

Abstract
Despite high quality standards and continual process improvements in manufacturing, host cell protein (HCP) process impurities remain a substantial risk for biological products. Even at low levels, residual HCPs can induce a detrimental immune response compromising the safety and efficacy of a biologic. Consequently, advanced-stage clinical trials have been cancelled due to the identification of antibodies against HCPs. To enable earlier and rapid assessment of the risks in Chinese Hamster Ovary (CHO)-based protein production of residual CHO protein impurities (CHOPs), we have developed a web tool called CHOPPI, for CHO Protein Predicted Immunogenicity. CHOPPI integrates information regarding the possible presence of CHOPs (expression and secretion) with characterizations of their immunogenicity (T cell epitope count and density, and relative conservation with human counterparts). CHOPPI can generate a report for a specified CHO protein (e.g., identified from proteomics or immunoassays) or characterize an entire specified subset of the CHO genome (e.g., filtered based on confidence in transcription and similarity to human proteins). The ability to analyze potential CHOPs at a genomic scale provides a baseline to evaluate relative risk. We show here that CHOPPI can identify clear differences in immunogenicity risk among previously validated CHOPs, as well as identify additional “risky” CHO proteins that may be expressed during production and induce a detrimental immune response upon delivery. We conclude that CHOPPI is a powerful tool that provides a valuable computational complement to existing experimental approaches for CHOP risk assessment and can focus experimental efforts in the most important directions. Biotechnol. Bioeng. 2014;111: 2170–2182.
Keywords: biologic, CHO, host cell protein, immunogenicity, T cell epitope, immunoinformatics
Introduction
Recombinant protein therapeutics have revolutionized the treatment of a wide variety of illnesses, with over 200 different biopharmaceuticals licensed and generating nearly 100 billion dollars in global sales (Walsh, 2010). Genetically engineered host cells are generally used to manufacture these biologics (Jayapal et al., 2007). Chinese Hamster Ovary (CHO) cells are one of the most common such systems; for example, 70% of recently approved therapeutic glycoproteins are produced in CHO cells (Higgins, 2010). A key advantage of CHO cells is their human-compatible post-translational modifications, particularly glycosylation, leading to improved therapeutic efficacy and protein longevity as well as increased safety (Omasa et al., 2010). Moreover, methods for cell transfection, gene amplification, and clone selection are well characterized in CHO cells, as are techniques to volumetrically scale production of complex therapeutics (Kim et al., 2012).
Like all other such processes, CHO-based protein production faces the potential problem of impurities in the final product that can lead to undesired effects upon administration. One particular concern is host cell proteins (HCPs). These are also known as “hitchhiker” proteins, which are synthesized in the cell and not fully removed during purification. In general, quite a few residual HCPs may remain in the final product despite close monitoring and high standards throughout downstream processing (Champion et al., 2005). Unfortunately, even at low levels HCPs may induce a detrimental immune response, contributing to the overall immunogenicity of the product (Champion et al., 2005); consequently, detection of anti-HCP antibodies following exposure to the therapeutic product has resulted in the cancellation of advanced clinical trials (Ipsen, 2012). In order to assess and mitigate the immunogenicity risk posed by HCPs in a particular protein production process, two questions must be addressed: what HCPs may be present, and how likely they are to stimulate a detrimental immune response.
Substantial work is done during downstream processing in order to identify and eliminate most impurities. Detection is typically performed with standard immunoassay and proteomic methods, such as enzyme-linked immunosorbent assays (ELISAs) and Western blotting. More recent techniques, such as differential gel electrophoresis and liquid chromatography combined with mass spectrometry, have also been used to identify CHO proteins in greater detail (Doneanu et al., 2012; Jin et al., 2010). However, HCPs are a complex and a heterogeneous group of impurities, with substantial differences in isoelectric point, structure, molecular mass, and hydrophobicity properties. Host expression system, subcellular localization of expression, culture condition, purification process, and the target protein being produced all affect HCP composition and abundance, and some protein products may interact in a covalent fashion with specific HCPs (Pezzini et al., 2011). Thus, HCPs may differ from one product to another even when manufactured using the same cell line; therefore, attention must always be given to monitoring residual HCPs.
While identification of residual protein impurities is important, additional analysis is required to characterize the resulting potential for a detrimental immune response upon delivery to patients. One powerful technique for immunogenicity analysis relies on immunoinformatics tools, which have been shown to make reliable predictions useful for and validated within the design of both biotherapeutics (Koren et al., 2007; Moise et al., 2012; Osipovitch et al., 2012) and vaccines (Gregory et al., 2009; Moise et al., 2011; Moss et al., 2011). Of particular relevance to HCP-driven immunogenicity is the T cell pathway, in which an antigen-presenting cell processes a foreign protein into constituent peptides, some of which (the “epitopes”) are recognized by major histocompatibility complex (MHC) class II proteins and brought to the cell surface for inspection by T cells. The formation of a ternary MHC: epitope: T cell receptor complex drives the initial naïve response and can stimulate subsequent B cell activation and maturation. Thus, much immunoinformatics research has been directed toward highly-reliable prediction of putative T cell epitopes (De Groot and Martin, 2009; Wang et al., 2008), and the EpiMatrix system is one heavily validated method based on peptide: MHC binding profiles (De Groot et al., 1997, 2004). In addition to identifying individual epitopes within a protein, EpiMatrix can then also assess the overall immunogenicity risk of a protein according to its epitope density relative to benchmark proteins (De Groot and Martin, 2009; Koren et al., 2007). Publication of the CHO genome in 2011 (Xu et al., 2011) made it possible to examine the entire genome for potentially immunogenic proteins, using well-validated epitope prediction tools (Gutierrez et al., 2012).
The immunogenicity of a CHO protein may be mitigated by the fact that it is quite similar to a human protein. However, some believe that “any protein is potentially immunogenic” (Worobec and Rosenberg, 2004) and proteins full of T cell epitopes are even more so, particularly when delivered in an unnatural fashion. Cases of severe adverse immune responses to autologous proteins have been published (Casadevall et al., 2002; Koren et al., 2002). Moreover, there are a number of animal models in which imperfect homology between an antigen and a host-origin protein contributes to the development of antibody responses to the protein (Inaba et al., 2009; Prasad et al., 2012). The response against the non-homologous epitope can then “spread” to other epitopes, even if they are host-like (Grosenbaugh et al., 2011).
The JanusMatrix method (Moise et al., 2013) provides a means to balance these concerns, refining epitope-based immunogenicity analysis with a characterization of the “human-ness” of the predicted epitopes. In particular, JanusMatrix evaluates the potential for cross-reactivity of predicted epitopes with counterpart peptides in the human genome. Putative epitopes with little homology to the human genome are more likely to drive a T effector response, while epitopes that are conserved are more likely to be tolerated by the human immune system (He et al., 2014). The epitope-level cross-reactivity analysis of JanusMatrix thereby complements the characterization of protein-level homology, enabling identification of more worrisome epitopes that are unique to the HCP.
This paper presents a new tool called CHOPPI (for CHO Protein Predicted Immunogenicity), available at http://www.immunome.org/CHOPPI, that unifies the two aspects of immunogenicity risk posed by CHO proteins (CHOPs) that may remain as process-related impurities: the potential presence of CHOPs (expression and secretion), and their potential immunogenicity (T cell epitope content predicted by EpiMatrix and human-ness assessed by JanusMatrix). Figure 1 illustrates the CHOPPI tool. In the “CHOP query” scenario a CHOP has already been identified experimentally (e.g., by immunoassays or proteomics), and CHOPPI is queried using its amino acid sequence, name, or accession number (entered in the text box in panel a), leading to a list of matches for further analysis (panel b). In a second scenario, called “genome filter,” CHOPPI searches a CHO genome for CHOPs meeting user-adjustable criteria regarding expression, secretion, and human homology (options in panel a), again leading to a list of possible CHOPs for inspection (panel c). Finally, validated CHOPs from the literature can be directly browsed (panel d). In all cases, drilling down on a particular protein yields a report (panel e) in which the protein is characterized in terms of both potential presence in the product and potential immunogenicity in humans, with a genomic-scale analysis providing a baseline for comparison.
