. 7D). These results were consistent with the phosphorylation data and suggested that higher phosphorylation of the receptor results in a more-rapid uncoupling from the Gq proteins.
Fig. 7.

Effect of EFR3B knockdown on Gq activation kinetics in HEK293 cells expressing wild-type or tail-deleted AT1a receptor. (A) Schematic of the principles of monitoring Gq activation using FRET. Upon excitation, the mTq fluorescent protein engineered into the Gq α-subunit will transfer its energy to the mutated YFP Venus, which is attached to the γ-subunit of the βγ complex in the tightly associated heterotrimer. After activation of the AT1a receptor, the dissociation of the heterotrimer will result in a decrease in direct energy transfer, which can be detected as an increased emission from mTq and a decreased emission from Venus (see (Goedhart et al., 2011) for details). (B) FRET measurements in HEK293-AT1 cells expressing the wild-type AT1a receptors were stimulated with increasing concentrations of AngII. (C) HEK293 cells expressing the wild-type AT1a receptors were treated for 3 days either with control siRNA (grey trace) or with EFR3B siRNA (red trace). Means±s.e.m. are shown for 250 and 216 cells in control and EFR3B-depleted cells, respectively, that were acquired in three independent knockdown experiments (note that the error bars are too small to be visible in the red trace). (D) FRET experiments as described in B, except that HEK293 cells expressing the truncated AT1a receptors were used. Means±s.e.m. are shown for 53 and 91 cells in control and EFR3B-depleted cells, respectively, that were acquired in two independent knockdown experiments. (E) cAMP changes in HEK293-AT1 cells that had been stimulated with isoproterenol. Cells were treated with siRNA against EFR3A/B or control siRNA and then transfected with a FRET-based cAMP sensor (Klarenbeek and Jalink, 2014) to monitor changes in cAMP levels. A decreased FRET means an increase in cAMP. Means±s.e.m. are shown of 50-52 cells for EFR3AB or control siRNA treated cells. These were obtained in two separate knockdown experiments and using six independent dishes for each group. The difference between the two curves at time points later than at 240 sec is statistically significant (P<0.0001; using either non-paired t-test or the non-parametric Mann–Whitney test).
Gs-coupled β-adrenergic cAMP responses are also affected by EFR3 depletion
To determine whether the rapid desensitization in EFR3-depleted cells similarly affects Gs-coupled receptors, we investigated the effects of EFR3 depletion on the cAMP responses of HEK293 cells to endogenous β-adrenergic stimulation by isoproterenol. For these measurements cells were transfected with a FRET-based cAMP sensor based on exchange protein directly activated by cAMP (EPAC) (Klarenbeek and Jalink, 2014). As shown in Fig. 7E, isoproterenol (10−5 M) evoked a rapid elevation of cAMP (reflected as a decreased FRET signal) that slowly started to decline after 5 min of stimulation. In cells treated with EFR3A/B siRNA, the amplitude of the cAMP response was similar but there was an enhanced decay in the response (Fig. 7E). This difference was statistically significant, but it was not as pronounced as those observed with the AT1 receptor.
DISCUSSION
These studies were designed to explore the importance of the EFR3 proteins in the regulation of signaling from PLC-activating GPCRs. Studies in yeast have clearly demonstrated that Efr3 is critical for the organization of the PI4KA orthologue, Stt4 into active signaling complexes (Baird et al., 2008). Since our previous studies suggested that PI4KA is the kinase that supplies the plasma membrane with PtdIns4P for PtdIns(4,5)P2 synthesis (Balla et al., 2005; Bojjireddy et al., 2014), we set out to investigate whether the mammalian EFR3 proteins EFR3A and EFR3B play any role(s) in maintaining the PtdIns4P and PtdIns(4,5)P2 supply during strong agonist activation by affecting the function of PI4KA. An Efr3 homologue has already been identified in Drosophila and its mutation was responsible for the Rolling blackout (Rbo) phenotype (Huang et al., 2004). Rbo inactivation is lethal in flies, but Rbo mutants show a rapidly diminishing light response in the retina and a phenotype that is somewhat similar to the dynamin mutant Shibire (Vijayakrishnan et al., 2010). Rbo was believed to be an integral membrane protein with features of a lipase (Huang et al., 2004). When we expressed the human EFR3A and EFR3B proteins either tagged with a HA-epitope or with GFP at the C-terminus, we found the protein exclusively localized at the plasma membrane. However, expression of an N-terminally truncated form was found in the cytosol, our attention was drawn to a cysteine-rich region characteristic of these proteins. We suspected that these proteins were palmitoylated, which we were able to confirm with 3H-palmitate labeling of the immunoprecipitated proteins. A mutant protein lacking the cysteines was found in the cytosol, which indicates that the protein is not an integral membrane protein. While our experiments were in progress, a similar conclusion was reached in a study from the DeCamilli laboratory (Nakatsu et al., 2012).
