

Abstract
Netupitant is a new, selective NK1 receptor antagonist under development for the prevention of chemotherapy-induced nausea and vomiting. Two studies were conducted to evaluate the brain receptor occupancy (RO) and disposition (ADME) of netupitant in humans. Positron emission tomography (PET) imaging with the NK1 receptor-binding–selective tracer [11C]-GR205171 was used to evaluate the brain penetration of different doses of netupitant (100, 300, and 450 mg) and to determine the NK1-RO duration. A NK1-RO of 90% or higher was achieved with all doses in the majority of the tested brain regions at Cmax, with a long duration of RO. The netupitant minimal plasma concentration predicted to achieve a NK1-RO of 90%, C90%, in the striatum was 225 ng/mL; after administration of netupitant 300 mg, concentrations exceeded the C90%. In the ADME study, a single nominal dose of [14C]-netupitant 300 mg was used to assess its disposition. Absorption was rapid and netupitant was extensively metabolized via Phase I and II hepatic metabolism. Elimination of >90% was predicted at day 29 and was principally via hepatic/biliary route (>85%) with a minor contribution of the renal route (<5%). In conclusion, these studies demonstrate that netupitant is a potent agent targeting NK1 receptors with long lasting RO. In addition, netupitant is extensively metabolized and is mainly eliminated through the hepatic/biliary route and to a lesser extent via the kidneys.
Keywords: NEPA, netupitant, palonosetron, metabolism, receptor occupancy
Nausea and vomiting represents one of the most feared side effects of chemotherapy.1,2 Chemotherapy-induced nausea and vomiting, CINV, is described as acute, when it occurs in the first 24 hours, and as delayed when it occurs from 25 to 120 hours after chemotherapy administration.3,4 Patient quality of life can be severely impaired when CINV is not correctly prevented and this can affect the completion of the chemotherapy plan.2,3,5 Serotonin and substance P have been identified as the major neurotransmitters involved in the regulation of CINV. It has been shown that serotonin plays a predominant role in the acute phase through the activation of the 5-HT3 receptors, while the delayed phase is mainly mediated by the substance P through the activation of the NK1 receptors.2,6,7
Several medical associations including the American Society of Clinical Oncology (ASCO), the National Comprehensive Cancer Network (NCCN), and the Multinational Association of Supportive Cancer Care (MASCC)/European Society of Medical Oncology (ESMO), have developed international antiemetic guidelines aimed at identifying the most effective antiemetic prophylaxis associated with a higher control of nausea and vomiting. To date, the combination of an NK1 receptor antagonist (RA) with a 5-HT3 RA and a corticosteroid is considered by all major associations as the recommended antiemetic regimen for patients receiving highly emetogenic chemotherapy.8–10
Although recent progress has been made into defining new treatments and regimens for CINV, delayed CINV and, in particular, delayed nausea, are a persistent problem.4,11–13 The development of new antiemetics with long lasting activity is therefore warranted by the medical community.
Netupitant (2-(3,5-Bis-trifluromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide) is a new, highly-selective antagonist of the human NK1 receptor14 which is being developed as an oral fixed dose combination with palonosetron, (NEPA, 300 mg netupitant plus 0.5 mg palonosetron). Palonosetron is a clinically superior15–18 and pharmacologically distinct 5-HT3 RA.19,20 A synergistic effect between netupitant and palonosetron has recently been described by Stathis et al.21 NEPA provides a unique single-dose treatment for acute and delayed CINV targeting the two major emetic pathways. Netupitant, as NK1 RA, is predicted to be active especially in the central nervous system2,6,7 and therefore the evaluation of the brain penetration and duration of RO is relevant in the antiemetic development program. Netupitant distribution, metabolism and excretion were evaluated in the second study.
Materials and Methods
Positron Emission Tomography (PET) Study
Study Design and Subjects
This was a single dose, randomized, open-label, PET study investigating the degree of occupancy of NK1 receptors in specific regions of the human brain after administration of oral doses of netupitant in healthy male subjects. The study was performed in accordance with the principles stated in the Declaration of Helsinki and subsequent revisions. The study protocol and informed consent form were approved by an independent ethics committee before enrollment of any subject into the study.
The study included six male subjects randomized to receive 100, 300, or 450 mg of netupitant (each dose was to be administered to two subjects). The site screened the volunteers according to the inclusion and exclusion criteria outlined in the study protocol. All subjects signed a document of informed consent before entering the trial. Subjects were in good health as assessed by clinical examination, were nonsmokers or had refrained from smoking and other nicotine containing products for the last 6 months before dose administration, and had no history of drug or alcohol abuse.
The subjects attended a screening visit, a PET baseline visit, a treatment period with up to 5 post-dose PET scans and a follow-up visit. All subjects were males and their mean age was 22.3 years (range: 20–25 years) and the mean BMI was 23.9 kg/m2 (range: 21–28 kg/m2).
Subjects were admitted to the investigational site on day 1. Urine drug screen, alcohol breath test and recording of blood pressure and pulse rate were performed before dose administration. Netupitant was administered as a single oral dose (100, 300, or 450 mg capsule formulation according to the subject's treatment allocation). Subjects fasted overnight (for approximately 10 hours) before dose administration and continued to fast until completion of the first post-dose PET scan. Subjects also had to fast 4 hours before each of the additional 4 post-dose PET scans. Samples for the determination of netupitant plasma concentrations were collected pre-dose, post dose at 1, 2, 3, 4, 4.5, 5, 5.5, and 12 hours and immediately before (6, 24, 48, 72, and 96 hours) and after the PET scan (7, 25, 49, 73, and 97 hours). [11C]-GR205171 was administered as an intravenous bolus injection at baseline, and at approximately 6, 24, 48, 72, and 96 hours after dosing with netupitant. The injections were followed by a 60-minute PET scan.
