

Abstract
Assessing the impact of natural enemies of plant and animal pathogens on their host's population dynamics is needed to determine the role of hyperparasites in affecting disease dynamics, and their potential for use in efficient control strategies of pathogens. Here, we focus on the long-term study describing metapopulation dynamics of an obligate pathogen, the powdery mildew (Podosphaera plantaginis) naturally infecting its wild host plant (Plantago lanceolata) in the fragmented landscape of the Åland archipelago (southwest Finland). Regionally, the pathogen persists through a balance of extinctions and colonizations, yet factors affecting extinction rates remain poorly understood. Mycoparasites of the genus Ampelomyces appear as good candidates for testing the role of a hyperparasite, i.e. a parasite of other parasites, in the regulation of their fungal hosts' population dynamics. For this purpose, we first designed a quantitative PCR assay for detection of Ampelomyces spp. in field-collected samples. This newly developed molecular test was then applied to a large-scale sampling within the Åland archipelago, revealing that Ampelomyces is a widespread hyperparasite in this system, with high variability in prevalence among populations. We found that the hyperparasite was more common on leaves where multiple powdery mildew strains coexist, a pattern that may be attributed to differential exposure. Moreover, the prevalence of Ampelomyces at the plant level negatively affected the overwinter survival of its fungal host. We conclude that this hyperparasite may likely impact on its host population dynamics and argue for increased focus on the role of hyperparasites in disease dynamics.
Keywords: disease, hyperparasite, metapopulation, molecular detection, plant pathogen, regulation
Introduction
Pathogens are frequently cited as important drivers of population dynamics of their hosts (Anderson & May 1991; Tompkins et al. 2002). Well-known examples include demographic cycles driven, at least partly, by parasites in the red grouse (Hudson et al. 1998; New et al. 2009) and the Soay sheep (Gulland et al. 1993). Highly virulent parasites may lead to a dramatic decrease of their host population (Harding et al. 2002), ultimately driving some populations to extinction (Vredenburg et al. 2010). Such disease-induced extinctions are not predicted by simple models of parasite dynamics where parasites should always go extinct before their hosts (Anderson & May 1991). However, small pre-epidemic host population size and the presence of alternative hosts (reservoirs) may lead to the extinction of a species by its parasites (de Castro & Bolker 2005). Hence, while the effect of pathogens on their host population dynamics appears highly plausible, empirical knowledge of whether and how parasites regulate host population size in natural populations remains limited, with most evidence being indirect (Schmid-Hempel 2011). Moreover, to date research on the effect of pathogens on host population dynamics has mostly focused on vertebrate hosts (Schmid-Hempel 2011).
Many species inhabit fragmented landscapes and may persist regionally as a metapopulation, that is an assemblage of spatially delimited local populations interconnected by migration and experiencing limited lifespan. The regional persistence of the metapopulation depends on a balance between local extinctions and colonizations (Levins 1969; Hanski 1999). The development of the metapopulation theory has been accelerated in the face of habitat fragmentation and the need for conservation applications for species inhabiting spatially fragmented landscapes. In this context, diseases are frequently mentioned as factors increasing extinction risk when enhancing migration levels through establishment of corridors (Simberloff & Cox 1987; Hess 1994). A modelling study confirmed that increasing migration within a host-pathogen metapopulation may lead to extinction of the host population under certain conditions (Harding et al. 2012). The impact of pathogens on their host′s metapopulation dynamics was also highlighted in a recent study focusing on infectious disease in coral reefs (Sokolow 2009). In spite of these two recent studies, theoretical and empirical assessment of the relevance of diseases as drivers of metapopulation dynamics remains surprisingly rare in the ecological literature.
The fungal pathogen Podosphaera plantaginis, a powdery mildew (Erysiphales, Ascomycota), persists as a metapopulation in the fragmented dry meadows of its host plant Plantago lanceolata in the Åland archipelago (Laine & Hanski 2006; Jousimo et al. 2014). Approximately 4000 patches of the host plant have been surveyed annually since 2001, representing one of the few long-term studies of plant pathogens (see also Antonovics 2004; Smith et al. 2011). This pathosystem is characterized by a very low prevalence at the metapopulation scale (proportion of infected meadows remained generally lower than 7%) and very high local turnover (Jousimo et al. 2014). Approximately 40% of the local pathogen populations go extinct every winter (Tack & Laine 2014), rendering it critical that we delineate the factors affecting off-season survival of the pathogen population to understand regional dynamics. To this purpose, Tack & Laine (2014) used reciprocal transplant experiments to explore the interplay between different factors (namely the pathogen population of origin and overwintering site) in determining infection intensity in the following season, but the precise mechanisms underlying these effects have not yet been elucidated. Pathogens may themselves be the target of parasitism, a phenomenon known as hyperparasitism. The striking impact that hyperparasites can have upon plant pathogens has been previously demonstrated by extensive studies of dsRNA viruses infecting Cryphonectria parasitica, the fungus responsible for chestnut blight (Milgroom & Cortesi 2004). The hyperparasite was shown to modulate the pathogen virulence and consequently the host plant population size (Davelos & Jarosz 2004) and to shape the pathogen diversity (Brusini et al. 2011). Between-population variability in hyperparasite prevalence was also evidenced, as well as between-strain differences in the hyperparasite effect on its mycohost (Robin et al. 2010). However, the potential impact of hyperparasites on the natural metapopulation dynamics of any plant pathogen has never been investigated.
The most widespread and oldest known natural enemies of powdery mildews are Ampelomyces spp. (Kiss 2008). Ampelomyces spp. are intracellular fungal mycoparasites, that is fungi parasitizing other fungi. Their hyphae grow inside the mycelia of their hosts, killing the powdery mildew hyphae by degeneration of the cell content. Asexual fruiting bodies of Ampelomyces, called pycnidia, are produced in the hyphae, conidiophores and immature chasmothecia (i.e. sexual resting structures, syn: cleistothecia) of their fungal hosts (see Fig. S1, Supporting information, and Kiss 2008). In other systems, it has been shown that a fraction of the powdery mildew overwintering structures are destroyed by Ampelomyces every year in the field (Kiss 1998; Füzi 2003). As the overwintering success in P. plantaginis depends on the abundance of these resting structures (Tack & Laine 2014), parasitism by Ampelomyces may have critical consequences on the dynamics of this pathosystem.