Figure 1.
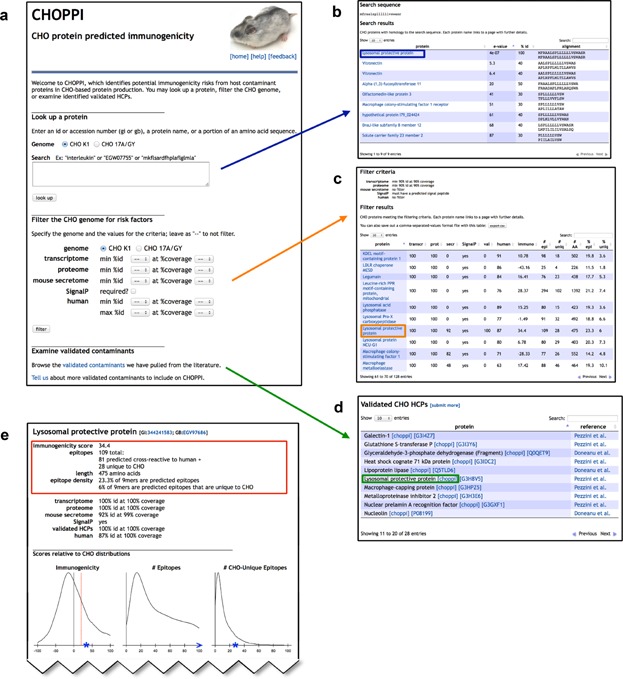
The CHOPPI web tool. CHOPPI allows two types of searches, “CHOP query” and “genome filter,” along with the ability to browse a set of validated CHOPs. These all lead into individual protein reports. (a → b) CHOP query. In this example, a fragment of an amino acid sequence is used as a query and CHO homologs to the query protein are identified. (a → c) Genome filter. In this example, the CHO genome is filtered for proteins with homology matches to both a CHO transcriptome and a CHO proteome, along with presence of a predicted signal peptide. The analysis of the filtered proteins is presented in a table. As with the CHOP query, an individual protein can then be examined. (a → d) The dataset of experimentally identified CHOPs. (one of {b, c, or d} → e) Individual protein report linked from the previous panel (boxed in each). The page summarizes both predicted epitope content (EpiMatrix whole-protein immunogenicity score, along with number and density of epitopes and CHO-unique epitopes), and predicted presence in protein production (homology matches against a CHO transcriptome, a CHO proteome, a mouse secretome, a set of validated CHOPs, and/or appearance of a predicted signal peptide). Immunogenicity evaluations are plotted relative to distributions over the whole genome.
In a first test of the CHOPPI tool, we characterized a set of validated CHOPs identified in experimental recombinant protein preparations and found that while some appeared to pose little immunogenicity risk, others had patterns of epitope content indicative of the increased potential for an adverse response in patients. Using the CHOPPI tool, we also identified a number of other CHOPs that, if they happened to hitchhike into a particular preparation or during a particular process, could pose a substantial immunogenic risk due to large and/or relatively CHO-unique (i.e., dissimilar to human) epitope content. Finally, we characterized trends over the entire CHO-K1 genome, both to calibrate the evaluation of individual proteins of interest as well as to assess the expected risk over all possible CHOPs. We found that there is reason to be concerned about the potential immunogenicity of some CHO proteins—there are a number of CHO proteins predicted to be highly immunogenic and that could appear during production. At the same time, CHOPPI can provide guidance regarding CHO proteins that are of lower risk according to these criteria. These results illustrate that by integrating genomic-scale predictions of CHO protein immunogenicity risk, CHOPPI provides an important new resource for assessing impurity risks. While the assessment is purely computational and predictive in nature, it can guide further experimental efforts (e.g., detection and immunogenicity assays) and ultimately the development of effective and safe biotherapeutics for human use.
Materials and Methods
Evaluating the immunogenicity risk of CHOPs involves two considerations: which proteins are likely to be present from a process, and how likely they are to be immunogenic. CHOPPI allows a user to select proteins of interest for possible presence either by simply specifying them or by filtering a CHO genome according to criteria relevant to process-related impurities. CHOPPI then assesses the immunogenicity of selected proteins by reporting predicted T cell epitope content according to EpiMatrix, as well as by comparing them to their human homologs and comparing their predicted epitopes to predicted epitopes within the human genome (Fig. 1).
CHOP Selection
The “CHOP query” usage of CHOPPI allows a user to specify a protein of interest, perhaps one identified through experimental techniques (Fig. 1a and b). The protein can be specified by accession number, name, or an amino acid sequence fragment. When an amino acid sequence is used as a query, CHOPPI uses BLAST [version 2.2.27+ (Camacho et al., 2009)], to search for homologs in a specified CHO genome; E-value and percent identity are reported. This application of CHOPPI enables users to determine the potential immunogenicity of a single CHOP or set of CHOPs, using the CHOPPI toolkit. See below for additional information on the CHOP query tool.
The “genome filter” usage (Fig. 1a and c) starts with a specified CHO genome and filters it according to user-specified criteria evaluating potential for expression and secretion, along with similarity to human proteins. The possible filters (Fig. 2) include matching to a CHO transcriptome, CHO proteome, mouse secretome, and human genome, and testing for a predicted signal peptide. Homology matching is based on results from BLAST according to user-specified sequence identity and coverage thresholds. Note that, except for the transcriptome, all sequences are amino acid sequences, previously translated and annotated from the relevant genomic data. This application of CHOPPI enables the identification of potentially immunogenic proteins that might be suitable for targeted downregulation in CHO protein production, if reduced immunogenicity is a desired outcome. See below for further illustration of the genome filter tool.
Figure 2.
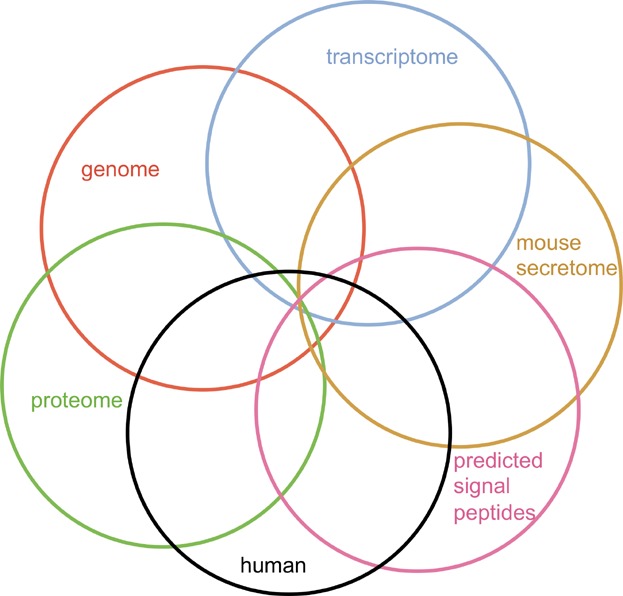
Filtering the CHO genome. CHOPPI enables selection of CHO protein subsets by homology matching with a CHO transcriptome, a human proteome, a mouse secretome, and the human genome, along with testing for the presence of a predicted signal peptide.