Concomitant medications and adverse events (AEs) were recorded during the treatment period. AEs were assessed by the study personnel via periodic observation and queries to the study subjects. Causality and intensity of AEs was determined by the Principal Investigator at the study site. The AE reporting period for this trial began upon randomization and ended at the follow-up visit. During the course of the study, all AEs, irrespective of the relatedness to the investigational medicinal product, were to be recorded on the AE pages of the Case Report Form (CRF). The investigator was responsible for ensuring that correct information concerning all AEs was included on the appropriate CRF pages.
The subjects were to be discharged from the investigational site when the 24 hours PET scan had been completed and then had to return for additional PET scans, PK samples, and safety assessments up to 4 days post-dose.
Radioactive Agent
The compound GR205171 is a highly selective, high affinity NK1 RA developed by GlaxoSmithKline.22 The ligand [11C]-GR205171 has been evaluated as a potential PET tracer to characterize NK1 receptor binding23 and this tracer has been extensively used at Uppsala Imanet in clinical studies, both related to drug development and scientific research.24 An analogue of GR205171 has also been labelled with 18F, called [18F]-SPA-RQ,25 and was used in clinical studies to determine central NK1 receptor occupancy (RO) after multiple dosing of the NK1 RA aprepitant26 as well as NK1 receptor distribution and quantification and age and gender effects on NK1 receptor availability27,28 in the human brain. The radioligand [11C]-GR205171 was synthesized by Uppsala Imanet AB, according to previously published methods23 and local quality control procedures. Tracer production was performed with the permission of the Medical Products Agency (MPA) and the Swedish Radiation Protection Institute.
Pharmacokinetic Analysis
The following pharmacokinetic (PK) parameters were derived for netupitant: Cmax, the maximum observed plasma concentration; AUC0–t, area under the plasma concentration versus time curve from time zero to the last quantifiable sampling time point (t); and tmax, the time at which the maximum plasma concentration was observed. The PK calculations, based on plasma concentrations and actual sampling times, were performed by means of a non-compartmental analysis using model 200 (extravascular administration) in WinNonlin Professional Edition Version 4.1.b (Pharsight Corporation, Mountain View, CA).
PET Scanning
The PET measurements were carried out at Uppsala Imanet AB. The subject was placed on the couch of the PET-camera, with the subject's head suitably fixed to prevent excessive movement. The head of the subject was placed in the scanner with transaxial planes orientated in parallel to the orbito-metal line, using pen markings to position the subject in the scanner to ensure consistency within scans. An indwelling catheter was inserted in the arm of the subject (cubital vein) to inject [11C]-GR205171 intravenously.
Dynamic PET data were acquired in 3D mode for all subjects using either of the two identical ECAT EXACT HR+ (Siemens/CTI) scanners, which have a 15.5 cm axial field of view and generate 63 transaxial planes with a distance of 2.46 mm. The tomographs have a reconstructed spatial resolution of about 5–6 mm after image reconstruction. A transmission scan correcting for attenuation of emitted radiation by skull and tissues was acquired during 10 minutes using three retractable 68Ge line sources. The injected radioactivity was not >400 MBq (max 20 µg cold substance) per investigation.
Data were acquired over 60 minutes divided into 17 successive time frames (4 × 1 minutes, 3 × 2 minutes, and 10 × 5 minutes). The pre-selected camera protocol programs were started simultaneously with the intravenous bolus injection of the PET-tracer. The raw data collected was corrected for decay, scatter, random coincidences, and (measured) photo attenuation and reconstructed using a filtered back projection algorithm with a Hanning filter. A time series of tomographic images was generated and these images show the quantitative distribution and time course of the tracer in contiguous planes.
The 6 PET scans were performed at the following times: baseline visit (within 7 days before dose administration), and 6, 24, 48, 72, and 96 hours post dosing.
Following the reconstruction of the data, dynamic PET images were converted to Scanditronix/IDA format using software developed by Uppsala Imanet (Uppsala Universitets PET-centrum). Images were realigned to correct for positioning between baseline and post dosing scans and for movement during scans.29 Realignment of the data allowed the same outlined regions to be used for all scans.
Regions of interests (ROIs) were delineated bilaterally to represent, lateral and medial temporal cortex, striatum, occipital cortex, frontal cortex anterior cingulate and cerebellum (2–4 consecutive slices). For the sets of ROIs, dynamic time-activity curves (TACs) data were generated for each of the PET scans. Although [11C]-GR205171 is a reversible ligand of the NK1 receptors, the binding could be considered as irreversible during a time window of up to 60 minutes.23,26 Therefore the TAC data of the tracer baseline and all post dosing tracer experiments were analyzed using a reference Patlak model.30 This model approach is based on the presence of a region without specific binding of the radioligand (reference region). In this case the cerebellum is assumed to have no receptors that can bind either [11C]-GR205171 or netupitant.23,26,27,31,32 The free distribution volume of [11C]-GR205171 is assumed to be the same in the reference region (cerebellum) and in any other ROI.
The percent of NK1 receptor occupancy (NK1-RO, %) was then calculated as the relative difference between the net uptake rate (Ki) estimated from the reference Patlak model for the baseline (Kibase) and post-treatment (KiPD) experiments:
Pharmacodynamic/Pharmacokinetic Relationship (PD/PK)
The possible relationship between the netupitant plasma concentration and % of RO in the different brain regions (lateral and medial temporal cortex, striatum, occipital cortex, and frontal cortex and anterior cingulate) was evaluated through exploratory PK/PD modeling using WinNonlin Professional Edition Version 4.1.b (Pharsight Corporation). The data were fitted to the Sigmoid Emax model (model 105):
where Emax is the maximal RO at the NK1 receptors, ConcPET is the mean plasma concentration of netupitant (mean of concentration measured immediately before and after a scan) at the PET scans, EC50 is the plasma concentration at which 50% of Emax is induced and γ is a slope parameter reflecting the shape (including the slope) of the curve.
