

ABSTRACT
The mechanism that coordinates activities of different adhesion receptors is poorly understood. We investigated this mechanism by focusing on the nectin-2 and E-cadherin adherens junction receptors. We found that, cadherin was not required for the basic process of nectin junction formation because nectin-2 formed junctions in cadherin-deficient A431D cells. Formation of nectin-2 junctions in these cells, however, became regulated by cadherin as soon as E-cadherin was re-expressed. E-cadherin recruited nectin-2 into adherens junctions, where both proteins formed distinct but tightly associated clusters. Live-cell imaging showed that the appearance of E-cadherin clusters often preceded that of nectin-2 clusters at sites of junction assembly. Inactivation of E-cadherin clustering by different strategies concomitantly suppressed the formation of nectin clusters. Furthermore, cadherin significantly increased the stability of nectin clusters, thereby making them resistant to the BC-12 antibody, which targets the nectin-2 adhesion interface. By testing different E-cadherin–α-catenin chimeras, we showed that the recruitment of nectin into chimera junctions is mediated by the actin-binding domain of α-catenin. Our data suggests that E-cadherin regulates assembly of nectin junctions through α-catenin-induced remodeling of the actin cytoskeleton around the cadherin clusters.
KEY WORDS: Cadherin, Nectin, α-catenin, Actin, Adhesion
INTRODUCTION
Two adhesion receptors, cadherin and nectin, form specific intercellular adhesive structures called adherens junctions that are crucial for the general organization, dynamics and strength of cell–cell contacts (Franke, 2009; Gumbiner et al., 1988; Perez-Moreno and Fuchs, 2006; Takai et al., 2008). These proteins can form junctions independently from one another; nectin junctions assemble in cadherin-deficient cells (Takahashi et al., 1999), and cadherin junctions are formed, albeit with reduced kinetics, in nectin-inactivated cells (Sato et al., 2006; Tanaka-Okamoto et al., 2011; Indra et al., 2014). In adherens junctions, cadherin and nectin are located in separate, yet closely associated, adhesive clusters (Indra et al., 2013). How these two different and independent adhesion receptors are co-recruited into the same adhesive structure is not yet known.
The major principles of nectin and cadherin assembly into the adherens junctions appear to be similar. The cadherin extracellular N-terminal domains self-associate, resulting in the formation of cadherin adhesive clusters (Brasch et al., 2012; Troyanovsky, 2012). Nectin adhesion is also mediated by nectin–nectin interactions through the N-terminal adhesive interface (Harrison et al., 2012; Samanta et al., 2012). Another similarity is that both cadherin and nectin junctions depend on the actin cytoskeleton. Actin filaments are essential for cadherin adhesion because they maintain and regulate cadherin cluster stability (Adams and Nelson., 1998; Mège et al., 2006; Hong et al., 2013). Although the role of the actin cytoskeleton in nectin adhesion is less understood, it has been shown that nectin interacts with the actin-binding protein afadin (Tachibana et al., 2000; Ozaki-Kuroda et al., 2002), and that actin depolymerization disrupts nectin junctions (Yamada et al., 2004). Given that both nectin and cadherin clusters in the adherens junctions interact with actin filaments and because these filaments have been shown to maintain the stability of both clusters, it is tempting to speculate that the actin cytoskeleton couples these two types of clusters. Another possibility is that the clusters are coupled through the cadherin-associated protein α-catenin because one of the α-catenin central domains, the M3 domain, interacts directly with the nectin-associated protein afadin (Tachibana et al., 2000; Pokutta et al., 2002).
It is not clear which of these two mechanisms results in the co-recruitment of cadherin and nectin into adherens junctions. It has been shown that the lack of α-catenin in epithelial cells disconnects the nectin and cadherin junctions (Tachibana et al., 2000). Experiments with cadherin–α-catenin chimeras have mapped the α-catenin region sufficient for cadherin–nectin colocalization to just two α-catenin domains – its afadin-binding domain (M3 domain) and its actin-binding domain (αABD). No further attempts were made to determine whether the indirect α-catenin–afadin interactions through actin filaments or the direct interactions through the α-catenin M3 domain are required for cadherin and nectin cluster association.
Another unanswered question is how cadherin and nectin coordinate their adhesive activities. Inhibitors of nectin adhesion or afadin knockdown slow the formation of cadherin adhesive clusters in a Ca2+-switch assay (Sato et al., 2006; Lorger and Moelling, 2006) or between two cells upon coming into contact with one another (Indra et al., 2014). However, this defect in the kinetics of adherens junction formation does not affect the general appearance of cell–cell adhesion contacts, including the ultrastructural features of adherens junctions (Zhadanov et al., 1999; Sawyer et al., 2009; Tanaka-Okamoto et al., 2011). Therefore, the available data show that nectin facilitates the formation of adherens junctions, but it is involved neither in the basic mechanisms of their assembly nor in their structural organization. No previous studies have addressed the alternative question regarding the role of cadherin in nectin junction formation. Indeed, the fact that nectin assembles junctions in cadherin-deficient cells does not exclude the possibility of a contribution of cadherin to nectin-based adhesive processes, particularly, to the organization and stability of the nectin adhesive structures. In order to fill this gap, we investigated the E-cadherin-induced changes of nectin-2 junctions. This study clearly shows that cadherin adhesive interactions regulate nectin cluster assembly and that this regulation is mediated by the actin cytoskeleton.
RESULTS
E-cadherin modulates the formation of nectin-2 junctions
To explore the relationship between cadherin and nectin adhesion, we first characterized nectin junctions in cadherin-deficient A431D cells. Using various anti-nectin antibodies, we found that these cells formed nectin-2 junctions (Fig. 1A–C). Structured illumination microscopy showed that these junctions, referred to below as linear nectin junctions, assembled in the form of a fine line that demarcated the apicolateral borders of each cell (Fig. 1C). These same junctions also recruited afadin (Fig. 1B). In agreement with published data, formation of the nectin junctions was independent of Ca2+ (supplementary material Fig. S1A) and they did not contain any components of cadherin adhesive structures, including vinculin or α-catenin (supplementary material Fig. S1B).
Fig. 1.

Nectin junctions depend on cadherin. (A) Representative image of anti-nectin-2-stained A431D cells. (B) The left column shows A431D cells double-stained for nectin-2 (Nt) and afadin (Af). Nectin forms apicolateral rings of wavy linear junctions that are associated with afadin. The set of images to the right shows A431D cells double-stained for nectin-2 (Nt) and α-catenin (αC). (C) Structured-illumination microscopy of nectin junctions in A431D cells stained for nectin-2. The apical cell region is shown. (D) A431D cells stably expressing EcadDn stained for nectin-2 (Nt). The boxed area is zoomed in the inset and is shown in two colors: red, nectin-2 (Nt), and green, EcadDn (Ec). Note that upon E-cadherin expression, nectin is localized at the apical adherens junctions. The images at the bottom show that apical adherens junctions (Ec, green) recruit vinculin (Vin, red). (E) EcadDn-expressing A431D cells were cultured for 20 min in the presence of function-blocking anti-cadherin antibody SHE78-7 (SHE) or in low Ca2+ medium (Low Ca) and then double-stained using mouse anti-cadherin (Ec, green) and rabbit anti-nectin-2 (Nt, red) antibodies. Note that both treatments severely affected both nectin and cadherin junctions. Higher magnifications of the selected regions (indicated by arrows) are shown in the insets. (F) Structured-illumination microscopy of nectin junctions in EcadDn- (EcDn) and cis-EcadDn (cis-EcDn)-expressing A431D cells double stained for nectin-2 (Nt) and E-cadherin (Ec). Note that the nectin-positive junctions were perpendicular to the cell–cell contact and associated with the apical cadherin junctions. Also note that mutation of the cadherin cis interface significantly disturbed cadherin junctional localization. Scale bars: 30 µm (A,B,E), 10 µm (D), 5 µm (C,F).
