

Abstract
Unlike the other MAP3Ks, MEKK1 (encoded by Map3k1) contains a PHD motif. To understand the role of this motif, we have created a knockin mutant of mouse Map3k1 (Map3k1mPHD) with an inactive PHD motif. Map3k1mPHD ES cells demonstrate that the MEKK1 PHD controls p38 and JNK activation during TGF-β, EGF and microtubule disruption signalling, but does not affect MAPK responses to hyperosmotic stress. Protein microarray profiling identified the adaptor TAB1 as a PHD substrate, and TGF-β- or EGF-stimulated Map3k1mPHD ES cells exhibit defective non-canonical ubiquitination of MEKK1 and TAB1. The MEKK1 PHD binds and mediates the transfer of Lys63-linked poly-Ub, using the conjugating enzyme UBE2N, onto TAB1 to regulate TAK1 and MAPK activation by TGF-β and EGF. Both the MEKK1 PHD and TAB1 are critical for ES-cell differentiation and tumourigenesis. Map3k1mPHD/+ mice exhibit aberrant cardiac tissue, B-cell development, testis and T-cell signalling.
Keywords: differentiation, signalling, stem cell, tumourigenesis, ubiquitin
Introduction
Mitogen-activated protein kinases (MAPKs) are involved in numerous cellular processes including cell death, proliferation, embryonic stem (ES) cell differentiation, migration and lymphocyte development (Chen et al, 2001; Xia & Karin, 2004; Karin & Gallagher, 2005; Raman et al, 2007). MEK Kinase 1 (MEKK1) is a member of the MAPK Kinase (MAP2K) Kinase (MAP3K) family that can regulate c-Jun N-terminal kinase (JNK) and p38 by phosphorylation of their upstream MAP2Ks (MAP2K4 and MAP2K7) activation loop (Weston & Davis, 2002; Karin & Gallagher, 2005). Map3k1ΔKD mice, lacking the MEKK1 kinase domain, have demonstrated the importance of MEKK1 signalling in B-cell germinal centre formation, cell cycle progression, antibody production, tumour necrosis factor (TNF) receptor (TNFR)-dependent JNK activation and keratinocyte migration (Xia et al, 2000; Zhang et al, 2003; Gallagher et al, 2007). CD4+ T cells from Map3k1ΔKD mice display an elevated production of T helper (Th) 2 cell cytokines, and mechanistically MEKK1 triggers JNK1-dependent negative regulation of the Homologous to the E6-AP Carboxyl Terminus (HECT) E3 Ubiquitin (Ub) ligase Itch (Gao et al, 2004; Gallagher et al, 2006). Ablation of the Map3k1 in ES cells has revealed a role for MEKK1 in MAPK activation by epidermal growth factor (EGF), LPA, cold shock, microtubule disruption and hyperosmotic stress (Gibson et al, 1999; Yujiri et al, 1999).
Lys48-linked Ub chains can modify protein targets for degradation by the 26S proteasome, while non-canonical Ub conjugation controls protein–protein interactions and modifies the biochemical activity of the target protein (Hochstrasser, 1996; Kravtsova-Ivantsiv & Ciechanover, 2012). Recent research has demonstrated that both Lys63-linked and linear poly-Ub chains are critical for the control of nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) and MAPK activation in cells (Matsuzawa et al, 2008; Karin & Gallagher, 2009; Ikeda et al, 2010; Walczak et al, 2012). Conjugation of Ub to target proteins requires the concentrated activities of a Ub-activating enzyme (E1), a Ub-conjugating enzyme (E2) and an E3 Ub ligase (Gao & Karin, 2005; Kravtsova-Ivantsiv & Ciechanover, 2012).
In addition to functioning as a protein kinase, MEKK1, uniquely among the MAP3Ks, exhibits E3 Ub ligase activity (Lu et al, 2002; Witowsky & Johnson, 2003). This is achieved by the plant homeodomain (PHD), present within the MEKK1 amino-terminal regulatory region, that closely resembles a really interesting new gene (RING) finger (Lu & Hunter, 2009). Overexpression of MEKK1 in cell lines was shown to negatively regulate ERK expression in response to osmotic stress and c-Jun following EGF receptor (EGFR) signalling (Lu et al, 2002; Xia et al, 2007). In addition, MEKK1 also undergoes PHD-dependent auto-ubiquitination (Lu et al, 2002; Witowsky & Johnson, 2003; Gallagher et al, 2007). Following T-cell receptor (TCR) engagement, MEKK1 is modified by Lys63-linked poly-Ub and this correlates with p38 and JNK activation (Matsuzawa et al, 2008; Wang et al, 2008; Enzler et al, 2009). In addition, full-length MEKK1 may also be targeted and cleaved by caspases in response to some forms of cellular stress (Cardone et al, 1997).
Transforming growth factor-β (TGF-β) activated kinase 1 (TAK1, encoded by Map3k7) was identified as a MAP3K that becomes activated following a variety of mitogenic stimuli, including TGF-β and bone morphogenetic protein (Yamaguchi et al, 1995). TAK1 interacts with and is activated by TAK1-binding proteins (TABs) (Shibuya et al, 1996; Takaesu et al, 2000). TAB1 is distinct among the TABs in containing a protein phosphatase 2C (PP2C)-like region and can bind TRAF6, p38 and TAK1 (Shibuya et al, 1996; Ge et al, 2002; Kang et al, 2006). Along with protein–protein interactions with TABs, non-canonical ubiquitination is a critical component in TAK1 activation (Wang et al, 2001). TAB1 can activate TAK1 by overexpression of its C-terminal 68 residues (Shibuya et al, 1996; Ono et al, 2001). TAB2 and TAB3 can be recruited to Lys63-linked poly-Ub chains by their zinc finger (ZnF) motifs (Kanayama et al, 2004). Genetic analysis has demonstrated that both TAK1 and TAB1 are critical for mammalian embryogenesis (Shim et al, 2005). MAPK signalling from the CD40 cytokine receptor requires both MEKK1 and TAK1, though what interplay occurs between these MAP3Ks, and why MEKK1, but not TAK1, contains a PHD motif remains to be clarified (Matsuzawa et al, 2008).
To better assess the role of the PHD in MAPK signalling, we have utilised gene targeting to create the Map3k1mPHD allele and found that an intact MEKK1 PHD is necessary for p38 and JNK activation by TGF-β and EGF in response to microtubule disruption in ES cells. We also found that the MEKK1 PHD mediates the Lys63-linked poly-ubiquitination of the adaptor TAB1 following cytokine stimulation at key lysines, thereby controlling the interaction between full-length TAB1 and TAK1, and activates TAK1, JNK and p38. The MEKK1 PHD is also required for TAB1-dependent ES-cell differentiation and tumour development in mice. Map3k1mPHD/+ mice have cardiac fibrosis and muscle damage, reduced B-cell development beyond the pro-B-cell stage, condensed and fewer numbers of Leydig cells and reduced Itch activation in T cells.
Results
Gene targeting of the PHD
To design a loss-of-function mutation within the MEKK1 PHD, the mouse MEKK1 PHD amino acid sequence was analysed by Phyre2 software, and its structure was modelled upon the known structure of the Deltex 2 RING E3 Ub ligase (Fig 1A) (Kelley & Sternberg, 2009). Sequence alignment of the RING motifs from MEKK1 (residues 437–490), TRAF6 (residues 69–107) and Deltex 2 (residues 407–470) revealed conserved cysteine and isoleucine residues at positions 438 and 440, respectively, of MEKK1, within the RING structure that binds E2 Ub-conjugating enzymes (UBEs) (Yin et al, 2009). We hypothesised that mutation of MEKK1 residues C438 and I440 into alanine residues (the MEKK1 mPHD mutant) would disrupt the function of the PHD as an E3 Ub ligase.
Figure 1. Gene targeting and analysis of the MEKK1 PHD.

- Molecular modelling of MEKK1 PHD. The mouse MEKK1 PHD sequence (residues 437–490) was submitted to the Phyre2 server to produce the WT MEKK1 PHD model. The side chain of residues 438 and 440 was then manually truncated to an alanine to create the mutant MEKK1 PHD model. As a comparison the structure of the mouse Deltex 2 RING (residues 407–470) is shown. The amino acid sequence alignment compares MEKK1 PHD, TRAF6 RING and Deltex 2 RING, with the conserved residues corresponding to MEKK1 residues 438 and 440 indicated by asterisks.
- The MEKK1 mPHD (C438A, I440A) mutant is unable to undergo auto-ubiquitination. HEK 293 cells were transiently transfected with WT MEKK1-Myc or MEKK1 C438A, I440A-Myc and Ub-HA as indicated. After 48 h the cells were lysed and analysed by immunoblotting (IB) with anti-HA antibody. MEKK1 phosphorylation was detected with anti-active MEKK1 (phospho T1381) and anti-phospho S67 antibodies. Anti-Myc antibody was used to immunoprecipitate (IP) MEKK1 or to detect total MEKK1. Anti-tubulin antibody was used as a loading control.