The data sources for CHOP query and genome filter are as follows:
CHO genome: All CHO proteins used in CHOPPI are derived from translated, annotated genomic sequence information. CHOPPI currently includes information from two genome projects, one for the commonly used CHO-K1 cell line (Xu et al., 2011) and the other for the 17A/GY strain (Brinkrolf et al., 2013). Others (e.g., Lewis et al., 2013) will be incorporated as protein-level information becomes available. We note that the CHOPPI analysis is all in terms of the basal cell lines. The CHOPPI sequence database can be extended in future iterations to include modifications that are present in any derivative cell line that is being employed in a particular process.
CHO transcriptome: Gene expression is, in part, tissue-specific, and thus genes expressed in the ovary cells, and not just present in the genome, are most likely to be present in CHO cell cultures. CHOPPI thus allows CHOPs from the genome to be filtered down to those with homology matches in the CHO-K1 transcriptome (Becker et al., 2011). CHOPs are homology-matched to translated transcripts via the “tblastn” form of BLAST, recognizing that proteins spanning contigs will be unfortunately dropped from the results.
Proteome: To further focus on proteins that have been translated, we have compared the genes against the sequences (both proteins and glycoproteins) identified by the proteomic analysis (Baycin-Hizal et al., 2012). This extensive proteomic study employed a combination of gel electrophoresis, multidimensional liquid chromatography, and solid phase extraction of glycoproteins, followed by tandem mass spectrometry, in order to identify a total of 6,164 proteins at a false discovery rate of 0.02. The deposited identified proteins were homology-matched to the genomes in CHOPPI via “blastp.”
Mouse secretome: Any expressed protein may be extracted as a hitchhiker, but secreted proteins may be considered even more likely to be present in CHO cell supernatant irrespective of target therapeutic protein. In lieu of a CHO secretome, CHOPPI characterizes potential secretion of CHOPs by homology matching to a set of secreted mouse proteins obtained from two databases, LOCATE and UniProt (Sprenger et al., 2008; UniProt Consortium, 2012).
Predicted signal peptides: As an alternative or complement to filtering against the relatively small and homology-derived set of secreted proteins, secretion status may be predicted by the presence of a signal peptide in the CHOP. In order to identify which proteins from the CHO genome contain predicted signal peptides, CHOPPI employs SignalP version 4.1, which has been shown to be a very accurate predictor; for example, it obtained a Matthews Correlation Coefficient of 0.874 in cross-validated signal peptide identification within a eukaryote dataset (Petersen et al., 2011).
Validated CHOPs: While only a relatively small number of CHOPs have been previously identified experimentally, CHOPPI provides the ability to restrict analysis to those proteins. The set of CHOPs that is integrated into the CHOPPI tool will be updated when additional CHOPs are identified; a web form is provided for users to alert CHOPPI administrators to additional data. For the current application, the CHO genome was homology-matched to a set of CHOPs previously identified in biotherapeutic protein products (Doneanu et al., 2012; Pezzini et al., 2011). We filtered the listed proteins to those whose names were unambiguous, and matched them against the genome using stringent homology thresholds of 90% identity at 90% coverage to ensure the best matches. The matches were further processed in order to obtain a high-quality set of 26 CHOPs: sequence matches with mismatched names were eliminated (e.g., “actin cytoplasmic 1” was taken while “actin cytoplasmic 2” was dropped) and longer matches were chosen over corresponding shorter ones (e.g., for “elongation factor 2”). Furthermore, perlecan was also eliminated because it is only a small fragment of the alternative match “basement membrane-specific heparan sulfate proteoglycan core protein.” A full list of the resulting 26 CHOPs, including their GenBank accession numbers, is provided in the Supplementary Material and on the CHOPPI website.
Human genome: A search for relevant CHOPs may be focused on those more or less similar to human proteins. If a particular human protein is relevant, then a CHOP query will find it via BLAST. If the general extent of human homology is important, then a genome filter can specify both an upper bound (i.e., only proteins with at most that much homology) and/or a lower bound (i.e., at least that much homology). An upper bound is useful to find CHOPs that are relatively divergent from human counterparts and thus pose general immunogenicity risk, while a lower bound is useful to find those that are relatively human-like, but may be distinct at the level of individual T cell epitopes, another form of risk as further discussed below. The human proteins considered are from the UniProt Reviewed database (UniProt Consortium, 2012).
Immunogenicity Analysis
Immunogenicity analysis of selected CHOPs is based on EpiMatrix, a “pocket profile” method that assesses interactions of peptides with the binding pockets of the most prevalent MHC types in the human population (McMurry et al., 2005; Moise et al., 2011; Moss et al., 2011). CHOPPI uses EpiMatrix to predict binding against a common, representative set of eight class II HLA alleles (DRB1*0101, 0301, 0401, 0701, 0801, 1101, 1301, and 1501) that “cover” the genetic backgrounds of most humans worldwide (Southwood et al., 1998). It thresholds EpiMatrix scores to select as epitopes only those peptides scoring in the top 5%; this threshold has been shown to identify peptides that are highly likely to bind the HLA molecule as predicted (De Groot et al., 1997). CHOPPI also employs the EpiMatrix whole-protein immunogenicity score to characterize the epitope density of a target protein relative to a background distribution (Diamond, 2003). In general, proteins having higher epitope densities (EpiMatrix whole-protein score >20) tend to be more immunogenic, while low-density proteins (< −20) tend to be immunologically inert (De Groot and Martin, 2009; Hai et al., 2009). EpiMatrix has been extensively applied and validated in the development of vaccines and therapeutic proteins as discussed in the Introduction Section, and benchmark tests have shown it to have an average sensitivity of 77% for HLA class II predictions, 5–17% better than that of a variety of other methods (De Groot and Martin, 2009).
T cells that respond to autologous epitopes may be deleted in the course of immune system maturation (thymic-dependent tolerance), and peripheral regulatory T cells that recognize autologous epitopes may actively suppress the immune response (peripheral tolerance). Thus, selected epitopes in CHOPs that are similar to autologous peptides may be actively tolerizing or at least non-immunogenic. Consequently, the raw epitope count and EpiMatrix whole protein score may overestimate immunogenicity by counting CHOP epitopes that are actually “human-like.” To help correct for this, CHOPPI also identifies the number of CHO-unique epitopes in a protein (i.e., those that are more likely to induce a detrimental immune response). Recognizing that due to T cell receptor (TCR) cross-reactivity of a CHOP epitope, the homology with human epitopes need not be exact in order to reduce immunogenicity risk, we employ the JanusMatrix tool that evaluates TCR cross-reactivity (Moise et al., 2013). A JanusMatrix-identified cross-reactive CHO/human epitope pair must have identical TCR-facing residues (positions 2, 3, 5, 7, and 8 for class II) but can have variations of the MHC-facing residues (positions 1, 4, 6, and 9) as long as they are still predicted by EpiMatrix to bind the same MHC allele. Cross-reactive pairs are thus likely to be presented by the same MHC to the same TCR. We have shown that a JanusMatrix-based immunogenicity score is different for T effector epitopes versus those that are T regulatory or null, with statistical significance (He et al., 2014). The CHO-unique epitope count thus assesses how many peptides are likely to contribute to a detrimental immune response, as they are not cross-reactive with the human genome.
Web Tool
The CHOPPI web tool is available from http://www.immunome.org/CHOPPI. It is free for academic users, subject to a registration step to ensure appropriate use. For adaptations to other host cell lines and for use by commercial entities, readers are referred to the contact information on the above website.