In order to analyze the influence of E-cadherin on nectin junctions, we re-expressed E-cadherin in A431D cells. As we previously reported, expression of Dendra2-tagged E-cadherin (EcadDn) results in the formation of radially oriented adherens junctions in A431D cells (Hong et al., 2010; Harrison et al., 2011). These junctions recruited nectin-2, thereby transforming the linear nectin junctions typical for the parental A431D cells into radial ones (Fig. 1D,F). Significant reorientation of nectin junctions by E-cadherin was also confirmed by quantification of the junctional angle frequency distribution (supplementary material Fig. S2A). As in many other cell types (Indra et al., 2013), the nectin-containing adherens junctions were also positive for vinculin (Fig. 1D). A striking loss of the linear organization of nectin junctions upon cadherin expression suggests that cadherin can control the distribution of nectin adhesive structures.
We then studied whether nectin junction organization in E-cadherin-expressing cells depends on the adhesive properties of E-cadherin. First, we suppressed cadherin adhesion in the EcadDn-expressing A431D cells using either the function-blocking anti-E-cadherin antibody SHE78-7 (Fig. 1E, SHE) or by placing the cells into low-Ca2+ medium (Fig. 1E, Low Ca). Notably, both of these approaches failed to restore the linear phenotype typical for nectin junctions in parental A431D cells. Instead, blocking E-cadherin adhesive activity by either strategy dramatically reduced the number of nectin junctions. The remaining junctions showed a much more diffuse appearance compared with the control cells and were still associated with the sites of highest E-cadherin concentration. Using another cell line (DLD-1, supplementary material Fig. S3), we verified that the Ca2+ dependency of nectin junctions in cadherin-expressing cells is a general phenomenon.
As an alternative approach to studying the role of cadherin in nectin junctions, we used adhesion-incompetent cadherin mutants. The first mutant was W2A-EcadDn, in which the point mutation W2A abolished the key cadherin trans-dimerization interface. Although this mutant was unable to form definitive junctions, it was still found concentrated at the cell–cell contacts of A431D cells (Fig. 2A, W2A), apparently, because of its second trans-X-dimerization interface (Harrison et al., 2010; Hong et al., 2011). Strikingly, similar to upon Ca2+ depletion and application of the function-blocking cadherin antibody, the expression of the W2A E-cadherin mutant dramatically reduced nectin junction abundance in A431D cells; only a few very weak junctions could be found in these cells (Fig. 2A, W2A, inserts).
Fig. 2.

Nectin-2 localization depends on cadherin adhesion properties. (A) A431D cells stably transfected to express EcadDn (Ec) or its mutants bearing point mutations inactivating either its adhesive (W2A) or cis-binding (Cis) interfaces. Cells were double-stained for cadherin (left column) and nectin-2 (Nt, right column). Higher magnifications of the selected regions (indicated by arrows) are shown in the insets. Scale bars: 30 µm. (B) High magnifications of the junctions produced in A431D cells expressing the cis-EcadDn mutant. The cells were double-stained for the mutant (Cis, green) and for nectin-2 (Nt), vinculin (Vin) or actin (Act). Green and red channels are shown separately for the boxed regions. Note that the cells contact one another through numerous actin-rich filopodia co-recruiting nectin and vinculin. Scale bars: 10 µm.
The second E-cadherin mutant was the cis-interface mutant cis-EcadDn. The mutation of this interface impairs cadherin lateral clustering, thereby resulting in the formation of very unstable adhesive structures (Harrison et al., 2011; Hong et al., 2013). In agreement with the previous data, we found that this mutant was unable to form distinguishable adherens junctions, and, similar to the W2A mutant, was found concentrated in cell–cell contact areas (Fig. 2A, cis). Careful inspection of the cis-EcadDn-expressing cells showed that the mutant was especially enriched along the filopodia-like protrusions that radially extend from the apical surface of many cells toward the centers of their neighbors. These cadherin-enriched radial protrusions, which often reached several micrometers in length, were the primary sites of nectin junction formation in these cells (Fig. 1F; Fig. 2B). Similar to the apical adherens junctions in EcadDn-expressing cells, the nectin-containing junctions that formed along these protrusions were aligned with the actin bundles and recruited vinculin (Fig. 2B). Therefore, the adhesive structures produced by the cis-interface E-cadherin mutant, despite substantial abnormalities, were still able to co-recruit nectin and vinculin. Taken together, these results show that cadherin adhesive activity is essential for nectin adhesion in cadherin-expressing cells.
E-cadherin forms junctions faster than nectin
As an alternative strategy to study the coupling between nectin and cadherin adhesion, we examined the cadherin and nectin junction formation in A431 cells co-expressing EcadDn and mCherry-tagged nectin-2 (N2mCH). As we have shown previously, these cells form typical adherens junctions consisting of cadherin (E- and P-cadherins) and nectin-2 clusters (Indra et al., 2013). We have also shown that once these cells touch one another, cadherin and nectin clusters always appear at the same sites (Indra et al., 2014). In half of the observed formations of intercellular contacts, cadherin and nectin clusters appeared simultaneously, in other half nectin clusters formed few seconds later (one such example is presented in Fig. 3A).
Fig. 3.
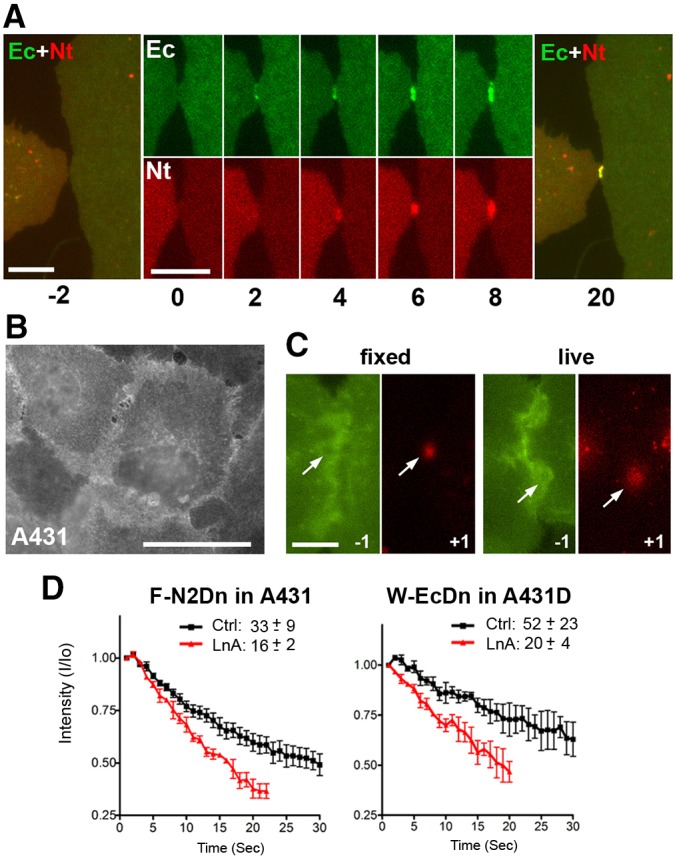
Kinetics of cadherin and nectin junction assembly. (A) A431 cells co-expressing EcadDn (Ec, green) and N2mCh (Nt, red) were imaged in 1-s intervals and with 1-s-long acquisition. Time (s) is shown below the frames. Time 0 is the moment when the cells touch one another. Note that cadherin clusters as fast as 2 s after time 0. Nectin clusters become visible only 2 s later. Merged images of the cells 2 s before (−2) and 20 s after (20) contact formation are also shown. Scale bars: 6 µm. (B) A431 cells expressing an adhesion-incompetent mutant of nectin-2, F136D-N2Dn. The mutant is randomly distributed along the cell surface. Scale bars: 30 µm. (C) Circular areas (diameter ∼2.5 µm) of F136D-N2Dn-expressing cells were photoconverted from green to red fluorescence. The green image taken 1 s before photoconvesion and the first red image taken 1 s after photoconversion are shown. The left pair of images was obtained from prefixed cells and the right pair from live cells. Note that the photoconverted red spot is much larger in live cells. Scale bar: 10 µm. (D) Graphs showing the change in intensity of the photoconverted red fluorescence over time (s) in the entire laser-irradiated area in the control conditions (Ctrl) and 10 min after Latrunculin A applications (LnA). The time needed for 50% decrease of the red fluorescence (t1/2) is shown for each case. The photoconversion experiments were repeated ten times with cells in each condition. Results are mean±s.d.