The equation was fitted to the corresponding occupancy values and plasma concentrations for all subjects simultaneously. Each data point was treated as a separate observation and one EC50 value for each brain region was estimated for NK1 receptors. One Emax value for each brain region was estimated. For anterior cingulate and lateral temporal, medial temporal and frontal cortex, the upper boundary for Emax was set to 100%.
The concentration giving RO of at least 90% (C90%) was calculated from the Sigmoid Emax equation, and the dose corresponding to this concentration was estimated.
Drug Metabolism (ADME) Study
Study Design and Subjects
This was a single dose open-label study investigating the mass balance recovery, pharmacokinetics, metabolite profile of a nominal dose of 300 mg [14C]-Netupitant.
The clinical study was performed at Quotient Clinical Ltd. (Nottingham, UK) in accordance with Good Clinical Practice and the principles of the 1996 version of the Declaration of Helsinki. The protocol was reviewed and approved by the Independent Ethics Committee and the Medicine and Healthcare Products Regulatory Agency (MHRA). The proposed radioactive dose was approved by the Administration of Radioactive Substances Advisory Committee (ARSAC). Written consent was obtained from all subjects before any protocol-specific procedures.
The study included six healthy white male subjects, between the ages of 32 and 56 years and with a mean body mass index of 25.5 kg/m2. Five subjects completed the study (one subject withdrew consent for personal reasons at day 8, and was included in the PK population). Subjects were in good health as assessed by clinical examination, nonsmokers, with no history of drug or alcohol abuse within the past 2 years, and were on no other medications at the time of the study and with no prescribed medications within 14 days of the study commencing.
Subjects were fasted overnight before netupitant administration. All subjects received a single oral nominal dose of 300 mg of [14C]-netupitant (up to 2.22 MBq) suspension.
After oral administration, blood samples for the measurement of total radioactivity in whole blood and plasma were collected at pre-dose and 1, 2, 3, 4, 4.5, 5, 5.5, 6, 8, 12, 24, 48, 72, 96, 120, 144, 168, 192, 240, 288, and 336 hours after netupitant administration. Urine samples were collected pre-dose, 0–4, 8–12, 12–24, 48–72, 72–96, 96–120, 120–144, 144–168, 168–192, 192–216, 240–264, 264–288, 288–312, 312–336 hours post-dose. Additional collections between 672 and 696 hours post-dose were performed. At the end of each collection period, samples were combined per subject and the net weight was recorded. Fecal samples were collected at pre-dose, 0–24, 24–48, 48–72, 72–96, 96–120, 120–144, 144–168, 168–192, 192–216, 216–240, 240–264, 264–288, 288–312, and 312–336 hours post-dose. Additional collections between 456 and 480 hours and between 672 and 696 hours post-dose were performed.
Radioactive Agent
[14C]-netupitant was manufactured and supplied by Quotient Chemistry & Metabolism, Rushden. The formulation for clinical use, [14C]-netupitant oral suspension, was manufactured by Quotient Clinical Ltd., in accordance with the investigational medicinal product (IMP) dossier and the approved batch record. The IMP was provided as an oral suspension containing a nominal dose of 300 mg [14C]–netupitant with not >2.2 MBq (60 µCi) of radioactivity; each dosage unit for administration was supplied in an amber glass bottle with a screw-cap lid.
Assay of Total Radioactivity
Radioactivity in whole blood and fecal homogenate samples was determined after combustion in oxygen using an automatic sample oxidiser. The combustion products were absorbed into CarboSorb E+ and mixed with the scintillator cocktail PermaFluor E+ for measurement of radioactivity. Radioactivity in liquid samples was quantified directly by liquid scintillation counting, LSC, using a liquid scintillation counter with automatic external standard quench correction. Samples were mixed with scintillant and counted. Detected counts per minute were converted to disintegrations per minute (dpm) using quench correction. The quench curves were prepared using standards purchased from Perkin Elmer Life and Analytical Sciences and were prepared from stock solutions that were calibrated against National Institute of Standards and Technology Reference Materials. The validity of the curves was checked throughout the experiments. The low limit of quantification using LSC was taken as twice the background dpm value for samples of the same type.
Netupitant, M1, M2, and M3 Quantification
Concentrations of netupitant and its major metabolites M1, M2, and M3, were determined using a simultaneous, validated liquid chromatography-tandem mass spectrometry (LC–MS/MS) method.33 The lower limit of quantification (LLOQ) was approximately 2 ng/mL for netupitant and its metabolites. Precision and accuracy were reported to be 5–8% and −16% to 7%, respectively, for all analytes.
Pharmacokinetic Analysis
Actual blood collection times and the actual dose administered were used for all pharmacokinetic calculations. Standard noncompartmental methods were used to derive pharmacokinetic parameters using WinNonlin (version 5.1; Pharsight). Maximum plasma concentration (Cmax) and time of Cmax (tmax) were taken directly from the pharmacokinetic concentration–time data. Area under the plasma concentration–time curve (AUC) was calculated using the linear trapezoidal method, to the last sampling time t (AUC0–t), and extrapolated to infinity (AUC0–inf) by the addition of the concentration at the last sampling time divided by the terminal elimination rate constant, λz. Half-life was determined as ln (2) divided by λz. Total radioactivity concentrations are reported in terms of mass equivalents of netupitant per mL for plasma and urine, and per gram for whole blood.
Mass balance and cumulative recovery of total radioactivity in urine, feces, and total (urine and feces) were calculated.
For all subjects, incomplete mass balance recovery from excreta (73%) was observed at the end of the planned clinical residency period (336 hours). The cumulative urine and fecal recovery data were therefore used to extrapolate the excretion profile. Additional 24 hours collection periods at day 20–21 and at day 29–30 were needed to follow the complete profile of netupitant excretion.