To support rapid CHOPPI analysis, an SQLITE (http://www.sqlite.org) database was constructed to store and index the annotated CHO genomes, homology matches across the input databases, and immunogenicity analyses. The NCBI BLAST executable, version 2.2.27+ (Camacho et al., 2009), was used to match the protein products from the CHO genomes, the contigs from the CHO transcriptome, the proteins from the CHO proteome, mouse secretome, the human genome, and the validated CHOP sequences. Homology matches at discrete sets of different percent identity and percent coverage thresholds were stored in the database. EpiMatrix immunogenicity scores, epitope counts, and JanusMatrix-based CHO-unique epitope counts were computed for each protein in the CHO genome, and stored in the database.
The CHOPPI web tool is implemented in PHP. A CHOP query request looks up a protein in the database by name or accession, or invokes BLAST to find it by amino acid sequence. A genome filter request processes a form specifying homology filters, performs the appropriate query to retrieve matches from the preprocessed database, and renders the resulting dataset in either HTML tables (sortable by column) or CSV format. Individual protein reports summarize the immunogenicity analyses and detail the homology matches with links to relevant NCBI, UniProt, and LOCATE entries. Immunogenicity scores are also plotted, using the “Raphael” Javascript package (http://raphaeljs.com), relative to a background distribution over the CHO genome.
Data Analysis and Visualization
Additional figures were developed for the Results section, in order to present some of the findings obtained from the CHOPPI site.
To characterize genomic-scale immunogenicity patterns, the EpiMatrix immunogenicity score and epitope distributions over sets of proteins (the whole genome, transcriptome, proteome, etc.) were analyzed and plotted in R (http://www.r-project.org). For large sets, smoothed histograms were generated and visualized via kernel density estimates computed by the “density” function. Cumulative distributions were generated by the “ecdf” function. Coarse-grain histograms were produced by the “hist” function.
To illustrate the patterns of cross-reactivity between particular CHO proteins and human proteins, Cytoscape (Shannon et al., 2003) was used to generate graphical representations of networks of cross-reactive epitopes (Moise et al., 2013). In these networks, CHO and human proteins, along with their constituent epitopes (restricted to cross-reactive ones on the human side) are represented with different symbols. Edges connect cross-reactive CHOP and human epitopes, as well as proteins with their constituent epitopes.
Results and Discussion
We applied CHOPPI to a variety of CHOP queries and genome filters in order to demonstrate its effectiveness at characterizing the immunogenic risk posed by CHOPs. We first used CHOPPI to assess the set of 26 validated CHOPs. We then used it to filter the CHO genome by different criteria in order to characterize immunogenicity patterns in genomic subsets. Finally, we selected and further detailed some CHOPs in different immunogenicity risk classes.
Illustration of the CHOP Query Tool: Validated CHOPs
Figure 3a, c, and e illustrates the immunogenicity analysis of the 26 validated CHOPs derived from the literature; a detailed spreadsheet is in the Supplementary Material and an interactive version is available on the CHOPPI website. We see that the validated CHOPs have a range of EpiMatrix immunogenicity scores and epitope content. Notably, four of these proteins have EpiMatrix immunogenicity scores exceeding 20, which is considered to be higher risk for immunogenicity (“risky”), yielding the secondary peak in the distribution in Figure 3a: annexin A1 (24.9), glutathione S-transferase P (25.2), lysosomal protective protein (34.4), and collagen alpha-1(III) chain (35.8). The collagen chain is quite large, with 1,051 amino acids and a total of 228 predicted epitopes (Fig. 3c). Of those 228 epitopes, 60 are unique to CHO, yielding the secondary peak in Figure 3e. For reasons discussed above, we consider epitopes that are unique to CHO to be higher risk than epitopes that are conserved with the human genome.
Figure 3.
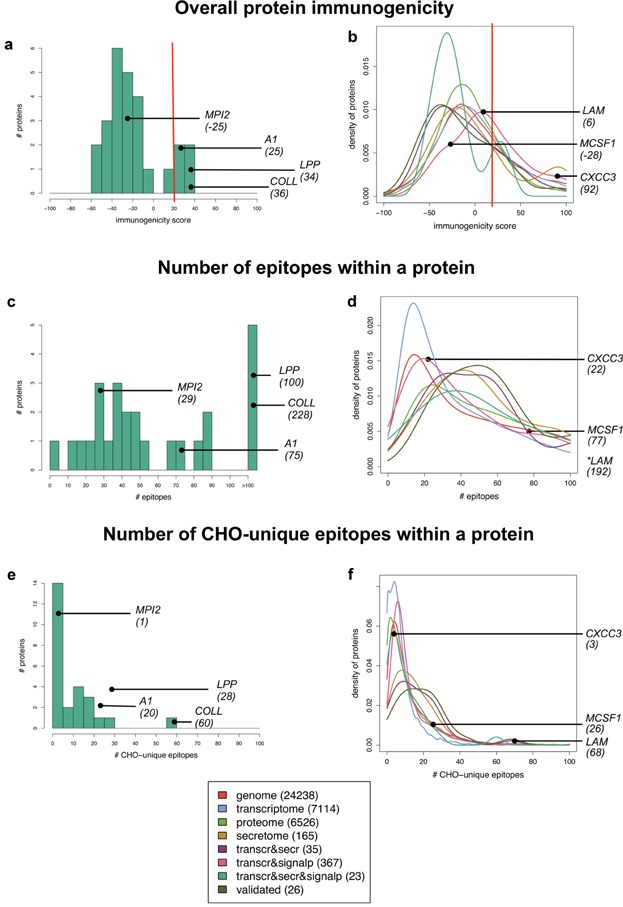
Immunogenicity evaluations over different sets of CHOPs. The left panels (a, c, and e) characterize the validated CHOPs with histograms. Examples of validated CHOPs and their scores are shown: collagen (COLL), lysosomal protective protein (LPP), annexin A1 (A1), and metalloproteinase inhibitor 2 (MPI2). The right panels (b, d, and f) characterize various genome-filtered subsets with density estimates (smoothed histograms). Note that the y-axes of the density plots are differently scaled. Subsets: CHO genome, CHO transcriptome (transcr), CHO proteome, mouse secretome (secr), intersection of transcr with secr, intersection with predicted CHOPs containing predicted signal peptides (SignalP), intersection with secr and SignalP, and validated CHOPs (repeated from the left panels). Examples of potential immunogenic proteins and their scores (in parenthesis) are shown: C-X-C motif chemokine 3 (CXCC3), macrophage colony-stimulating factor 1 (MCSF1), and lysosomal alpha-mannosidase (LAM). a and b: Overall protein immunogenicity, truncated to show proteins scoring between −100 and 100 on the EpiMatrix immunogenicity scale. A red line indicates the level at which a protein is considered to be higher risk (EpiMatrix score of 20). c and d: Number of epitopes within a protein, truncated to 100. e and f: Number of epitopes within a protein that are not human-like, according to cross-reactivity analysis using JanusMatrix, truncated to 100.
Lysosomal protective protein is also fairly large, containing 109 predicted epitopes over its 475 total amino acids, with 28 of those epitopes being unique to CHO. The other two are smaller but still have substantial epitope content: 45 epitopes (14 unique) in the 221 amino acids of glutathione S-transferase P and 75 epitopes (20 unique) in the 346 amino acids of annexin A1.
Figure 4 (left) illustrates the cross-reactivity patterns of two of these relatively risky proteins (lysosomal protective protein and annexin A1), along with a contrasting protein (metalloproteinase inhibitor 2) that has low overall immunogenicity and only one CHO-unique epitope (of 29 epitopes). As noted above, the two riskier proteins (lysosomal protective protein and annexin A1) both have sizable fractions of CHO-unique epitopes, clustered toward the center with the source protein. Epitopes that have cross-reactive partners are fairly dispersed—each is relatively unique, with few shared ninemers in the human genome. The lower-risk protein (metalloproteinase inhibitor 2) clearly has a big group of epitopes with cross-reactive partners, though each has only one or two matches in the human genome.