- Strategy for generating Map3k1mPHD knockin mice.
- Targeted ES cells were genotyped by Southern blotting to confirm the in-frame insertion of the mPHD mutation into Map3k1 exon 7.
- MEKK1 expression is similar between WT and Map3k1mPHD ES cell clones. ES cell clones were lysed and analysed by IB using the indicated antibodies.
- MAPK stability is not critically dependent on the PHD. ES cell clones were left unstimulated or stimulated for up to 8 h with 500 mM sorbitol in the presence or absence of 25 μM MG132. Lysates were analysed by IB with the indicated antibodies.
Data information: Results are representative of three independent experiments. Source data are available online for this figure.
To test whether the MEKK1 mPHD retains E3 Ub ligase function, we transfected HEK 293 cells with full-length MEKK1 PHD and mPHD. The experiment demonstrated that MEKK1 auto-ubiquitination by the PHD was significantly reduced relative to the WT motif (Fig 1B). Relative MEKK1 phosphorylation at residues S67 and T1381 (which both reflect MEKK1 activation) was similar between MEKK1 WT and mPHD, indicating that the PHD motif is not involved in the mechanism of overexpressed MEKK1 activation and auto-phosphorylation, as previously reported (Supplementary Fig S1A) (Gallagher et al, 2002; Lu et al, 2002; Matsuzawa et al, 2008). To understand the mechanism by which MEKK1 acts as an E3 Ub ligase, we performed in vitro ubiquitination assays testing E2 conjugating enzymes that can act in concert with UBE1 and the MEKK1 PHD (Supplementary Fig S1B). The MEKK1 PHD underwent strong auto-ubiquitination in the presence of UBE2D2, UBE2D3 or UBE2N:UBE2V1 (Supplementary Fig S1B) and predominantly formed Lys63-linked poly-Ub. Conversely, a relatively small amount of linear Ub chains were generated in ubiquitination assays with the MEKK1 PHD (Supplementary Fig S1C). Analysis of deubiquitinating enzymes (DUBs) (Reyes-Turcu et al, 2009) that can act as deubiquitinating peptidases for auto-ubiquitinated MEKK1 identified Ub-specific proteases (USPs) 2, 7 and 8 (Supplementary Fig S1D). Yeast two-hybrid analysis demonstrated that residues 1–719 of the MEKK1 amino-terminal regulatory domain bind to UBE2N and that this interaction is abolished by the mPHD mutation, which is located within this fragment (Supplementary Fig S1E).
To examine the physiological consequences of the MEKK1 mPHD mutation in mammalian biology, we generated Map3k1mPHD knockin mice, utilising a targeting vector containing a loxP-flanked neomycin resistance cassette and a mutated Map3k1 exon 7 to insert the mPHD mutation into the Map3k1 locus on chromosome 13 (Fig 1C). Map3k1mPHD ES cell clones were genotyped by Southern blotting and genomic PCR for the in-frame insertion of the mPHD mutation into mouse chromosome 13 (Fig 1D). Map3k1mPHD ES cells were found to express full-length MEKK1 at the same amount as the WT protein (Fig 1E).
It was reported that overexpression of the MEKK1 PHD motif can negatively regulate ERK2 expression following hyperosmotic stress (Lu et al, 2002). To test MEKK1-dependent negative regulation of ERK2, we treated Map3k1mPHD ES cells with sorbitol over 8 h (Fig 1F). ERK stability was unaltered in Map3k1mPHD relative to WT ES cells (Fig 1F), and the protein stability of p38 and JNK was also unchanged in Map3k1mPHD ES cells (Fig 1F).
Impaired TGF-β, EGF and nocodazole-induced JNK and p38 activation in Map3k1mPHD ES cells
We next tested whether MAPK activation was altered between Map3k1mPHD and WT ES cells stimulated with TGF-β, EGF, hyperosmotic stress and the microtubule-disrupting agent nocodazole (Fig 2A–D). Microarray profiling of pluripotent Map3k1mPHD and WT ES cells revealed normal expression of TGF-β receptors (TGFβRs) (Supplementary Fig S7A and B and Supplementary Table S1). Map3k1mPHD ES cells exhibited impaired JNK and p38, but unaltered ERK, activation after TGF-β stimulation (Fig 2A). SMAD expression and phosphorylation, however, were unaffected in Map3k1mPHD relative to WT ES cells (Supplementary Fig S2A). Similarly, microarray profiling revealed normal expression of EGF family receptors (EGFRs) (Supplementary Table S1), but Map3k1mPHD ES cells have significantly impaired JNK and p38 activation after EGF stimulation (Fig 2B). ERK, JNK and p38 activation were all unaltered in Map3k1mPHD ES cells treated with sorbitol to induce hyperosmotic stress (Fig 2C). We did note, however, that full-length MEKK1 protein expression was unstable following treatment with sorbitol, with its expression significantly reduced after 30 min, and after several hours, MEKK1 expression was no longer detected (Supplementary Fig S2B). Although the kinetics of MEKK1 degradation were altered in Map3k1mPHD relative to WT cells, mutation of the MEKK1 PHD was insufficient to prevent its degradation after a few hours (Supplementary Fig S2B). Map3k1mPHD ES cells show impaired ERK, JNK and p38 activation after incubation with nocodazole (Fig 2D). From these experiments, we can propose a new model whereby the MEKK1 PHD controls downstream MAPK activation following cytokine stimulation and microtubule disruption, but is not critical for MAPK activation in response to hyperosmotic stress (Supplementary Fig S2C).
Figure 2. Map3k1mPHD ES cells exhibit defective JNK and p38 activation following TGF-β, EGF and nocodazole stimulation.

A–D WT and Map3k1mPHD ES cells were kept on low serum and stimulated with (A) TGF-β (10 ng/ml), (B) EGF (100 ng/ml), (C) sorbitol (500 mM) or (D) nocodazole (0.5 μg/ml) for 10, 30 and 60 min or left unstimulated. Cells were lysed and analysed by IB using the indicated antibodies.
Data information: Results are representative of three independent experiments.
Source data are available online for this figure.
Identification of novel PHD substrates
Since the defects present in Map3k1mPHD ES cells demonstrate that the MEKK1 PHD is not essential for MAPK stability, but is important for JNK and p38 activation, we used protein microarray profiling of 9,400 full-length human proteins in ubiquitination reactions to identify new MEKK1 PHD substrates (Fig 3A and B). Bioinformatics analysis of the protein array data revealed 55 proteins as potential substrates for a ubiquitination reaction comprising UBE2N:UBE2V1 (Supplementary Table S2), and 82 proteins as potential substrates for a ubiquitination reaction containing UBE2N:UBE2V1 and the MEKK1 PHD (Supplementary Table S3). To compare the results from the two array screens, a heat map of the data sets was created using GeneSpring software (Fig 3C).
Figure 3. Protein array screening for MEKK1 PHD substrates.

A Schematic illustrating the protein array screen for MEKK1 PHD substrates using UBE1, UBE2N:UBE2V1 and MEKK1 PHD in a ubiquitination assay.
B Ubiquitination assays were performed comprising UBE1, UBE2N:UBE2V1 and MEKK1 PHD or UBE1 and UBE2N:UBE2V1 in the presence of biotin-Ub and profiled using Protoarray Human Protein Microarrays v.5.
C A heat map comparing hits between UBE1 + UBE2N:UBE2V1 and UBE1 + UBE2N:UBE2V1 + MEKK1 PHD reactions.
D–H Ubiquitination of (D) TAB1, (E) TNIP1, (F) TNIP2, (G) TRAF2, and (H) STAM1 by MEKK1. HEK 293 cells were transfected as indicated, lysates prepared and analysed by IP and IB with the indicated antibodies.
Data information: Results are representative of three independent experiments.
Source data are available online for this figure.
Pathway analysis of the hits from the protein array screen identified TAB1 as a critical component of the TGF-β signal transduction pathway (Supplementary Fig S3A), where we identified defective MAPK signalling in Map3k1mPHD ES cells (Fig 2A). In addition to TAB1, a number of the protein array hits, including TNF receptor-associated factor 2 (TRAF2), TNFAIP3 interacting protein 1 (TNIP1), TNFAIP3 interacting protein 2 (TNIP2) and Signal-Transducing Adaptor Molecule 1 (STAM1), are also classified as signal transduction adaptors and were selected along with TAB1 as possible PHD substrates that might mediate MEKK1 PHD-dependent TGF-β or EGF MAPK activation in ES cells, and to validate our Ub substrate screening in orthogonal assays.