Results
PET Study
Three netupitant doses (100, 300, and 450 mg) were administered to six healthy subjects. The pharmacokinetics (PK) of the three different doses of netupitant were evaluated to identify the minimal dose of netupitant requested to reach the 90% RO (C90%). The [11C]-GR205171 tracer was used as sensitive and specific tracer to evaluate the level of RO before and after netupitant administration.
The mean Cmax and mean AUC0–t of netupitant varied from 185.9 to 615.4 ng/mL and from 4,016 to 14,638 h ng/mL, respectively, after single oral dosing of 100–450 mg. The exposure parameters for netupitant, mean AUC0–t and Cmax, were proportional to dose and were three times higher in the 300 mg group when compared to the 100 mg group and 1.1 times higher in the 450 mg group compared to the 300 mg dose group. The mean plasma concentrations at 6, 24, 48, 72, and 96 hours are also reported. There was no trend for a consistent change in median tmax, which ranged from 5.56 to 5.74 hours (Figure1).
Figure 1.
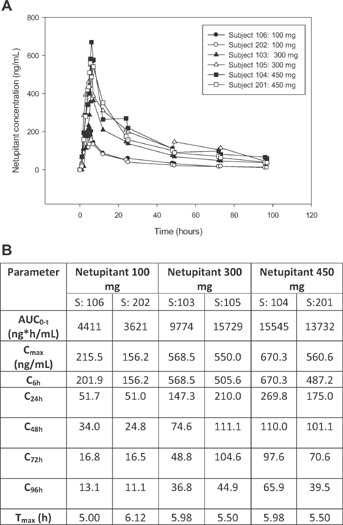
(A) Individual concentrations of netupitant for each dose group and (B) summary (mean, SD) of netupitant pharmacokinetic parameters and netupitant plasma concentration at 6 (C6h), 24 (C24h), 48 (C48h), 72 (C72h), 96 hours (C96h).
Based on the previous PK studies, netupitant was expected to achieve maximum concentrations at approximately 6 hours after administration and this was confirmed in the present study. The PET scan at 6 hours demonstrated that the mean NK1-RO of 90% or higher was achieved for occipital and frontal cortex for all investigated doses as well as for striatum (for 300 and 450 mg netupitant) and anterior cingulate (for 100 and 450 mg netupitant) (Figure2).
Figure 2.
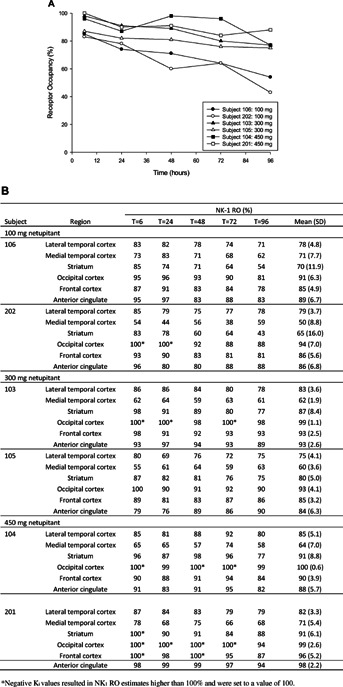
(A) Individual netupitant NK1 receptor occupancy (NK1-RO) in the striatum at different time and doses; (B) Individual netupitant NK1-RO in all tested brain regions at 6, 24, 48, 72, and 96 hours. Data are mean (SD), CV%.
To evaluate the duration of netupitant receptor binding, the tracer was administered at 24, 48, 72, and 96 hours after the antiemetic administration. The data interpretation was performed through the Patlak's graphical analysis using the cerebellum as reference region which showed a good fit of the linear model to the data during the time interval 15–60 minutes after administration of [11C]-GR205171.
At baseline conditions the average estimated net uptake rate for [11C]-GR205171 reflected differences in NK1 receptor density in the various brain regions. These values decreased in the following order: striatum > occipital cortex > frontal cortex and anterior cingulate > lateral temporal cortex > medial temporal cortex > cerebellum. In general, those results are in line with results from autoradiography studies in human post mortem brain27 as well as results from clinical studies [26, unpublished data Uppsala Imanet] thus confirming specificity of the binding.
A comparison of the results by dose groups (100, 300, and 450 mg) showed a general but low increase in NK1-RO with increasing dose. There was also a dose dependent NK1-RO decline over time, which was more pronounced in the striatum compared to the other regions of interest. Netupitant 300 mg demonstrated to have a long lasting RO especially in the anterior cingulate, 89.5%, frontal cortex, 89.5%, and occipital cortex, 94%, up to 96 hours after administration (Figure2).
The relationship between the NK1-RO and netupitant plasma concentrations at 6, 24, 48, 72, and 96 hours after administration of netupitant was evaluated for all brain regions. The maximum NK1-RO (Emax) was estimated to be higher than 90% in four of the six studied brain regions, that is, striatum, occipital cortex, frontal cortex and anterior cingulate. The highest Emax was obtained for the occipital cortex (98.9%, CV: 1.4%), whereas the medial temporal cortex had the lowest Emax (66.0%, CV: 8.8%). The EC50 determined for striatum was 10.2 ng/mL (CV: 14.5%). The EC50 values for the cortex areas were generally estimated with low precision (CV higher than 50%) and should therefore be interpreted with caution (Figure3).
Figure 3.
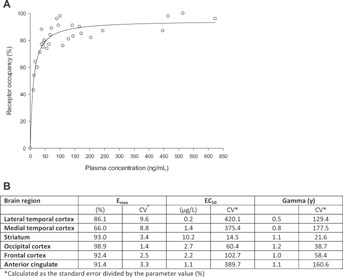
(A) Relationship between NK1 receptor occupancy (%) and netupitant plasma concentrations (PD/PK) in striatum at 6, 24, 48, 72, and 96 hours after administration of netupitant; (B) Summary of PK/PD variables estimated from the sigmoid Emax model in all brain regions.