Figure 4.
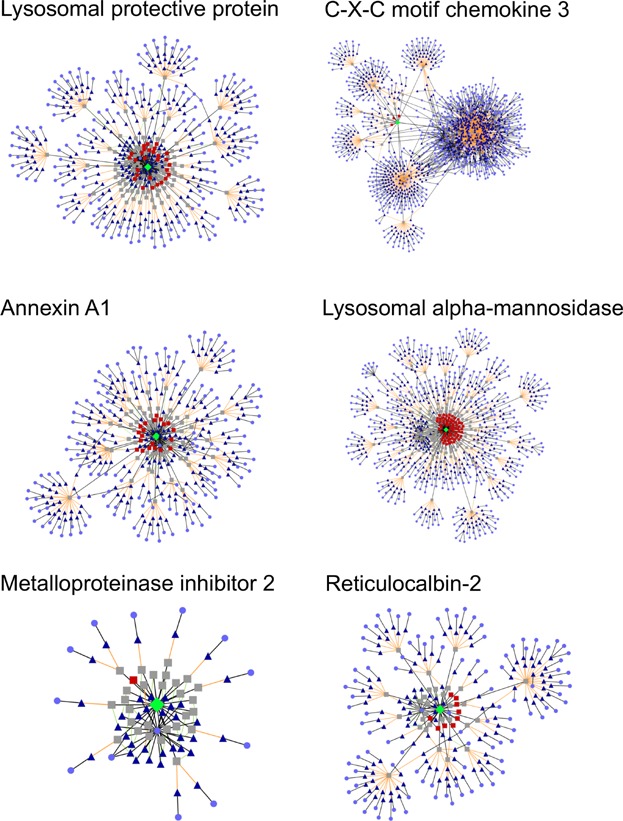
Cross-reactivity analysis of example CHOPs. Left: Validated CHOPs and (right) CHOPPI-identified immunologically risky CHOPs. In the epitope network visualizations, CHOPs are shown as green diamonds (one per network), their epitopes as either red squares (no cross-reactive epitopes) or gray squares (one or more), their cross-reactive partners in the human genome as blue triangles, and the source human proteins as light purple circles.
Genomic-Scale Analysis
We used CHOPPI's genome filter to identify CHOPs that are likely to be expressed, secreted, or both. To predict expression and secretion, by homology matching the CHO genome against the CHO transcriptome, CHO proteome, and mouse secretome, it is necessary to establish required levels of sequence identity and coverage. We considered three different alternatives: 50% identity covering at least 50% of the sequence (relatively loose matches), to 70% identity at 70% coverage (moderate), and 90% identity at 90% coverage (tight). We also employed the SignalP-based secretion filter. Table I summarizes the number of proteins from the 24,238 in the CHOPPI-processed CHO genome, meeting different transcription and secretion criteria. It shows that the different criteria produce different sets of proteins, making trade-offs between possible false positives (including CHOPs that will not actually be present in recombinant therapeutic preparations) and false negatives (excluding CHOPs actually present). We note that at higher identity and coverage cutoffs, a number of secreted proteins from mouse did not have homologs in CHO.
Table I.
Subsets of CHO-K1 genes (24,238 processed in CHOPPI) that are predicted to be transcribed and/or secreted
| Subset | 50% id. at 50% cov. | 70% id. at 70% cov. | 90% id. at 90% cov. |
|---|---|---|---|
| Transcriptome | 10,157 (41.91%)a | 7,114 (29.35%) | 4,536 (18.71%) |
| Proteome | 8,626 (35.59%) | 6,526 (26.92%) | 5,018 (20.70%) |
| Mouse secretome | 288 (1.19%) | 165 (0.68%) | 46 (0.19%) |
| Transcriptome and mouse secretome | 76 (0.31%) | 35 (0.14%) | 5 (0.02%) |
| Transcriptome and SignalP | 571 (2.36%) | 367 (1.51%) | 251 (1.04%) |
| Transcriptome and mouse secretome and SignalP | 36 (0.15%) | 23 (0.09%) | 2 (0.01%) |
Summary of the number of proteins from the CHO genome that are predicted to be transcribed and/or secreted. Different identity (id.) and coverage (cov.) percentages produced different sets of proteins. 70% identity at 70% coverage was used for subsequent results.
Percentage of the CHO-K1 genes processed in CHOPPI represented in the subset.
For the remaining results, we adopted the moderate criteria of 70% identity at 70% coverage as a reasonable balance. Figure 3b, d, and f illustrates these immunogenicity score distributions with estimated density plots (essentially smoothed histograms as described in the Data Analysis and Visualization Section). The Supplementary Material includes corresponding cumulative distributions from which one can infer what fraction of each dataset falls above or below various thresholds. We now discuss the overall characteristics of the plots; the following subsection details some of the interesting risky CHOPs highlighted on the plots.
Whole-Protein Immunogenicity Score
To assess the overall predicted immunogenicity of the filtered proteins, we first evaluated each CHO protein on the EpiMatrix whole-protein immunogenicity scale, which is calibrated such that a score above 20 indicates a relatively high-risk protein and a score below −20 indicates a protein that is predicted to be non-immunogenic. Figure 3b shows the distribution of EpiMatrix immunogenicity scores over the different protein sets. Most of the proteins in each subset have medium or low epitope density and thus do not pose a clear immunogenicity risk. But clearly, each subset includes some proteins with a high risk of immunogenicity (EpiMatrix immunogenicity score above 20); this is further illustrated by the cumulative distribution (see Supplementary Material). The tails of the distributions are fairly long (even including secondary peaks in the secreted proteins), yielding sizable numbers of proteins with high overall EpiMatrix immunogenicity scores.
Epitope Content
Focusing on the epitope content of individual proteins, Figure 3d shows that in each set, proteins tend to have 20–40 epitopes (i.e., ninemers predicted to bind at least one MHC allele). Clearly, this depends on protein length; for example, for abnormal spindle-like microcephaly-associated protein, 587 epitopes are predicted for this 1,831 amino acid protein (not shown). As with the overall protein immunogenicity score, the genomic-scale analysis by CHOPPI enables the user to see that each set of proteins includes some with substantial numbers of epitopes.
CHO-Unique Epitope Content
While the overall EpiMatrix immunogenicity score and individual epitope count are indicative of the potential for immunogenicity, these scores do not incorporate any adjustment for conservation with autologous (human) proteins and thus may overestimate immunogenic risk. Figure 3f complements these assessments by illustrating the numbers of CHO-unique epitopes, that is, those that lack cross-reactivity with autologous proteins and thus are most likely to induce a detrimental immune response. Due to the overall homology between CHOPs and their human protein counterparts, many of the CHO epitopes do indeed have homologous partners and can be predicted to have low immunogenicity and unlikely to break peripheral tolerance. However, we still see a number of proteins in any genomic subset with a substantial number of unique epitopes (even by the relaxed JanusMatrix evaluation that allows variation in the MHC-facing residues). The potential for these slightly dissimilar proteins to cross-react and induce immune responses under the right inflammatory circumstances can be predicted from published experience, for example, with autoimmune disease models in which the immunogen is often the human version of the murine autoimmune disease target such as in the use of human thyroid stimulating hormone receptor in the induction of a murine Graves' disease model. More specifically, it is well known that a single epitope difference can trigger an immune response against a self protein that then spreads to other epitopes (Inaba et al., 2013; McLachlan et al., 2012; You and Chatenoud, 2006).