The impact of hyperparasites on the dynamics of natural pathogen populations is poorly understood. The effect of Ampelomyces mycoparasites on natural populations of powdery mildews has been scarcely documented, despite the commercialization of Ampelomyces as a biocontrol agent of powdery mildews (AQ10™ Biofungicide; Ecogen Inc., Langhorne, PA, USA; Kiss et al. 2004). The aim of this study was to document the prevalence of Ampelomyces within the metapopulation of P. plantaginis and to test whether the hyperparasite has an impact on extinction rates of its host.
For this purpose, we developed a molecular screening test to detect Ampelomyces spp. in field-collected mixed samples containing both the powdery mildew and the host plant DNA. We chose to amplify the ITS region as it, the reference DNA region for fungal species identification (Schoch et al. 2012), by quantitative polymerase chain reaction (qPCR) method. We then applied this detection method to a global sampling within the Åland metapopulation. We use these data to assess whether the level of multiple genotype infection (i.e. when multiple powdery mildew strains infect the same host leaf) affects the probability that the sample contains an Ampelomyces infection. Assuming that each powdery mildew strain has a certain probability of carrying Ampelomyces, leaves infected by multiple powdery mildew strains would be more likely to harbour Ampelomyces than leaves infected by a single powdery mildew strain. Secondly, we tested for a negative impact of the prevalence of Ampelomyces on the key life history stage of P. plantaginis – overwintering. Our results revealed that taking the hyperparasite into account might be important for understanding its phytopathogenic hosts' population dynamics.
Materials and methods
Study system
Podosphaera plantaginis is an obligate pathogen specific to the ribwort plantain Plantago lanceolata (Plantaginaceae). This powdery mildew species has repeated cycles of asexual reproduction throughout the summer with the production of wind-dispersed conidia. It survives host dormancy in winter as sexual resting structures called chasmothecia (Glawe 2008).
Within the Åland archipelago (southwest Finland), populations of P. lanceolata are typically small (<1 ha) and fragmented, occurring mainly on dry meadows. Since 2001, the occurrence of P. plantaginis has been systematically surveyed in approximately 4000 populations of P. lanceolata throughout the Åland archipelago. The presence/absence of this pathogen in each P. lanceolata meadow is recorded by a group of students every year in early September, when the clonal summer spread of the pathogen has terminated. Podosphaera plantaginis is the only powdery mildew species known to infect P. lanceolata within the Åland islands (A.-L. Laine, unpublished data). Infection of leaves by powdery mildew is easily visible due to the white-greyish mycelial growth on the leaf surface. Upon discovery, an infected leaf has always been collected and dried, and the presence of the powdery mildew has been confirmed by subsequent microscopy of these samples (see also Laine & Hanski 2006).
Molecular characterization of the first field observations of Ampelomyces
In September 2011, potentially infected leaves were collected in 292 meadows during the September survey, of which 282 were confirmed to support P. plantaginis. Obvious signs of pycnidia typical of Ampelomyces sp. were found in four of the 282 infected leaves collected. The location of these four observations is indicated in Fig.1.
Fig 1.
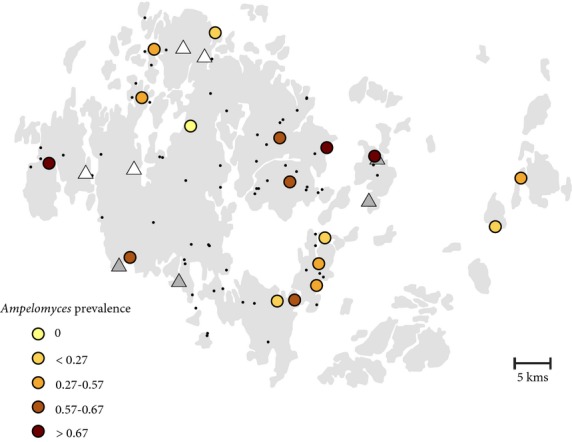
Map of the study sites, indicating the location of (i) the initial observation of pycnidia in 2011 (four populations indicated with white triangles), (ii) the populations sampled in 2013 (four sites indicated with grey triangles), (iii) the populations having sufficient sample size (>22 samples) to estimate Ampelomyces prevalence (the colour gradient indicates the prevalence level) and (iv) all the sampled populations (black dots).
We sequenced the ITS regions of these four samples exhibiting pycnidia to confirm our visual identification of Ampelomyces and to identify their Ampelomyces lineage. For this purpose, we extracted DNA of 1 cm² samples of the dried infected Plantago leaves bearing pycnidia using E.Z.N.A. plant DNA kit (Omega Bio-Tek, Doraville, GA, USA). We amplified the ITS region according to the nested PCR protocol described in Ito & Takamatsu (2010), using primers P3 (5′-GCCGCTTCACTCGCCGTTAC-3′) and ITS5 (5′-GGAAGTAAAAGTCGTAACAAGG-3′) in a first amplification, followed by a second PCR using ITS5 and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′). The PCR products were purified using Exosap-IT (GE Healthcare, Buckinghamshire, UK). Sequencing of the purified PCR products was performed by the Institute for Molecular Medicine Finland (FIMM, Helsinki, Finland) laboratory. We compared the obtained sequences to the GenBank nucleotide database (http://www.ncbi.nlm.nih.gov/genbank/).
In September 2013, a small-scale field survey was conducted in Åland to detect again the presence of Ampelomyces mycoparasites in powdery mildew colonies. Mycoparasites were collected from four sample sites (Fig.1), and seven strains were isolated and maintained in culture as described in Liang et al. (2007). The ITS sequences were determined in these seven newly isolated Åland strains as described above.
