

Abstract
Background and purpose
To assess the long-term safety and efficacy of pramipexole as a once-daily (q.d.) extended-release oral formulation in early or advanced Parkinson's disease (PD).
Methods
In two double-blind (DB) studies of early PD and one of advanced PD, active-treatment arms received pramipexole immediate release (IR) or extended release (ER), with exposure lasting up to 33 weeks. In open-label (OL) extensions that followed immediately, subjects took ER q.d. for up to 80 weeks, with dosage adjustment permitted (range 0.375–4.5 mg q.d.).
Results
Of 590 subjects completing an early-PD DB study, 511 entered the early-PD OL extension; 408 completed it. Reported adverse events (AEs) with incidence ≥10.0% were somnolence (15.1%), peripheral edema (11.7%) and back pain (10.6%). Of 465 subjects completing the advanced-PD DB study, 391 entered the advanced-PD OL extension; 329 completed it. Reported AEs with incidence ≥10.0% were dyskinesia (27.4%) and somnolence (13.6%). Impulse control disorders were identified by semi-structured interview in 13 subjects (1.4% of 902). In exploratory analyses, adjusted mean Unified Parkinson's Disease Rating Scale (UPDRS) Parts II + III scores (excluding ex-placebo recipients) remained substantially improved from DB baseline scores prior to pramipexole introduction, at −6.6 and −6.3 points amongst ex-DB-ER and ex-DB-IR recipients after 113 weeks of pramipexole (33 DB plus 80 OL) in early PD, and −11.5 and −9.1 after up to 113 weeks (up to 33 DB plus 80 OL) in advanced PD.
Conclusions
These results support the long-term safety and efficacy of pramipexole ER in early and advanced PD. AEs were typical for dopaminergic medications, and UPDRS scores suggested sustained symptomatic benefit.
Keywords: once daily, oral formulation, pramipexole, unified Parkinson's disease rating scale
Introduction
Pramipexole extended release (ER) is a once-daily (q.d.) dopamine agonist with demonstrated efficacy in both early and advanced Parkinson's disease (PD). For use in early PD, supporting studies include a 33-week, double-blind (DB) trial 1 in which subjects were randomized to pramipexole ER q.d., pramipexole immediate release (IR) three times daily (t.i.d.) or placebo. In another trial 2 lasting 11–13 weeks, the safety and tolerability of an overnight switch from pramipexole IR to pramipexole ER in early PD was evaluated. For this purpose, non-fluctuating patients on pramipexole IR t.i.d., alone or in combination with levodopa, were randomly switched overnight to DB treatment with pramipexole ER q.d. or pramipexole IR t.i.d. For use in advanced PD, supportive evidence derives from a 33-week DB trial 3 in which subjects with motor fluctuations on levodopa were randomized to the addition of pramipexole ER q.d., pramipexole IR t.i.d. or placebo.
Subjects completing one or another of these three studies were invited to enroll immediately in a long-term open-label (OL) extension study, one for early and another for advanced PD (Table1). In all three initial studies, DB treatment was administered t.i.d. – specifically, IR or placebo as morning, afternoon and evening doses, plus placebo or ER as a morning dose (double-dummy design). In both of the extensions, the OL treatment was pramipexole ER, taken q.d. This report describes the results of the extensions.
Table 1.
Study-drug treatments during each OL extension and its preceding DB trial(s)
| PD phase studied | Preceding DB trial | OL extension | Planned treatment duration (total) | ||
|---|---|---|---|---|---|
| Duration | Treatments | Duration (transfer + maintenance) | Treatment | ||
| Early | 33 weeks | ER q.d. + IR placebo t.i.d. | 6 + 74 weeks | ER q.d. | 113 weeks |
| IR t.i.d. + ER placebo q.d. | |||||
| ER placebo q.d. + IR placebo t.i.d. | |||||
| 11–13 weeksa | ER q.d. + IR placebo t.i.d. | 0 + 72 weeks | ER q.d. | 83–85 weeksa | |
| IR t.i.d. + ER placebo q.d. | |||||
| Advanced | Up to 33 weeks | ER q.d. + IR placebo t.i.d. | 6 + 74 weeks | ER q.d. | Up to 113 weeks |
| IR t.i.d. + ER placebo q.d. | |||||
| ER placebo q.d. + IR placebo t.i.d. | |||||
DB, double-blind; ER, extended-release pramipexole; IR, immediate-release pramipexole; OL, open-label; PD, Parkinson's disease; q.d., once daily; t.i.d., three times daily.
Not including IR pre-treatment for ≥3 months.