To determine whether the supply of PtdIns4P and PtdIns(4,5)P2 might be limited in cells depleted in EFR3A and EFR3B, we stimulated HEK293-AT1 cells with AngII, and measured cytoplasmic Ca2+ responses and the kinetics of PtdIns4P and PtdIns(4,5)P2 in myo-[3H]inositol-or [32P]phosphate-labeled cells during stimulation. These experiments showed a highly consistent decrease in the sustained phase of the Ca2+ increase after AngII stimulation in EFR3-depleted cells, but no indication of a reduction of the PtdIns4P and PtdIns(4,5)P2 pools. We reasoned that our failure to show such lipid depletion might be related to the inability of the AT1 receptors to evoke a robust sustained PLC activation due to the known desensitization of the receptor. Therefore, we turned to cells that express a truncated AT1 receptor that shows no internalization and desensitization, with the idea that stimulation of the mutated receptor would pose a bigger challenge for the PtdIns(4,5)P2 synthesizing machinery (Olivares-Reyes et al., 2001). However, contrary to our expectation, EFR3 knockdown failed to affect the Ca2+ signal in these cells, suggesting that the effects of EFR3 depletion on the Ca2+ responses observed with the wild-type AT1 receptor were not caused by either a short supply of inositol lipid precursors or an alteration of Ca2+ handling of the cells. Instead, these results indicated that EFR3s controls the receptor itself through a process that is linked to the tail region of the receptor that is lacking in the deletion mutant. Since this region contains the phosphorylation sites, next we examined the phosphorylation status of the receptor and found that EFR3 depletion caused an increased basal phosphorylation and an enhanced phosphorylation response following stimulation. In parallel experiments we also determined that AT1 receptor internalization and the subsequent trafficking were not inhibited in EFR3-depleted cells. These findings together pointed to a possible defect in the ability of the receptor to maintain Gq activation in EFR3-depleted cells caused by hyperphosphorylation and uncoupling from the G-proteins, in other words, a more rapid desensitization. This was studied directly by using a FRET-based Gq activation sensor (Adjobo-Hermans et al., 2011) and the results showed that EFR3-depletion, indeed, yielded faster inactivation of the wild-type receptor but not the tail-deleted mutant.
Taken all these findings together, we assume that there is a defect in the resensitization of the receptors from their desensitized state in EFR3-depleted cells. This would also explain the rapidly diminishing light responses of the Rbo fly and their increased pool of PtdIns(4,5)P2 due to the inability of the photoreceptor to couple transducin and activate PLC (Huang et al., 2004). There is limited knowledge on how phosphorylated GPCRs are dephosphorylated and return to their active state. In Drosophila, a Ca2+-activated protein phosphatase, a product of the rdgC gene, was found to be responsible for the dephosphorylation of the photoreceptor (Steele et al., 1992; Vinós et al., 1997). Mammalian homologues of rdgC have been identified as PPEF1 and PPEF2 (Montini et al., 1997; Sherman et al., 1997), but mice deficient in these proteins had no problems with GPCR desensitization or reactivation, either in the retina or other tissues (Ramulu et al., 2001). In mammalian cells several phosphatases can dephosphorylate GPCRs, but it is generally agreed that PP2A dephosphorylates β2-receptors and, perhaps, other GPCRs as well (Vasudevan et al., 2011). A special ‘latent’ pool of PP2A residing in internal membranes was postulated to be sufficient to act on β2-receptors (Yang et al., 1988) and it was suggested that β-adrenergic receptors have to be internalized and recycled to regain their coupling competence (Yu et al., 1993). However, other studies showed that GRK-mediated GPCR phosphorylations could be reversed at the plasma membrane (Iyer et al., 2006). Since EFR3s themselves do not show internalization we assume that their function is linked to the plasma membrane. It should also be noted that the higher level of receptor phosphorylation still subsides after a 20-minute stimulation. This suggests either a delayed dephosphorylation response or the existence of more than one mechanism responsible for receptor dephosphorylation at different stages of receptor trafficking. No signs of a defect in AT1 receptor internalization or subsequent trafficking were observed in the present study. The notable decrease in the AT1aR-GFP signal at the plasma membrane relative to the cytoplasm in EFR3-depleted cells may suggest a problem with the return of the phosphorylated and internalized receptors to the plasma membrane. However, flow cytometry did not indicate decreased levels of receptors on the cell surface after EFR3A/B knockdown, and did not find accumulation of receptor-containing vesicles beneath the plasma membrane at light-microscopy level.