Overexpressed TAB1, TRAF2, TNIP1, TNIP2 and STAM1 proteins were then purified from HEK 293 cells and examined in ubiquitination assays with UBE1, UBE2N:UBE2V1 and MEKK1 PHD or MEKK1 mPHD. In all cases, enhanced poly-Ub modification was detected when the MEKK1 PHD was used as an E3 Ub ligase, whereas the MEKK1 mPHD mutant was unable to enhance the poly-Ub of any of the proteins tested, confirming the protein array screening by an orthogonal assay approach (Supplementary Fig S3B–F). We then examined whether MEKK1 acts as an E3 Ub ligase towards these proteins in cells by cotransfection of TAB1, TNIP1, TNIP2, TRAF2 and STAM1 with MEKK1 and HA-Ub into HEK 293 cells. Immunoprecipitation and Western analysis indicated that poly-Ub of all five proteins was strongly enhanced by coexpression with MEKK1 (Fig 3D–H).
PHD-dependent TAB1 Lys63-linked Ub is critical for EGF and TGF-β signalling
We next analysed the MEKK1 PHD as an E3 Ub ligase in EGF and TGF-β signalling. Pre-treatment of TGF-β-stimulated ES cells with small molecule inhibitors of TGFβR, UBE2N or TAK1 inhibited JNK and p38 activation (Fig 4A). Similarly, pre-treatment of EGF-stimulated ES cells with inhibitors of EGFR, UBE2N or TAK1 inhibited EGF-mediated JNK and p38 activation (Fig 4B). Next, WT and Map3k1mPHD ES cells were stimulated with TGF-β and endogenous MEKK1, TRAF2, TAB1, TNIP1, TNIP2 and STAM1 were immunoprecipitated and then immunoblotted with a Lys63-linked Ub antibody. While we detected Lys63-linked Ub upon MEKK1 and TAB1 in TGF-β stimulated WT ES cells, no Lys63-linked Ub was detected upon TRAF2, TNIP1, TNIP2 and STAM1 (Fig 4C–F and unpublished observations). The Lys63-linked Ub of MEKK1 and TAB1 was strongly reduced in Map3k1mPHD ES cells (Fig 4C and F). An intact MEKK1 PHD is critical for the Lys63-linked ubiquitination of TAB1 (Supplementary Fig S4A), and MEKK1 and TAB1 coimmunoprecipitated when coexpressed in HEK 293 cells (Supplementary Fig S4B).
Figure 4. MEKK1 PHD dependence of TGF-β-stimulated TAK1 and MAPK signalling.

A WT ES cells were rested in low serum conditions and stimulated for 10 min with TGF-β (10 ng/ml) in the presence or absence of DMSO (control), SB431542, NSC697923, (5Z)-7-Oxozeaenol (Oxozeaenol) or left unstimulated. Lysates were made and analysed by IB using the indicated antibodies.
B WT ES cells were rested in low serum and stimulated for 10 min with EGF in the presence or absence of DMSO (control), AG-490, NSC697923, (5Z)-7-Oxozeaenol (Oxozeaenol) or left unstimulated. Lysates were prepared and analysed as above.
C–F WT or Map3k1mPHD ES cells kept in low serum were stimulated or not for 10 min with TGF-β (10 ng/ml). Lysates were prepared and IP and IB performed using the indicated antibodies (* indicates a non-specific band).
G WT or Tab1−/− ES cells were analysed as above by the indicated antibodies before and after TGF-β (10 ng/ml) stimulation.
H HEK 293 cells were transfected with the indicated constructs and lysates made. IP and IB were performed using the indicated antibodies.
I Tab1−/− ES cells were transfected with TAB1 or mTAB1 as indicated, rested in low serum and stimulated or not for 10 min with TGF-β (10 ng/ml). Lysates were made and analysed as above.
Data information: Results are representative of three experiments.
Source data are available online for this figure.
To understand the role TAB1 plays in EGF and TGF-β signalling in ES cells, we generated Tab1−/− ES cells, utilising a targeting vector containing a loxP-flanked neomycin resistance cassette and a mutated Tab1 exon 1 to insert a stop codon and disrupt the Tab1 coding sequence on chromosome 15 (Supplementary Fig S4C and D). Tab1−/− ES cells displayed defective TAK1 and MAPK activation following TGF-β or EGF stimulation (Fig 4G and Supplementary Fig S4E), indicating for the first time that TAB1 is critical for EGF signalling. To map MEKK1-dependent ubiquitination sites within TAB1, we utilised a sequential series of TAB1 deletion mutants and found that TAB1 residues 1–373 were strongly ubiquitinated, whereas 1–313 were weakly ubiquitinated and 1–273 were barely ubiquitinated (Fig 4H and Supplementary Fig S4F). Subsequent mutation of lysines within TAB1 residues 273–373, which contain part of the PP2C-like region of TAB1, at residues K294A, K319A, K335A and K350A blocked the MEKK1-mediated ubiquitination of TAB1, as well as the binding of TAB1 to TAK1 (Supplementary Fig S4G and H). Add-back of WT, but not mTAB1 (K294A, K319A, K335A and K350A), into Tab1−/− ES cells restored TAK1 and MAPK activation by TGF-β (Fig 4I). TAB1 immunoprecipitates with endogenous MEKK1 in ES cells stimulated by TGF-β (Supplementary Fig S4I). To determine whether endogenous TAK1 is activated in a manner dependent upon the MEKK1 PHD, Map3k1mPHD ES cells were stimulated with TGF-β and showed defective TAK1 phosphorylation relative to WT cells (Supplementary Fig S4J).
The MEKK1:TAB1 signalling complex recruits TAB2 by its ZnF motif
Although not a MEKK1 PHD motif substrate (Fig 3), TAB2 can bind poly-Ub via its ZnF motif (Kanayama et al, 2004). Thus, we tested whether TAB2 binds TAB1 coexpressed with MEKK1 by its ZnF motif. TAB2, but not a TAB2 mutant lacking the ZnF motif, purified with TAB1 coexpressed with, and also ubiquitinated by, MEKK1 (Fig 5A). TAK1 activation by TGF-β is critically regulated by UBE2N and TGFβR activity (Fig 5B). To identify TAK1 activation in vitro by the MEKK1 PHD:TAB1 complex, we utilised a two-step approach comprising a ubiquitination assay followed by a kinase assay (Supplementary Fig S5A). The MEKK1 PHD, but not MEKK1 mPHD, was able to ubiquitinate TAB1 and Ub-modified TAB1 potentiated TAK1 activation (Supplementary Fig S5B). TAB1, but not mTAB1, was ubiquitinated by MEKK1 and enhanced TAK1 activation by TAB1 (Supplementary Fig S5C). The MEKK1 PHD ubiquitinates TAB1 with Lys63-linked Ub to potentiate TAK1 activation, and TAB2 can be recruited to this signalling complex in a manner dependent upon its ZnF motif (Fig 5C).
Figure 5. TAB2 interacts with TAB1 ubiquitinated by the MEKK1 PHD motif.
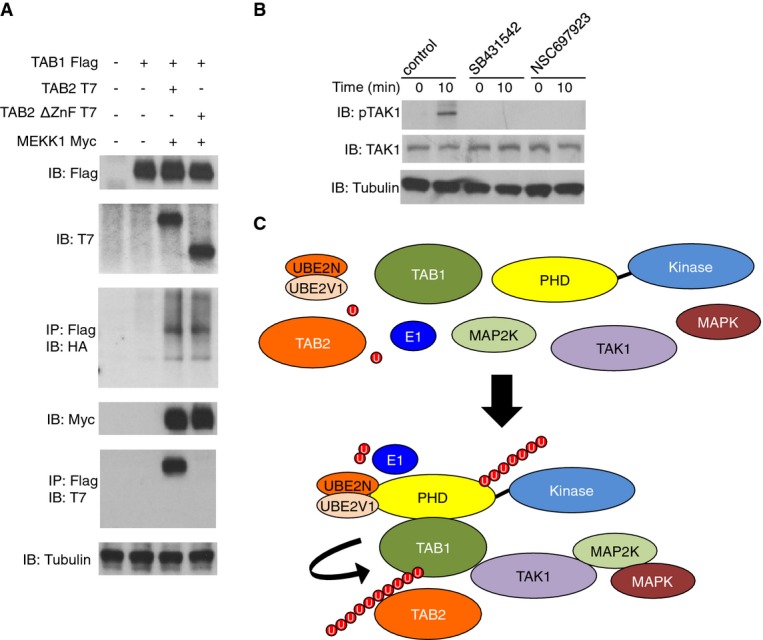
- HEK 293 cells were transiently transfected as indicated along with HA-Ub. 48 h later cells were lysed and analysed by IP and IB using the indicated antibodies.
- WT ES cells were rested in low serum conditions and stimulated for 10 min with TGF-β (10 ng/ml) in the presence or absence of DMSO (control), SB431542, NSC697923 or left unstimulated. Lysates were made and analysed by IB using the indicated antibodies.
- Schematic diagram showing the formation of the MEKK1, TAK1 and TABs signalling complex.
Source data are available online for this figure.