Patients and methods
Study subjects
For the OL extension study of early PD, each of the initial two DB trials 1,2 had required that all subjects had PD at Hoehn and Yahr 4 stage 1–3, diagnosed within the previous 5 years at an age ≥30 years. In one of these DB trials – of ER or IR versus placebo for 33 weeks 1 – all subjects had needed initiation or augmentation of dopaminergic therapy. Previous levodopa exposure of <3 months' total duration was permitted if it had been discontinued ≥8 weeks prior to randomization. Previous dopamine-agonist exposure was also permitted if it had been discontinued ≥4 weeks prior to randomization. In the other DB trial – of a switch to ER versus continued IR in early PD 2 – subjects had already been under IR treatment for ≥3 months, with a stable, optimized dosage ≥0.5 mg t.i.d. for the 4 weeks preceding randomization. During this trial, pramipexole exposure lasted 11–13 weeks (comprising 2–4 weeks of OL IR run-in followed by 9 weeks of DB IR or ER maintenance); hence total pramipexole exposure, including the IR pre-treatment, was at least 23–25 weeks. Concomitant levodopa was permitted, provided the dosage had been stable for ≥4 weeks.
For the OL extension study of advanced PD, the preceding DB trial – of ER or IR versus placebo for up to 33 weeks 3 – had required that all subjects have PD at Hoehn and Yahr stage 2–4 during ‘on’ time, diagnosed ≥2 years before entry, and were being treated with levodopa (plus a dopa-decarboxylase inhibitor) at an optimized dosage unchanged for ≥4 weeks. Subjects were also required to be experiencing motor fluctuations (≥2 h of daily ‘off’ time, as documented by patient diary on two consecutive days). Subjects were not permitted any dopamine agonists within the previous 4 weeks.
In the OL extensions, as in the preceding DB trials, patients were excluded for secondary or atypical parkinsonism and for medical or psychiatric conditions capable of impeding the patient's trial participation.
Study designs
In the extension study of early PD, completers of the preceding 33-week trial 1 underwent 6-week blinded down-titration of their DB pramipexole or placebo, administered q.d. during this transfer phase (after IR recipients were switched overnight to ER), and concomitant optimization of OL ER. They then entered a 74-week OL ER maintenance phase. The planned total treatment duration was therefore 113 weeks. Completers of the preceding 11- to 13-week early-PD trial 2 were switched to OL ER from DB ER or IR and entered a 72-week OL maintenance phase (planned total treatment duration 83–85 weeks, not including their IR pre-treatment). In the extension study of advanced PD, completers of the preceding up-to-33-week trial 3 underwent a 6-week transfer from DB ER, IR or placebo to OL ER (as described above), followed by 74 weeks of OL maintenance (planned total treatment duration up to 113 weeks). During the extension studies, the maintenance dosage of OL ER could be adjusted (range 0.375–4.5 mg q.d.), and concomitant PD drugs were allowed at stable dosages (as they had been in the DB trials).
Safety measures
Throughout each OL extension, safety was assessed by the incidence of adverse events (AEs) and of withdrawals due to AEs. To this end, subjects were asked to spontaneously report AEs and were queried about AEs at all on-site visits. For subjects from the 33-week study of early PD 1 and the up-to-33-week study of advanced PD 3, the visits were scheduled to occur weekly during the 6-week transfer phase and then at weeks 2, 8, 14, 20, 26, 38, 50, 62 and 74 of OL ER maintenance, with a follow-up visit at week 75. For subjects from the switch study in early PD 2, the visits were scheduled for weeks 6, 12, 18, 24, 36, 48, 60 and 72 of OL ER maintenance, with follow-up at week 73.
The incidence of impulse control disorders was assessed by a modified Minnesota Impulsive Disorders Interview (mMIDI) 5 consisting of items for pathological gambling, compulsive buying and compulsive sexual behavior. Subjects from the 33-week and the up-to-33-week studies 1,3 completed the mMIDI at maintenance weeks 26, 74 and 75, and subjects from the switch study 2 did so at maintenance weeks 4, 72 and 73. At all other visits, all subjects were asked whether they had experienced pathological gambling, compulsive buying or compulsive sexual behavior, and all visits included an open-ended question concerning ‘any other abnormal behaviors or urges’, which were recorded as AEs.
Daytime somnolence was assessed using the Epworth Sleepiness Scale (ESS) 6. Subjects from the 33-week and the up-to-33-week studies 1,3 completed it at week 3 of the transfer phase and weeks 2, 26, 50 and 74 of maintenance treatment. Subjects from the switch study 2 completed it at maintenance weeks 24, 48 and 72.