Recent reports have shown that EFR3 and TTC7 proteins are responsible for the recruitment of PI4KA to the plasma membrane in yeast (Baird et al., 2008) and also in mammalian cells (Nakatsu et al., 2012), a finding we were able to confirm (N.B. and T.B., unpublished observation). The question then naturally arises whether the rapid desensitization of the receptor in EFR3-depleted cells is related to a defect in the function of PI4KA. We found no indication of a problem regarding maintenance of the PtdIns(4,5)P2 pool in EFR3-depleted cells, even when stimulated through a non-desensitizing AT1R. However, it is important to emphasize that these results do not argue against the role of EFR3s in the control of PI4KA function. They merely suggest that, at the level of EFR3 knockdown we achieved, PI4KA can still supply the necessary phosphoinositides. It should be also noted that it has proven to be extremely difficult to achieve PI4KA knockdowns that would limit PtdIns4P production at the plasma membrane (Balla et al., 2008; Balla et al., 2005). This, however, also suggests that the process that controls the resensitization of the AT1R is more sensitive to EFR3 depletion than the PI4KA-mediated maintenance of the PtdIns(4,5)P2 pool. Furthermore, PI4KA-depleted cells had shown no signs of rapid desensitization of the AT1a receptor in our previous studies (Korzeniowski et al., 2009).
We also examined, whether the same phenomenon is observed when the cells are stimulated through a Gs-coupled receptor and found that the cAMP response to isoproterenol in HEK293 cells that express endogenous β–receptors is also affected by a knockdown of EFR3A/B, although the effect was not as pronounced as the one observed with the AT1a receptor. This may be caused by the fact that we did not directly measure the coupling of Gs but only cAMP levels, and the latter might not reflect the extent of desensitization so directly. More studies will be needed to extend the generality of this phenomenon to other GPCRs and to address the exact mechanism by which receptor resensitization occurs. Nevertheless, the cAMP studies also suggest that the observed phenomenon is not related to changes of phosphoinositide, as evoked by Gq-coupled receptors.
In summary, the present results identify a hitherto unrecognized and unexpected function of EFR3 proteins in mammalian cells, namely their importance in the control of GPCR responsiveness by affecting receptor phosphorylation. AT1Rs in EFR3-depleted cells are hyperphosphorylated and uncouple from Gq proteins more rapidly after stimulation, but they maintain their ability to internalize. These findings highlight the caveats in our knowledge of the mechanism(s) that control GPCR dephosphorylation and resensitization, and should facilitate further studies to uncover the molecular details of these processes, as well as the role of the EFR3 proteins within.
MATERIALS AND METHODS
Materials
All chemicals were of the highest analytical grade. Angiotensin II (human octapeptide; AngII) was from Bachem (Torrance, CA). [γ32P]ATP (6000 Ci/mmol) was purchased from Perkin-Elmer. Myo-[3H]inositol (30–80 Ci/mmol) was from Amersham and American Radiolabeled Chemicals (St Louis, MO). The monoclonal anti-hemagglutinin (HA) antibody (Ab) (HA.11) was from Covance, the anti-FLAG and the EFR3A antibodies were from Sigma (St Louis, MO). Antibodies against tubulin and actin were from Sigma Aldrich and Cell Signaling, respectively.