Pluripotent Map3k1mPHD ES cells exhibit a defective gene expression signature
To identify intrinsic gene expression defects within pluripotent Map3k1mPHD ES cells, WT and Map3k1mPHD ES cell cDNA were analysed using GeneChip Mouse Gene 1.0 ST arrays. A heat map of the gene expression profiles was generated with GeneSpring software (Fig 6A). 56 genes were found to be downregulated, and 12 genes were upregulated more than twofold in Map3k1mPHD cells compared to WT ES cells grown in the presence of serum and LIF (Supplementary Fig S6A and Supplementary Table S1). A few of these genes, namely Acta1, Ddx3Y, Dusp4, Dusp14, Nnat, Otx2, Tec, TGFB2, Nes, Nuak1, Runx1 and Tagin, were selected, and their mRNA expression levels confirmed by orthogonal real-time PCR profiling (Fig 6B). Bioinformatics analysis of these hits revealed that they belong to 22 different classes (including cytoskeletal proteins, signalling molecules, cytokines and transcription factors) and are implicated in seven different molecular functions, including the TGF-β signalling pathway (Supplementary Fig S6B). Importantly, the pluripotency genes Nanog and Oct4 were unaltered between WT, Tab1−/− and Map3k1mPHD ES cells growing in serum and LIF, indicating that the MEKK1 PHD is not critical for maintaining ES cells in a pluripotent state (Supplementary Fig S6C). WT, Tab1−/− and Map3k1mPHD ES cells had significantly reduced expression of Nanog and Oct4 following 9 days of culture under conditions that induce ES cell differentiation (Supplementary Fig S6C). There were no significant differences in the proliferation of WT, Tab1−/− and Map3k1mPHD ES cells (Supplementary Fig S6D).
Figure 6. Altered gene expression in Map3k1mPHD ES cells.

- Heat map showing differential gene expression between pluripotent WT and Map3k1mPHD ES cells.
- Confirmation of selected microarray hits by real-time PCR. (
 ) WT and (
) WT and ( ) Map3k1mPHD ES cells RNAs were analysed by real-time PCR. The average relative expression (± SEM) of the indicated mRNA from three independent experiments was statistically analysed, where appropriate, by two-tailed Student's t-test (*P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001).
) Map3k1mPHD ES cells RNAs were analysed by real-time PCR. The average relative expression (± SEM) of the indicated mRNA from three independent experiments was statistically analysed, where appropriate, by two-tailed Student's t-test (*P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001).
Source data are available online for this figure.
Bioinformatics analysis demonstrated that there is no significant difference in the expression of TGFβRs and EGFRs between WT and Map3k1mPHD ES cells (Supplementary Fig S7A). Orthogonal real-time PCR analysis demonstrated that there was no gene expression difference in TGFβR expression between WT, Tab1−/− and Map3k1mPHD ES cells (Supplementary Fig S7B). When grown in the presence of serum and LIF, there is a small reduction in the amount of phospho-MEKK1 in Tab1−/− and Map3k1mPHD ES cells (Supplementary Fig S7C).
The MEKK1 PHD and TAB1 signalling are critical for ES-cell differentiation
We performed embryoid body (EB) formation assays with WT, Tab1−/− and Map3k1mPHD ES cells to determine whether the MEKK1 PHD motif and TAB1 control ES-cell differentiation (Doetschman et al, 1985; Dang et al, 2002; Wu et al, 2010). EBs were formed by WT, Map3k1mPHD and Tab1−/− ES cells, and also by ES cells treated with p38 or TGFβR inhibitors (Fig 7A and unpublished observations). By contrast, long-term treatment of WT ES cells with UBE2N, EGFR or JNK inhibitors reduced ES-cell viability, preventing them from forming EBs in long-term culture (unpublished observations). To assess whether Map3k1mPHD and Tab1−/− ES cells exhibit defective ES-cell differentiation, days 6 and 9 EBs were analysed by real-time PCR analysis using neuroectoderm (Nestin, Pax6 and Mash1), endoderm (Mixl1, Gata6 and Gata4) and mesoderm (Brachyury) markers (Fig 7B–D and Supplementary Fig S8A–C). On day 6 post-differentiation, Map3k1mPHD and Tab1−/− ES cells displayed significantly elevated expression of the neuroectoderm gene markers Nestin, Pax6 and Mash1 (Fig 7B), while, by contrast, at day 9, the mesoderm gene marker Brachyury was significantly reduced in both Map3k1mPHD and Tab1−/− relative to WT ES cells (Supplementary Fig S8C). Chemical inhibition of p38 or TGFβR also elevated expression of the neuroectoderm gene markers at day 6 and reduced day 9 Brachyury expression (Fig 7E and Supplementary Fig S8D).
Figure 7. Map3k1mPHD ES cells exhibit an altered differentiation pattern.

WT, Map3k1mPHD and Tab1−/− ES cells were plated under differentiation conditions without LIF for 6 or 9 days.
A Pictures of EBs were taken using an Olympus light microscope after 9 days of differentiation and analysed using Image Pro-Software at 40× magnification. Scale bar is 250 μm.
B–D ( ) WT, (
) WT, ( ) Map3k1mPHD and (
) Map3k1mPHD and ( ) Tab1−/− ES cells were plated under differentiation conditions for 6 days, and their RNAs analysed by real-time PCR with primers specific for (B) neuroectoderm, (C) endoderm and (D) mesoderm genes.
) Tab1−/− ES cells were plated under differentiation conditions for 6 days, and their RNAs analysed by real-time PCR with primers specific for (B) neuroectoderm, (C) endoderm and (D) mesoderm genes.
E WT ES cells were differentiated for 6 days in the presence of ( ) DMSO, (
) DMSO, ( ) SB203580 or (
) SB203580 or ( ) SB431542, and their RNAs analysed by real-time PCR with primers specific for neuroectoderm genes.
) SB431542, and their RNAs analysed by real-time PCR with primers specific for neuroectoderm genes.
F Tab1−/− ES cells were transfected with ( ) CMV, (
) CMV, ( ) CMV TAB1 or (
) CMV TAB1 or ( ) CMV mTAB1 and used alongside (
) CMV mTAB1 and used alongside ( ) WT ES cells in differentiation assays for 6 days, mRNA was extracted and their RNAs analysed by real-time PCR with primers specific for the neuroectoderm gene Mash1.
) WT ES cells in differentiation assays for 6 days, mRNA was extracted and their RNAs analysed by real-time PCR with primers specific for the neuroectoderm gene Mash1.
Data information: The average relative expression (± SEM) of the indicated gene mRNA from three independent experiments was statistically analysed, where appropriate, by two-tailed Student's t-test (*P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001).
Source data are available online for this figure.
To assess whether changes in ES cell neuroectoderm and mesoderm gene markers were due to altered TAB1 ubiquitination, Tab1−/− ES cells were transfected with TAB1 or mTAB1 (Supplementary Fig S8E) and EB assays performed (Fig 7F, Supplementary Figs S8F and S9). Tab1−/− ES cells reconstituted with TAB1, but not mTAB1, expressed similar levels of neuroectoderm and mesoderm markers as WT ES cells (Fig 7F, Supplementary Figs S8F and S9).
Analysis of Map3k1mPHD and Tab1−/− ES-cell tumourigenicity
We tested whether transplanted Map3k1mPHD and Tab1−/− ES cells, when injected into immunodeficient NOD.CB17-Prkdcscid/lcrCrl recipient mice, exhibited defective teratoma formation. While WT transplanted ES cells were able to form large tumours within 5 weeks of transplantation, Map3k1mPHD or Tab1−/− ES cells produced tumours of much smaller size and mass (Fig 8A–C). Add-back of TAB1, but not mTAB1, into Tab1−/− ES cells restored tumor development to WT levels (Fig 8D). Histological analysis of Map3k1mPHD and Tab1−/− ES-cell tumours revealed a deficit in the formation of cartilage-like tissues (Fig 8E).
Figure 8. Regulation of ES-cell tumourigenesis by the MEKK1 PHD.
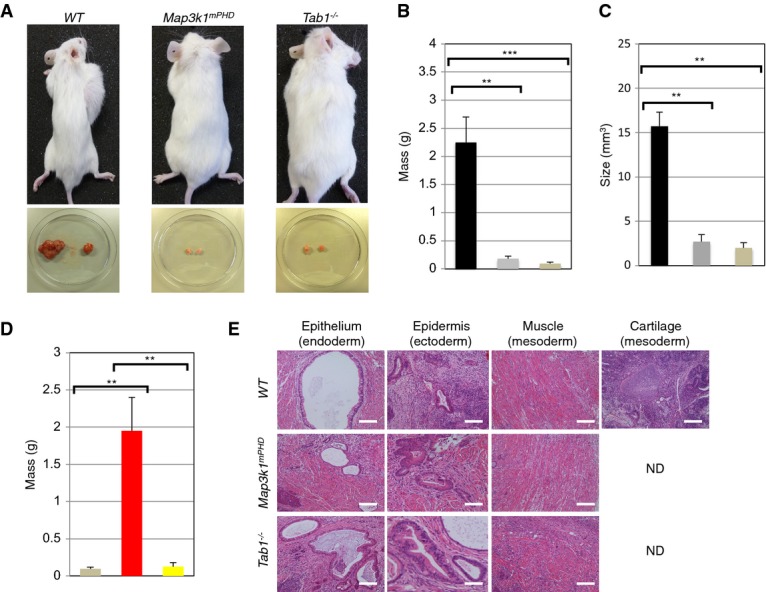
- Analysis of WT, Map3k1mPHD and Tab1−/− ES-cell teratoma formation in NOD.CB17-Prkdcscid/lcrCrl recipient mice.