Efficacy measures
In each of the DB trials, pramipexole efficacy had been assessed primarily by a combined score on Parts II + III of the Unified Parkinson's Disease Rating Scale (UPDRS) 7. In each extension study, sustained pramipexole efficacy [necessarily excluding subjects who had previously been receiving placebo (ex-placebo recipients)] was assessed at end-point – 80 or 72 OL weeks, depending on the prior DB trial – as an exploratory analysis, again using UPDRS Parts II + III. For subjects with advanced PD, the Part II score was the average for ‘on’ and ‘off’ time and Part III was scored during best ‘on’ time. Exploratory assessments at end-point also included a UPDRS II + III responder rate (defined as a ≥20% improvement from baseline in the preceding DB trial), change in OL ER dosage and change in levodopa dosage. In advanced PD, analyses also included an ‘off’ time responder rate (defined as a ≥20% decrease from DB baseline in ‘off’ time as a percentage of the subject's waking day).
Statistical analyses
Because the extension studies had no randomized groups, no statistical sample-size need was calculated and all findings are descriptive. Nevertheless, mean changes in UPDRS II + III score were adjusted by analysis of covariance (ancova), with country and DB treatment as fixed effects and DB baseline as covariate. All analyses were of observed cases.
Ethics and good clinical practice
All studies were conducted in accordance with their protocols, with good clinical practice and with the provisions of the Declaration of Helsinki and its amendments. Before subjects were enrolled, each study's protocol, informed consent form and all protocol amendments were approved by local Institutional Review Boards or Independent Ethics Committees. Each study's nature and purpose were explained to its subjects, who provided written informed consent before any study procedures. The studies were registered with ClinicalTrials.gov with identifiers NCT00601523 and NCT00577460.
Results
Disposition of subjects
Of 441 subjects completing the 33-week DB trial in early PD 1, 368 (83.4%) entered the OL extension. Amongst them, 291 (79.1% of 368) completed the extension. Of 149 subjects completing the 11- to 13-week DB trial in early PD 2, 143 (96.0%) entered the OL extension. Amongst them, 117 (81.8% of 143) completed the extension. Subjects participated in the early-PD extension at 105 centers in 15 countries, chiefly European (65.2% of subjects), beginning in January to November 2008. Of 465 subjects completing the DB trial in advanced PD 3, 391 (84.1%) entered its OL extension. Amongst them, 329 (84.1% of 391) completed the extension. Subjects participated at 70 centers in 14 countries, Asian and European (50.9% and 49.1% of subjects, respectively), beginning in January to November 2008. Disposition of subjects throughout both extension studies is summarized in Fig.1, grouped by preceding DB trial.
Figure 1.
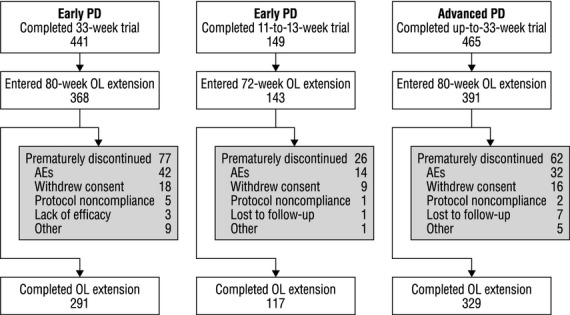
Disposition of subjects. AEs, adverse events; OL, open-label; PD, Parkinson's disease.
Baseline characteristics
The characteristics of all OL ER recipients at OL baseline are summarized in Table2, grouped by preceding DB trial.
Table 2.