The slight delay of nectin clustering relative to cadherin cluster formation indicates that a cadherin-specific signal is required for nectin to initiate its clustering. Alternatively, cadherin junctions might form first due to an earlier arrival of cadherin to the intercellular contacts. To examine the second possibility, we compared the surface diffusion of the extrajunctional E-cadherin and nectin-2 molecules. To this end, we expressed the nectin-2 mutant, F136D-N2Dn, in A431 cells. In addition to the green-to-red photoswitchable protein Dendra2, this mutant harbored the point mutation F136D, which inactivates the adhesive nectin interface and results in a random distribution of nectin on the cell surface (Harrison et al., 2012; see also Fig. 3B). We reasoned that in the absence of adhesive interactions, the surface diffusion rate of this mutant corresponded to that of extrajunctional nectin-2 and, therefore, its lateral motility reflects the nectin-2 diffusion toward the sites of junction assembly. The mutant was photoconverted in a small spot (2.5 µm in diameter, Fig. 3C), and the red fluorescence intensity of this spot and its area were monitored over time. Our measurements showed that the photoconverted area doubled during the first second after photoconversion (Fig. 3C), and its red fluorescence decay (t1/2) was about 30 s (Fig. 3D). Latrunculin A treatment of these cells accelerated the red fluorescence decay (t1/2 = 15 s), indicating that extrajunctional nectin-2 molecules interact with or are constrained by actin filaments.
Then we performed the same experiments with the cadherin W2A-EcadDn mutant. This cadherin mutant, like the F136D nectin-2 mutant, could not assemble adhesive dimers or intercellular junctions (Fig. 2A). Photoconversion experiments with this mutant (Fig. 3D) clearly showed that its surface motility was significantly slower than that of nectin-2 (t1/2≈50 s). Latrunculin treatment, however, attenuated any differences in the rate of surface diffusion between nectin and cadherin adhesion-incompetent mutants. This result is consistent with previous experiments showing that cadherin extrajunctional molecules interact with the actin cytoskeleton (Borghi et al., 2012). In addition, our results show that interactions with actin slow down E-cadherin diffusion significantly more than that of nectin-2. Therefore, the late arrival of nectin-2 into adherens junctions cannot be explained by its diffusion rate. Taken together with the cadherin inactivation experiments presented above, these live-imaging experiments suggest that a nascent cadherin cluster might provide an initial impulse for nectin junction assembly.
E-cadherin increases the stability of nectin junctions
Cadherin might influence the assembly of the nectin junctions by changing the structural organization of the nectin clusters. In order to assess the structural features of nectin clusters, we studied their behavior in the presence of the anti-nectin-2 monoclonal antibody BC-12. The epitope for this antibody had been mapped to the nectin-2 N-terminal domain (Lopez et al., 2000), which forms the adhesive interface of this protein (Harrison et al., 2012). Therefore, we expected that this antibody would specifically destabilize nectin-2 junctions. Indeed, the addition of 10 µg/ml of this antibody to the culture medium of A431D cells completely disrupted nectin-2 junctions within 20 min (Fig. 4, A431D). Remarkably, the same concentration of BC-12 antibody added to EcadDn-expressing A431D cells was unable to induce the dissolution of the nectin-2 clusters even though the antibody was still able to bind nectin-2 (Fig. 4, EcDn). This difference (quantified in supplementary material Fig. S2B) suggests that the nectin-2 clusters in adherens junctions are much more stable than the same clusters formed without cadherin.
Fig. 4.

E-cadherin reinforces the nectin junctions. Parental A431D cells (A431D) and their derivatives expressing EcadDn (EcDn) or cis-EcadDn (cis-EcDn) were incubated for 20 min with BC-12 antibody (10 µg/ml) at 37°C (37°C) and then double-stained using anti-mouse antibody (Nt), detecting localization of BC-12–nectin-2 complexes, and rabbit anti-Dendra antibody (Dn), detecting recombinant Dendra-tagged E-cadherin. The control cells (4°C) were incubated with BC-12 antibody (20 min, 10 µg/ml) on ice. Higher magnifications of the selected regions (indicated by arrows) are shown in the insets. Scale bars: 10 µm. Note that BC-12 antibody disintegrates nectin junctions in A431D cells but is unable to do so in presence of cadherin.
Then, using the same assay, we studied the stability of the nectin-2 junctions in the cis-EcadDn-expressing A431D cells. We expected that unstable cis-EcadDn mutant clusters would be unable to shield nectin clusters from the BC-12 antibody, but we observed that nectin-2 junctions were in fact also BC-12 resistant (Fig. 4, cis-EcDn). Formation of such BC-12-resistant nectin-2 junctions in cells expressing the cis-interface E-cadherin mutant suggests that nectin does not directly associate with cadherin. Rather, cadherin adhesive clusters, including the unstable clusters formed by the cis-interface mutant, establish the sites that reinforce the nectin clusters. One possibility is that such nectin-assembly-promoting sites are formed through cadherin-based modifications of the actin cytoskeleton.
Actin-binding domain of α-catenin recruits nectin-2 into adherens junctions
The experiments described above demonstrate that nectin-2 is targeted to the sites of cadherin adhesive interactions. If cadherin cannot form junctions, the assembly of nectin-2 junctions is also disturbed. Previous work suggested, at least partially, that the cadherin–nectin association in adherens junctions is facilitated by α-catenin (Tachibana et al., 2000). In order to understand how α-catenin mediates the formation of proximal cadherin and nectin junctions, we took a reductionist approach by constructing a set of E-cadherin–α-catenin chimeras (Fig. 5A). It has been previously shown that a chimera consisting of a β-catenin-uncoupled E-cadherin module followed by the C-terminal α-catenin region (amino acids 509–906) forms nectin-associated cadherin junctions in cadherin-deficient L-cells (Tachibana et al., 2000). However, the interpretation of those results was not straightforward because the α-catenin-derived region of that chimera consisted of two functionally different domains, the M3 domain and αABD. Furthermore, the E-cadherin module contained a binding site for p120-catenin, a protein that itself had been suggested to interact with afadin (Hoshino et al., 2005; Birukova et al., 2012). Therefore, in order to exclude any effects of p120-catenin, we reinvestigated the nectin-targeting activity of cadherin–α-catenin chimeras using the cadherin module EcΔDn, which lacks both the p120- and β-catenin-binding sites (Hong et al., 2011, Fig. 5A). We first verified that nectin was not associated with the junctions produced by EcΔDn itself: indeed, even the highest expression level of EcΔDn neither changed the linear organization of nectin junctions in A431D cells nor resulted in nectin–cadherin cluster association (Fig. 5B).
Fig. 5.

