

Abstract
The phase 3 ODYSSEY OPTIONS studies (OPTIONS I, NCT01730040; OPTIONS II, NCT01730053) are multicenter, multinational, randomized, double‐blind, active‐comparator, 24‐week studies evaluating the efficacy and safety of alirocumab, a fully human monoclonal antibody targeting proprotein convertase subtilisin/kexin type 9, as add‐on therapy in ∼ 650 high‐cardiovascular (CV)‐risk patients whose low‐density lipoprotein cholesterol (LDL‐C) levels are ≥100 mg/dL or ≥70 mg/dL according to the CV‐risk category, high and very high CV risk, respectively, with atorvastatin (20–40 mg/d) or rosuvastatin (10–20 mg/d). Patients are randomized to receive alirocumab 75 mg via a single, subcutaneous, 1‐mL injection by prefilled pen every 2 weeks (Q2W) as add‐on therapy to atorvastatin (20–40 mg) or rosuvastatin (10–20 mg); or to receive ezetimibe 10 mg/d as add‐on therapy to statin; or to receive statin up‐titration; or to switch from atorvastatin to rosuvastatin (OPTIONS I only). At week 12, based on week 8 LDL‐C levels, the alirocumab dose may be increased from 75 mg to 150 mg Q2W if LDL‐C levels remain ≥100 mg/dL or ≥70 mg/dL in patients with high or very high CV risk, respectively. The primary efficacy endpoint in both studies is difference in percent change in calculated LDL‐C from baseline to week 24 in the alirocumab vs control arms. The studies may provide guidance to inform clinical decision‐making when patients with CV risk require additional lipid‐lowering therapy to further reduce LDL‐C levels. The flexibility of the alirocumab dosing regimen allows for individualized therapy based on the degree of LDL‐C reduction required to achieve the desired LDL‐C level.
Introduction
Statins are first‐line therapy for reducing atherosclerotic cardiovascular disease (ASCVD) risk.1 Several international guidelines recommend high‐risk and very high‐risk patients achieve low‐density lipoprotein cholesterol (LDL‐C) levels <100 mg/dL and <70 mg/dL, respectively.2, 3, 4 However, many patients fail to reach expected or recommended LDL‐C levels on statin therapy. For example, a 2012 cross‐sectional survey reported that approximately two‐thirds of high‐risk patients on statin monotherapy for >90 days did not achieve LDL‐C levels <100 mg/dL, and only an estimated quarter of these high‐risk patients had LDL‐C levels <70 mg/dL.5 Surveys from other countries show similar results.6, 7, 8, 9 A European study found that of dyslipidemic patients receiving lipid‐lowering drugs, fewer than half achieved LDL‐C levels <100 mg/dL.6 In addition, studies have shown that approximately 80% of patients with heterozygous familial hypercholesterolemia (heFH) do not achieve LDL‐C levels <100 mg/dL from a statin.10, 11
In clinical practice, patients are typically started on a statin of moderate intensity, where the daily dose is expected to lower LDL‐C levels by approximately 30% to <50%.12, 13 However, the recent 2013 American College of Cardiology/American Heart Association Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults recommended high‐intensity statin therapy that lowers LDL‐C by ≥50% for higher‐risk patients, such as those with clinical ASCVD, diabetes mellitus (with an estimated 10‐year ASCVD risk ≥7.5%), and for LDL‐C ≥190 mg/dL.1
If patients fail to reach target LDL‐C levels or achieve the expected therapeutic response on their initial statin therapy, the treating clinician currently has the options of increasing the statin dose or switching to a more effective LDL‐C–lowering statin; and for those unable to tolerate high‐intensity statins, nonstatin therapy may be added to further lower LDL‐C (most commonly ezetimibe).1 Given the degree to which hypercholesterolemia remains poorly controlled, even with statin therapy, high–CV‐risk patients may benefit from more intensive cholesterol‐lowering therapy options.1, 5, 14
Alirocumab (formerly REGN727/SAR236553; Sanofi‐Regeneron) is a fully human monoclonal antibody currently in phase 3 development for LDL‐C lowering. In phase 2 trials in patients receiving concomitant statin or statin + ezetimibe therapy (NCT01288443, NCT01266876, and NCT01288469), alirocumab significantly reduced mean LDL‐C levels by up to 72.4% and also reduced levels of apolipoprotein B, total cholesterol, non–high‐density lipoprotein cholesterol (non–HDL‐C), lipoprotein(a), and triglycerides (TG), and moderately increased HDL‐C and apolipoprotein A‐1 levels.15, 16, 17 The most common treatment‐emergent adverse event (TEAE; defined as an adverse event (AE) occurring in the time period from first study treatment dosing to 70 days following the last study treatment dosing) in phase 2 trials was injection‐site reaction, which was generally of mild intensity and short duration.15, 16, 17
The OPTIONS I and II studies (NCT01730040 and NCT01730053, respectively) will compare the lipid‐lowering efficacy and safety of alirocumab with various therapeutic choices in patients at high CV risk on atorvastatin 20 mg or 40 mg daily or rosuvastatin 10 mg or 20 mg daily and who had LDL‐C levels above prespecified levels of ≥70 mg/dL or ≥100 mg/dL. The OPTIONS studies are part of the 14‐study ODYSSEY alirocumab phase 3 program that includes more than 23 500 patients in >2000 centers globally; this also comprises a large CV outcomes trial evaluating the long‐term impact of alirocumab and lower levels of LDL‐C on the occurrence of CV events in 18 000 patients after a recent (<52 weeks) acute coronary syndrome event. Here we describe the OPTIONS I and II study designs and discuss how these studies may inform clinical management.