Plasmids and cells
The GFP-tagged β-arrestin2 (Barak et al., 1997) was kindly provided by Marc Caron (Duke University, Durham, NC). The Gq FRET construct (Goedhart et al., 2011) was a kind gift from Joachim Goedhart and Theodorus W. Gadella Jr. (University of Amsterdam). The HEK293-AT1 cells stably express the rat AT1a angiotensin receptor (Balla et al., 2005). HEK293-AT1-GFP cells stably express the rat AT1a angiotensin receptor fused to GFP (AT1aR-GFP) (Hunyady et al., 2002), and the HEK-AT1Δ cell line stably expresses the rat AT1a angiotensin receptor with a stop at Y319 (Hunyady et al., 1994). EST clones for EFR3A (EHS1001-213247615) and EFR3B (MHS6278-202802246) (IHS1382-8680872, containing the N-terminal EFR3B piece missing from clone MHS6278-202802246) were from Open Biosystems. The coding sequence of EFR3A, EFR3B short form and long form were subcloned into the pCDNA3.1(+) vector with an HA-tag at C-termini. For localization studies EFR3A and 3B were also subcloned into pEGFP-N1 and mRFP-N1 plasmids. Palmitoylation sites EFR3A-GFP (amino acids 5-CCCC-11) and EFR3B-GFP (amino acids 4-CGCC-9) were mutated to EFR3A (5-SSSS-11) and EFR3B (4-SGSS-9) by site directed mutagenesis using the Quikchange mutagenesis kit from Promega.
Transfection of cells
Cells (50,000 cells/well) were plated onto 25-mm-diameter circular glass coverslips treated with Poly-lysine in 6-well plates and plasmid DNAs (0.5–1 µg/well) were transfected with the indicated constructs (Szentpetery et al., 2009) using Lipofectamine2000 reagent (Invitrogen) and OPTI-MEM (Invitrogen) following the manufacturer's instructions. For siRNA treatment, cells were either cultured on coverslips in 6-well dishes for imaging or 12-well dishes for metabolic labeling. Cells were treated with 100 nM siRNA using oligofectamine. Experiments were carried out with the knockdown cells 3 days post siRNA treatment.
Live-cell imaging
After 20–24 hrs of transfection, cells were washed on the glass coverslips with a modified Krebs-Ringer solution, containing 120 mM NaCl, 4.7 mM KCl, 1.2 mM CaCl2, 0.7 mM MgSO4, 10 mM glucose, 10 mM Na-HEPES pH 7.4 and the coverslip was placed into a metal chamber (Atto, Invitrogen) that was mounted on a heated stage (35°C). Cells were incubated in 1 ml of the Krebs-Ringer buffer and the stimulating agents were dissolved and added in 200 µl warm buffer removed from the cells. Cells were examined by using inverted microscopes. Confocal images were obtained with a Zeiss LSM 510-META laser confocal microscope (Carl Zeiss MicroImaging, Inc.) using a 63×oil-immersion objective equipped with an objective heater (Bioptech).
Cytoplasmic Ca2+ measurements
HEK-AT1 cells (4×105 cells) cultured on coverslips were treated with siRNA as described above. Cells were loaded with 3 µM Fura 2/AM at room temperature for 45 min in HEPES-buffered M199-Hanks' salt solution containing 200 µM sulfinpyrazone and 0.06% pluronic acid. Ca2+ measurements were performed in modified Krebs-Ringer solution (see above) containing 200 µM sulfinpyrazone. Single-cell Ca2+ measurements were carried out at room temperature using an Olympus IX70 inverted microscope equipped with Lamda-DG4 illuminator and a MicroMAX-1024BFT digital camera with appropriate filters. Data were acquired using MetaFluor software (Molecular Devices).
Analysis of myo-[3H]inositol- or [32P]phosphate-labeled lipids
HEK293-AT1 cells plated on 12-well plates (at a density of 30,000 cells/ml) were labeled with myo-[3H]inositol (20 µCi/ml) in 1 ml of inositol-free DMEM supplemented with 2% dialyzed FBS for 24 h or with 2 µCi/ml o-[32P]phosphate for 3 h in phosphate-free DMEM supplemented with 2% dialyzed FBS. Cells were stimulated with AngII (100 nM) for the indicated times or left unstimulated. Reactions were terminated by the addition of ice-cold perchloric acid (5% final concentration), and cells were kept on ice for 30 min. Cells were scraped off the plates and frozen and thawed once; they were then centrifuged and the cell pellet was processed to extract the phosphoinositides by an acidic chloroform–methanol extraction followed by thin-layer chromatography (TLC) essentially as described previously (Balla et al., 2008; Nakanishi et al., 1995). TLC plates were sprayed with EN3HANCE™ solution (Perkin-Elmer) and subjected to autoradiography (in the case of 3H-labeling) or analyzed without spraying using a PhosphorImager (for the 32P-labeled samples). Autographic films were exposed several times to find the correct exposure time for densitometric analysis.