Design of a molecular detection test for Ampelomyces spp
To screen our field samples that consisted of a mix of plant, powdery mildew and possibly Ampelomyces DNA, we aimed at designing a primer pair specific for Ampelomyces, and amplifying different Ampelomyces lineages (see below), that would not amplify the fungal host P. plantaginis, nor the host plant P. lanceolata. The forward primer (AQ-F264, 5′-GATGAAGAACGCAGC GAAAT-3′) is located in a conserved region of the ITS sequence: 100% identity with P. plantaginis and 95% with P. lanceolata. The reverse primer (AQ-R462, 5′-GCT GCCAATTGCTTTGAGAT-3′) is located in a hypervariable region; however, it presents 60% identity with ITS sequence of the host plant and the powdery mildew fungal host. This partial overlap of the Ampelomyces-specific AQ-F264/AQ-R462 primers and the ITS sequence of P. plantaginis could not be avoided due to the diversity of ITS sequences determined in different Ampelomyces lineages (Pintye et al. 2012). However, the qPCR method developed in this work did reliably distinguish the Ampelomyces-specific amplifications from false-positive results by setting up a proper cut-off value (see below).
Each qPCR reaction was performed in a final volume of 10 μL containing 5 μL SYBRGreen PCR Master Mix (Applied Biosystems, Madrid, Spain), 0.5 μL of each primer at 5 μM and 1 μL DNA sample. Assays were carried out using Bio-Rad CFX-496 (Hercules, CA, USA) qPCR machine with the following conditions: initial denaturation of 10 min at 95 °C, followed by 40 cycles of denaturation 30 s at 95 °C, annealing 30 s at 60 °C and extension 30 s at 72 °C; final extension lasted 5 min at 72 °C and the melting curve analysis from 45 °C to 95 °C by 0.5 °C increment every 5-s. The software Bio-Rad CFX Manager was used to visualize the results.
As the within-species diversity of Ampelomyces within Åland has not yet been studied, we assessed the generality of our detection method by testing six different Ampelomyces strains (A1, AQ10, B124-a, GYER, TP1 and Vitis32, see details in Table S1, Supporting information) belonging to four different lineages of the five described to date (Pintye et al. 2012). These strains were grown on Czapek-Dox medium supplemented with 2% malt. DNA was extracted using the same method as previously described. Ten-fold serial dilutions of the DNA isolated from these strains were used to establish standard curves.
To assess the reliability of the newly designed method and to determine a qPCR cycle threshold with which to score the binary response of infected or uninfected samples, we generated Ampelomyces-infected P. plantaginis under laboratory conditions. Detached P. lanceolata leaves from a single maternal lineage were placed onto moist filter paper in 9-cm petri dishes and inoculated with single conidial chains of Åland-derived strains of P. plantaginis following the protocol described by Nicot et al. (2002). After 9 days of growth at 21 °C at 16 h:8 h, D:L mildew lesions were inoculated by spraying 0.069 ± 0.004 mL of either a suspension of Ampelomyces spores in water (5 × 105 spores/mL) or an equal volume of water directly onto the infected leaves. The Ampelomyces strains used in these mycoparasitic tests were isolated in Åland in September 2013 (see above). After 19 days, Ampelomyces treated lesions were observed under a dissecting microscope and scored on a four-category scale for presence of pycnidia: ‘none’ = no pycnidia observed, ‘few’ = one or two patches of very few pycnidia, ‘moderate’ = pycnidia covering <75% of Podosphaera infection, and ‘many’ = pycnidia covering more than 75% of Podosphaera infection. Control leaves infected with just P. plantaginis were also checked to confirm the absence of Ampelomyces contamination. Both treated and control leaves were prepared for DNA extraction following the same protocol as described for the large-scale field sampling (see below). The qPCR procedure was applied in triplicate for each treated sample (n = 20), for nine P. plantaginis-only controls and for five infection-free leaves. An additional 36 P. plantaginis-only controls were screened once. We tested for a correlation between the observed categories of pycnidia coverage and the result of the molecular test.
Finally, we used an in vitro method assessing the reliability of the newly designed molecular detection test through the use of plasmids containing the ITS sequence obtained from most Ampelomyces strains isolated in Aland (see the detailed methodology in Appendix S1, Supporting information).
Molecular procedures applied to a large-scale field sampling
In late September 2011, 282 Plantago populations were found to be infected by the powdery mildew during the survey of the entire meadow network. We chose 98 of these powdery mildew populations, located throughout the Åland archipelago (Fig.1), for sample collection. In each population, five infected plants were randomly selected for sampling. Sampled plants were labelled using coloured wooden sticks, and plant positions were recorded using GPS. One leaf per plant was collected in most populations, whereas 17 populations were sampled more intensively with five infected leaves collected from four plants and ten infected leaves collected from one plant.
Powdery mildew-infected Plantago leaves collected in the field were first placed in a falcon tube. After being transported to the laboratory in a cool box, samples were prepared by collecting a 1-cm2 piece of infected leaf as well as all the fungal material that could be scraped off the leaf with a scalpel into a 1.5-mL microcentrifuge tube kept at −80 °C. Both plant and fungal DNA were thus extracted jointly at the Institute of Biotechnology (University of Helsinki), using E.Z.N.A. plant DNA kit as described in Tollenaere et al. (2012). The molecular detection test described above was performed three times on each of these samples. We converted the cycle threshold (Ct) values of the qPCR assay into a binary response variable (positive vs. negative samples) using a cut-off value (Ct = 24) determined with the data obtained from experimentally infected samples (see the results section). For 25 randomly selected positive samples, we amplified the ITS region using conventional PCR with the primers AQ-F264 and AQ-R462, followed by one sequencing reaction (partial sequencing only with the same methodology described above) to ensure the amplicon was derived from Ampelomyces DNA.
The collected samples consisted of all the fungal material found on one Plantago leaf but whether powdery mildew infection on that leaf consists of a unique strain or a mix of various strains may affect the pattern of infection by Ampelomyces. We consequently carried out a SNP (Single Nucleotide Polymorphism) assay specific for P. plantaginis in these same samples, allowing us to infer whether infection of the leaf is due to a unique strain or various strains of powdery mildew (Tollenaere et al. 2012). As P. plantaginis is haploid, we considered the samples to be coinfected by various mildew strains if two alleles were detected in at least one SNP locus (see details in Tollenaere et al. 2012).