The actin-binding domain of α-catenin is sufficient for nectin–cadherin junction association. (A) Schematic representation of the E-cadherin–α-catenin chimeras. Each chimera includes a different α-catenin region fused with the EcΔDn module consisting of dendra2, Dn (green) and the tailless cadherin, EcΔ (brown), which lacks any known cadherin intracellular binding sites. The C-terminal portion of α-catenin (amino acids 506–906), encompassing the M3 domain and αABD, is present in the EcΔDn–α506 chimera. Two other chimeras, EcΔDn–M3 and EcΔDn–HαABD, incorporate either one of these domains. (B–D) Double immunostaining of A431D cells expressing EcΔDn (B), EcΔDn–α506 (C), EcΔDn–M3 (D), EcΔDn–HαABC (E). All cells were stained with rabbit anti-dendra2 (Dn) to detect the chimeras and mouse anti-nectin-2 (Nt), mouse anti-afadin (Af), or Alexa-Fluor-555–phalloidin (Act). Higher magnifications of the boxed regions (shown in the merged images) are shown either in the insets or in the separate rows. Some of the radial and polymorphic non-radial junctions are indicated by arrows and arrowheads, respectively. Scale bars: 20 µm (5 µm for the zoomed areas).
Then, we analyzed A431D cells expressing EcΔDn fused with the α-catenin region 506–906 (EcΔDn–α506). This chimera, in a sharp contrast to the parental EcΔDn chimera, produced numerous well-defined junctions, many of which were aligned with the radial actin filament bundles (Fig. 5C, Act, arrows). These radial junctions were the only sites of nectin junction assembly in these cells (Fig. 5C, Dn+Nt). Notably, the chimera junctions that showed non-radial arrangement, were also devoid of nectin-2 (Fig. 5C, arrowheads). This preferential association of nectin junctions with radial cadherin–actin structures resulted in the complete loss of the linear nectin-2 organization that is typical for A431D cells.
To determine whether the M3 domain or the αABD domain was responsible for the nectin-2 colocalization with the EcΔDn–α506 junctions, we tested chimeras that contained either the M3 domain, EcΔDn–M3, or the αABD, EcΔDn–HαABD. Interestingly, both of these chimeras showed a clear association with afadin, although the characteristics of the nectin structures they induced were very different. Similar to EcΔDn, the EcΔDn–M3 chimera neither formed discernable junctional structures nor changed the linear character of nectin junctions (Fig. 5D, Dn+Nt). However, the cell–cell contact areas enriched with the EcΔDn–M3 chimera exhibited diffuse afadin staining, which was never observed in the control A431D cells (Fig. 5D, Dn+Af). This suggests that the weak binding of the M3 domain of the chimera to afadin might have been sufficient to drag afadin to the sites of cell–cell contacts, but was unable to link the chimera to nectin molecules in the junctions.
By contrast, the second truncated chimera, EcΔDn–HαABD, produced large, polymorphic and extraordinarily stable junctions, most of which showed no particular orientation (Hong et al., 2013; Fig. 5E, arrowheads). Strikingly, nectin-2 was unable to form any junctions in the areas of these polymorphic junctional structures (Fig. 5E, Dn+Nt, arrowheads). Nectin-2 was recruited only into the specific population of EcΔDn–HαABD junctions (Fig. 5E, Dn+Nt) that were characterized by a radial organization and association with radial actin bundles (Fig. 5E, Dn+Act, arrows). These radial junctions were particularly common at the sites of tri-cellular contacts or at the margins of cell colonies. These radial junctions were, therefore, very similar to the nectin-2-containing junctions formed by the EcΔDn–α506 chimera.
αABD is unique in its ability to promote nectin junctions
Taken together, the experiments with the EcΔDn–α-catenin chimeras showed a remarkable feature of αABD: once it is present, the chimera forms a variety of actin-associated junctional structures, but only one of these structures provides a platform for nectin junction assembly. Concomitantly, this domain inhibits formation of nectin junctions in other places. Is this function unique to αABD? In order to answer this question, we first investigated whether nectin-2 is targeted into the junctions produced by the EcΔDn–UtrABD chimera in A431D cells. This chimera, in which actin cytoskeleton binding is mediated by the actin-binding domain of utrophin (UtrABD, see Fig. 6A), produced junctions very similar in morphology and dynamics to those produced by EcΔDn–HαABD (Hong et al., 2013). Staining for nectin-2 showed that the UtrABD-based junctions, regardless of their orientation, were devoid of nectin-2 (Fig. 6C). Surprisingly, the expression of the EcΔDn–UtrABD chimera also dramatically reduced the number of linear nectin junction. This interesting effect prompted us to construct and to test another chimera, EcΔDn–Lact, in which actin binding is mediated by the actin-binding peptide Lifeact (Riedl et al., 2008). This chimera, like all actin-binding cadherin chimeras tested so far, formed prominent actin-associated adhesive structures (Fig. 6D). Remarkably, this chimera also dramatically suppressed nectin-2 junction formation in A431D cells, and, as in case of EcΔDn–UtrABD expression, the remaining nectin clusters were uncoupled from the chimera junctions. Finally, in an attempt to facilitate association of the UtrABD- or Lifeact-based junctions with the nectin junctions, we added the afadin-binding domain M3 N-terminal to the actin-binding domains in the corresponding chimeras (Fig. 6A). However, this addition was neither able to promote formation of nectin junctions nor to couple them with the chimera junctions (Fig. 6E).
Fig. 6.
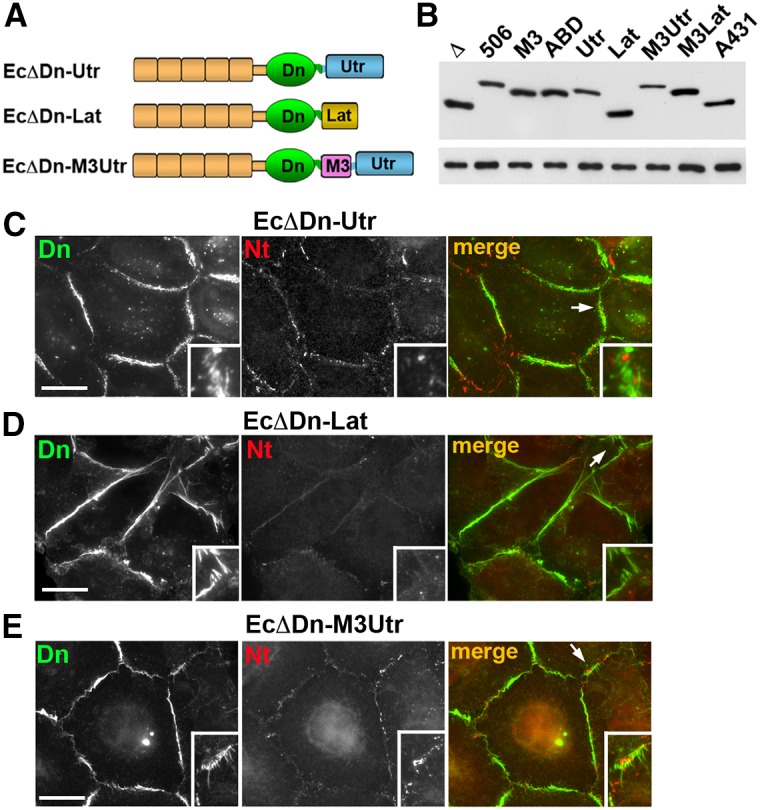
Replacement of the αABD with other actin-binding domains abolishes cadherin–nectin junction association. (A) Schematic representation of non-αABD cadherin chimeras tested. In all cases the cadherin module, EcΔDn, was the same as in the cadherin–α-catenin chimeras (see Fig. 5). The αABD was replaced with the actin-binding domain of utrophin (Utr) in the EcΔDn–Utr chimera or with the actin-binding peptide Lifeact (Lat) in the EcΔDn–Lat chimera. The EcΔDn–M3Utr chimera contained the α-catenin M3 domain in front of the Utr. (B) Immunoblot analysis of the stable cell lines expressing different chimeras: EcΔDn (Δ), EcΔDn-α506 (506), EcΔDn-M3 (M3), EcΔDn-HαABD (ABD), EcΔDn-Utr (Utr), EcDDn-Lat (Lat), EcΔDn-M3Utr (M3Utr), EcΔDn-LatUtr (LatUtr); and the control A431 cells expressing endogenous E-cadherin (A431). Equal amounts of total cell lysates were analyzed using anti-E-cadherin SHE78-7 (upper panel) and anti-tubulin (lower panel) antibodies. (C–E) Double immunostaining of A431D cells expressing EcΔDn-Utr (C), EcΔDn-Lat (D), and EcΔDn-M3Utr (E) using rabbit anti-dendra2 (Dn, green) antibody, to detect the chimeras, and mouse anti-nectin-2 (Nt, red) antibody, to detect nectin junctions. Higher magnifications of the selected regions (indicated by arrows in the merged images) are shown in the insets. Scale bars: 20 µm. Note that the cell–cell contacts enriched with the chimera junctions are mostly devoid of nectin junctions. If present, nectin junctions do not colocalize with the chimeras.