Analysis of incorporation of [3H]-palmitate
The procedure described by Linder et al. was followed (Linder et al., 1995). Briefly, COS-7 cells were transiently transfected with pGFP-N1-EFR3A or pEGFP-N1-EFR3B. After 24 hrs, cells were metabolically labeled with [3H]-palmitate (0.3 mCi/ml) for 4 hrs. Prior to lysis, cells were washed once with ice-cold PBS pH 7.4, lysates were immunoprecipitated with GFP-trap (ChromoTek GmbH, Germany) overnight and washed three times with lysis buffer. Proteins were resolved using SDS-PAGE, and the gel was incubated with EN3HANCE solution for 2 hrs, dried and subjected to autoradiography. pEGFP-N1-PI4K2A was used as a positive control in this experiment.
Analysis of receptor phosphorylation
HEK293-AT1 cells (ctrl siRNA- or EFR3AB siRNA-treated) in 6-well plates were metabolically labeled with 100 µCi/mL ortho-32PO4 for 4 hrs at 37°C in phosphate-free Dulbecco's modified Eagle's medium (DMEM) containing 2% dialyzed FBS. Cells were stimulated with 100 nM AngII for the indicated time points and lysed in ice-cold lysis buffer [50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.25% sodium deoxycholate, 1% NP-40, 1 mM DTT, 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride, 10 µg/ml aprotinin and 10 µg/ml leupeptin]. Cell lysates were incubated on ice for 15 min and centrifuged at 16,000 g. Supernatants were further incubated with a mixture of anti-HA and anti-FLAG tag antibodies and protein-G–agarose beads overnight at 4°C. Immunoprecipitates were further washed three times with lysis buffer and incubated in sample buffer for 1 h at 48°C followed by SDS-PAGE; the gel was dried and subjected to analysis using a PhosphorImager.
FRET analysis of Gq activation and cAMP changes
To explore the kinetics of the Gαq or cAMP Förster resonance energy transfer (FRET) reporter, the appropriate HEK293 cells were cultured on glass coverslips and transfected with the pGβ-2A-YFP-Gγ2-IRES-Gαq-mTq construct (kind gift from Joachim Goedhart and Theodorus Gadella, University of Amsterdam) (Goedhart et al., 2011) or the EPAC-based cAMP FRET sensor (Klarenbeek and Jalink, 2014) (kindly provided by Dr Kees Jalink, The Netherlands Cancer Institute, Amsterdam) for 24 h. Coverslips were placed into a metal Attofluor cell chamber (Invitrogen) and the cells were incubated in 1 ml modified Krebs-Ringer buffer (see above) at room temperature. Cells were stimulated with AngII (0.1–100 nM) (for Gq activation) or isoproterenol (10−5 M) (for cAMP) (stimuli, dissolved in 200 µl buffer removed from the cells, were added with proper mixing). FRET measurements were performed on an Olympus IX70 inverted microscope equipped with a Lamda-DG4 illuminator (Sutter Instruments, Novato, CA) and a MicroMAX-1024BFT digital camera (Roper Scientific, Trenton, NJ). A beam-splitter (DualView, Photometrics) with the appropriate filter sets was used to obtain a time-lapse of cyan and yellow fluorescent protein (CFP and YFP, respectively) images. Data acquisition and processing was performed by using MetaFluor software (Molecular Devices), where YFP:CFP ratios were calculated after background-subtraction and normalized to the initial pre-stimulatory ratio values. For the EFR3A and EFR3B siRNA experiments, HEK293-AT1 cells were treated with the respective siRNAs (100 nM) for 3 days before transfection with the pGβ-2A-YFP-Gγ2-IRES-GαqmTq or the EPAC-FRET construct.