- Mass (g) of (
 ) WT, (
) WT, ( ) Map3k1mPHD and (
) Map3k1mPHD and ( ) Tab1−/− tumours formed in the above mouse strain 5 weeks post-transplantation. The average mass (± SEM) of tumours from 3 independent experiments was statistically analysed, where appropriate, by two-tailed Student's t-test (**P ≤ 0.01; ***P ≤ 0.001).
) Tab1−/− tumours formed in the above mouse strain 5 weeks post-transplantation. The average mass (± SEM) of tumours from 3 independent experiments was statistically analysed, where appropriate, by two-tailed Student's t-test (**P ≤ 0.01; ***P ≤ 0.001). - The average size (± SEM) of (
 ) WT, (
) WT, ( ) Map3k1mPHD and (
) Map3k1mPHD and ( ) Tab1−/− tumours from 3 independent experiments was statistically analysed, where appropriate, by two-tailed Student's t-test (**P ≤ 0.01).
) Tab1−/− tumours from 3 independent experiments was statistically analysed, where appropriate, by two-tailed Student's t-test (**P ≤ 0.01). - Tab1−/− ES cells were transfected with (
 ) CMV, (
) CMV, ( ) CMV TAB1 or (
) CMV TAB1 or ( ) CMV mTAB1 expression vectors and their tumourigenic potential was analysed as above. Average results (SEM) from three independent experiments were statistically analysed by two-tailed Student's t-test (**P ≤ 0.01).
) CMV mTAB1 expression vectors and their tumourigenic potential was analysed as above. Average results (SEM) from three independent experiments were statistically analysed by two-tailed Student's t-test (**P ≤ 0.01). - Analysis of WT, Map3k1mPHD and Tab1−/− ES-cell teratomas. Tumours were extracted and analysed by H&E staining (ND indicates tissue not detected). Pictures were taken using an Olympus light microscope, and pictures were analysed using Image Pro-Software at 40× magnification. Scale bar is 70 μm.
Data information: Results are representative of three independent experiments.
Source data are available online for this figure.
Analysis of the Map3k1mPHD mutation in mice
Map3k1mPHD mice are non-viable due to early embryonic lethality, so we analysed Map3k1mPHD/+ heterozygote mice to understand the developmental function of the MEKK1 PHD motif. Map3k1mPHD/+ mice have significantly enlarged testes and hearts (Fig 9A and B). Since Mekk1−/− mice exhibit cardiac abnormalities (Minamino et al, 1999, 2002), we analysed cardiac tissue from Map3k1mPHD/+ mice. H&E staining revealed extensive fibrosis and cardiac muscle damage relative to WT mice, and cardiac enlargement (Fig 9C). Since Map3k1ΔKD mice have minor abnormalities in the testis, we analysed testis morphology and spermatogenesis of male Map3k1mPHD/+ mice (Warr et al, 2011). H&E staining showed condensed and reduced numbers of Leydig cells (Fig 9C).
Figure 9. Analysis of Map3k1mPHD/+ mice.

- Testis and heart were extracted from WT and Map3k1mPHD/+ mice.
- Quantitation of the mass (g) of testis and heart tissues from () WT and () Map3k1mPHD/+ mice. The average mass (± SEM) of testis and heart from 3 independent experiments was statistically analysed, where appropriate, by two-tailed Student's t-test (*P ≤ 0.05).
- Testis and heart were extracted from WT and Map3k1mPHD/+ mice. H&E-stained cardiac and testis tissue sections were prepared from WT and Map3k1mPHD/+ mice. Pictures were taken using an Olympus light microscope, and pictures were analysed using Image Pro-Software at 40× magnification. Scale bar is 50 μm. Arrows indicate Leydig cells in the testis.
- Bone marrow was harvested from WT, Map3k1mPHD/+, Map3k1+/ΔKD and Map3k1ΔKD mice. Cells were stained with antibodies for the B-cell markers B220 and IgM and analysed by FACS.
- Splenocytes from WT and Map3k1mPHD/+ mice were stained with antibodies for the T-cell markers CD4 and CD8 and analysed by FACS.
- T cells were purified from WT and Map3k1mPHD/+ mice and costimulated with anti-CD3 and anti-CD28 antibodies for 10, 30 and 60 min or left unstimulated. Lysates were made, and IB was performed using the indicated antibodies.
Data information: Results are representative of three experiments.
Source data are available online for this figure.
Since Map3k1ΔKD mice have defects in B- and T-cell signalling (Gao et al, 2004; Gallagher et al, 2007), we analysed lymphocytes from Map3k1mPHD/+ mice (Fig 9D and E, and Supplementary Fig S10A). Bone marrow cells from Map3k1mPHD/+ mice were stained with antibodies for mature B-cell markers (B220 and IgM), pro-B-cell markers (IL-7R and CD34), pre-B-cell markers (CD45 and CD38) and immature B-cell markers (CD45 and IgM). Map3k1mPHD/+ mice exhibited reduced numbers of pre-B-cells, immature B cells and mature B cells (Fig 9D and Supplementary Fig S10A), in contrast to Map3k1+/ΔKD or Map3k1ΔKD mice, which were similar in numbers to WT (Fig 9E). Thymocytes were analysed for the T-cell markers CD4 and CD8 to determine whether the MEKK1 PHD domain is also essential for T-cell development. No significant differences between the numbers of WT, Map3k1+/ΔKD, Map3k1ΔKD and Map3k1mPHD/+ single-positive or double-positive thymocytes or total numbers of mature T cells were observed, and the size of the thymus is normal in Map3k1mPHD/+ mice (Fig 9E and Supplementary Fig S10B and C). To analyse whether MEKK1 might be important for MEKK1-dependent regulation of HECT E3 Ub ligase Itch (Gallagher et al, 2006; Enzler et al, 2009), purified T cells from WT and Map3k1mPHD/+ mice were costimulated by anti-CD3 and anti-CD28 antibodies. Itch phosphorylation was significantly reduced in Map3k1mPHD/+ relative to WT T cells (Fig 9F).
Discussion
We report that TAB1 is a substrate for non-canonical ubiquitination mediated by the MEKK1 PHD and that this modification enhances MAPK activation by EGF and TGF-β. The kinase activity of MEKK1 is well documented for its role in the regulation of MAP2Ks, and detailed analysis of Map3k1ΔKD mice has demonstrated important roles for MEKK1 in lymphocyte effector responses, keratinocyte migration and eyelid fusion (Xia et al, 2000; Zhang et al, 2003; Gao et al, 2004; Gallagher et al, 2007). Consistent with previous reports, mutation of the MEKK1 PHD reduced its auto-ubiquitination, but had comparatively little impact upon MEKK1 kinase activity (Lu et al, 2002; Witowsky & Johnson, 2003). Yet, Map3k1mPHD ES cells were defective in MAPK activation in response to TGF-β, EGF and microtubule disruption by nocodazole. Thus, the MEKK1 PHD has important signalling functions that are not identical to those of the kinase domain.
Protein microarray screening identified several novel targets for the E3 Ub ligase activity of the MEKK1 PHD. From these targets, we focused on TAB1, since it is a critical adaptor in the TGF-β signalling pathway, and confirmed it as a MEKK1 PHD target protein that was ubiquitinated in WT, but not Map3k1mPHD, cells following TGF-β or EGF stimulation. TAB2 and TAB3 were not substrates for the MEKK1 PHD motif in our protein array analysis. Analysis of Tab1−/− ES cells revealed that TAB1 was required for EGF- and TGF-β-stimulated TAK1 and MAPK activation. Mapping of the TAB1 residues subjected to ubiquitination by the MEKK1 PHD motif identified TAB1 lysines 294, 319, 335 and 350 whose replacement with alanines abolished TAB1 ubiquitination. TAB1 is known to bind and activate TAK1 or it may signal independently of TAK1 by binding p38 MAPK (Shibuya et al, 1996; Wang et al, 2001; Ge et al, 2002; Kang et al, 2006). MEKK1 binds and ubiquitinates TAB1 via its PHD domain to enhance the molecular interaction between TAB1 and TAK1, and the K294A, K319A, K335A and K350A substitutions also diminished the binding of TAB1 to TAK1 and inhibited TAK1 phosphorylation after EGF and TGF-β stimulation. TAB2, although not a MEKK1 PHD substrate, can be recruited to the TAB1:MEKK1 complex, and this interaction is dependent upon the presence of an intact Ub binding ZnF motif within TAB2 (Kanayama et al, 2004). Recruitment of TAB2 to the MEKK1:TAB1 signalling complex may facilitate further downstream signalling. Our results reveal that TAB1 ubiquitination is the major conduit for the signalling function of the MEKK1 PHD from TGFβRs. The interdependency between the MEKK1 PHD and TAK1 also explains why either genetic disruption of the MEKK1 PHD or chemical inhibition of TAK1 kinase activity leads to loss of MAPK activation following EGF or TGF-β stimulation of ES cells. Our results suggest that UBE2N is critical for TAK1 and MAPK activation in response to TGF-β. We also suggest that TRAF2 is not critical for TGF-β signalling, unlike its important roles in TNF and CD40 signal transduction (Karin & Gallagher, 2009). Indeed, TRAF6, but not TRAF2, is critical for TGFβR-dependent MAPK activation (Yamashita et al, 2008).