Linking the prevalence of Ampelomyces with its fungal host population dynamics
Firstly, we tested whether Ampelomyces prevalence increased with the level of coinfection (as estimated from the SNP genotyping of P. plantaginis samples described above). This analysis was performed on the whole data set (808 samples from 424 plants from 98 populations). Specifically, we modelled for each plant the proportion of leaves with Ampelomyces as a function of the fraction of leaves showing multiple genotypes of the powdery mildew (i.e. whether a unique strain or a mix of various strains was detected using SNP markers). As the response variable is a proportion, we used a binomial distribution and logit link. To account for variation among populations, we included population identity as a random effect. We used the framework of generalized linear mixed models as implemented in procedure glimmix in sas 9.3 (Littell et al. 2006).
Secondly, we assessed the impact of the prevalence of Ampelomyces hyperparasite on the overwintering of its fungal host from September 2011 to July 2012 at two spatial scales: a 1-m2 quadrat and the population level. For both analyses, we used Ampelomyces prevalence and the fraction of leaves with chasmothecia (resting structures) in autumn 2011 as the explanatory variables of overwintering success. We included the latter variable as previous work has shown that the overwintering success of P. plantaginis can be affected by the abundance of chasmothecia (Tack & Laine 2014).
To analyse the impact of Ampelomyces prevalence on the overwintering of its fungal host at a small spatial scale, we relocated the plants (n = 78 plants in 16 of the 17 more intensively sampled populations) that had been marked as infected in September 2011 in early July 2012 at the onset of within season transmission (Soubeyrand et al. 2014; Ovaskainen & Laine 2006; for details on the July survey, please see Tack & Laine 2014). Powdery mildew overwintering success was inferred by recording the fraction of infected plants within the 1-m2 quadrat surrounding the marked locations of plants recorded to be infected in autumn 2011. In autumn 2011, we visually scored the presence–absence of chasmothecia on ten randomly selected leaves (or at least five if the plant had <10 leaves) of the same five plants that were selected for the Ampelomyces sampling. The fraction of leaves bearing chasmothecia was estimated out of the total number of infected leaves at the plant level; an additional population-level estimate was derived by averaging the estimate across the five plants (see also Tollenaere & Laine 2013 and Tack & Laine 2014). We used the procedure glimmix (described above) to model the fraction of infected plants in quadrats in July 2012 as a function of the level of Ampelomyces (fraction of leaves infected) and chasmothecia (fraction of infected leaves with chasmothecia) on the focal sampled plant in the previous autumn. To account for variation among populations, we included population identity as a random effect.
To analyse the impact of Ampelomyces on overwintering at the population scale, we recorded the abundance of P. plantaginis in July 2012 using a categorical scale (for details on the methods, please see Tack & Laine 2014) in all 98 studied populations: (i) no infected plants, (ii) 1–10 infected plants, (iii) 10–100 infected plants, and iv) more than 1000 infected plants. This categorical scale facilitates the logistics of surveying infection across a large number of populations (Tack & Laine 2014). Mildew abundance in July 2012 was then used as the response variable for a weighted generalized linear model (glm) performed using r software (R Core Team 2013). The explanatory variables were the prevalence of Ampelomyces and the proportion of chasmothecia in the previous autumn at the population level, both estimated as described above. We used the quasi-Poisson family in the final model because of overdispersion of the data.
Results
ITS sequencing of Ampelomyces strains from Åland
The ITS sequences determined in the four infected dried Plantago leaves bearing Ampelomyces pycnidia, collected in Åland in September 2011, as well as those determined in six of seven strains isolated in the archipelago in 2013, were all identical (GenBank Accession nos: KC494280 and KM066091–KM066097) and approximately 97–98% similar to the ITS sequences of the Ampelomyces strains belonging to the ‘Podosphaera (sect. Sphaerotheca) clade’ identified by Park et al. (2010). The seventh strain isolated in 2013 belonged to the same clade but differed by a single SNP from the other Ampelomyces strains isolated in Åland.
Design of the molecular detection test
We obtained successful amplification with all the six Ampelomyces strains presented in the Table S1 using the newly designed primer pair AQ-F264/AQ-R462. However, limited growth on the plate and subsequent low DNA concentration prevented the use of the strain B124-a for the calibration curve. The standard curves were similar for the five remaining strains with slopes varying between −2.58 and −3.31 (Fig. S2, Supporting information). Correlation coefficients were high (0.966 < r < 0.990) indicating low interassay variability. Although there may be some differences of efficacy between the different Ampelomyces strains, the designed molecular test is likely to be applicable to any Ampelomyces spp.
When applying the molecular detection test to experimentally infected samples (Fig S3, Supporting information), we found that lesions showing visible evidence of Ampelomyces infection (categories: ‘few’, ‘moderate’, ‘lots’) had significantly lower Ct values than either of the control groups (‘none’, ‘control’) (Tukey's HSD, P < 0.006 in all cases). There was no significant difference in Ct values between ‘control’ samples and those that were exposed to, but did not grow, Ampelomyces (‘none’) (Tukey's HSD: P = 0.95). Based on these data, we set the Ct cut-off value for Ampelomyces presence/absence to 24 cycles, which is between the highest Ct obtained for samples bearing pycnidia and the lowest Ct found in samples not showing infection. Furthermore, 24 cycles is significantly lower than the amplification threshold of either control group (‘control’: one-sample t-test, t = 5.7101, d.f. = 4, P = 0.004, ‘none’: one-Sample t-test; t = 2.848, d.f. = 4, P = 0.046) and thus serves a suitable cut-off for determining the presence of established infections despite the potential for nontarget amplification mentioned above (Fig S3, Supporting information).
When the plasmid construction containing the ITS region of an Ampelomyces strain isolated in Åland was used in a dilution series prepared in DNA extracted from powdery mildew-infected P. lanceolata leaves, the standard curves showed that the lowest number of ITS copies leading to reliable amplifications was 104/μL (Average Ct = 24.08). The qPCR cut-off, Ct = 24, used for field samples, is very close to the cycle threshold for 104/μL. Detection of 103 or less ITS copies was not reliable due to the effect of the DNA extracted from Ampelomyces-free powdery mildew-infected P. lanceolata leaves. Therefore, this experiment also confirmed Ct = 24 to be a robust threshold for detecting Ampelomyces in field-collected samples.