Due to the precision of the EC50 and γ estimates, further calculations were only performed for the striatum. Based on the sigmoid Emax equation, the netupitant C90% in striatum was 225 ng/mL. As shown in Figure1, among the three tested netupitant doses, the lowest reaching and exceeding the C90% was the 300 mg and has been therefore investigated in further studies. As estimate, the minimal netupitant dose achieving the C90% was to be between the 100 and 300 mg.
Netupitant, given as single doses of 100–450 mg, was safe and well tolerated. There were four treatment emergent AEs (headache, nausea, nasopharyngitis and epistaxis), which were reported by four different subjects and only one of these events (nausea in one subject dosed with 450 mg netupitant) was considered to be possibly treatment-related.
Drug Metabolism Study (ADME)
After oral administration of a 300 mg [14C]-netupitant nominal dose to six healthy male subjects, netupitant was rapidly absorbed. Individual peak plasma netupitant concentrations, ranging from 99.2 to 517 ng/mL, were observed at 2–5.5 hours post-dose (tmax). Netupitant was shown to undergo extensive metabolism, forming both phase I and phase II metabolites (Figure 4, Phase I metabolites only shown). Phase I metabolites observed included those formed through N-demethylation (mono and bis), mono and di-hydroxylation, N-oxidation, desaturation, N-formylation, oxidation to a carbonyl group and to a carboxylic acid. Intermediate metabolites in the 1-methylpiperazine degradation pathway to the further oxidized 6-amino-pyridinyl derivatives were also observed. Phase II metabolites included those formed by glucuronidation and conjugation to a hexose (C6 sugar) group. A glucuronic acid conjugate of the acid ½ molecule of netupitant was also observed in urine.
Figure 4.
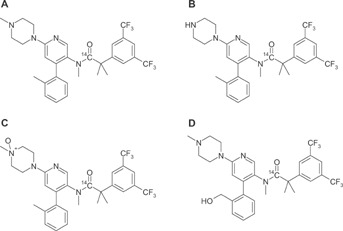
Molecular structures of: (a) netupitant, (b) M1, (c) M2, and (d) M3.
Total radioactivity in whole blood was detectable in sporadic samples in five of the six subjects. Since concentrations were below the limit of detection at the majority of time points, the parameters derived from these data cannot be interpreted (maximum concentrations of total radioactivity in subjects exhibiting detectable concentrations occurred at a median tmax of 5.5 hours).
Figure 5 presents the mean total radioactivity concentration time profile. Mean plasma netupitant/plasma radioactivity ratios ranged from 0.13 to 0.49 over 96 hours post-dose. The ratios were time dependent with values decreasing gradually beyond 24 hours post-dose. Exposure (AUC0–336) and Cmax geometric mean values for netupitant were approximately 34% and 41% of the exposure and Cmax geometric mean values for total radioactivity; this difference is attributed to metabolite species. A difference between the lower limit of detection of total radioactivity measured by LSC and the limit of quantification of netupitant analyzed by LC–MS/MS resulted in netupitant concentrations that were measurable at later sampling time points compared to total radioactivity.
Figure 5.
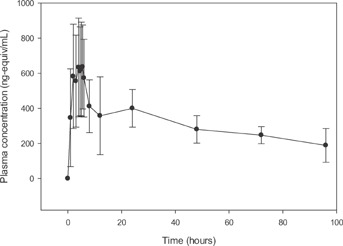
Mean plasma total radioactivity concentration versus time profile for 300ï¿1/21/2mg [14C]Netupitant.
Further analysis of the plasma samples for the metabolites M1, M2, and M3 indicated that metabolites were detectable 1 hour after netupitant administration. On average, exposure to these metabolites was equivalent to 29%, 14%, and 33%, respectively, of the systemic exposure to netupitant; thus, these results confirm that M1, M2, and M3 are the major plasma circulating metabolites of netupitant since they each account for >10% of parent-drug related exposure.
Approximately half the administered dose of radioactivity was recovered in urine and feces within 120 hours of dosing. At 336 hours total radioactivity recovered in all samples accounted for approximately 70% of the dose. Since recovery of radioactivity was not complete and excretion of radioactive material was still present, two supplementary sample collections were performed between 456–480 hours and 672–696 hours.
Including the extrapolated values for the periods 336–456 hours and 480–672 hours (based on the assumption that the excretion was proceeding at a steadily decreasing rate), the total drug-related material to have been excreted by 696 hours post-dose via the feces was estimated to be 86.49% and a mean of 4.75% of drug-related material was estimated to have been excreted in the urine for the same time period. These data indicate that the hepatic/biliary route, rather than renal clearance, is the major elimination route for the parent drug and its metabolites.
In this study one subject reported a total of 5 AEs, all of which were mild in severity and had resolved by the end of the study. Four of these events (abdominal pain, diarrhea, dyspepsia and nausea) were considered either possibly or probably related to the investigational medicinal product and one event (nasopharyngitis) was considered unlikely to be related. In general there were no clinically significant findings in clinical laboratory assessments, vital signs parameters, ECG measurements or physical examinations. The treatment was well tolerated.
Overall Safety
Across studies, all AEs were mild in intensity (headache, nausea, dyspepsia, abdominal pain, and diarrhea). In general there were no clinically significant findings in clinical laboratory assessments, vital sign parameters, ECG measurements, or physical examinations. The treatments were well tolerated.
Discussion
CINV still represents an issue impacting patient quality of life, especially in the delayed phase.4,11–13 Netupitant is a new, highly-selective antagonist of the human NK1 receptor under development as a fixed dose combination with palonosetron (NEPA, netupitant, 300 mg, and palonosetron, 0.5 mg). NEPA is the only antiemetic targeting two critical pathways associated with CINV: the serotonin and substance P mediated mechanisms.2,6,7 The use of NEPA represents a convenient single dose capsule for the prevention of nausea and vomiting in both the acute and delayed phases and would represent a valid alternative for CINV prevention.