Interestingly, the MEKK1 PHD and TAB1 repress neuroectoderm marker expression and enhance long-term mesoderm gene marker expression as ES cells differentiate into EBs. p38α is known to play a role in ES-cell neuroectoderm and mesoderm differentiation (Gaur et al, 2010; Barruet et al, 2011). Similarly, Jnk1 and Jnk2 double deficiency in ES cells reveals a role for the JNK MAPKs in promoting ES-cell differentiation (Xu & Davis, 2010). To date, no compound Jnk and p38α ES-cell mutations have been reported that would mimic the effects of the MEKK1 PHD or TAB1 ablation. Transplantation of Tab1−/− or Map3k1mPHD ES cells into NOD.CB17-Prkdcscid/lcrCrl mice results in tumours of altered tissue composition, reduced size and mass. Importantly, reintroduction of TAB1, but not lysine mutated TAB1, into Tab1−/− ES cells was able to restore normal ES-cell differentiation and tumour formation, indicating that the TAB1 lysines ubiquitinated by the MEKK1 PHD are critical for its function in ES-cell differentiation and tumour formation.
The analysis of Map3k1mPHD mice has been complicated by their early embryonic lethality, which is a more severe phenotype than the partial lethality observed in Map3k1ΔKD mice (Bonnesen et al, 2005). Alterations in the regulation of the Ub-proteasome system in combination with aberrant MAPK activation in ES cells provide an explanation for this more severe phenotype (Lu et al, 2002). However, Map3k1mPHD/+ mice are viable, and their analysis revealed defects in B-cell development, TCR signal transduction, cardiac tissue and testis development. These results reinforce the critical importance of the MEKK1 PHD in mammalian biology.
Materials and Methods
ES cell gene targeting and mice
Map3k1 kinase-deficient mice (Map3k1ΔKD) were generated as previously described (Gao et al, 2004). To create Map3k1mPHD gene knockin mice, a targeting vector containing a loxP-flanked neomycin resistance cassette and a mutated exon 7 was inserted into the Map3k1 locus on chromosome 13. Targeted Map3k1mPHD/+ and Map3k1mPHD ES cells were generated according to standard procedures (Gossler et al, 1986) and genotyped by Southern blotting or genomic PCR (Ledermann, 2000; Xia et al, 2000). Four independently generated Map3k1mPHD knockin ES cell clones were injected into C57BL/6 blastocysts and the resulting transgenics were genotyped by PCR (Zhang et al, 2003). To create Tab1−/− gene knockout ES cells, a targeting vector containing a loxP-flanked neomycin resistance cassette and a mutated exon 1 was inserted in the Tab1 locus on chromosome 15. Targeted ES cells were generated according to standard procedures (Gossler et al, 1986) and genotyped by Southern blotting or genomic PCR (Ledermann, 2000). NOD.CB17-Prkdcscid/lcrCrl mice were purchased from Charles River Laboratories. All mice were bred and maintained under pathogen-free conditions in conventional barrier protection in accordance with the guidelines of the Home Office and Imperial College London.
Reagents and antibodies
Phospho-SMAD antibody sampler kit (9963) was from Cell Signaling. Mouse anti-TRAF2 (sc-7346), rabbit anti-STAM (sc-33588), goat anti-TAB1 (sc-6052), rabbit anti-MEKK1 (sc-252) and rabbit anti-Abin1 (or TNIP1) (sc-134660) antibodies were from Santa Cruz Biotechnology. Mouse anti-HA (32-6700), mouse anti-Myc (R950-25) and mouse anti-Ub (13-1600) antibodies were from Invitrogen. Rabbit anti-p38 (9212), rabbit anti-pp38 (9211), rabbit anti-SAPK/JNK (9252), rabbit anti-Itch (12117), rabbit anti-TAK1 (4505), rabbit anti-pTAK1 (4508), rabbit anti-pSAPK/JNK (9251) and rabbit anti-Lys63-linked Ub (5621) antibodies were from Cell Signaling. Rabbit anti-pItch (AB10050) antibody was purchased from Merck Millipore. Mouse anti-p-ERK (M8159), rabbit anti-ERK (M5670), mouse anti-tubulin (T5168), mouse anti-T7 (T8823) and rabbit anti-Flag (F7425) antibodies were from Sigma. Anti-CD3 (555273) and anti-CD28 (347690) antibodies were from BD Biosciences. pCMV-Myc mouse MEKK1 and pCMV-Myc mouse MEKK1 C438A, I440A plasmids were purchased from GenScript. pCMV6-XL4 TRAF2, pCMV6-XL4 TNIP2 and pCMV6-Myc TNIP1 were from Origene. pCMV TAB1 and pCMV mTAB1 were purchased from GenScript. Rabbit anti-pMEKK1 was custom made by ThermoScientific. Rabbit anti-phospho MEKK1 was generated as previously described (Matsuzawa et al, 2008). TGF-β and EGF (100-35) were purchased from Peprotech. Chemical inhibitors include AG-490 (EGFR inhibitor, Santa Cruz Biotechnology), NSC697923 (UBE2N inhibitor, Millipore), (5Z)-7-Oxozeaenol (TAK1 inhibitor, Sigma), SB431542 (TGFβR inhibitor, Sigma) and SB203580 (p38 MAPK inhibitor, Sigma). Nocodazole and sorbitol were purchased from Sigma.
Cells and cell culture conditions
HEK 293 cells were maintained in DMEM (22320, Invitrogen) supplemented with 10% FBS (SH3007003, Thermo Scientific) and antibiotics in a humidified atmosphere at 37°C. Cells were passaged every 2–3 days when approaching full confluence. T cells were isolated from mouse splenocytes using CD4 Microbeads (130-049-201, Miltenyi Biotec) according to the manufacturer's instructions (Gallagher et al, 2007). Mouse ES cells were grown in mESC medium (Knockout™ DMEM supplemented with 10% FBS, non-essential amino acids solution, l-glutamine, recombinant human LIF and 2-mercaptoethanol) in a humidified atmosphere at 37°C. Cells were passaged every 2 days.
EB formation assays
ES cells were plated in low attachment 6-well plates in mESC medium with 5% FBS and no LIF at a density of 1 × 105 cells per well (Doetschman et al, 1985; Dang et al, 2002; Wu et al, 2010). They were collected 6 or 9 days later for RNA extraction.
Tumourigenesis assays
Pluripotent ES cells were injected subcutaneously into the flank of NOD.CB17-Prkdcscid/lcrCrl mice (1 × 106 cells in PBS/animal) (Dressel et al, 2008). Mice were monitored daily for 5 weeks before the animals were culled and tumours extracted.
Yeast two-hybrid
Y190 yeast were transformed and grown as previously described (Gallagher et al, 2007).
Transfection
HEK 293 cells were plated in 6-well plates at a density of 1 × 106 cells per well. The following day cells were transfected with Lipofectamine 2000 (11668-019, Invitrogen) or Jet Prime (114-07, Polyplus) transfection reagents according to the manufacturer's instructions. Cells were collected and lysed 48 h later. ES cells were transfected using Xfect Mouse Embryonic Stem Cell Transfection Reagent (631321, Clontech) according to the manufacturer's protocols.
Ubiquitination and kinase assays
TRAF2, TNIP1, TNIP2, TAB1 and STAM1 were overexpressed in HEK 293 cells, immunoprecipitated, washed extensively and protein eluted. Subsequently, they were incubated for 1 h at 37°C with the ubiquitination assay enzymes E1 (100 nM), UBE2N:UBE2V1 (0.36 μM) or UBE2D2 (0.5 μM), Ub or no K Ub (150 μM) and ATP, with or without WT MEKK1 PHD (100 ng) or MEKK1 mPHD (100 ng) (Matsuzawa et al, 2008). All ubiquitination assay reagents were from Boston Biochem. Kinase assays were performed as previously described (Gallagher et al, 2002).