Ampelomyces detection within the Åland archipelago
When applying the molecular test to the large-scale field sampling (951 samples), a straight shape instead of the expected sigmoid for the qPCR assay (evolution of the fluorescence through time) was obtained for 126 samples (13.2% of the data set). This suggests possible inhibition of the reaction, and these samples were removed before further analyses. The powdery mildew SNP assay was applied to all the samples. SNP genotyping assay worked for all the samples except 12 (1.3% of the data set), which were consequently removed from all the analyses as a negative result for such samples may be due to poor DNA quality. A total of 813 samples were finally used in this study.
We performed three replicates on each sample and found repeatability of 88.1%. We considered as positive the samples having Ct lower cycle threshold than 24 for all the amplifications performed.
Among the 813 analysed samples, 362 (44.5%) were positive for Ampelomyces. Between populations, prevalences varied between 0 and 89% in the 17 powdery mildew populations having sufficient sample size (minimum 22 samples, see Table S2, Supporting information). No spatial structure in Ampelomyces prevalence was evident at the metapopulation scale (Fig.1).
Relationship between Ampelomyces prevalence and the fungal host population dynamics
There was a strong positive relationship between Ampelomyces prevalence and the proportion of powdery mildew infections that were attributed to multiple strains (F1,325 = 5.68 and P = 0.02; Fig.2). We also detected significant variation in Ampelomyces prevalence among populations (log-likelihood ratio test for the random effect ‘population’:  = 81.43 and P < 0.001).
= 81.43 and P < 0.001).
Fig 2.
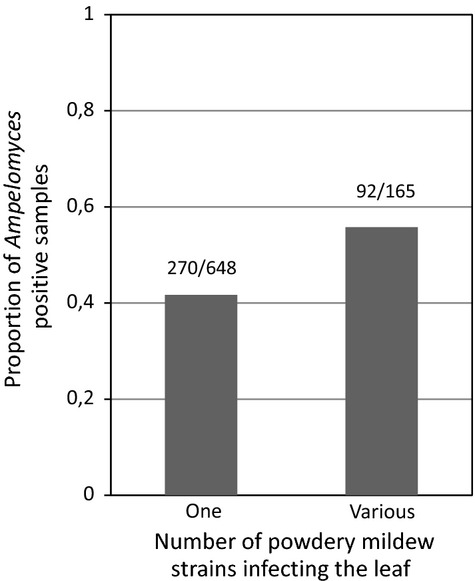
Relationship between the powdery mildew genetic diversity (one vs. multiple strains) and the probability of infection by Ampelomyces at the leaf level.
At the population scale (96 analysed populations), Ampelomyces prevalence in autumn did not significantly affect powdery mildew overwintering success measured as the abundance of P. plantaginis in the following spring (t = −0.462, P = 0.645). At the small spatial scale (1-m2 quadrat), we detected a significant negative impact of Ampelomyces prevalence in autumn 2011 on the overwintering success of the powdery mildew (F1,60 = 4.22 and P = 0.04; Fig.3). We did not detect an effect of chasmothecia on mildew overwintering (F1,60 = 0.47 and P = 0.50).
Fig 3.
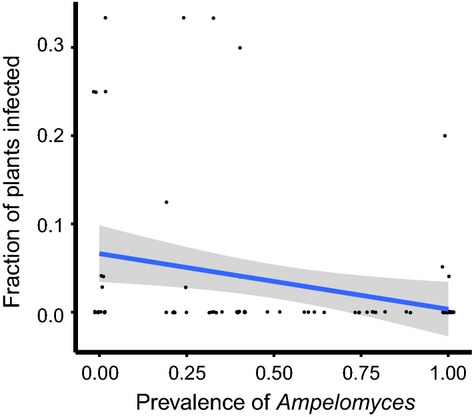
The impact of the prevalence of the hyperparasite Ampelomyces in autumn 2011 on the overwintering success (fraction of infected plants in July 2012) of its powdery mildew host Podosphaera plantaginis at a small spatial scale (1-m2 quadrat). Values are slightly jittered horizontally to prevent overplotting. The linear regression line and its 95% confidence interval (as implemented in stat_smooth, package ggplot2 within r software) are for visual illustration.
Discussion
Although microscopic observation allows Ampelomyces detection, molecular tools can improve the feasibility and accuracy of large-scale screening of fungal pathogens. For this purpose, we aimed to design a molecular detection test of Ampelomyces spp. using a quantitative molecular assay based on species-specific ITS amplification. Although some lower-quality samples had to be removed from the data set, the molecular test was successful for the majority of our samples (86.8%), and repeatability over three replicates was fairly good (88.1%). The newly developed molecular test was applied to experimentally infected samples revealing a good correlation between the qPCR results and microscopic observations of pycnidia (presence and abundance), thus verifying its utility in screening samples collected from nature.
Furthermore, amplification worked well for the six selected strains tested, despite high genetic variability in their ITS sequences (Pintye et al. 2012), suggesting that the test could be applied to any known lineage of Ampelomyces. This allows the test to be applied widely to assess Ampelomyces prevalence within any powdery mildew species, even without previous investigation of the genetic variability of the mycoparasite. Notably, different Ampelomyces lineages could coexist within the positive samples detected. Many more Ampelomyces strains from P. plantaginis samples within the Åland archipelago would have to be characterized genetically to get a comprehensive picture of the genetic diversity of this mycoparasite in the region. However, the data presented here (four pycnidial samples collected in 2011 and isolation of seven strains in 2013) suggest little genetic diversity as all the ITS sequences determined in these samples were highly similar (10 of 11 were identical) and closely related to those identified in the ‘Podosphaera (sect. Sphaerotheca) clade’ (Park et al. 2010). Preliminary data (partial sequencing of 25 randomly selected positive leaf samples, data not shown) are in accordance with this, as only one sample of 25 belonged to another Ampelomyces clade (Park et al. 2010). In contrast, Pintye et al. (2012) found high diversity of Ampelomyces strains in the grape powdery mildew and Kiss et al. (2011) showed the coexistence of different ITS haplotypes of Ampelomyces on the same powdery mildew-infected host plants. Further investigations on the genetic diversity of Ampelomyces from Åland would also require the use of rapidly evolving genetic markers (e.g. microsatellites, Kiss et al. 2011).