Flow cytometry
Measurement of AngII internalization rates by flow cytometry was performed on HEK293-AT1 cells at sub-confluent density that had been treated with EFR3 A/B or control siRNA duplexes. Two days following the second knockdown treatment cells were incubated with 1 µM Alexa-Fluor-488-conjugated AngII for 5 min, washed with the acid wash solution (150 mM NaCl and 50 mM acetic acid, pH 4.0) in order to remove the surface-bound ligand, and assayed by live-cell flow cytometry.
Analysis of total and surface AT1aR levels was performed in HEK19 cells stably expressing HA-AT1aR-GFP that had been treated for EFR3 A/B or control knockdown prior to trypsinization, pelleting and fixation in 4% PFA. Cells were next immunostained with a mouse antibody against the extracellular HA epitope under the non-permeabilizing conditions. Secondary antibody staining was performed using the PerCP-conjugated goat anti-mouse secondary antibody. Data acquisition was performed on Becton Dickinson FACScan cytometer (Franklin Lakes, NJ) using CELLQuest software, processing 10,000 cells per sample.
Quantitative RT-PCR
Total RNA from various mouse tissues (replicate tissues from three mice) were isolated using RNeasy Kit (Qiagen), and cDNA was synthesized using Omniscript reverse transcriptase and random hexamers according manufacturers instructions (Qiagen). Real-time quantitative PCR (RT-qPCR) was performed in duplicates using SYBR green (Bio-rad). Primers used for RT-qPCR were EFR3A-F: 5′-CAGCTGACAAAGAAGAGAAC-3′, EFR3B-R: 5′-ACTGAGCCTGGATAGAATAC-3′ and EFR3B-F: 5′-GAGCATGAGGAGTGCATGTTCCAG-3′, EFR3B-R: 5′- GAAACCTGTGGATACCTGAAGGAGGG-3′. RT-qPCR results for the mRNA levels of each gene were normalized to level of 18S rRNA.
Acknowledgments
The plasmid for the simultaneous expression of the three Gq subunits was kindly provided by Joachim Goedhart and Theodorus W. Gadella Jr. (Swammerdam Institute for Life Sciences, University of Amsterdam, The Netherlands) and the EPAC-based cAMP sensor by Kees Jalink (The Netherlands Cancer Institute, Amsterdam). Confocal imaging was performed at the Microscopy and Imaging Core of the National Institute of Child Health and Human Development (NICHD), NIH, with the kind assistance of V. Schram and J. T. Russell.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
N.B. performed Ca2+, lipid labeling, palmitoylation, receptor phosphorylation and cAMP experiments and contributed to planning experiments and writing the manuscript. M.L.G.-H. performed Ca2+ measurements and the internalization experiments. N.R.R. performed the Gq FRET measurements. M.J. performed the flow cytometric analysis of AT1 receptor quantifications. T.B. designed the experiments, prepared figures and wrote the manuscript.
Funding
This research was supported by the Intramural Research Program of the NICHD, National Institutes of Health (NIH). N.R.R. was supported by a grant from the Joe Kolk Foundation sponsoring her visit to the NIH as part of her undergraduate program from the Swammerdam Institute for Life Sciences, University of Amsterdam, The Netherlands. Deposited in PMC for release after 12 months.