Overall Human Homology
To further evaluate the effects of homology, we identified those proteins from the transcriptome set that also have global sequence-level (not necessarily epitope-level) homology to a human protein. Such proteins thus are likely to be expressed and are overall quite similar to human proteins, but they may still have segments that are immunologically risky. Figure 5 illustrates the distributions of the percent of epitopes that are CHO-unique, over the proteins in a set with 70% identity to human at 70% coverage, as well as in a set with 90% identity at 90% coverage. Strikingly, even though the overall human homology of these CHOPs is quite high, there are substantial numbers of epitopes in some proteins that do not appear in any human protein, even in a relaxed JanusMatrix cross-reactivity evaluation. In the 70% at 70% set, 298 proteins have at least 40% of their epitopes as unique to CHO, while even in the 90% at 90% set, 13 proteins have at least 40% CHO-unique epitopes.
Figure 5.
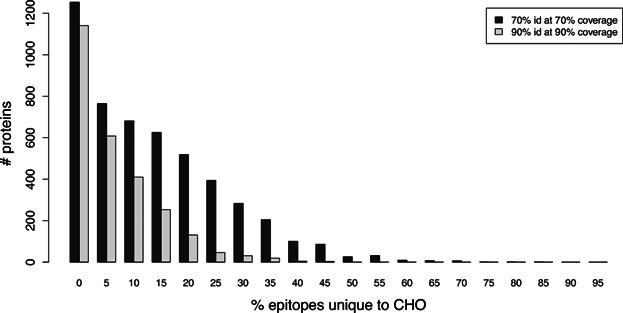
CHOP cross-reactivity with human. CHO-unique epitope content within CHOPs that have matches in both the CHO transcriptome (70% identity at 70% coverage) and the human genome (either with 70% identity at 70% coverage or with 90% at 90%). CHOPs that are more cross-reactive with human proteins at the protein and epitope level are less likely to induce a T effector response. Conversely, even those CHOPs that are highly conserved may have one or more CHO-unique epitopes that could contribute to anti-CHOP immune response (with potential for spreading to the more conserved epitopes).
Example Risky CHOPs
For further analysis (described below) we focused on the set of transcribed, secreted proteins as representatives of those likely to be found in CHO cultures. A detailed spreadsheet with the CHOPPI analysis of all 35 proteins is provided in the Supplementary Material and an interactive version is available on the CHOPPI site. Several examples, highlighted on the graphs in Figure 3b, d, and f, illustrate the immunogenicity risks posed by some of the proteins in the tails of the distributions. These risky proteins were not selected from the set above that was pre-filtered to have high human homology, but we note that they do all have at least 60% identity with a human homolog and yet retain novel epitopes that pose an immunogenicity risk.
High EpiMatrix Immunogenicity Scores and Epitope Content
Of the 35 proteins, eight were identified as having EpiMatrix whole-protein immunogenicity scores above 20 (Fig. 3b), indicating a stronger immunogenicity risk. Of these, C-X-C motif chemokine 3 (CXCC3) has a score of 92, due to the presence of 22 epitopes (Fig. 3d) within only 101 amino acids total length. However, only three of these epitopes are unique to CHO (Fig. 3f) according to the JanusMatrix analysis, and thus we would expect that the chemokine might not be as immunogenic as indicated by the raw EpiMatrix immunogenicity score. There are similar stories for pigment epithelium-derived factor (score of 62 with four CHO-unique epitopes among 14 epitopes in 58 amino acid residues) and leukemia inhibitory factor (score of 56 with five CHO-unique epitopes among 30 in 158 residues) (not shown). However, we see a slightly higher level of CHO-unique epitopes in gamma-glutamyl hydrolase (score of 41 with 25 unique epitopes among 58 in 271 residues) and metalloproteinase inhibitor 1 (score of 40 with 16 unique epitopes among 40 in 203 residues) (not shown).
High CHO-Unique Epitope Content
Sorting the proteins by their numbers of CHO-unique epitopes leads to the observation that the heavy tail in this distribution (Fig. 3f) is due to large proteins such as lysosomal alpha-mannosidase (LAM; 68 CHO-unique epitopes of 192, within 1,009 residues total), sialate O-acetylesterase (43 of 102 in 550), and complement C1r-A subcomponent (34 of 102 in 705). The middle of the distribution has a number of cathepsins (cathepsin B with 27/52 unique, D with 21/76, and L1 with 18/52), along with a wide range of other proteins such as a macrophage colony-stimulating factor 1 (MCSF1), an extracellular matrix protein, and granulins. Sorting instead on the percentage of epitopes that are CHO-unique (not shown) reveals a few additional proteins with relatively high CHO-unique epitope density, including plasminogen activator inhibitor 1 (11% of epitopes are CHO-unique), lactadherin (8.3%), and metalloproteinase inhibitor 1 (8.2%). The relatively high CHO-unique epitope content of metalloproteinase inhibitor 1 stands in contrast to that of its homolog metalloproteinase inhibitor 2 (38% sequence identity over a 167-residue aligned fragment), which as discussed above only has a single CHO-unique epitope. Epitope content is a function of the particular constituent peptides, and can thus be quite different even among overall similar proteins, highlighting the need for protein-specific analysis of immunogenicity risk along with expression/secretion risk.
High CHO-Unique Epitope Content, Despite High Overall Human Identity
Recall that the distribution of epitope conservation in CHO proteins with close human homologs (Fig. 5) reveals a number of CHOPs with high relative CHO-unique epitope content. Even in the extremely homologous set of 90% identity at 90% coverage, we are able to identify some proteins with substantial potential for immunogenicity, by having a sizable CHO-unique percentage of epitopes within a high overall number of epitopes. For example, calreticulin has a 94% identical human homolog, yet the CHO version has five CHO-unique epitopes among the 43 that are identified in this protein, over its 417 residues. Reticulocalbin-2 has nine CHO-unique epitopes out of a total of 39 despite having a 92% identical homolog in the human genome. And connective tissue growth factor has a 91% identical homolog, still leaving 7 of 31 unique epitopes in only 348 amino acids. These proteins thus have the potential for driving a detrimental immune response with their “foreign” epitopes, potentially even spreading to the remaining more “human” epitopes once the attention of the immune system is raised.
Epitope-level analysis of one protein from each of these categories was conducted: CXCC3 representing the high whole-protein immunogenicity group, LAM the high CHO-unique group, and reticulocalbin-2 the high-unique though high-human identity group. Figure 4 (right) presents visualizations of the networks of cross-reactive epitopes. Differences between protein CXCC3 and LAM are noticeable. The network for CXCC3 shows high conservation with human proteins, and there are few CHO-unique epitopes near the CXCC3 node. In contrast, the LAM epitope network shows several such epitopes, which are concentrated around LAM. We also note that CXCC3 has fewer predicted epitopes in general than LAM, but because CXCC3 has more cross-reactive epitopes with human proteins, its branches are more populated. Moreover, the CXCC3 network has more connections due to the presence of cross-reactive epitopes in multiple human proteins. In addition, one epitope from CXCC3 is cross-reactive with numerous (637) human epitopes, further populating the network. Analogous high cross-reactivity with the human genome was observed in Tregitopes and an HCV regulatory epitope (Moise et al., 2013). Thus, CXCC3 might induce a regulatory immune response in humans. Finally, reticulocalbin-2 looks much like the riskier validated proteins annexin A1 and lysosomal protective protein, with a cluster of CHO-unique epitopes near the protein and the cross-reactive epitopes dispersed away from it.