Our study revealed that Ampelomyces is a very common parasite within P. plantaginis populations, as 363 of the 813 (44.5%) samples analysed were found to be infected. Data from 17 intensively sampled populations (with at least 22 samples per population) indicated that Ampelomyces was present in the vast majority of the populations (only one of 17 had no positive sample). While the sampling was not optimal for detecting within-population variability (i.e. we measured multiple leaves on a few plants for each population), we did note striking variability in hyperparasite frequency between powdery mildew populations, which ranged between 0 and 89% (Table S2). Although the four initial samples of Ampelomyces were collected in the northern part of the Åland archipelago, subsequent analysis did not detect any spatial pattern in Ampelomyces prevalence (Fig.1). Very few studies have documented the prevalence of Ampelomyces in field-collected samples of powdery mildew hosts. Kiss (1998) documented the prevalence of Ampelomyces in field-collected samples representing 27 powdery mildew species infecting 41 host plant genera in Hungary and Romania and found Ampelomyces spp. in 4.3–68.8% of the studied samples, depending on the fungal host genera considered. This study also reported that the intensity of mycoparasitism (proportion of powdery mildew structures infected within sample) ranged from 0.15 to 65% in powdery mildew mycelia (Kiss 1998), while it remained lower than 4% in chasmothecia from grapevine powdery mildew populations from northern Italy (Angeli et al. 2009). The methodologies used in these studies were, however, very different from the one applied in the present study, and the results can therefore not easily be compared with each other.
We found that the dynamics of the powdery mildew host and the hyperparasite Ampelomyces were interrelated. First, the probability of an infected leaf sample to contain also Ampelomyces differed depending on whether the powdery mildew infection was caused by a single strain or multiple strains (i.e. coinfection). While disentangling the cause and effect may be difficult, this result supports the prediction that leaves infected by multiple powdery mildew genotypes have a higher probability to be infected by a powdery mildew strain bearing Ampelomyces, as a consequence of higher exposure. Secondly, we found an effect of Ampelomyces presence/absence on the overwintering success of the powdery mildew in the immediate surroundings (1-m2 quadrat) of an infected plant. Off-season survival and re-initiation of the epidemics is a critical but poorly understood stage of many plant pathogens, and high stochasticity is often invoked (Soubeyrand et al. 2009). The factors classically involved in the seasonal disease spread (pathogen genotype, host genotype and the environment) were recently shown to affect the overwintering stage in the pathosystem studied here (Tollenaere & Laine 2013; Tack & Laine 2014). Our results show that a hyperparasite may significantly affect the overwintering success of its phytopathogenic host, as we found a significant effect of the presence/absence of Ampelomyces on overwinter survival of the powdery mildew at the local scale (immediate neighbours of focal plants). However, no effect of Ampelomyces was detected on the overwintering success at the population level. This suggests that occurrence of Ampelomyces is highly aggregated within the powdery mildew populations, having a highly localized effect on its host. Other variables, such as climate, are likely to be important determinants of extinction at the population scale. However, the aggregated sampling scheme may also have contributed to the lack of a significant effect at the population scale. More intensive sampling across multiple populations would help resolve whether Ampelomyces could be a driver of extinctions at the population level in the P. plantaginis metapopulation dynamics.
A significant effect of the hyperparasite on its host's overwintering would be a case of local disease-induced extinction, a phenomenon that could more easily be explained with the presence of alternative hosts according to theoretical studies (de Castro & Bolker 2005). In our system, we frequently observed other plant species infected by powdery mildew in the P. lanceolata infected meadows (C. Tollenaere, pers. com.), and consequently, various powdery mildew species possibly hosting Ampelomyces are likely to co-occur with P. plantaginis. Some host specificity has been suggested to maintain Ampelomyces lineages (Park et al. 2010), but it has also been clearly shown that the same haplotypes of these hyperparasites can infect various powdery mildew species in the field (Kiss et al. 2011), and this may be the case in our system too. Testing other possible fungal hosts for presence/absence and diversity of Ampelomyces would determine the importance of other mycohosts for the interaction between P. plantaginis and Ampelomyces in the Åland metapopulation.
To conclude, we argue that the understanding of host-pathogen metapopulation dynamics may need to extend the scale of the study system into a tri-trophic spatial network and take into account the role of natural enemies of plant and animal pathogens. In addition to the fundamental knowledge gained on the regulation of natural population dynamics (see also Lafferty et al. 2008), studying such tri-(or more) trophic interactions in wild populations may lead to important applications such as the prevention of disease spread through biological control (for example Nuss 1992).
Acknowledgments
We are grateful to Hanna Susi, Elisa Metsovuori, Coong Lo and Aki Suzuki for their participation in sample collection and to Claire Morandin, Kalle Trontti and Caroline Tatard for their help with the design of the quantitative PCR assay. We thank Krista Raveala and Anna Huber for assistance in the laboratory work and Evgeniy Meyke for producing the map. The manuscript benefitted from helpful comments from three anonymous reviewers. This work was supported by funding from the Academy of Finland (Grant Nos 250444, 136393, 133499), European Research Council (PATHEVOL; 281517) to A-LL and the Hungarian Scientific Research Fund (OTKA, Grant NN 100415). G. M. Kovács acknowledges the support of a János Bolyai Research Scholarship of the Hungarian Academy of Sciences (MTA).
Biography
C.T. and A.L.L. designed the research. C.T., S.R.P., A.J.M.T. and L.K. contributed to field sampling. Genetic characterization of a subset of samples was performed by B.P. and M.Z.N. C.T. and B.P. designed the molecular detection test that was validated through experimental inoculations by S.R.P., and using an in vitro method designed by M.Z.N., G.M.K. and L.K. B.P. and H.S.M. applied the molecular test to the field sampling, producing data that were analyzed by C.T. and A.J.M.T. C.T. drafted the manuscript and coordinated the writing. All authors read, corrected and approved the final manuscript.
Data accessibility
DNA sequences: GenBank Accession nos: KC494280 and KM066091–KM066097. Data sets used for analyses at the leaf, plant and population scale: Dryad doi:10.5061/dryad.mv332.