DISCUSSION
In this study, we analyzed the relationship between two transmembrane adhesive receptors in epithelial cells, E-cadherin and nectin-2. These two receptors are key components of adherens junctions, where they form separate but closely associated adhesive clusters interconnected by an actin bundle (Takai et al., 2008; Indra et al., 2013). It has been suggested that cadherin and nectin co-recruitment into adherens junctions is under the control of nectin. This idea stems from the fact that nectin assembles into junctions in the absence of cadherin, whereas cadherin junction formation is impaired upon nectin inactivation (Sato et al., 2006). According to this hypothesis, nectin–afadin complexes interact with cadherin–catenin complexes at cell–cell contact sites, and this interaction inhibits endocytosis of cadherin thereby stabilizing cadherin at sites of adhesion and allowing cadherin to cluster (Takai et al., 2008). However, numerous experimental data, summarized by Chen and Gumbiner (Chen and Gumbiner, 2006), have shown that nectin and afadin are not required for cadherin to produce junctions, though they do accelerate junction formation. Direct imaging of clathrin-based endocytosis has also shown that this process is not responsible for the adherens junction instability in the afadin-depleted cells (Indra et al., 2014).
In order to strictly confine both cadherin and nectin to the adherens junctions, two independent processes – assembly of cadherin junctions and assembly of nectin junctions – should be interdependent and tightly coupled. In our study, we sought to uncover the mechanism of this coupling. We confirmed that nectin-2 assembles into junctions in cells lacking cadherin, but we also showed that the nectin junctions are controlled by E-cadherin when E-cadherin is present. Our work further suggests that cadherin exerts its effect on nectin through the actin cytoskeleton.
As has been reported for two other cadherin-deficient cell lines, L and CHO cells (Momose et al., 2002; Struyf et al., 2002), cadherin-deficient A431D cells exhibit prominent, linearly organized nectin junctions that are stable in low Ca2+ medium. In order to reveal the role of cadherin in nectin-mediated adhesion, we analyzed nectin-2 in cadherin-reconstituted A431D cells. We observed that in these cells, nectin junctions undergo a notable conversion: their linear arrangement is replaced with a radial one. This transformation is caused by the alignment of nectin-2 clusters along the radial E-cadherin-containing adherens junctions. E-cadherin expression also changes the stability of nectin junctions in response to the BC-12 antibody, which recognizes an epitope close to the nectin-2 adhesive interface: this antibody disrupts nectin-2 clusters in the A431D cells but not in their E-cadherin-expressing counterparts. The leading role of cadherin in nectin organization is also evident from the fact that all our attempts to restore the linear distribution of nectin junctions in the E-cadherin-expressing cells by E-cadherin inactivation were unsuccessful. Specifically, we found that the suppression of E-cadherin function either by low Ca2+ medium or by a function-blocking antibody led to the disassembly of nectin junctions instead of restoring their linear distribution. Similarly, the formation of nectin junctions was severely suppressed in A431D cells that expressed an adhesion-incompetent W2A E-cadherin mutant. Another E-cadherin mutant, lacking the cis interface, dramatically changed the morphology of nectin junctions, but was also unable to return them to the linear state. Remarkably, the A431D cells expressing this cadherin mutant could not produce stable adherens junctions, but the nectin junctions in these cells were BC-12-resistant. Taken together, these data clearly show that cadherin is not needed for the basic nectin clustering process, but, if cadherin is present, this process is significantly modified and is controlled by cadherin clustering. It should be mentioned that the surprising dependency of nectin junctions on Ca2+ ions (despite the Ca2+ independence of nectin adhesion) and on cadherin adhesive activity could be observed on figures in some reports (Asakura et al., 1999; Tanaka et al., 2003; Lorger and Moelling, 2006), but this phenomenon was never investigated in depth or discussed.
The leading role of cadherin in the coordination of cadherin and nectin adhesive systems is also evident in our time-lapse experiments: once the cells touch one another, the assembly of the initial nectin clusters proceeds in seconds but never precedes the formation of the cadherin clusters. These two events either occur simultaneously, or cadherin clustering occurs a few seconds earlier. Such a small temporal difference in the formation of the nascent cadherin and nectin junctions could not possibly be observed in the previous experiments with fixed cells (Asakura et al., 1999). The analyses of nectin-2 and E-cadherin diffusion in the plasma membrane showed that the more rapid assembly of E-cadherin clusters is not caused just by a faster arrival of E-cadherin into the junctions, because E-cadherin lateral diffusion was approximately two times slower than that of nectin-2.
The simplest mechanism for cadherin-mediated control of nectin adhesion is that these two proteins form cis- or trans-heterocomplexes in which the nectin adhesive interface is blocked, and the interface only becomes active upon cadherin trans-dimerization. However, there is no evidence that the E-cadherin and nectin-2 extracellular regions interact with one another. The experiments reported here with A431D cells expressing the EcadΔDn deletion mutant provide additional arguments against this possibility. Even the cells expressing the highest level of this mutant, which retain the intact extracellular region of E-cadherin, exhibited neither the nectin-2 and cadherin cluster colocalization nor the changes in the linear nectin-2 or afadin organization that is typical for the wild-type A431D cells. Furthermore, the M3 domain of α-catenin, which directly interacts with afadin (Tachibana et al., 2000; Pokutta et al., 2002) was not sufficient to couple cadherin and nectin adhesion: addition of this domain to the EcadΔDn module was unable to promote nectin–cadherin co-clustering. It is, therefore, possible that afadin binding to the M3 domain is incompatible with afadin binding to nectin. Such mutually exclusive interactions might facilitate the segregation of cadherin and nectin clustering processes. In fact, direct probing of α-catenin domains for cadherin–nectin coupling activity using cadherin–α-catenin chimeras mapped this activity to the αABD.