References
- Adjobo-Hermans M. J., Goedhart J., van Weeren L., Nijmeijer S., Manders E. M., Offermanns S., Gadella T. W., Jr (2011). Real-time visualization of heterotrimeric G protein Gq activation in living cells. BMC Biol. 9, 32 10.1186/1741-7007-9-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audhya A., Emr S. D. (2002). Stt4 PI 4-kinase localizes to the plasma membrane and functions in the Pkc1-mediated MAP kinase cascade. Dev. Cell 2, 593–605 10.1016/S1534-5807(02)00168-5 [DOI] [PubMed] [Google Scholar]
- Baird D., Stefan C., Audhya A., Weys S., Emr S. D. (2008). Assembly of the PtdIns 4-kinase Stt4 complex at the plasma membrane requires Ypp1 and Efr3. J. Cell Biol. 183, 1061–1074 10.1083/jcb.200804003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla A., Tuymetova G., Tsiomenko A., Várnai P., Balla T. (2005). A plasma membrane pool of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase type-III alpha: studies with the PH domains of the oxysterol binding protein and FAPP1. Mol. Biol. Cell 16, 1282–1295 10.1091/mbc.E04-07-0578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla A., Kim Y. J., Varnai P., Szentpetery Z., Knight Z., Shokat K. M., Balla T. (2008). Maintenance of hormone-sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4-kinase IIIalpha. Mol. Biol. Cell 19, 711–721 10.1091/mbc.E07-07-0713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak L. S., Ferguson S. S., Zhang J., Caron M. G. (1997). A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J. Biol. Chem. 272, 27497–27500 10.1074/jbc.272.44.27497 [DOI] [PubMed] [Google Scholar]
- Berridge M. J. (1984). Inositol trisphosphate and diacylglycerol as second messengers. Biochem. J. 220, 345–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojjireddy N., Botyanszki J., Hammond G., Creech D., Peterson R., Kemp D. C., Snead M., Brown R., Morrison A., Wilson S. et al. (2014). Pharmacological and genetic targeting of pPI4KA reveals its important role in maintaining plasma membrane PtdIns4p and PtdIns(4,5)p2 levels. J. Biol. Chem. 289, 6120–6132 10.1074/jbc.M113.531426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedhart J., van Weeren L., Adjobo-Hermans M. J., Elzenaar I., Hink M. A., Gadella T. W., Jr (2011). Quantitative co-expression of proteins at the single cell level – application to a multimeric FRET sensor. PLoS ONE 6, e27321 10.1371/journal.pone.0027321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F. D., Matthies H. J., Speese S. D., Smith M. A., Broadie K. (2004). Rolling blackout, a newly identified PIP2-DAG pathway lipase required for Drosophila phototransduction. Nat. Neurosci. 7, 1070–1078 10.1038/nn1313 [DOI] [PubMed] [Google Scholar]
- Hunyady L., Bor M., Balla T., Catt K. J. (1994). Identification of a cytoplasmic Ser-Thr-Leu motif that determines agonist-induced internalization of the AT1 angiotensin receptor. J. Biol. Chem. 269, 31378–31382. [PubMed] [Google Scholar]
- Hunyady L., Baukal A. J., Gaborik Z., Olivares-Reyes J. A., Bor M., Szaszak M., Lodge R., Catt K. J., Balla T. (2002). Differential PI 3-kinase dependence of early and late phases of recycling of the internalized AT1 angiotensin receptor. J. Cell Biol. 157, 1211–1222 10.1083/jcb.200111013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer V., Tran T. M., Foster E., Dai W., Clark R. B., Knoll B. J. (2006). Differential phosphorylation and dephosphorylation of beta2-adrenoceptor sites Ser262 and Ser355,356. Br. J. Pharmacol. 147, 249–259 10.1038/sj.bjp.0706551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarenbeek J., Jalink K. (2014). Detecting cAMP with an EPAC-based FRET sensor in single living cells. Methods Mol. Biol. 1071, 49–58 10.1007/978-1-62703-622-1_4 [DOI] [PubMed] [Google Scholar]
- Korzeniowski M. K., Popovic M. A., Szentpetery Z., Varnai P., Stojilkovic S. S., Balla T. (2009). Dependence of STIM1/Orai1-mediated calcium entry on plasma membrane phosphoinositides. J. Biol. Chem. 284, 21027–21035 10.1074/jbc.M109.012252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder M. E., Kleuss C., Mumby S. M. (1995). Palmitoylation of G-protein alpha subunits. Methods Enzymol. 250, 314–330 10.1016/0076-6879(95)50081-2 [DOI] [PubMed] [Google Scholar]
- Luttrell L. M., Lefkowitz R. J. (2002). The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 115, 455–465. [DOI] [PubMed] [Google Scholar]