Subjects' characteristics at OL baseline (all treated subjects)
| Early PD | Advanced PD | ||
|---|---|---|---|
| Duration of preceding study | 33 weeks | 11–13 weeksa | Up to 33 weeks |
| N in OL extension | 368 | 143 | 391 |
| Sex, n (% of group) | |||
| Males | 204 (55.4) | 81 (56.6) | 213 (54.5) |
| Females | 164 (44.6) | 62 (43.4) | 178 (45.5) |
| Age (years), mean (SD) | 62.2 (9.2) | 64.0 (9.0) | 61.6 (9.7) |
| Race, n (% of group) | |||
| White | 222 (60.3) | 139 (97.2) | 192 (49.1) |
| Asian | 146 (39.7) | 4 (2.8) | 199 (50.9) |
| PD duration (years), mean (SD) | 1.6 (1.1) | 3.6 (2.1) | 7.0 (4.2) |
| Hoehn and Yahr stage, n (% of group)b | |||
| 1–1.5 | 147 (39.9) | 59 (41.3) | 2 (0.5) |
| 2–3 | 221 (60.1) | 84 (58.7) | 351 (89.8) |
| 4–5 | 0 (0.0) | 0 (0.0) | 37 (9.5) |
| Missing | 0 (0.0) | 0 (0.0) | 1 (0.3) |
| UPDRS score, mean (SD) | |||
| Part IIc | 5.2 (3.7) | 6.8 (3.7) | 8.8 (6.0)d |
| Part III | 13.8 (8.5) | 14.6 (8.2) | 19.7 (12.3) |
| Parts II+III | 19.0 (11.6) | 21.4 (11.0) | 28.6 (17.4)d |
| ‘Off’ time, mean (SD) | |||
| % of waking day | – | – | 23.6 (17.0)e |
| Hours/day | – | – | 3.7 (2.8)e |
OL, open-label; PD, Parkinson's disease; SD, standard deviation; UPDRS, Unified Parkinson's Disease Rating Scale.
Not including IR pre-treatment for ≥3 months
in advanced PD, assessed during ‘on’ time
in advanced PD, ‘on’ time/‘off ’ time average
n = 389
n = 386.
AE patterns
AE incidence during the OL extensions is summarized in Table3. AE types are listed by the Medical Dictionary for Drug Regulatory Activities (Version 13) preferred term.
Table 3.
Summary of adverse events (all treated subjects)
| Early PD | Advanced PD | |
|---|---|---|
| N | 511 | 391 |
| n (% of group) with | ||
| Any AE | 420 (82.2) | 324 (82.9) |
| Mild | 185 (36.2) | 171 (43.7) |
| Moderate | 187 (36.6) | 106 (27.1) |
| Severe | 49 (9.6) | 47 (12.0) |
| Any study-drug-related AEa | 201 (39.3) | 191 (48.8) |
| Any serious AE | 74 (14.5) | 39 (10.0) |
| Any AE leading to study-drug discontinuation | 50 (9.8) | 31 (7.9) |
| Death | 8 (1.6) | 4 (1.0) |
| Study-drug-related deatha | 0 (0.0) | 0 (0.0) |
| Reported in ≥5% of subjects, n (% of group)b | ||
| Somnolence | 77 (15.1) | 53 (13.6) |
| Edema peripheral | 60 (11.7) | 18 (4.6) |
| Back pain | 54 (10.6) | 19 (4.9) |
| Nausea | 47 (9.2) | 24 (6.1) |
| Nasopharyngitis | 37 (7.2) | 15 (3.8) |
| Dizziness | 35 (6.8) | 29 (7.4) |
| Cataract | 32 (6.3) | 26 (6.6) |
| Fall | 29 (5.7) | 28 (7.2) |
| Insomnia | 25 (4.9) | 27 (6.9) |
| Hallucination | 19 (3.7) | 23 (5.9) |
| Dystonia | 4 (0.7) | 20 (5.1) |
| Dyskinesia | 2 (0.4) | 107 (27.4) |
AE, adverse event; PD, Parkinson's disease.
Investigator-defined
in either the early-PD or the advanced-PD group.
In early PD, 82.2% of subjects reported one or more AEs. The most frequent types (incidence ≥5.0%) were somnolence, peripheral edema, back pain, nausea, nasopharyngitis, dizziness, cataract and fall. Amongst AEs classified as severe, the types reported in more than one subject were myocardial infarction, in four subjects (1.1%); pneumonia, in three subjects (0.8%); and back pain, fall and psychotic disorder, each in two subjects (0.5%). In advanced PD, 82.9% of subjects reported AEs. The most frequent types were dyskinesia, somnolence, dizziness, fall, insomnia, cataract, nausea, hallucination and dystonia. Amongst AEs classified as severe, the types reported in more than one subject were dyskinesia, in nine subjects (2.3%); fall and headache, each in three subjects (0.8%); and dizziness, dyspnea, hypotension, somnolence and tremor, each in two subjects (0.5%).
In early PD, 39.3% of subjects reported one or more AEs considered to be treatment related. The most frequent types (incidence ≥2.0%) were somnolence, in 65 subjects (12.7%); peripheral edema, in 36 subjects (7.0%); nausea, in 34 subjects (6.7%); dizziness, in 17 subjects (3.3%); hallucination, in 14 subjects (2.7%); fatigue, in 13 subjects (2.5%); and vomiting, in 10 subjects (2.0%). In advanced PD, 48.8% of subjects reported AEs considered to be treatment related. The most frequent types were dyskinesia, in 77 subjects (19.7%); somnolence, in 43 subjects (11.0%); hallucination, in 17 subjects (4.3%); nausea, in 16 subjects (4.1%); insomnia, in 13 subjects (3.3%); dizziness, in 11 subjects (2.8%); dystonia, in 10 subjects (2.6%); dizziness postural, in nine subjects (2.3%); and hypersomnia and peripheral edema, each in eight subjects (2.0%).