In the first study (PET Study), the measure of the brain NK1-RO occupancy was obtained through PET analysis. In this study the anterior cingulate, the frontal, occipital, medial, and temporal cortex and the striatum were identified as the brain regions of interest to be analyzed. Among these, the striatum represents the area with the highest expression of NK1 receptor in human34 and therefore was used in the present study to establish the relationship between netupitant concentration and NK1-RO. Although the role of these regions in the modulation of the emetic reflex is not known, the RO represents a valuable tool for establishing the degree and duration of binding of netupitant and therefore the results of all regions were reported in the present study. A direct link between duration of NK1-RO and efficacy of the NK1 RA in the acute or delayed CINV phases has not been established, however, based on the literature, 90% of NK1-RO at Tmax in the striatum was identified as the threshold for the antagonistic activity of netupitant.26,32,34 This value was also predicted to be associated with the maximal antiemetic and antidepressant efficacy of aprepitant26,35,36 the first NK1 RA available on the market. Ninety percent of RO at 6 hours (corresponding approximately to the Cmax) after the 300 mg netupitant dose was reached in the frontal and occipital cortex and in the striatum, 86% of RO was reported for the anterior cingulate, 83% and 58.8% for the lateral and medial temporal cortex, respectively. Netupitant 300 mg demonstrated a long lasting RO especially in the anterior cingulate, 89.5%, frontal cortex, 89.5%, and occipital cortex, 94%, up to 96 hours after administration. Based on the sigmoid Emax model, netupitant was shown to have a C90% of 225 ng/mL. Based on this data the 300 mg netupitant represented the lowest dose among the ones tested reaching the 90% of RO in the striatum and was identified as the potential dose to be associated to palonosetron 0.5 mg. The validity of the PET analysis in the early development phase was confirmed in the NEPA phase II dose ranging clinical study were the 300 mg netupitant dose was selected as the preferred dose to be associated with palonosetron 0.5 mg and in the pivotal phase III studies where the high efficacy and safety of NEPA were confirmed.37–39
A comparison of these study results with those presented in the literature for aprepitant, fosaprepitant, and casopitant demonstrates similarities in terms of region of interest and results. A level of 90% of RO was reached by aprepitant in the striatum, 24 hours after the last dose, following 14 days of treatment with 100 and 300 mg daily oral doses.26 The RO of the IV formulation of aprepitant, fosaprepitant, was recently compared to the RO of a single 165 mg dose of aprepitant over 120 hours in Van Laer et al.32 through the use of the [18F]-MK-0999 radioactive tracer. Both compounds demonstrated a high level of RO in the striatum at Cmax (over 90%) and a RO between 41% and 75% for fosaprepitant and 37% and 76% for aprepitant after 120 hours. In another study by Zamuner et al.40 the [11C]-GR205171 was used as tracer to evaluate the percent of RO after the administration of different single doses of casopitant, a new NK1 RA whose development was recently interrupted for safety issues. As reported by the authors, the 90% of RO was achieved in the striatum at a plasma concentration above 10 ng/mL.
A Tmax of approximately 6 hours was confirmed in the ADME study. Netupitant 300 mg is rapidly absorbed after the administration and undergoes extensive metabolism, forming Phase I and II metabolites. The main metabolites are M1 (N-demethylated netupitant), M2 (netupitant N-oxide), and M3 (monohydroxy netupitant), which on average, account for 29%, 14%, and 33% of the netupitant plasma exposure. In animal models, M1, M2, and M3 have been shown to have pharmacological activity, suggesting a possible contribution in the PET study results. The elimination of more than 90% of drug-related radioactivity was predicted at day 29 and was mainly through the feces.
In conclusion, the present studies demonstrate that netupitant is a potent agent targeting NK1 receptors with a high degree and a long lasting RO. Netupitant 300 mg is rapidly absorbed and has a long terminal half-life. Netupitant is extensively metabolized and is mainly eliminated through the hepatic/biliary route (85% of drug-related radioactivity) while the renal elimination accounted for <5%.
Netupitant, given as single doses of 100–450 mg, was safe and well tolerated.
Acknowledgments
The authors wish to acknowledge the contribution of Dr. A. McEwen, Quotient Chemistry & Metabolism, Rushden, UK for his contribution to the elucidation of netupitant metabolism. We also thank Silvia Olivari Tilola, Helsinn Healthcare SA, and Helen Pentikis, SAJE Consulting, for their contribution to the manuscript preparation and Silvia Sebastiani, Helsinn Healthcare SA, Jennifer Vanden Burgt, pharmaceutical consultant, and Norman Nagle from EISAI, New Jersey, USA for critically reviewing the manuscript.