The cadherin–α-catenin chimera EcΔDn–HαABD, the α-catenin-derived portion of which is limited only to αABD, formed intercellular junctions in A431D cells. The cytoplasmic side of these junctions reorganized actin filaments into bundles. Intriguingly, only chimera junctions that were associated with the radial actin bundles recruited nectin and permitted nectin junction assembly. Nectin junctions were absent in the areas of cell–cell contacts where the chimera formed non-radial junctions. Nectin junction formation was also dramatically suppressed in A431D cells in which cell–cell adhesion was mediated by cadherin chimeras containing the utrophin or Lifeact actin-binding units, even in combination with the afadin-binding M3 domain. Therefore, αABD-initiated radial actin bundles are unique structures: only these structures could promote nectin junction assembly. Importantly, the larger chimera EcΔDn–α506, which encompasses both the M3 domain and αABD, produced very similar junctions, but the fraction of radial nectin-2-incorporating junctions was significantly increased. Additional work, which is currently underway in our laboratory, is needed to understand how the M3 domain stimulates the αABD-mediated formation of these actin bundles, and whether actin-binding domains of other proteins associated with adherens junctions (such as vinculin) could also support the assembly of nectin junctions. It would also be interesting to understand the role of mechanical forces in the formation of the radial actin bundles co-assembling cadherin and nectin clusters.
The observation that the non-radial αABD-based junctions, as well as the UtrABD- or Lifeact-mediated junctions, were devoid of nectin-2 was surprising. Two closely opposed plasma membranes at sites of such junctions would be expected to work as an efficient diffusion trap for nectin adhesive dimers and, in fact, ultimately enhance nectin junction assembly. Therefore, the lack of nectin-2 adhesive clusters around the UtrABD- or Lifeact-based junctions implies that these sites suppress nectin-2 adhesive functions. Similarly, cell–cell contacts in cells expressing the W2A E-cadherin mutant were unable to form nectin junctions, whereas they were able to trap the cadherin mutant. Taken together, these data suggest that the recruitment of nectin-2 into the radial EcΔDn–αABD junctions is a very specific process.
The association of nectin junctions with specific cadherin-anchored actin structures shows that the actin cytoskeleton could be a key intermediary in cadherin–nectin coupling. Considering the important role of the actin cytoskeleton in clustering of the adhesive receptors, one might propose that only specific actin structures promote clustering of a particular subpopulation of adhesion receptors. This idea is consistent with our observation that the extrajunctional nectin molecules interact with cortical actin (Fig. 3D) and that nectin junctions require actin for their assembly (Yamada et al., 2004).
Recent advances in understanding cadherin adhesion can explain how particular actin structures can enhance or inhibit cell–cell adhesion. It has been found that cadherin-based cell–cell adhesion is driven by trans- and cis-inter-cadherin interactions that cooperatively reinforce one another (Wu et al., 2010; Harrison et al., 2011). Importantly, the cis-interactions in the resulting adhesive lattice are insufficiently strong to maintain the junction integrity. The junction stability is maintained only if its cis-interactions are reinforced by the tethering of cadherin molecules to the actin cytoskeleton (Hong et al., 2013). Therefore, an actin bundle stabilizes the cadherin adhesive lattice if it binds cadherin in a way that is compatible with cadherin–cadherin cis-interactions. In contrast, the actin bundle should prevent cadherin clustering if the distance between its cadherin-binding sites clashes with the length of the inter-cadherin cis-bond. Such compatibility could be achieved through specific alignment of actin filaments in the bundle, a specific conformation of the individual filaments, or recruitment of accessory proteins. Although the exact structure of the nectin adhesive cluster is still unclear, it also apparently combines trans- and cis-nectin–nectin interactions (Momose et al., 2002; Takai et al., 2008). One might propose that actin bundles induced by αABD are ‘nectin cluster-compatible’, whereas the bundles formed by UtrABD or Lifeact are not. Understanding the exact structure of nectin junctions and actin bundles associated with cadherin could shed light on these mechanisms.
In summary, our results present evidence for a new mechanism that temporally and spatially couples two independent adhesive processes, cadherin- and nectin-mediated adhesion. This coupling confines nectin junction assembly to the sites of cadherin junctions. The actin-binding domain of α-catenin appears to be instrumental for such a close positioning of two junctional structures. Our results suggest that this domain, once it is clustered in the junctions, remodels actin filaments in a way that enhances their nectin-clustering activity (Fig. 7). Adherens junctions have been shown to reorganize the actin cytoskeleton into a large variety of cell-type-specific actin structures (Perez-Moreno and Fuchs, 2006; Meng and Takeichi, 2009; Yonemura et al., 2011; Brieher and Yap, 2013). In theory, some of these cadherin-initiated actin structures could function as scaffolds for the clustering of other adhesive proteins, including nectin. Such actin-based coupling of cadherin adhesion with the assembly of other adhesive receptors might contribute to the global organization of the epithelial junctional complex.
Fig. 7.
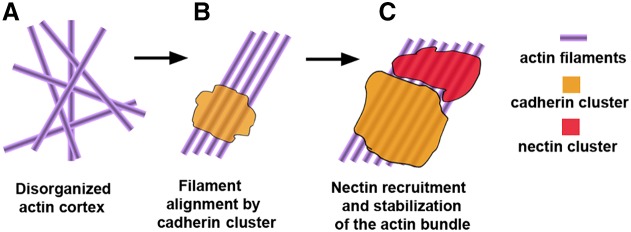
Hypothetical mechanism of cadherin and nectin cluster assembly. (A) Actin filaments are disorganized in the actin cortex. (B) Filaments are specifically aligned owing to the adhesive clustering of the cadherin–catenin complex. (C) The resulting cadherin-associated actin filament structure becomes the substrate for nectin–afadin clustering. Nectin–afadin clusters could reinforce the actin structure and facilitate further clustering of cadherin.
MATERIALS AND METHODS
Plasmids
The plasmids, which all were based on the vector pRcCMV (Invitrogene, Carlsbad, CA), encoding EcΔDn (in previous works EcDendra-Δ748-KL), full-length E-cadherin (EcDn), its W2A and cis mutants and F136D-N2Dn, were as described previously (Hong et al., 2010, Harrison et al., 2011; Harrison et al., 2012). The plasmid encoding the chimeric proteins EcΔDn–HαABD and EcΔDn–UtrABD were as described previously (Hong et al., 2013). New plasmids encoding chimeric proteins presented in Figs 5 and 6 were created using EcΔDn as a template. The maps of EcΔDn–α-catenin and its mutants are presented in Fig. 5. The original plasmid was published in Troyanovsky et al. (Troyanovsky et al., 2011). All plasmid inserts were completely sequenced before use.
Cell culture and transfection
Transfection and culture of A431D, A431 and DLD-1 cells were performed as described previously (Hong et al., 2010; Troyanovsky et al., 2011). In brief, cells were grown in DMEM with 10% fetal bovine serum (FBS). Cells were transfected using Lipofectamine 2000 (Invitrogene, Grand Island, NY) according to the manufacturer's instructions. After Geneticin selection (0.5 mg/ml), the cells were sorted for transgene expression by FACS, and only moderately expressing cells were used. All cell sublines expressed similar levels of the recombinant proteins (Fig. 6B).
Immunofluorescence microscopy
The following antibodies were used: mouse anti-E-cadherin, clones HECD1 and SHE78-7 (Zymed Laboratories, South San Francisco, CA); rabbit anti-Dendra2 (Evrogen, Moscow, Russia); mouse anti-β-catenin (BD Biosciences, San Jose, CA); mouse anti-vinculin and rabbit anti-afadin (Sigma, St Louis, MO); goat anti-α-catenin and mouse anti-nectin-2, clones BC-12 and R2.525 (Santa Cruz Inc. Dallas, TX). Alexa-Fluor-555–phalloidin and Latrunculin A were purchased from Invitrogen. For the Ca2+-switch assay, cells were cultivated in a low Ca2+ medium (20 µM Ca2+) for the indicated time. The concentrations of BC-12 antibody in the function-blocking experiments are indicated in the text.