Conclusion
CHOPPI provides a powerful new resource for computationally predicting the immunogenicity risk of CHO proteins, whether previously identified by experimental techniques or selected from a genomic analysis according to factors affecting whether the CHOP would be hitchhiking along during the purification process of a therapeutic product. We show by examination of validated CHOPs, as well as by discovery of new potentially risky CHOPs, that CHOPPI reveals important aspects of CHOP risk, including the potential to be present in cell culture medium, and whether the protein contains many unique CHO epitopes, which could contribute to the stimulation of a detrimental immune response. CHOPPI enhances currently available experimental methods by summarizing and detailing the contributions of individual T cell epitopes to immunogenic risk, and accounts for both epitope density and similarity to autologous proteins. An important take-away message is that the CHO genome contains many proteins that are substantially dissimilar to the human genome from the T cell epitope standpoint, and thus inherently pose some risk of triggering anti-self immune responses. This finding supports the current FDA and EMA perspectives on CHOP; that lower levels of CHOPs are important for drug safety (EMA, 2012; FDA, 2012).
We intend to continue expansion and generalization of CHOPPI's capabilities. We will incorporate additional genomic, transcriptomic, proteomic, and secretomic information as these databases continue to be generated. We will also continually expand the set of validated CHOPs identified within CHOPPI. CHOPPI provides a general-purpose architecture for HCP analysis, and we may apply it to other cell lines that are commonly used to produce protein therapeutics.
CHOPPI's results are all predictive in nature, based on computational analyses. A key next step is the experimental validation of CHOPPI predictions. We intend to experimentally test immunogenicity of internally identified CHOPs as well as support others in doing so by making this resource available (http://www.immunome.org/CHOPPI). In general, CHOPPI provides a valuable resource for focusing follow-up experimental characterization and immunogenicity studies on the possible impurities of greatest predicted importance and relevance.
The authors gratefully acknowledge the thoughtful review of A.J. Vincelli of EpiVax and Denise Krawitz, Valerie Quarmby, Martin Vanderlaan, and Patty Siguenza of Genentech, with regards to the usefulness of this approach to CHO protein analysis. The URI and EpiVax research effort devoted to the development of this tool was supported by grant number U19AI082642-01 from the National Institutes of Health (NIH) and the National Institute of Allergy and Infectious Diseases (NIAID). CBK was supported in part by grant IIS-0905206 from the National Science Foundation.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
References
- Baycin-Hizal D, Tabb DL, Chaerkady R, Chen L, Lewis NE, Nagarajan H, Sarkaria V, Kumar A, Wolozny D, Colao J, Jacobson E, Tian Y, O'Meally RN, Krag SS, Cole RN, Palsson BO, Zhang H, Betenbaugh M. Proteomic analysis of Chinese hamster ovary cells. J Proteome Res. 2012;11:5265–5276. doi: 10.1021/pr300476w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker J, Hackl M, Rupp O, Jakobi T, Schneider J, Szczepanowski R, Bekel T, Borth N, Goesmann A, Grillari J, Kaltschmidt C, Noll T, Pühler A, Tauch A, Brinkrolf K. Unraveling the Chinese hamster ovary cell line transcriptome by next-generation sequencing. J Biotechnol. 2011;156:227–235. doi: 10.1016/j.jbiotec.2011.09.014. [DOI] [PubMed] [Google Scholar]
- Brinkrolf K, Rupp O, Laux H, Kollin F, Ernst W, Linke B, Kofler R, Romand S, Hesse F, Budach WE, Galosy S, Müller D, Noll T, Wienberg J, Jostock T, Leonard M, Grillari J, Tauch A, Goesmann A, Helk B, Mott JE, Pühler A, Borth N. Chinese hamster genome sequenced from sorted chromosomes. Nat Biotechnol. 2013;31:694–695. doi: 10.1038/nbt.2645. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: Architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian J-J, Martin-Dupont P, Michaud P, Papo T, Ugo V, Teyssandier I, Varet B, Mayeux P. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346:469–475. doi: 10.1056/NEJMoa011931. [DOI] [PubMed] [Google Scholar]
- Champion K, Madden H, Dougherty J, Shacter E. Defining your product profile and maintaining control over it, Part 2. BioProcess Int. 2005;3:52–57. [Google Scholar]
- Diamond B. Speculations on the immunogenicity of self proteins. Dev Biol. 2003;112:29–34. [PubMed] [Google Scholar]
- Doneanu CE, Xenopoulos A, Fadgen K, Murphy J, Skilton SJ, Prentice H, Stapels M, Chen W. Analysis of host-cell proteins in biotherapeutic proteins by comprehensive online two-dimensional liquid chromatography/mass spectrometry. MAbs. 2012;4:24–44. doi: 10.4161/mabs.4.1.18748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMA. Draft guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: Quality issues. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/05/WC500127960.pdf.
- FDA. Draft guidance for industry scientific considerations in demonstrating biosimilarity to a reference protein product. 2012. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf.
- Gregory SH, Mott S, Phung J, Lee J, Moise L, McMurry JA, Martin W, De Groot AS. Epitope-based vaccination against pneumonic tularemia. Vaccine. 2009;27:5299–5306. doi: 10.1016/j.vaccine.2009.06.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot AS, Bishop EA, Khan B, Lally M, Marcon L, Franco J, Mayer KH, Carpenter CCJ, Martin W. Engineering immunogenic consensus T helper epitopes for a cross-clade HIV vaccine. Methods. 2004;34:476–487. doi: 10.1016/j.ymeth.2004.06.003. [DOI] [PubMed] [Google Scholar]
- De Groot AS, Jesdale BM, Szu E, Schafer JR, Chicz RM, Deocampo G. An interactive Web site providing major histocompatibility ligand predictions: Application to HIV research. AIDS Res Hum Retroviruses. 1997;13:529–531. doi: 10.1089/aid.1997.13.529. [DOI] [PubMed] [Google Scholar]
- De Groot AS, Martin W. Reducing risk, improving outcomes: Bioengineering less immunogenic protein therapeutics. Clin Immunol. 2009;131:189–201. doi: 10.1016/j.clim.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Grosenbaugh DA, Leard AT, Bergman PJ, Klein MK, Meleo K, Susaneck S, Hess PR, Jankowski MK, Jones PD, Leibman NF, Johnson MH, Kurzman ID, Wolchok JD. Safety and efficacy of a xenogeneic DNA vaccine encoding for human tyrosinase as adjunctive treatment for oral malignant melanoma in dogs following surgical excision of the primary tumor. Am J Vet Res. 2011;72:1631–1638. doi: 10.2460/ajvr.72.12.1631. [DOI] [PubMed] [Google Scholar]
- Gutierrez AH, Moise L, De Groot AS. Of [hamsters] and men. Hum Vaccines Immunother. 2012;8:1172–1174. doi: 10.4161/hv.22378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai S-H, McMurry JA, Knopf PM, Martin W, De Groot AS. Immunogenicity screening using in silico methods: Correlation between T-cell epitope content and clinical immunogenicity of monoclonal antibodies. In: An Z, editor. Therapeutic Monoclonal Antibodies. Hoboken, NJ: John Wiley & Sons, Inc; 2009. pp. 417–437. [Google Scholar]
- He L, De Groot AS, Gutierrez AH, Martin WD, Moise L, Bailey-Kellogg C. Integrated assessment of predicted MHC binding and cross-conservation with self reveals patterns of viral camouflage. BMC Bioinformatics. 2014;15:S1. doi: 10.1186/1471-2105-15-S4-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins E. Carbohydrate analysis throughout the development of a protein therapeutic. Glycoconj J. 2010;27:211–225. doi: 10.1007/s10719-009-9261-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba H, Moise L, Martin W, De Groot AS, Desrosiers J, Tassone R, Buchman G, Akamizu T, De Groot LJ. Epitope recognition in HLA-DR3 transgenic mice immunized to TSH-R protein or peptides. Endocrinology. 2013;154:2234–2243. doi: 10.1210/en.2013-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba H, Pan D, Shin Y-H, Martin W, Buchman G, De Groot LJ. Immune response of mice transgenic for human histocompatibility leukocyte Antigen-DR to human thyrotropin receptor-extracellular domain. Thyroid Off J Am Thyroid Assoc. 2009;19:1271–1280. doi: 10.1089/thy.2008.0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipsen. Ipsen's partner Inspiration Biopharmaceuticals announces hold of phase III clinical trials evaluating IB1001 for the treatment and prevention of hemophilia B. 2012. http://www.ipsen.com/wp-content/uploads/2013/03/PR_IB1001-Clinical-Hold_EN_0.pdf.