In early PD, 74 subjects (14.5%) had AEs classified as serious (SAEs). The types seen in at least three subjects (0.6%) were fall, in eight subjects (1.6%); myocardial infarction, in five subjects (1.0%); PD, in four subjects (0.8%); and femoral-neck fracture, hallucination, osteoarthritis and pneumonia, each in three subjects (0.6%). Six subjects' SAEs were considered to be treatment related: hallucination, in two subjects (0.4%), and hepatitis, PD, psychotic disorder and a traffic accident associated with sleep attack, each in one subject (0.2%). In advanced PD, 39 subjects (10.0%) had SAEs. The types seen in at least two subjects (0.5%) were fall, in five subjects (1.3%); pneumonia, in three subjects (0.8%); and cataract, contusion, femoral-neck fracture and hyponatremia, each in two subjects (0.5%). Five subjects' SAEs were considered to be treatment related: hypertensive encephalopathy, hypotension, pathological gambling, psychotic disorder, and hallucination and paramnesia, each in one subject (0.3%).
In early PD, 50 subjects (9.8%) had AEs leading to study-drug discontinuation. The types reported in at least three subjects (0.6%) were hallucination, in five subjects (1.0%); peripheral edema, in four subjects (0.8%); and abdominal pain upper, myocardial infarction, nausea, PD, pneumonia and vomiting, each in three subjects (0.6%). In advanced PD, 31 subjects (7.9%) had AEs leading to study-drug discontinuation. The only types reported in more than one subject were hallucination, in five subjects (1.3%); hypotension, in two subjects (0.5%); and peripheral edema, in two subjects (0.5%).
Of the eight deaths in early PD and four in advanced PD, none was considered to be treatment related. In early PD, five deaths were from cardiac causes, one from cerebrovascular accident, one from pulmonary embolism and one by drowning. In advanced PD, two deaths had cerebrovascular causes, one had cardiac and cardiovascular causes, and one resulted from pneumonia, septic shock and multi-organ failure.
Impulse control disorders
In early PD, abnormal behaviors were identified by mMIDI in four ER recipients (0.8%), including three cases of compulsive buying and one of pathological gambling. Questioning yielded positive responses in another nine subjects (1.8%). As AEs, pathological gambling was reported in four subjects (0.8%), and compulsive sexual behavior, compulsive shopping, hypersexuality and libido increased were each reported in two (0.4%).
In advanced PD, abnormal behaviors were identified by mMIDI in nine ER recipients (2.3%), including three cases of compulsive buying, three of compulsive sexual behavior, two of pathological gambling, and one of both compulsive buying and compulsive sexual behavior. Questioning yielded positive responses in another nine subjects (2.3%). As AEs, compulsive sexual behavior and compulsive shopping were each reported in four subjects (1.0%), pathological gambling in three (0.8%) and libido increased in one (0.3%).
Daytime somnolence
In early PD, the proportion of subjects with an ESS score >10 was 21.2% at OL baseline and 24.2% at end of treatment. As reported AEs, four subjects (0.8%) experienced sudden onset of sleep and three (0.6%) experienced sleep attacks. In advanced PD, the proportion of subjects with an ESS score >10 was 26.6% at OL baseline and 33.7% at end of treatment. As reported AEs, three subjects (0.8%) experienced sleep attacks and none experienced sudden onset of sleep.
Sustained efficacy
The characteristics of OL ER recipients at OL baseline, excluding those who received placebo in a preceding DB trial, are summarized in Table4.
Table 4.