For immunofluorescence, cells were grown for 2 days on glass coverslips and were fixed and permeabilized either with methanol-acetone or with 3% formaldehyde with 1% Triton X-100, as described previously (Troyanovsky et al., 2006; Indra et al., 2013). Both fixation protocols produced the same results with respect to the subcellular distribution of the cadherin chimeras. The actin cytoskeleton was studied using a formaldehyde-based protocol. Wide-field images were taken using an Eclipse 80i Nikon microscope (Plan Apo 100×/1.40 objective lens) and a digital camera (CoolSNAP EZ; Photometrics, Tucson, AZ). The images were then processed using Nikon's NIS-Elements software.
Live-cell imaging and data processing
These experiments were performed essentially as described previously (Hong et al., 2010 and Hong et al., 2013). In brief, a cell suspension (∼105 cells) was plated into a homemade chamber built on cover glass. The next day, the culture medium was replaced with imaging medium (L-15 plus 10% FBS) and the chamber was imaged with an Eclipse Ti-E microscope (Nikon, Melville, NY) at 37°C controlled with Nikon's NIS-Elements software. The microscope was equipped with an incubator chamber, a CoolSNAP HQ2 camera (Photometrics), Plan Apo 60×/1.40 and Plan Apo VC 100×/1.40 lenses and halogen and mercury light sources. Time-lapse images (Fig. 3A) were taken in both FITC and TRITC channels simultaneously using a beam splitter using halogen light that minimized phototoxicity and photobleaching. To analyze cadherin junctional turnover, we used a junctional Dendra photoconversion assay (Hong et al., 2010) in which the point of interest (φ = 2.5 µm) was photoconverted by a 100-ms-long exposure to the 405-nm wavelength laser. Time-lapse images were then taken in the red channel in 1 s intervals with 1 s (Fig. 3C) of image acquisition time.
All images were saved as Tiff files and processed using ImageJ software (National Institutes of Health). In the Dendra photoconversion assay, the red fluorescence intensity was normalized in such a way that 0 and 1 corresponded to the background and the initial (immediately after activation) values. The background value was obtained from the image taken right before the photoconversion. The time course of the intensity change was produced from ten sets of independent experiments. Mean±s.d. values were calculated for each time point.
Quantitative analysis of nectin-2 staining was performed using ImageJ. An area of 30×30 pixels at the cell junction was selected (n>10), and the integrated density of fluorescence intensity of the selected area was measured. The data were corrected for background. Intensities of the nectin junctions at 37°C were normalized to their intensities at 4°C, and the results were used to calculate the mean±s.d.
Supplementary Material
Acknowledgments
We are grateful to L. Shapiro, B. Honig, O. Harrison (Columbia University, New York, NY) and M. Seeger (University of Wisconsin, Madison, WI) for helpful discussion and valuable comments on the manuscript. Sequencing and Flow Cytometry were performed at the Northwestern University Genetic and Flow Cytometry Core Facilities. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
R.T., I.I., C.S., S.H. and S.T. designed and performed experiments; R.T., I.I., C.S. and S.T. analyzed data; and S.T. wrote the manuscript
Funding
The work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health [grant numbers AR44016 and AR057992]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.161588/-/DC1
References
- Adams C. L., Nelson W. J. (1998). Cytomechanics of cadherin-mediated cell-cell adhesion. Curr. Opin. Cell Biol. 10, 572–577 10.1016/S0955-0674(98)80031-8 [DOI] [PubMed] [Google Scholar]
- Asakura T., Nakanishi H., Sakisaka T., Takahashi K., Mandai K., Nishimura M., Sasaki T., Takai Y. (1999). Similar and differential behaviour between the nectin-afadin-ponsin and cadherin-catenin systems during the formation and disruption of the polarized junctional alignment in epithelial cells. Genes Cells 4, 573–581 10.1046/j.1365-2443.1999.00283.x [DOI] [PubMed] [Google Scholar]
- Birukova A. A., Fu P., Wu T., Dubrovskyi O., Sarich N., Poroyko V., Birukov K. G. (2012). Afadin controls p120-catenin-ZO-1 interactions leading to endothelial barrier enhancement by oxidized phospholipids. J. Cell. Physiol. 227, 1883–1890 10.1002/jcp.22916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghi N., Sorokina M., Shcherbakova O. G., Weis W. I., Pruitt B. L., Nelson W. J., Dunn A. R. (2012). E-cadherin is under constitutive actomyosin-generated tension that is increased at cell-cell contacts upon externally applied stretch. Proc. Natl. Acad. Sci. USA 109, 12568–12573 10.1073/pnas.1204390109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasch J., Harrison O. J., Honig B., Shapiro L. (2012). Thinking outside the cell: how cadherins drive adhesion. Trends Cell Biol. 22, 299–310 10.1016/j.tcb.2012.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieher W. M., Yap A. S. (2013). Cadherin junctions and their cytoskeleton(s). Curr. Opin. Cell Biol. 25, 39–46 10.1016/j.ceb.2012.10.010 [DOI] [PubMed] [Google Scholar]
- Chen X., Gumbiner B. M. (2006). Crosstalk between different adhesion molecules. Curr. Opin. Cell Biol. 18, 572–578 10.1016/j.ceb.2006.07.002 [DOI] [PubMed] [Google Scholar]
- Franke W. W. (2009). Discovering the molecular components of intercellular junctions – a historical view. Cold Spring Harb. Perspect. Biol. 1, a003061 10.1101/cshperspect.a003061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner B., Stevenson B., Grimaldi A. (1988). The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J. Cell Biol. 107, 1575–1587 10.1083/jcb.107.4.1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison O. J., Bahna F., Katsamba P. S., Jin X., Brasch J., Vendome J., Ahlsen G., Carroll K. J., Price S. R., Honig B. et al. (2010). Two-step adhesive binding by classical cadherins. Nat. Struct. Mol. Biol. 17, 348–357 10.1038/nsmb.1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison O. J., Jin X., Hong S., Bahna F., Ahlsen G., Brasch J., Wu Y., Vendome J., Felsovalyi K., Hampton C. M. et al. (2011). The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure 19, 244–256 10.1016/j.str.2010.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison O. J., Vendome J., Brasch J., Jin X., Hong S., Katsamba P. S., Ahlsen G., Troyanovsky R. B., Troyanovsky S. M., Honig B. et al. (2012). Nectin ectodomain structures reveal a canonical adhesive interface. Nat. Struct. Mol. Biol. 19, 906–915 10.1038/nsmb.2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S., Troyanovsky R. B., Troyanovsky S. M. (2010). Spontaneous assembly and active disassembly balance adherens junction homeostasis. Proc. Natl. Acad. Sci. USA 107, 3528–3533 10.1073/pnas.0911027107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S., Troyanovsky R. B., Troyanovsky S. M. (2011). Cadherin exits the junction by switching its adhesive bond. J. Cell Biol. 192, 1073–1083 10.1083/jcb.201006113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S., Troyanovsky R. B., Troyanovsky S. M. (2013). Binding to F-actin guides cadherin cluster assembly, stability, and movement. J. Cell Biol. 201, 131–143 10.1083/jcb.201211054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino T., Sakisaka T., Baba T., Yamada T., Kimura T., Takai Y. (2005). Regulation of E-cadherin endocytosis by nectin through afadin, Rap1, and p120ctn. J. Biol. Chem. 280, 24095–24103 10.1074/jbc.M414447200 [DOI] [PubMed] [Google Scholar]