Supporting Information
Additional supporting information may be found in the online version of this article.
Characteristics of the six Ampelomyces strains used to test the newly designed molecular detection method.
Table S2. Prevalence of Ampelomyces observed in the 17 powdery mildew populations presenting at least 22 samples.
Illustrations on the study system.
Fig. S2. Calibration curves obtained for five different Ampelomyces strains (see Table S1 for details on the strains).
Fig. S3. Results of the molecular detection test applied on powdery mildew samples experimentally infected by Ampelomyces.
Supplementary experiment assessing in vitro the reliability of Ct = 24 as a cut-off value.
References
- Anderson R, May R. Infectious Diseases in Humans: Dynamics and Control. Oxford: Oxford University Press; 1991. [Google Scholar]
- Angeli D, Pellegrini E, Pertot I. Occurrence of Erysiphe necator chasmothecia and their natural parasitism by Ampelomyces quisqualis. Phytopathology. 2009;99:704–710. doi: 10.1094/PHYTO-99-6-0704. [DOI] [PubMed] [Google Scholar]
- Antonovics J. Long-term study of a plant-pathogen metapopulation. In: Hanski I, Gaggiotti O, editors. Ecology, Genetics and Evolution of Metapopulations. Oxford: Elsevier Academic Press; 2004. pp. 471–488. [Google Scholar]
- Brusini J, Robin C, Franc A. Parasitism and maintenance of diversity in a fungal vegetative incompatibility system: the role of selection by deleterious cytoplasmic elements. Ecology Letters. 2011;14:444–452. doi: 10.1111/j.1461-0248.2011.01602.x. [DOI] [PubMed] [Google Scholar]
- de Castro F, Bolker B. Mechanisms of disease-induced extinction. Ecology Letters. 2005;8:117–126. [Google Scholar]
- Davelos AL, Jarosz AM. Demography of American chestnut populations: effects of a pathogen and a hyperparasite. Journal of Ecology. 2004;92:675–685. [Google Scholar]
- Füzi I. Natural parasitism of Uncinula necator cleistothecia by Ampelomyces hyperparasites in the south-western vineyards of Hungary. Acta Phytopathologica et Entomologica Hungarica. 2003;38:53–60. [Google Scholar]
- Glawe DA. The powdery mildews: a review of the world's most familiar (yet poorly known) plant pathogens. Annual Review of Phytopathology. 2008;46:27–51. doi: 10.1146/annurev.phyto.46.081407.104740. [DOI] [PubMed] [Google Scholar]
- Gulland FM, Albon SD, Pemberton JM, Moorcroft PR, Clutton-Brock TH. Parasite-associated polymorphism in a cyclic ungulate population. Proceedings of the Royal Society of London. Series B: Biological Sciences. 1993;254:7–13. doi: 10.1098/rspb.1993.0119. [DOI] [PubMed] [Google Scholar]
- Hanski I. Metapopulation Ecology. Oxford: Oxford University Press; 1999. [Google Scholar]
- Harding KC, Harkonen T, Caswell H. The 2002 European seal plague: epidemiology and population consequences. Ecology Letters. 2002;5:727–732. [Google Scholar]
- Harding KC, Begon M, Eriksson A, Wennberg B. Increased migration in host-pathogen metapopulations can cause host extinction. Journal of Theoretical Biology. 2012;298:1–7. doi: 10.1016/j.jtbi.2011.12.009. [DOI] [PubMed] [Google Scholar]
- Hess GR. Conservation corridors and contagious-disease – a cautionary note. Conservation Biology. 1994;8:256–262. [Google Scholar]
- Hudson PJ, Dobson AP, Newborn D. Prevention of population cycles by parasite removal. Science. 1998;282:2256–2258. doi: 10.1126/science.282.5397.2256. [DOI] [PubMed] [Google Scholar]
- Ito M, Takamatsu S. Molecular phylogeny and evolution of subsection Magnicellulatae (Erysiphaceae: Podosphaera) with special reference to host plants. Mycoscience. 2010;51:34–43. [Google Scholar]
- Jousimo J, Tack AJM, Ovaskainen O, et al. Ecological and evolutionary effects of fragmentation on infectious disease dynamics. Science. 2014;344:1289–1293. doi: 10.1126/science.1253621. [DOI] [PubMed] [Google Scholar]
- Kiss L. Natural occurence of Ampelomyces intracellular mycoparasites in mycelia of powdery mildew fungi. New Phytologist. 1998;140:709–714. doi: 10.1046/j.1469-8137.1998.00316.x. [DOI] [PubMed] [Google Scholar]
- Kiss L. Intracellular mycoparasites in action: interactions between powdery mildew fungi and Ampelomyces. In: Avery SV, Stratford M, Van West P, editors. Stress in Yeast and Filamentous Fungi. Dordrecht: British Mycological Society Symposia Series, Kluwer; 2008. pp. 37–52. [Google Scholar]
- Kiss L, Russell JC, Szentiványi O, et al. Biology and biocontrol potential of Ampelomyces mycoparasites, natural antagonists of powdery mildew fungi. Biocontrol Science and Technology. 2004;14:635–651. [Google Scholar]
- Kiss L, Pintye A, Kovács GM, et al. Temporal isolation explains host-related genetic differentiation in a group of widespread mycoparasitic fungi. Molecular Ecology. 2011;20:1492–1507. doi: 10.1111/j.1365-294X.2011.05007.x. [DOI] [PubMed] [Google Scholar]
- Lafferty KD, Allesina S, Arim M, et al. Parasites in food webs: the ultimate missing links. Ecology Letters. 2008;11:533–546. doi: 10.1111/j.1461-0248.2008.01174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine AL, Hanski I. Large-scale spatial dynamics of a specialist plant pathogen in a fragmented landscape. Journal of Ecology. 2006;94:217–226. [Google Scholar]
- Levins R. Some demographic and genetic consequences of environmental heterogeneity for biological control. Bulletin of the Entomological Society of America. 1969;71:237–240. [Google Scholar]
- Liang C, Yang J, Kovács GM, et al. Genetic diversity of Ampelomyces mycoparasites isolated from different powdery mildew fungi in China inferred from analyses of rDNA ITS sequences. Fungal Diversity. 2007;24:225–240. [Google Scholar]
- Littell RC, Milliken GA, Stroup WW, Wolfinger RD, Schabenberger O. SAS for Mixed Models. Cary, North Carolina: SAS Inst. Inc; 2006. 2nd edn. [Google Scholar]