- Montini E., Rugarli E. I., Van de Vosse E., Andolfi G., Mariani M., Puca A. A., Consalez G. G., den Dunnen J. T., Ballabio A., Franco B. (1997). A novel human serine-threonine phosphatase related to the Drosophila retinal degeneration C (rdgC) gene is selectively expressed in sensory neurons of neural crest origin. Hum. Mol. Genet. 6, 1137–1145 10.1093/hmg/6.7.1137 [DOI] [PubMed] [Google Scholar]
- Moore C. A., Milano S. K., Benovic J. L. (2007). Regulation of receptor trafficking by GRKs and arrestins. Annu. Rev. Physiol. 69, 451–482 10.1146/annurev.physiol.69.022405.154712 [DOI] [PubMed] [Google Scholar]
- Nakanishi S., Catt K. J., Balla T. (1995). A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proc. Natl. Acad. Sci. USA 92, 5317–5321 10.1073/pnas.92.12.5317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu F., Baskin J. M., Chung J., Tanner L. B., Shui G., Lee S. Y., Pirruccello M., Hao M., Ingolia N. T., Wenk M. R. et al. (2012). PtdIns4P synthesis by PI4KIIIα at the plasma membrane and its impact on plasma membrane identity. J. Cell Biol. 199, 1003–1016 10.1083/jcb.201206095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares-Reyes J. A., Smith R. D., Hunyady L., Shah B. H., Catt K. J. (2001). Agonist-induced signaling, desensitization, and internalization of a phosphorylation-deficient AT1A angiotensin receptor. J. Biol. Chem. 276, 37761–37768. [DOI] [PubMed] [Google Scholar]
- Ramulu P., Kennedy M., Xiong W. H., Williams J., Cowan M., Blesh D., Yau K. W., Hurley J. B., Nathans J. (2001). Normal light response, photoreceptor integrity, and rhodopsin dephosphorylation in mice lacking both protein phosphatases with EF hands (PPEF-1 and PPEF-2). Mol. Cell. Biol. 21, 8605–8614 10.1128/MCB.21.24.8605-8614.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter E., Ahn S., Shukla A. K., Lefkowitz R. J. (2012). Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52, 179–197 10.1146/annurev.pharmtox.010909.105800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman P. M., Sun H., Macke J. P., Williams J., Smallwood P. M., Nathans J. (1997). Identification and characterization of a conserved family of protein serine/threonine phosphatases homologous to Drosophila retinal degeneration C. Proc. Natl. Acad. Sci. USA 94, 11639–11644 10.1073/pnas.94.21.11639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele F. R., Washburn T., Rieger R., O'Tousa J. E. (1992). Drosophila retinal degeneration C (rdgC) encodes a novel serine/threonine protein phosphatase. Cell 69, 669–676 10.1016/0092-8674(92)90230-A [DOI] [PubMed] [Google Scholar]
- Szentpetery Z., Balla A., Kim Y. J., Lemmon M. A., Balla T. (2009). Live cell imaging with protein domains capable of recognizing phosphatidylinositol 4,5-bisphosphate; a comparative study. BMC Cell Biol. 10, 67 10.1186/1471-2121-10-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan N. T., Mohan M. L., Goswami S. K., Naga Prasad S. V. (2011). Regulation of β-adrenergic receptor function: an emphasis on receptor resensitization. Cell Cycle 10, 3684–3691 10.4161/cc.10.21.18042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayakrishnan N., Woodruff E. A., III and Broadie K. (2009). Rolling blackout is required for bulk endocytosis in non-neuronal cells and neuronal synapses. J. Cell Sci. 122, 114–125 10.1242/jcs.036673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayakrishnan N., Phillips S. E., Broadie K. (2010). Drosophila rolling blackout displays lipase domain-dependent and -independent endocytic functions downstream of dynamin. Traffic 11, 1567–1578 10.1111/j.1600-0854.2010.01117.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinós J., Jalink K., Hardy R. W., Britt S. G., Zuker C. S. (1997). A G protein-coupled receptor phosphatase required for rhodopsin function. Science 277, 687–690 10.1126/science.277.5326.687 [DOI] [PubMed] [Google Scholar]
- Wei H., Ahn S., Shenoy S. K., Karnik S. S., Hunyady L., Luttrell L. M., Lefkowitz R. J. (2003). Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. USA 100, 10782–10787 10.1073/pnas.1834556100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. D., Fong Y. L., Benovic J. L., Sibley D. R., Caron M. G., Lefkowitz R. J. (1988). Dephosphorylation of the beta 2-adrenergic receptor and rhodopsin by latent phosphatase 2. J. Biol. Chem. 263, 8856–8858. [PubMed] [Google Scholar]
- Yu S. S., Lefkowitz R. J., Hausdorff W. P. (1993). Beta-adrenergic receptor sequestration. A potential mechanism of receptor resensitization. J. Biol. Chem. 268, 337–341. [PubMed] [Google Scholar]