References
- 1.Sun CC, Bodurka DC, Weaver CB, et al. Rankings and symptom assessments of side effects from chemotherapy: insights from experienced patients with ovarian cancer. Support Care Cancer. 2005;13(4):219–227. doi: 10.1007/s00520-004-0710-6. [DOI] [PubMed] [Google Scholar]
- 2.Feyer P, Jordan K. Update and new trends in antiemetic therapy: the continuing need for novel therapies. Ann Oncol. 2011;22(1):30–38. doi: 10.1093/annonc/mdq600. [DOI] [PubMed] [Google Scholar]
- 3.Schwartzberg L. Chemotherapy-induced nausea and vomiting: state of the art in 2006. J Support Oncol. 2006;4:3–8. (2 Suppl 1): [PubMed] [Google Scholar]
- 4.Bloechl-Daum B, Deuson RR, Mavros P, Hansen M, Herrstedt J. Delayed nausea and vomiting continue to reduce patients' quality of life after highly and moderately emetogenic chemotherapy despite antiemetic treatment. J Clin Oncol. 2006;24(27):4472–4478. doi: 10.1200/JCO.2006.05.6382. [DOI] [PubMed] [Google Scholar]
- 5.Jordan K, Sippel C, Schmoll HJ. Guidelines for antiemetic treatment of chemotherapy-induced nausea and vomiting: past, present, and future recommendations. Oncologist. 2007;12(9):1143–1150. doi: 10.1634/theoncologist.12-9-1143. [DOI] [PubMed] [Google Scholar]
- 6.Hesketh PJ, Van Belle S, Aapro M, et al. Differential involvement of neurotransmitters through the time course of cisplatin-induced emesis as revealed by therapy with specific receptor antagonists. Eur J Cancer. 2003;39(8):1074–1080. doi: 10.1016/s0959-8049(02)00674-3. [DOI] [PubMed] [Google Scholar]
- 7.Rubenstein EB, Slusher BS, Rojas C, Navari RM. New approaches to chemotherapy-induced nausea and vomiting: from neuropharmacology to clinical investigations. Cancer J. 2006;12(5):341–347. doi: 10.1097/00130404-200609000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Roila F, Herrstedt J, Aapro M, et al. Guideline update for MASCC and ESMO in the prevention of chemotherapy and radiotherapy-induced nausea and vomiting: results of the Perugia consensus conference; Ann Oncol; 2010. pp. v232–v243. (Suppl 5): http://www.mascc.org. [DOI] [PubMed] [Google Scholar]
- 9.Basch E, Prestrud AA, Hesketh PJ, et al. Antiemetics: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol. 2011;29(31):4189–4198. doi: 10.1200/JCO.2010.34.4614. http://www.asco.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.NCCN Clinical Practice Guidelines in Oncology: Antiemesis Version 1. 2014. http://www.nccn.org.
- 11.Cohen L, de Moor CA, Eisenberg P, Ming EE, Hu H. Chemotherapy-induced nausea and vomiting: incidence and impact on patient quality of life at community oncology settings. Support Care Cancer. 2007;15(5):497–503. doi: 10.1007/s00520-006-0173-z. Epub 2006 Nov. 14. [DOI] [PubMed] [Google Scholar]
- 12.Roscoe JA, Morrow GR, Hickok JT, Mustian KM, Shelke AR. Biobehavioral factors in chemotherapy-induced nausea and vomiting. J Natl Compr Canc Netw. 2004;2(5):501–508. doi: 10.6004/jnccn.2004.0039. Review. [DOI] [PubMed] [Google Scholar]
- 13.Salsman JM, Grunberg SM, Beaumont JL, et al. Communicating about chemotherapy-induced nausea and vomiting: a comparison of patient and provider perspectives. J Natl Compr Canc Netw. 2012;10(2):149–157. doi: 10.6004/jnccn.2012.0018. [DOI] [PubMed] [Google Scholar]
- 14.Rizzi A, Campi B, Camarda V, et al. In vitro and in vivo pharmacological characterization of the novel NK(1) receptor selective antagonist Netupitant. Peptides. 2010;37(1):86–97. doi: 10.1016/j.peptides.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 15.Gralla R, Lichinitser M, Van Der Vegt S, et al. Palonosetron improves prevention of chemotherapy-induced nausea and vomiting following moderately emetogenic chemotherapy: results of a double-blind randomized phase III trial comparing single doses of palonosetron with ondansetron. Ann Oncol. 2003;14(10):1570–1577. doi: 10.1093/annonc/mdg417. [DOI] [PubMed] [Google Scholar]
- 16.Eisenberg P, Figueroa-Vadillo J, Zamora R, et al. Improved prevention of moderately emetogenic chemotherapy-induced nausea and vomiting with palonosetron, a pharmacologically novel 5-HT3 receptor antagonist: results of a phase III, single-dose trial versus dolasetron. Cancer. 2003;98(11):2473–2482. doi: 10.1002/cncr.11817. [DOI] [PubMed] [Google Scholar]
- 17.Saito M, Aogi K, Sekine I, et al. Palonosetron plus dexamethasone versus granisetron plus dexamethasone for prevention of nausea and vomiting during chemotherapy: a double-blind, double-dummy, randomised, comparative phase III trial. Lancet Oncol. 2009;10(2):115–124. doi: 10.1016/S1470-2045(08)70313-9. Epub 2009 Jan. 8. Erratum in: Lancet Oncol. 2010;11(3):226. [DOI] [PubMed] [Google Scholar]
- 18.Aapro MS, Grunberg SM, Manikhas GM, et al. A phase III, double-blind, randomized trial of palonosetron compared with ondansetron in preventing chemotherapy-induced nausea and vomiting following highly emetogenic chemotherapy. Ann Oncol. 2006;17(9):1441–1449. doi: 10.1093/annonc/mdl137. [DOI] [PubMed] [Google Scholar]
- 19.Rojas C, Li Y, Zhang J, et al. The antiemetic 5-HT3 receptor antagonist Palonosetron inhibits substance P-mediated responses in vitro and in vivo. J Pharmacol Exp Ther. 2010;335(2):362–368. doi: 10.1124/jpet.110.166181. Epub 2010 Aug. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rojas C, Slusher BS. Pharmacological mechanism of 5-HT3 and tachykinin NK-1 receptor antagonism to prevent chemotherapy-induced nausea and vomiting. Eur J Pharmacol. 2012;684(1–3):1–7. doi: 10.1016/j.ejphar.2012.01.046. Epub 2012 Mar. 9. [DOI] [PubMed] [Google Scholar]