Subjects' characteristics at OL baseline (all treated subjects, excluding ex-placebo recipients)
| Early PD | Advanced PD | ||
|---|---|---|---|
| Duration of preceding study | 33 weeks | 11–13 weeksa | Up to 33 weeks |
| N in OL extension | 292 | 143 | 262 |
| Sex, n (% of group) | |||
| Males | 165 (56.5) | 81 (56.6) | 142 (54.2) |
| Females | 127 (43.5) | 62 (43.4) | 120 (45.8) |
| Age (years), mean (SD) | 62.1 (9.2) | 64.0 (9.0) | 61.7 (9.8) |
| Race, n (% of group) | |||
| White | 174 (59.6) | 139 (97.2) | 126 (48.1) |
| Asian | 118 (40.4) | 4 (2.8) | 136 (51.9) |
| PD duration (years), mean (SD) | 1.7 (1.2) | 3.6 (2.1) | 7.2 (4.4) |
| Hoehn and Yahr stage, n (% of group)b | |||
| 1–1.5 | 117 (40.1) | 59 (41.3) | 3 (1.1) |
| 2–3 | 175 (59.9) | 84 (58.7) | 252 (96.2) |
| 4–5 | 0 (0.0) | 0 (0.0) | 6 (2.3) |
| Missing | 0 (0.0) | 0 (0.0) | 1 (0.4) |
| UPDRS score, mean (SD) | |||
| Part IIc | 5.0 (3.7) | 6.8 (3.7) | 8.6 (6.1)d |
| Part III | 13.2 (8.2) | 14.6 (8.2) | 18.6 (12.2) |
| Parts II + III | 18.2 (11.2) | 21.4 (11.0) | 27.2 (17.5)d |
| ‘Off’ time, mean (SD) | |||
| % of waking day | – | – | 21.5 (16.0)e |
| Hours/day | – | – | 3.4 (2.6)e |
| Levodopa users at OL baseline,n (% of group) | 18 (6.2) | 77 (53.8) | 261 (99.6) |
| Concomitant PD medication users during theOL extension, n (% of group) | 212 (72.6) | 129 (90.2) | 262 (100.0) |
OL, open-label; PD, Parkinson's disease; SD, standard deviation; UPDRS, Unified Parkinson's Disease Rating Scale.
Not including IR pre-treatment for ≥3 months
in advanced PD, assessed during ‘on’ time
in advanced PD, ‘on’ time/‘off ’ time average
n = 260
n = 261.
Of 292 early-PD patients from the preceding 33-week DB trial 1 (excluding 76 placebo recipients), 6.2% were taking levodopa at OL baseline. On UPDRS Parts II + III amongst 234 observed cases, mean changes in the ex-DB-ER and ex-DB-IR groups (adjusted for country, DB treatment and DB baseline) were −6.6 and −6.3 from DB baseline and +3.1 and +3.9 from OL baseline. Mean values at DB baseline and OL end-point (80 OL weeks) are displayed in Fig.2a.
Figure 2.
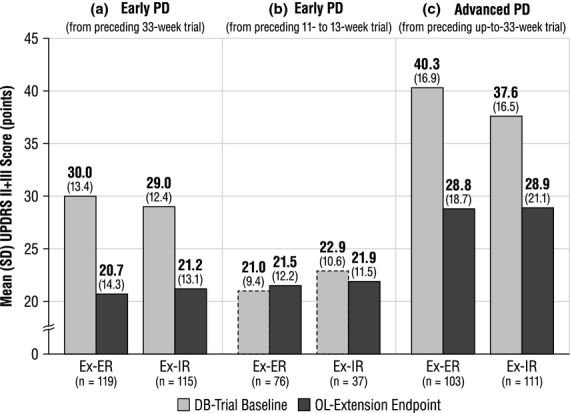
Mean (SD) UPDRS II + III scores at DB baseline and OL end-point (observed cases). (a) DB baseline UPDRS II + III scores from the 33-week early-PD trial prior to initiation of pramipexole according to the initial treatment assignment (light gray bars), and OL extension UPDRS II + III end-point scores with all subjects on pramipexole ER (dark gray bars). (b) DB baseline UPDRS II + III scores from the 11–13-week switch study according to switch assignment (dashed light gray bars). At DB baseline, subjects had been on pramipexole IR for ≥3 months. Dark gray bars illustrate UPDRS II + III scores at OL extension end-point with all subjects on pramipexole ER. (c) DB baseline UPDRS II + III scores from the up-to-33-week advanced PD trial prior to initiation of pramipexole according to the initial treatment assignment (light gray bars). All subjects had been experiencing motor fluctuations despite optimized levodopa. UPDRS Part II score was averaged for ‘on’ and ‘off’ time, and Part III was scored for ‘on’ time. Dark gray bars illustrate OL extension UPDRS II + III end-point scores with all subjects on pramipexole ER. DB, double-blind; ER, extended-release pramipexole; IR, immediate-release pramipexole; OL, open-label; PD, Parkinson's disease; SD, standard deviation; UPDRS II + III, Unified Parkinson's Disease Rating Scale Parts II + III.