- Milgroom MG, Cortesi P. Biological control of chestnut blight with hypovirulence: a critical analysis. Annual Review of Phytopathology. 2004;42:311–338. doi: 10.1146/annurev.phyto.42.040803.140325. [DOI] [PubMed] [Google Scholar]
- New LF, Matthiopoulos J, Redpath S, Buckland ST. Fitting models of multiple hypotheses to partial population data: investigating the causes of cycles in red grouse. American Naturalist. 2009;174:399–412. doi: 10.1086/603625. [DOI] [PubMed] [Google Scholar]
- Nicot PC, Bardin M, Dik AJ. Basic methods for epidemiological studies of powdery mildews: culture and preservation of isolates, production and delivery of inoculum, and disease assessment. In: Bélanger RR, Bushnell WR, Dik AJ, Carver LW, editors. The Powdery Mildews – a Comprehensive Treatise. St. Paul, Minnesota: The American Phytopathological Society; 2002. pp. 83–99. [Google Scholar]
- Nuss DL. Biological control of chestnut blight: an example of virus-mediated attenuation of fungal pathogenesis. Microbiology and Molecular Biology Reviews. 1992;56:561–576. doi: 10.1128/mr.56.4.561-576.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovaskainen O, Laine AL. Inferring evolutionary signals from ecological data in a plant-pathogen metapopulation. Ecology. 2006;87:880–891. doi: 10.1890/0012-9658(2006)87[880:iesfed]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Park MJ, Choi YJ, Hong SB, Shin HD. Genetic variability and mycohost association of Ampelomyces quisqualis isolates inferred from phylogenetic analyses of ITS rDNA and actin gene sequences. Fungal Biology. 2010;114:235–247. doi: 10.1016/j.funbio.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Pintye A, Bereczky Z, Kovács GM, et al. No indication of strict host associations in a widespread mycoparasite: grapevine powdery mildew (Erysiphe necator) is attacked by phylogenetically distant Ampelomyces strains in the field. Phytopathology. 2012;102:707–716. doi: 10.1094/PHYTO-10-11-0270. [DOI] [PubMed] [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. . URL http://www.R-project.org/ [Google Scholar]
- Robin C, Lanz S, Soutrenon A, Rigling D. Dominance of natural over released biological control agents of the chestnut blight fungus Cryphonectria parasitica in south-eastern France is associated with fitness-related traits. Biological Control. 2010;53:55–61. [Google Scholar]
- Schmid-Hempel P. Evolutionary Parasitology – The Intergrated Study of Infectionsm Immunology, Ecology and Genetics. New York: Oxford University Press; 2011. [Google Scholar]
- Schoch CL, Seifert KA, Huhndorf S, et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6241–6246. doi: 10.1073/pnas.1117018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simberloff D, Cox J. Consequences and costs of conservation corridors. Conservation Biology. 1987;1:63–71. [Google Scholar]
- Smith DL, Ericson L, Burdon JJ. Co-evolutionary hot and cold spots of selective pressure move in space and time. Journal of Ecology. 2011;99:634–641. [Google Scholar]
- Sokolow S. Effects of a changing climate on the dynamics of coral infectious disease: a review of the evidence. Diseases of Aquatic Organisms. 2009;87:5–18. doi: 10.3354/dao02099. [DOI] [PubMed] [Google Scholar]
- Soubeyrand S, Laine AL, Hanski I, Penttinen A. Spatiotemporal structure of host-pathogen interactions in a metapopulation. American Naturalist. 2009;174:308–320. doi: 10.1086/603624. [DOI] [PubMed] [Google Scholar]
- Soubeyrand S, Tollenaere C, Haon-Lasportes E, Laine A-L. Regression-based Ranking of pathogen strains with respect to their contribution to natural epidemics. PLoS One. 2014;9:e86591. doi: 10.1371/journal.pone.0086591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tack AJM, Laine AL. Ecological and evolutionary implications of spatial heterogeneity during the off-season for a wild plant pathogen. New Phytologist. 2014;202:297–308. doi: 10.1111/nph.12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollenaere C, Laine AL. Investigating the production of sexual structures in a plant pathogen reveals unexpected self-fertility and genotype-by-environment effects. Journal of Evolutionary Biology. 2013;26:1716–1726. doi: 10.1111/jeb.12169. [DOI] [PubMed] [Google Scholar]
- Tollenaere C, Susi H, Nokso-Koivisto J, et al. SNP Design from 454 sequencing of Podosphaera plantaginis transcriptome reveals a genetically diverse pathogen metapopulation with high levels of mixed-genotype infection. PLoS One. 2012;7:e52492. doi: 10.1371/journal.pone.0052492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompkins DM, Dobson AP, Arneberg P. Parasites and host population dynamics. In: Hudson PJ, Rizzoli A, Grenfell BT, Heesterbeek H, Dobson AP, et al., editors. The Ecology of Wildlife Diseases. Oxford: Oxford University Press; 2002. pp. 45–62. [Google Scholar]
- Vredenburg VT, Knapp RA, Tunstall TS, Briggs CJ. Dynamics of an emerging disease drive large-scale amphibian population extinctions. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9689–9694. doi: 10.1073/pnas.0914111107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characteristics of the six Ampelomyces strains used to test the newly designed molecular detection method.
Table S2. Prevalence of Ampelomyces observed in the 17 powdery mildew populations presenting at least 22 samples.
Illustrations on the study system.
Fig. S2. Calibration curves obtained for five different Ampelomyces strains (see Table S1 for details on the strains).
Fig. S3. Results of the molecular detection test applied on powdery mildew samples experimentally infected by Ampelomyces.
Supplementary experiment assessing in vitro the reliability of Ct = 24 as a cut-off value.
Data Availability Statement
DNA sequences: GenBank Accession nos: KC494280 and KM066091–KM066097. Data sets used for analyses at the leaf, plant and population scale: Dryad doi:10.5061/dryad.mv332.