- Jayapal KP, Wlaschin KF, Hu W-S, Yap MG. Recombinant protein therapeutics from CHO cells—20 years and counting. Chem Eng Prog. 2007;103:40–47. [Google Scholar]
- Jin M, Szapiel N, Zhang J, Hickey J, Ghose S. Profiling of host cell proteins by two-dimensional difference gel electrophoresis (2D-DIGE): Implications for downstream process development. Biotechnol Bioeng. 2010;105:306–316. doi: 10.1002/bit.22532. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kim Y-G, Lee GM. CHO cells in biotechnology for production of recombinant proteins: Current state and further potential. Appl Microbiol Biotechnol. 2012;93:917–930. doi: 10.1007/s00253-011-3758-5. [DOI] [PubMed] [Google Scholar]
- Koren E, De Groot AS, Jawa V, Beck KD, Boone T, Rivera D, Li L, Mytych D, Koscec M, Weeraratne D, Swanson S, Martin W. Clinical validation of the “in silico” prediction of immunogenicity of a human recombinant therapeutic protein. Clin Immunol. 2007;124:26–32. doi: 10.1016/j.clim.2007.03.544. [DOI] [PubMed] [Google Scholar]
- Koren E, Zuckerman LA, Mire-Sluis AR. Immune responses to therapeutic proteins in humans—Clinical significance, assessment and prediction. Curr Pharm Biotechnol. 2002;3:349–360. doi: 10.2174/1389201023378175. [DOI] [PubMed] [Google Scholar]
- Lewis NE, Liu X, Li Y, Nagarajan H, Yerganian G, O'Brien E, Bordbar A, Roth AM, Rosenbloom J, Bian C, Xie M, Chen W, Li N, Baycin-Hizal D, Latif H, Forster J, Betenbaugh MJ, Famili I, Xu X, Wang J, Palsson BO. Genomic landscapes of Chinese hamster ovary cell lines as revealed by the Cricetulus griseus draft genome. Nat Biotechnol. 2013;31:759–765. doi: 10.1038/nbt.2624. [DOI] [PubMed] [Google Scholar]
- McLachlan SM, Aliesky HA, Chen C-R, Chong G, Rapoport B. Breaking tolerance in transgenic mice expressing the human TSH receptor A-subunit: Thyroiditis, epitope spreading and adjuvant as a “double edged sword. PLoS ONE. 2012;7:e43517. doi: 10.1371/journal.pone.0043517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurry J, Sbai H, Gennaro ML, Carter EJ, Martin W, De Groot AS. Analyzing Mycobacterium tuberculosis proteomes for candidate vaccine epitopes. Tuberculosis (Edinb) 2005;85:95–105. doi: 10.1016/j.tube.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Moise L, Buller RM, Schriewer J, Lee J, Frey SE, Weiner DB, Martin W, De Groot AS. VennVax, a DNA-prime, peptide-boost multi-T-cell epitope poxvirus vaccine, induces protective immunity against vaccinia infection by T cell response alone. Vaccine. 2011;29:501–511. doi: 10.1016/j.vaccine.2010.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moise L, Gutierrez AH, Bailey-Kellogg C, Terry F, Leng Q, Abdel Hady KM, Verberkmoes NC, Sztein MB, Losikoff PT, Martin WD, Rothman AL, De Groot AS. The two-faced T cell epitope: Examining the host-microbe interface with JanusMatrix. Hum Vaccines Immunother. 2013;9:1577–1586. doi: 10.4161/hv.24615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moise L, Song C, Martin WD, Tassone R, De Groot AS, Scott DW. Effect of HLA DR epitope de-immunization of Factor VIII in vitro and in vivo. Clin Immunol. 2012;142:320–331. doi: 10.1016/j.clim.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss SF, Moise L, Lee DS, Kim W, Zhang S, Lee J, Rogers AB, Martin W, De Groot AS. HelicoVax: Epitope-based therapeutic Helicobacter pylori vaccination in a mouse model. Vaccine. 2011;29:2085–2091. doi: 10.1016/j.vaccine.2010.12.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omasa T, Onitsuka M, Kim W-D. Cell engineering and cultivation of Chinese hamster ovary (CHO) cells. Curr Pharm Biotechnol. 2010;11:233–240. doi: 10.2174/138920110791111960. [DOI] [PubMed] [Google Scholar]
- Osipovitch DC, Parker AS, Makokha CD, Desrosiers J, Kett WC, Moise L, Bailey-Kellogg C, Griswold KE. Design and analysis of immune-evading enzymes for ADEPT therapy. Protein Eng Des Sel PEDS. 2012;25:613–623. doi: 10.1093/protein/gzs044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Pezzini J, Joucla G, Gantier R, Toueille M, Lomenech A-M, Le Sénéchal C, Garbay B, Santarelli X, Cabanne C. Antibody capture by mixed-mode chromatography: A comprehensive study from determination of optimal purification conditions to identification of contaminating host cell proteins. J Chromatogr A. 2011;1218:8197–8208. doi: 10.1016/j.chroma.2011.09.036. [DOI] [PubMed] [Google Scholar]
- Prasad S, Kohm AP, McMahon JS, Luo X, Miller SD. Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9-23 epitope and involves functional epitope spreading. J Autoimmun. 2012;39:347–353. doi: 10.1016/j.jaut.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwood S, Sidney J, Kondo A, del Guercio MF, Appella E, Hoffman S, Kubo RT, Chesnut RW, Grey HM, Sette A. Several common HLA-DR types share largely overlapping peptide binding repertoires. J Immunol. 1998;160:3363–3373. [PubMed] [Google Scholar]
- Sprenger J, Lynn Fink J, Karunaratne S, Hanson K, Hamilton NA, Teasdale RD. LOCATE: A mammalian protein subcellular localization database. Nucleic Acids Res. 2008;36:D230–D233. doi: 10.1093/nar/gkm950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol. 2010;28:917–924. doi: 10.1038/nbt0910-917. [DOI] [PubMed] [Google Scholar]
- Wang P, Sidney J, Dow C, Mothe B, Sette A, Peters B. A systematic assessment of MHC Class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4:e1000048. doi: 10.1371/journal.pcbi.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worobec A, Rosenberg AS. A risk-based approach to immunogenicity concerns of therapeutic protein products, Part 1: Considering consequences of the immune response to a protein. BioPharm Int. 2004;17:22–26. [Google Scholar]
- Xu X, Nagarajan H, Lewis NE, Pan S, Cai Z, Liu X, Chen W, Xie M, Wang W, Hammond S, Andersen MR, Neff N, Passarelli B, Koh W, Fan HC, Wang J, Gui Y, Lee KH, Betenbaugh MJ, Quake SR, Famili I, Palsson BO, Wang J. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat Biotechnol. 2011;29:735–741. doi: 10.1038/nbt.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You S, Chatenoud L. Proinsulin: A unique autoantigen triggering autoimmune diabetes. J Clin Invest. 2006;116:3108–3110. doi: 10.1172/JCI30760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.