Of 143 early-PD patients from the preceding 11- to 13-week DB trial 2, 53.8% were taking levodopa at OL baseline. Amongst 113 observed cases, adjusted mean UPDRS II + III changes from DB baseline (at least 3 months after pramipexole initiation) were +0.5 in the ex-DB-ER group and −1.0 in the ex-DB-IR group. Mean changes from OL baseline were +2.6 and +0.2. Mean values at DB baseline and OL end-point (72 OL weeks) are displayed in Fig.2b.
Of 262 advanced-PD patients from the preceding 33-week DB trial 3 (excluding 129 placebo recipients), 99.6% were taking levodopa at OL baseline. Amongst 214 observed cases, adjusted mean UPDRS II + III changes in the ex-DB-ER and ex-DB-IR groups were −11.5 and −9.1 from DB baseline and +1.1 and +2.5 from OL baseline. Mean values at DB baseline and OL end-point (80 OL weeks) are displayed in Fig.2c.
OL extension findings for UPDRS II + III responder rate, ‘off’ time responder rate, OL ER dosage and levodopa usage and dosage are presented in Table S1.
Discussion
Results from these OL extension studies support the long-term safety and efficacy of pramipexole ER. During OL treatment for up to 80 weeks (in patients whose total exposure was up to 113 weeks), the AEs reported in early and in advanced PD had patterns typical for use of dopaminergic medications in an aging PD population, with somnolence and peripheral edema the most frequent AE types in early PD and dyskinesia and somnolence the most frequent types in advanced PD. By ESS self-ratings, however, daytime somnolence appeared to show little change during the studies (from a substantial prevalence at OL baseline). Overall, 9.8% of subjects (88 of 902) discontinued prematurely because of AEs. Here, too, the AE types were consistent with advancing age, PD and dopaminergic medication. By semi-structured interviews, impulse control disorders were identified in 1.4% of all subjects (13 of 902), with a comparably small number of additional cases uncovered by other forms of questioning.
Amongst the studies' measures of long-term ER efficacy, UPDRS II + III scores remained substantially improved after 80 weeks of OL treatment in both early and advanced PD, as compared with the DB baseline in subjects from the two DB trials in which the DB baseline value was obtained prior to introduction of pramipexole 1,3. Compared with OL baseline, UPDRS scores after 72–80 weeks were unchanged to mildly worse, as might be expected with disease progression.
With regard to both safety and efficacy, the extension studies' OL design and their recruitment of subjects from prior trials may be considered limitations to interpreting the data. During the prior trials, patients relatively intolerant of study drug and those with small subjective improvement may have dropped out or become disinclined to enroll in a further trial. Recruitment from prior trials may also have decreased the extension studies' incidence of AEs with a tendency to occur or be especially noticeable early in treatment. Nevertheless, an OL design permitted assessment of ER safety and efficacy for a duration of treatment essentially infeasible in a PD study with placebo control. The design also permitted a treatment-convenience analysis, published elsewhere 8, in which extension subjects were queried as to their preference between their ongoing q.d. dosing and their prior t.i.d. dosing. Ninety-four percent of 374 early-PD patients and 89% of 334 advanced-PD patients preferred q.d. dosing. Greater convenience may translate into better adherence and a better clinical response, although this has not been formally tested.
Pramipexole ER remains an important therapeutic option in early and advanced PD. The OL extension studies reported here support its long-term safety and efficacy.
Acknowledgments
The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors, were fully responsible for all content and editorial decisions, and were involved at all stages of manuscript development. The authors received no compensation related to the development of the manuscript. This work was supported by Boehringer Ingelheim Pharmaceuticals Inc. (BIPI). Writing, editorial support and/or formatting assistance was provided by Michael Feirtag, of the Curry Rockefeller Group, which was contracted and compensated by BIPI for these services.
This study was supported by Boehringer Ingelheim GmbH.
Disclosure of conflicts of interest
Dr Hauser has received honoraria or payments for consulting, advisory services, speaking services over the past 12 months from Abbott Laboratories, Allergan Inc., AstraZeneca, Ceregene Inc., Chelsea Therapeutics Inc., GE Healthcare, Impax Laboratories Inc., Ipsen Biopharmaceuticals Inc., Lundbeck, Med-IQ, Merck/MSD, Noven Pharmaceuticals Inc., Straken Pharmaceuticals Ltd, Targacept Inc., Teva Pharmaceutical Industries Ltd, Teva Neuroscience Inc., Upsher-Smith Laboratories, UCB Inc., UCB Pharma SA and XenoPort Inc. Dr Hauser's institution has received research support over the past 12 months from Abbott Laboratories, Addex Therapeutics, Allergan Inc., AstraZeneca, Chelsea Therapeutics Inc., GE Healthcare, Impax Laboratories Inc., Ipsen Biopharmaceuticals Inc., Merck/MSD, Merz, Michael J. Fox Foundation for Parkinson's Research, Schering-Plough, Teva Neuroscience Inc., UCB Inc. and Vita-Pharm. Dr Hauser has received royalties in the last 12 months from the University of South Florida. In addition, Dr Hauser has consulted in litigation with lawyers representing various current and former manufacturers of welding consumables.