Western blotting and immunoprecipitation
Cells were lysed in whole-cell lysis buffer (50 mM Tris pH 7.6, 150 mM NaCl and 1% Triton X-100) (Gao et al, 2004). For detection of ubiquitination, 20 mM N-ethylmaleimide (NEM) (E1271, Sigma) was added to the buffer. For immunoprecipitation, cells were lysed in buffer containing 20 mM Tris pH 7.6, 120 mM NaCl, 0.5 mM EDTA, 1.5 mM MgCl2 and 0.5% Triton X-100. All buffers were supplemented with protease and phosphatase inhibitors (Sigma). Following cell lysis, proteins were resolved in SDS polyacrylamide gels and transferred to PVDF membranes, blocked in 5% milk, incubated with specific primary and secondary antibodies and detected with ECL solution (32106, Pierce) or immunoprecipitated with 1–2 μg of the antibody of interest overnight at 4°C (Gao et al, 2004). Immunoprecipitates were captured with protein A/G Plus agarose beads (sc-2003, Santa Cruz). Beads were washed three times in lysis buffer without Triton X-100, and proteins were eluted in elution buffer (1858606, Pierce) or released by boiling in Laemmli Sample buffer (161-0737, Bio-Rad).
Protein purification
BL21 (DE3) E. coli cells were transformed with pGEX KG MEKK1 PHD and were grown in Luria Broth (LB) media at 37°C (Lu et al, 2002). At an OD600 of 0.6 protein production was induced with 1 mM IPTG. Cells were further grown for approximately 18 h at 18°C. After bacterial cell lysis, supernatant was applied to GST column (GE Healthcare) to capture MEKK1 PHD. Protein was eluted from the column with a buffer containing PBS, 50 mM reduced glutathione and 1 mM DTT, pH 7.8. Full-length His-MEKK1, His-MEKK1 mPHD, His-TAB1 and His-TAK1 were overexpressed and purified from Sf9 cells as previously described (Gallagher et al, 2002). Proteins expressed in HEK 293 cells were IP, bound to HiTrap Protein G columns (GE Healthcare Life Sciences), washed extensively and then eluted for further assays according to the manufacturer's protocols.
Proliferation assays
Proliferation was measured using a CellTrace CFSE Cell Proliferation kit (Life Technologies) according to the manufacturer's protocols.
Protein microarray
UBE1 (100 nM), UBE2N:UBE2V1 (500 nM) and purified MEKK1 PHD-GST (50 or 250 nM) were used in ubiquitination assays in the presence of biotin-Ub (100 μg/ml) (Invitrogen). Arrays probed with buffer only or UBE1 and UBE2N:UBE2V1 without MEKK1 PHD served as negative controls. Ubiquitination of the immobilised proteins on the arrays treated with UBE1, UBE2N:UBE2V1, and MEKK1 PHD-GST was evaluated by the Z-score and background subtracted signal values within the array relative to the control assays.
Real-time PCR
Total RNA was extracted using the RNeasy Midi kit (Qiagen) according to the manufacturer's instructions. RNA was converted to cDNA using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). cDNA was amplified using SYBR Green PCR Master Mix (Applied Biosystems) and primer pairs of interest (Supplementary Table S4) (Gao et al, 2004).
Affymetrix microarray global gene expression screening
Total RNA was converted to cRNA using the WT Expression kit (Ambion). Quality of cRNA was assessed with a 2100 Bioanalyser. cRNA was converted to second-strand cDNA using the WT Expression kit (Ambion). cDNA was fragmented and labelled using the GeneChip WT Terminal Labelling kit (Affymetrix). Labelled cDNA was hybridised to a GeneChip Mouse Gene 1.0 ST Array. GeneSpring software was used for data analysis and quality control. Probes were normalised by quantile normalisation among all microarray data.
Molecular modelling of the MEKK1 PHD
The amino acid sequence of mouse MEKK1 PHD (residues 432–485) was submitted to the Phyre2 server to produce the WT MEKK1 PHD model (Kelley & Sternberg, 2009). The side chain of residues 438 and 440 was then manually truncated to an alanine to create the mutant MEKK1 PHD model.
Bioinformatics
GeneSpring software was used to create heat maps of the Protoarray and Affymetrix data according to the software vendor's instructions. Ingenuity IPA and iReport were used according to the software vendor's protocols for bioinformatics analysis.
Accession numbers
ArrayExpress accession: E-MTAB-1679. IMEx accession: IM-22822.
Acknowledgments
We would like to thank Tony Hunter (Salk Institute for Biological Sciences, USA) for providing MEKK1 constructs, Lorna Fiedler (Imperial College London, UK) for TAB1 constructs, Jiahuai Han (University of Xiamen, China) for TAB1 constructs, Steve Ley (NIMR, UK) for Abin2 antibody, Kazuo Sugamura (Tokyo University, Japan) for STAM1 and STAM2 constructs, and Kunihiro Matsumoto (Nagoya University, Japan) and Jun Ninomiya-Tsuji (North Carolina State University, USA) for TAB2 constructs. Melanie Cobb (UTSW, USA) for rat MEKK1 and pCMV5 Myc constructs. We would like to thank Tony Hunter and Atsushi Matsuzawa (University of Tokyo, Japan) for insights and comments on the manuscript. Our research was supported by grants from the Wellcome Trust (WT090939MA) and Cancer Research UK (C26616/A12679) for EG, grants NIH AI043477 and ALR 257214 for MK and XW was supported by a Susan G. Komen for the Cure postdoctoral fellowship (KG111506).
Author contributions
NC, TS, XW, SA, MK and EG conceived aspects of the experimental design, performed experiments, interpreted the data and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Barruet E, Hadadeh O, Peiretti F, Renault VM, Hadjal Y, Bernot D, Tournaire R, Negre D, Juhan-Vague I, Alessi MC, Binetruy B. p38 mitogen activated protein kinase controls two successive-steps during the early mesodermal commitment of embryonic stem cells. Stem Cells Dev. 2011;20:1233–1246. doi: 10.1089/scd.2010.0213. [DOI] [PubMed] [Google Scholar]
- Bonnesen B, Orskov C, Rasmussen S, Holst PJ, Christensen JP, Eriksen KW, Qvortrup K, Odum N, Labuda T. MEK kinase 1 activity is required for definitive erythropoiesis in the mouse fetal liver. Blood. 2005;106:3396–3404. doi: 10.1182/blood-2005-04-1739. [DOI] [PubMed] [Google Scholar]
- Cardone MH, Salvesen GS, Widmann C, Johnson G, Frisch SM. The regulation of anoikis: MEKK-1 activation requires cleavage by caspases. Cell. 1997;90:315–323. doi: 10.1016/s0092-8674(00)80339-6. [DOI] [PubMed] [Google Scholar]
- Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. MAP kinases. Chem Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- Dang SM, Kyba M, Perlingeiro R, Daley GQ, Zandstra PW. Efficiency of embryoid body formation and hematopoietic development from embryonic stem cells in different culture systems. Biotechnol Bioeng. 2002;78:442–453. doi: 10.1002/bit.10220. [DOI] [PubMed] [Google Scholar]
- Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R. The in vitro development of blastocyst-derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. J Embryol Exp Morphol. 1985;87:27–45. [PubMed] [Google Scholar]
- Dressel R, Schindehutte J, Kuhlmann T, Elsner L, Novota P, Baier PC, Schillert A, Bickeboller H, Herrmann T, Trenkwalder C, Paulus W, Mansouri A. The tumorigenicity of mouse embryonic stem cells and in vitro differentiated neuronal cells is controlled by the recipients' immune response. PLoS ONE. 2008;3:e2622. doi: 10.1371/journal.pone.0002622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enzler T, Chang X, Facchinetti V, Melino G, Karin M, Su B, Gallagher E. MEKK1 binds HECT E3 ligase Itch by its amino-terminal RING motif to regulate Th2 cytokine gene expression. J Immunol. 2009;183:3831–3838. doi: 10.4049/jimmunol.0803412. [DOI] [PubMed] [Google Scholar]
- Gallagher ED, Xu S, Moomaw C, Slaughter CA, Cobb MH. Binding of JNK/SAPK to MEKK1 is regulated by phosphorylation. J Biol Chem. 2002;277:45785–45792. doi: 10.1074/jbc.M207702200. [DOI] [PubMed] [Google Scholar]
- Gallagher E, Gao M, Liu YC, Karin M. Activation of the E3 ubiquitin ligase Itch through a phosphorylation-induced conformational change. Proc Natl Acad Sci USA. 2006;103:1717–1722. doi: 10.1073/pnas.0510664103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher E, Enzler T, Matsuzawa A, Anzelon-Mills A, Otero D, Holzer R, Janssen E, Gao M, Karin M. Kinase MEKK1 is required for CD40-dependent activation of the kinases Jnk and p38, germinal center formation, B cell proliferation and antibody production. Nat Immunol. 2007;8:57–63. doi: 10.1038/ni1421. [DOI] [PubMed] [Google Scholar]
- Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu YC, Karin M. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science. 2004;306:271–275. doi: 10.1126/science.1099414. [DOI] [PubMed] [Google Scholar]