- Indra I., Hong S., Troyanovsky R., Kormos B., Troyanovsky S. (2013). The adherens junction: a mosaic of cadherin and nectin clusters bundled by actin filaments. J. Invest. Dermatol. 133, 2546–2554 10.1038/jid.2013.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indra I., Troyanovsky R., Troyanovsky S. M. (2014). Afadin controls cadherin cluster stability using clathrin-independent mechanism. Tissue Barriers 2, e28687 10.4161/tisb.28687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez M., Cocchi F., Menotti L., Avitabile E., Dubreuil P., Campadelli-Fiume G. (2000). Nectin2alpha (PRR2alpha or HveB) and nectin2delta are low-efficiency mediators for entry of herpes simplex virus mutants carrying the Leu25Pro substitution in glycoprotein D. J. Virol. 74, 1267–1274 10.1128/JVI.74.3.1267-1274.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorger M., Moelling K. (2006). Regulation of epithelial wound closure and intercellular adhesion by interaction of AF6 with actin cytoskeleton. J. Cell Sci. 119, 3385–3398 10.1242/jcs.03027 [DOI] [PubMed] [Google Scholar]
- Mège R. M., Gavard J., Lambert M. (2006). Regulation of cell-cell junctions by the cytoskeleton. Curr. Opin. Cell Biol. 18, 541–548 10.1016/j.ceb.2006.08.004 [DOI] [PubMed] [Google Scholar]
- Meng W., Takeichi M. (2009). Adherens junction: molecular architecture and regulation. Cold Spring Harb. Perspect. Biol. 1, a002899 10.1101/cshperspect.a002899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momose Y., Honda T., Inagaki M., Shimizu K., Irie K., Nakanishi H., Takai Y. (2002). Role of the second immunoglobulin-like loop of nectin in cell-cell adhesion. Biochem. Biophys. Res. Commun. 293, 45–49 10.1016/S0006-291X(02)00183-3 [DOI] [PubMed] [Google Scholar]
- Ozaki-Kuroda K., Nakanishi H., Ohta H., Tanaka H., Kurihara H., Mueller S., Irie K., Ikeda W., Sakai T., Wimmer E. et al. (2002). Nectin couples cell-cell adhesion and the actin scaffold at heterotypic testicular junctions. Curr. Biol. 12, 1145–1150 10.1016/S0960-9822(02)00922-3 [DOI] [PubMed] [Google Scholar]
- Perez-Moreno M., Fuchs E. (2006). Catenins: keeping cells from getting their signals crossed. Dev. Cell 11, 601–612 10.1016/j.devcel.2006.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokutta S., Drees F., Takai Y., Nelson W. J., Weis W. I. (2002). Biochemical and structural definition of the l-afadin- and actin-binding sites of alpha-catenin. J. Biol. Chem. 277, 18868–18874 10.1074/jbc.M201463200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J., Crevenna A. H., Kessenbrock K., Yu J. H., Neukirchen D., Bista M., Bradke F., Jenne D., Holak T. A., Werb Z. et al. (2008). Lifeact: a versatile marker to visualize F-actin. Nat. Methods 5, 605–607 10.1038/nmeth.1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta D., Ramagopal U. A., Rubinstein R., Vigdorovich V., Nathenson S. G., Almo S. C. (2012). Structure of Nectin-2 reveals determinants of homophilic and heterophilic interactions that control cell-cell adhesion. Proc. Natl. Acad. Sci. USA 109, 14836–14840 10.1073/pnas.1212912109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T., Fujita N., Yamada A., Ooshio T., Okamoto R., Irie K., Takai Y. (2006). Regulation of the assembly and adhesion activity of E-cadherin by nectin and afadin for the formation of adherens junctions in Madin-Darby canine kidney cells. J. Biol. Chem. 281, 5288–5299 10.1074/jbc.M510070200 [DOI] [PubMed] [Google Scholar]
- Sawyer J. K., Harris N. J., Slep K. C., Gaul U., Peifer M. (2009). The Drosophila afadin homologue Canoe regulates linkage of the actin cytoskeleton to adherens junctions during apical constriction. J. Cell Biol. 186, 57–73 10.1083/jcb.200904001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struyf F., Martinez W. M., Spear P. G. (2002). Mutations in the N-terminal domains of nectin-1 and nectin-2 reveal differences in requirements for entry of various alphaherpesviruses and for nectin-nectin interactions. J. Virol. 76, 12940–12950 10.1128/JVI.76.24.12940-12950.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana K., Nakanishi H., Mandai K., Ozaki K., Ikeda W., Yamamoto Y., Nagafuchi A., Tsukita S., Takai Y. (2000). Two cell adhesion molecules, nectin and cadherin, interact through their cytoplasmic domain-associated proteins. J. Cell Biol. 150, 1161–1176 10.1083/jcb.150.5.1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Nakanishi H., Miyahara M., Mandai K., Satoh K., Satoh A., Nishioka H., Aoki J., Nomoto A., Mizoguchi A. et al. (1999). Nectin/PRR: an immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with Afadin, a PDZ domain-containing protein. J. Cell Biol. 145, 539–549 10.1083/jcb.145.3.539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai Y., Ikeda W., Ogita H., Rikitake Y. (2008). The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annu. Rev. Cell Dev. Biol. 24, 309–342 10.1146/annurev.cellbio.24.110707.175339 [DOI] [PubMed] [Google Scholar]
- Tanaka Y., Nakanishi H., Kakunaga S., Okabe N., Kawakatsu T., Shimizu K., Takai Y. (2003). Role of nectin in formation of E-cadherin-based adherens junctions in keratinocytes: analysis with the N-cadherin dominant negative mutant. Mol. Biol. Cell 14, 1597–1609 10.1091/mbc.E02-10-0632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka-Okamoto M., Hori K., Ishizaki H., Itoh Y., Onishi S., Yonemura S., Takai Y., Miyoshi J. (2011). Involvement of afadin in barrier function and homeostasis of mouse intestinal epithelia. J. Cell Sci. 124, 2231–2240 10.1242/jcs.081000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanovsky S. (2012). Adherens junction assembly. Subcell. Biochem. 60, 89–108 10.1007/978-94-007-4186-7_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanovsky R. B., Sokolov E. P., Troyanovsky S. M. (2006). Endocytosis of cadherin from intracellular junctions is the driving force for cadherin adhesive dimer disassembly. Mol. Biol. Cell 17, 3484–3493 10.1091/mbc.E06-03-0190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanovsky R. B., Klingelhöfer J., Troyanovsky S. M. (2011). α-Catenin contributes to the strength of E-cadherin-p120 interactions. Mol. Biol. Cell 22, 4247–4255 10.1091/mbc.E11-03-0250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Jin X., Harrison O., Shapiro L., Honig B. H., Ben-Shaul A. (2010). Cooperativity between trans and cis interactions in cadherin-mediated junction formation. Proc. Natl. Acad. Sci. USA 107, 17592–17597 10.1073/pnas.1011247107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada A., Irie K., Fukuhara A., Ooshio T., Takai Y. (2004). Requirement of the actin cytoskeleton for the association of nectins with other cell adhesion molecules at adherens and tight junctions in MDCK cells. Genes Cells 9, 843–855 10.1111/j.1365-2443.2004.00768.x [DOI] [PubMed] [Google Scholar]
- Yonemura S. (2011). Cadherin-actin interactions at adherens junctions. Curr. Opin. Cell Biol. 23, 515–522 10.1016/j.ceb.2011.07.001 [DOI] [PubMed] [Google Scholar]
- Zhadanov A. B., Provance D. W., Jr, Speer C. A., Coffin J. D., Goss D., Blixt J. A., Reichert C. M., Mercer J. A. (1999). Absence of the tight junctional protein AF-6 disrupts epithelial cell-cell junctions and cell polarity during mouse development. Curr. Biol. 9, 880–888– S1–S2 10.1016/S0960-9822(99)80392-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.