Dr Schapira has received honoraria or consulting fees from Boehringer Ingelheim, Teva-Lundbeck, Novartis-Orion, GSK, UCB and Merck-Serono.
Dr Barone has received compensation for consulting services and/or research support from Boehringer Ingelheim, as well as for consulting services and symposia from Novartis, Schwarz Pharma/UCB, Merck-Serono, Solvay, General Electric and Lundbeck. He receives a salary from the University of Salerno, Italy.
Dr Mizuno has received compensation as an advisory board member from Boehringer Ingelheim Japan, FP Pharmaceutical Company, Otsuka Pharmaceutical Company, Abbott Pharmaceutical Company and Kyowa Hakko-Kirin Pharmaceutical Company. He has received funding to his institution for research from Boehringer Ingelheim Japan and Medtronics Japan.
Dr Rascol has received personal compensation for advisory boards: Eisai, Novartis, GSK, Lundbeck, Teva, Solvay, Pfizer, Servier and Kyowa; is a consultant for Boehringer Ingelheim, Novartis, GSK, Servier, Eisai, Solvay, Merck and Serono; and has received financial support for research activities from Servier, Eisai and Pfizer (unrestricted grant to the ParkMiP cohort of parkinsonian patients).
Drs Busse, Debieuvre and Fraessdorf are employees of Boehringer Ingelheim.
Dr Poewe has received personal compensation for consultancy and speaking from Abbott, AstraZeneca, Teva, Novartis, GSK, Boehringer Ingelheim, UCB, Orion Pharma, Merck Serono and Merz Pharmaceuticals.
Supporting Information
Additional Supporting Information may be found in the online version of this article
Responder rates and dosage changes (observed cases, excluding ex-placebo recipients).
References
- 1.Poewe W, Rascol O, Barone P, et al. Extended-release pramipexole in early Parkinson disease: a 33-week randomized controlled trial. Neurology. 2011;77:759–766. doi: 10.1212/WNL.0b013e31822affb0. [DOI] [PubMed] [Google Scholar]
- 2.Rascol O, Barone P, Hauser RA, et al. Efficacy, safety, and tolerability of overnight switching from immediate- to once daily extended-release pramipexole in early Parkinson's disease. Mov Disord. 2010;25:2326–2332. doi: 10.1002/mds.23262. [DOI] [PubMed] [Google Scholar]
- 3.Schapira AHV, Barone P, Hauser RA, et al. Extended-release pramipexole in advanced Parkinson's disease: a randomized controlled trial. Neurology. 2011;77:767–774. doi: 10.1212/WNL.0b013e31822affdb. [DOI] [PubMed] [Google Scholar]
- 4.Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology. 1967;17:427–442. doi: 10.1212/wnl.17.5.427. [DOI] [PubMed] [Google Scholar]
- 5.Grant JE, Levine L, Kim D, Potenza MN. Impulse control disorders in adult psychiatric inpatients. Am J Psychiatry. 2005;162:2184–2188. doi: 10.1176/appi.ajp.162.11.2184. [DOI] [PubMed] [Google Scholar]
- 6.Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep. 1991;14:540–545. doi: 10.1093/sleep/14.6.540. [DOI] [PubMed] [Google Scholar]
- 7.Fahn S, Elton RL. UPDRS Development Committee. Unified Parkinson's Disease Rating Scale. In: Fahn S, Marsden CD, Calne DB, Goldstein M, editors. Recent Developments in Parkinson's Disease. Florham Park, NJ: Macmillan; 1987. pp. 153–163. [Google Scholar]
- 8.Schapira AH, Barone P, Hauser RA, et al. Patient-reported convenience of once-daily versus three-times-daily dosing during long-term studies of pramipexole in early and advanced Parkinson's disease. Eur J Neurol. 2012;20:50–56. doi: 10.1111/j.1468-1331.2012.03712.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Responder rates and dosage changes (observed cases, excluding ex-placebo recipients).