- 21.Stathis M, Pietra C, Rojas C, Slusher BS. Inhibition of substance P-mediated responses in NG 108-15 cells by netupitant and palonosetron exhibit synergistic effects. Eur J Pharmacol. 2012;689(1–3):25–30. doi: 10.1016/j.ejphar.2012.05.037. [DOI] [PubMed] [Google Scholar]
- 22.Gardner CJ, Armour DR, Beattie DT, et al. GR205171: a novel antagonist with high affinity for the tachykinin NK1 receptor, and potent broad-spectrum anti-emetic activity. Regul Pept. 1996;65(1):45–53. doi: 10.1016/0167-0115(96)00071-7. [DOI] [PubMed] [Google Scholar]
- 23.Bergström M, Fasth K-J, Kilpatrick G, et al. Brain uptake and receptor binding of two [11C]-labelled high affinity NK1 antagonists, GR203040 and GR205171—PET studies in rhesus monkey. Neuropharmacology. 2000;39(4):664–670. doi: 10.1016/s0028-3908(99)00182-3. [DOI] [PubMed] [Google Scholar]
- 24.Michelgård A, Appel L, Pissiota A, et al. Symptom provocation in specific phobia affects the substance P neurokinin-1 receptor system. Biol Psychiatry. 2007;61(8):1002–1006. doi: 10.1016/j.biopsych.2006.07.003. Epub 2006 Sept. 1. [DOI] [PubMed] [Google Scholar]
- 25.Solin O, Eskola O, Hamill TG, et al. Synthesis and characterization of a potent, selective, radiolabeled substance-P antagonist for NK1 receptor quantitation: ([18F]SPA-RQ) Mol Imaging Biol. 2004;6(6):373–384. doi: 10.1016/j.mibio.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Bergström M, Hargreaves RJ, Burns DH, et al. Human positron emission tomography studies of brain neurokinin 1 receptor occupancy by aprepitant. Biol Psychiatry. 2004;55:1007–1012. doi: 10.1016/j.biopsych.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 27.Hietala J, Nyman MJ, Eskola O, et al. Visualization and quantification of neurokinin-1 (NK1) receptors in the human brain. Mol Imaging Biol. 2005;7(4):262–272. doi: 10.1007/s11307-005-7001-6. [DOI] [PubMed] [Google Scholar]
- 28.Nyman MJ, Eskola O, Kajander J, et al. Gender and age affect NK1 receptors in the human brain—a positron emission tomography study with [18F]SPA-RQ. Int J Neuropsychopharmacol. 2007;10(2):219–229. doi: 10.1017/S1461145706006572. Epub 2006 Mar. 30. [DOI] [PubMed] [Google Scholar]
- 29.Andersson JLR. A rapid and accurate method to realign PET scans utilizing image edge transformation. J Nucl Med. 1995;36:657–669. [PubMed] [Google Scholar]
- 30.Patlak CS, Blasberg R. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. Generalizations. J Cereb Blood Flow Metab. 1985;5:584–590. doi: 10.1038/jcbfm.1985.87. [DOI] [PubMed] [Google Scholar]
- 31.Wolfensberger SPA, van Berckel BNM, Airaksinen AJ, et al. First evaluation of [11C]R116301 as an in vivo tracer of NK1 receptors in man. Mol Imaging Biol. 2009;11(4):241–245. doi: 10.1007/s11307-009-0204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Laer K, De Hoon J, Bormans G, et al. Equivalent dyanamic human brain NK-1 receptor occupancy following single-dose IV fosaprepitant vs. oral aprepitant as assessed by PET imaging. Clin Pharmacol Ther. 2012;92(2):243–250. doi: 10.1038/clpt.2012.62. [DOI] [PubMed] [Google Scholar]
- 33.Helsinn Internal Report: Determination of Netupitant and Its Metabolites, M1, M2, and M3 Concentrations in Human Plasma and Urine Samples from Study NETU-09-21. December 2011.
- 34.Caberlotto L, Hurd YL, Murdock P, et al. Neurokinin 1 receptor and relative abundance of the short and long isoforms in human brain. Eur J Neurosci. 2003;17:1736–1746. doi: 10.1046/j.1460-9568.2003.02600.x. [DOI] [PubMed] [Google Scholar]
- 35.Hargreaves R. Imaging substance P receptors (NK1) in the living human brain using positron emission tomography. J Clin Psychiatry. 2002;63:18–24. (Suppl 11): [PubMed] [Google Scholar]
- 36.Keller M, Montgomery S, Ball W, et al. Lack of efficacy of the substance p (neurokinin1 receptor) antagonist aprepitant in the treatment of major depressive disorder. Biol Psychiatry. 2006;59(3):216–223. doi: 10.1016/j.biopsych.2005.07.013. Epub 2005 Oct. 24. [DOI] [PubMed] [Google Scholar]
- 37.Hesketh P, Rossi G, Rizzi G, et al. Efficacy of NEPA, a novel combination of netupitant (NETU) and palonosetron (PALO), for prevention of chemotherapy-induced nausea and vomiting (CINV) following highly emetogenic chemotherapy (HEC) J Clin Oncol. 2013;31 doi: 10.1093/annonc/mdu110. (suppl; abstr 9512J) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aapro M, Rossi G, Rizzi G, Palmas M, Grunberg S. Phase 3 study of NEPA, a fixed-dose combination of netupitant (NETU) and palonosetron (PALO), versus PALO for prevention of chemotherapy-induced nausea and vomiting (CINV) following moderately emetogenic chemotherapy (MEC) J.Clin Oncol. 2013;31 doi: 10.1093/annonc/mdu101. (suppl; abstr LBA9514) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jordan K, Rossi G, Rizzi G, Palmas M, Gralla RJ. NEPA, a fixed-dose combination of netupitant and palonosetron, for prevention of chemotherapy-induced nausea and vomiting (CINV) following repeated chemotherapy cycles: Results of a phase 3 trial. J Clin Oncol. 2013;31 (suppl; abstr e20716) [Google Scholar]
- 40.Zamuner S, Rabiner EA, Fernandes SA, et al. A pharmacokinetic PET study of NK1 receptor occupancy. Eur J Nucl Med Mol Imaging. 2012;39(2):226–235. doi: 10.1007/s00259-011-1954-2. Epub 2011 Oct. 13. [DOI] [PubMed] [Google Scholar]