- Gao M, Karin M. Regulating the regulators: control of protein ubiquitination and ubiquitin-like modifications by extracellular stimuli. Mol Cell. 2005;19:581–593. doi: 10.1016/j.molcel.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Gaur M, Ritner C, Sievers R, Pedersen A, Prasad M, Bernstein HS, Yeghiazarians Y. Timed inhibition of p38MAPK directs accelerated differentiation of human embryonic stem cells into cardiomyocytes. Cytotherapy. 2010;12:807–817. doi: 10.3109/14653249.2010.491821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge B, Gram H, Di Padova F, Huang B, New L, Ulevitch RJ, Luo Y, Han J. MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science. 2002;295:1291–1294. doi: 10.1126/science.1067289. [DOI] [PubMed] [Google Scholar]
- Gibson S, Widmann C, Johnson GL. Differential involvement of MEK kinase 1 (MEKK1) in the induction of apoptosis in response to microtubule-targeted drugs versus DNA damaging agents. J Biol Chem. 1999;274:10916–10922. doi: 10.1074/jbc.274.16.10916. [DOI] [PubMed] [Google Scholar]
- Gossler A, Doetschman T, Korn R, Serfling E, Kemler R. Transgenesis by means of blastocyst-derived embryonic stem cell lines. Proc Natl Acad Sci USA. 1986;83:9065–9069. doi: 10.1073/pnas.83.23.9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M. Protein degradation or regulation: Ub the judge. Cell. 1996;84:813–815. doi: 10.1016/s0092-8674(00)81058-2. [DOI] [PubMed] [Google Scholar]
- Ikeda F, Crosetto N, Dikic I. What determines the specificity and outcomes of ubiquitin signaling? Cell. 2010;143:677–681. doi: 10.1016/j.cell.2010.10.026. [DOI] [PubMed] [Google Scholar]
- Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15:535–548. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Kang YJ, Seit-Nebi A, Davis RJ, Han J. Multiple activation mechanisms of p38alpha mitogen-activated protein kinase. J Biol Chem. 2006;281:26225–26234. doi: 10.1074/jbc.M606800200. [DOI] [PubMed] [Google Scholar]
- Karin M, Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life. 2005;57:283–295. doi: 10.1080/15216540500097111. [DOI] [PubMed] [Google Scholar]
- Karin M, Gallagher E. TNFR signaling: ubiquitin-conjugated TRAFfic signals control stop-and-go for MAPK signaling complexes. Immunol Rev. 2009;228:225–240. doi: 10.1111/j.1600-065X.2008.00755.x. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kravtsova-Ivantsiv Y, Ciechanover A. Non-canonical ubiquitin-based signals for proteasomal degradation. J Cell Sci. 2012;125:539–548. doi: 10.1242/jcs.093567. [DOI] [PubMed] [Google Scholar]
- Ledermann B. Embryonic stem cells and gene targeting. Exp Physiol. 2000;85:603–613. [PubMed] [Google Scholar]
- Lu Z, Xu S, Joazeiro C, Cobb MH, Hunter T. The PHD domain of MEKK1 acts as an E3 ubiquitin ligase and mediates ubiquitination and degradation of ERK1/2. Mol Cell. 2002;9:945–956. doi: 10.1016/s1097-2765(02)00519-1. [DOI] [PubMed] [Google Scholar]
- Lu Z, Hunter T. Degradation of activated protein kinases by ubiquitination. Annu Rev Biochem. 2009;78:435–475. doi: 10.1146/annurev.biochem.013008.092711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa A, Tseng PH, Vallabhapurapu S, Luo JL, Zhang W, Wang H, Vignali DA, Gallagher E, Karin M. Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science. 2008;321:663–668. doi: 10.1126/science.1157340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T, Yujiri T, Papst PJ, Chan ED, Johnson GL, Terada N. MEKK1 suppresses oxidative stress-induced apoptosis of embryonic stem cell-derived cardiac myocytes. Proc Natl Acad Sci USA. 1999;96:15127–15132. doi: 10.1073/pnas.96.26.15127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T, Yujiri T, Terada N, Taffet GE, Michael LH, Johnson GL, Schneider MD. MEKK1 is essential for cardiac hypertrophy and dysfunction induced by Gq. Proc Natl Acad Sci USA. 2002;99:3866–3871. doi: 10.1073/pnas.062453699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K, Ohtomo T, Sato S, Sugamata Y, Suzuki M, Hisamoto N, Ninomiya-Tsuji J, Tsuchiya M, Matsumoto K. An evolutionarily conserved motif in the TAB1 C-terminal region is necessary for interaction with and activation of TAK1 MAPKKK. J Biol Chem. 2001;276:24396–24400. doi: 10.1074/jbc.M102631200. [DOI] [PubMed] [Google Scholar]
- Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya H, Yamaguchi K, Shirakabe K, Tonegawa A, Gotoh Y, Ueno N, Irie K, Nishida E, Matsumoto K. TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science. 1996;272:1179–1182. doi: 10.1126/science.272.5265.1179. [DOI] [PubMed] [Google Scholar]
- Shim JH, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS, Lee KY, Bussey C, Steckel M, Tanaka N, Yamada G, Akira S, Matsumoto K, Ghosh S. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005;19:2668–2681. doi: 10.1101/gad.1360605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, Ninomiya-Tsuji J, Matsumoto K. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell. 2000;5:649–658. doi: 10.1016/s1097-2765(00)80244-0. [DOI] [PubMed] [Google Scholar]
- Walczak H, Iwai K, Dikic I. Generation and physiological roles of linear ubiquitin chains. BMC Biol. 2012;10:23. doi: 10.1186/1741-7007-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- Wang H, Matsuzawa A, Brown SA, Zhou J, Guy CS, Tseng PH, Forbes K, Nicholson TP, Sheppard PW, Hacker H, Karin M, Vignali DA. Analysis of nondegradative protein ubiquitylation with a monoclonal antibody specific for lysine-63-linked polyubiquitin. Proc Natl Acad Sci USA. 2008;105:20197–20202. doi: 10.1073/pnas.0810461105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr N, Bogani D, Siggers P, Brixey R, Tateossian H, Dopplapudi A, Wells S, Cheeseman M, Xia Y, Ostrer H, Greenfield A. Minor abnormalities of testis development in mice lacking the gene encoding the MAPK signalling component, MAP3K1. PLoS ONE. 2011;6:e19572. doi: 10.1371/journal.pone.0019572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev. 2002;12:14–21. doi: 10.1016/s0959-437x(01)00258-1. [DOI] [PubMed] [Google Scholar]
- Witowsky JA, Johnson GL. Ubiquitylation of MEKK1 inhibits its phosphorylation of MKK1 and MKK4 and activation of the ERK1/2 and JNK pathways. J Biol Chem. 2003;278:1403–1406. doi: 10.1074/jbc.C200616200. [DOI] [PubMed] [Google Scholar]
- Wu J, Kubota J, Hirayama J, Nagai Y, Nishina S, Yokoi T, Asaoka Y, Seo J, Shimizu N, Kajiho H, Watanabe T, Azuma N, Katada T, Nishina H. p38 Mitogen-activated protein kinase controls a switch between cardiomyocyte and neuronal commitment of murine embryonic stem cells by activating myocyte enhancer factor 2C-dependent bone morphogenetic protein 2 transcription. Stem Cells Dev. 2010;19:1723–1734. doi: 10.1089/scd.2010.0066. [DOI] [PubMed] [Google Scholar]
- Xia Y, Makris C, Su B, Li E, Yang J, Nemerow GR, Karin M. MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc Natl Acad Sci USA. 2000;97:5243–5248. doi: 10.1073/pnas.97.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Karin M. The control of cell motility and epithelial morphogenesis by Jun kinases. Trends Cell Biol. 2004;14:94–101. doi: 10.1016/j.tcb.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Xia Y, Wang J, Xu S, Johnson GL, Hunter T, Lu Z. MEKK1 mediates the ubiquitination and degradation of c-Jun in response to osmotic stress. Mol Cell Biol. 2007;27:510–517. doi: 10.1128/MCB.01355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Davis RJ. c-Jun NH2-terminal kinase is required for lineage-specific differentiation but not stem cell self-renewal. Mol Cell Biol. 2010;30:1329–1340. doi: 10.1128/MCB.00795-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell. 2008;31:918–924. doi: 10.1016/j.molcel.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q, Lin SC, Lamothe B, Lu M, Lo YC, Hura G, Zheng L, Rich RL, Campos AD, Myszka DG, Lenardo MJ, Darnay BG, Wu H. E2 interaction and dimerization in the crystal structure of TRAF6. Nat Struct Mol Biol. 2009;16:658–666. doi: 10.1038/nsmb.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yujiri T, Fanger GR, Garrington TP, Schlesinger TK, Gibson S, Johnson GL. MEK kinase 1 (MEKK1) transduces c-Jun NH2-terminal kinase activation in response to changes in the microtubule cytoskeleton. J Biol Chem. 1999;274:12605–12610. doi: 10.1074/jbc.274.18.12605. [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang W, Hayashi Y, Jester JV, Birk DE, Gao M, Liu CY, Kao WW, Karin M, Xia Y. A role for MEK kinase 1 in TGF-beta/activin-induced epithelium movement and embryonic eyelid closure. EMBO J. 2003;22:4443–4454. doi: 10.1093/emboj/cdg440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.