Methods
Overview
The OPTIONS I and OPTIONS II clinical trials are phase 3, multinational, multicenter, randomized, double‐blind, active‐comparator, and parallel‐group studies that were conducted across North America, Europe, and Australia. OPTIONS I commenced October 2012, was conducted at 91 sites, and completed in June 2014. OPTIONS II commenced November 2012, was conducted at 90 sites, and also completed in June 2014.
The trials were performed in accordance with the ethical principles that have their origin in the Declaration of Helsinki and all applicable amendments laid down by the World Medical Assemblies and the International Conference on Harmonisation Good Clinical Practice Guidelines. Institutional review board or independent ethics committee approval of the protocols and informed consent forms were obtained from each study site, and written informed consent was obtained from all patients.
Study Objectives
The primary objective of OPTIONS I is to compare the LDL‐C–lowering efficacy and safety of adding alirocumab to the most commonly used doses of atorvastatin (20 mg or 40 mg) with the strategies of adding ezetimibe, doubling the atorvastatin dose, or switching from atorvastatin 40 mg to rosuvastatin 40 mg. Patients were receiving either atorvastatin 20 mg or 40 mg at baseline and had received this stable dose for ≥4 weeks.
OPTIONS II focuses on patients receiving rosuvastatin. Here, the LDL‐C–lowering efficacy and safety of adding alirocumab to rosuvastatin will be compared with adding ezetimibe or doubling the rosuvastatin dose. Patients were receiving either rosuvastatin 10 mg or 20 mg at baseline and had received the same dose for ≥4 weeks.
Both studies are evaluating these treatment options in high–CV‐risk patients whose LDL‐C levels were greater than or equal to the prespecified levels of 70 mg/dL or 100 mg/dL on their existing statin dose (Table 1). The levels were based on guidelines current at the time the protocols were finalized.12, 13 In both studies, patients could have been on other lipid‐lowering therapies (LLTs), except for ezetimibe or statins other than atorvastatin (for OPTIONS I) and rosuvastatin (OPTIONS II).
Table 1.
Key Inclusion and Exclusion Criteria
| Inclusion Criteria | |
|---|---|
| Patients with hypercholesterolemia (non‐FH or heFH) at high CV risk with LDL‐C levels above those prespecified and receiving: | |
| OPTIONS I | OPTIONS II |
| Atorvastatin (20 mg or 40 mg daily), with or without other LLT (excluding ezetimibe), for ≥4 weeks prior to screening visit | Rosuvastatin (10 mg or 20 mg daily), with or without other LLT (excluding ezetimibe), for ≥4 weeks prior to screening visit |
| Baseline Entry Criteria | |
| Inclusion criteria | |
| Patients with LDL‐C levels ≥70 mg/dL (≥1.81 mmol/L) at the screening visit with a history of documented CVD; patients with a history of documented CHD, non‐CHD CVD, or DM with target organ damage | |
| Patients with LDL‐C levels ≥100 mg/dL (≥2.59 mmol/L) at the screening visit in patients without history of documented CVD; patients must also have heFH, or have non‐FH, without CHD or non‐CHD CVD, but with calculated 10‐year fatal CVD risk SCORE ≥5%, or with moderate CKD, or with DM but no target organ damage | |
| Exclusion criteria | |
| Age <18 years | |
| Fasting serum TG >400 mg/dL (>4.52 mmol/L) during the screening period | |
| Currently taking ezetimibe, or had received ezetimibe, within 4 weeks of the screening visit | |
| Uncontrolled endocrine disease known to influence serum lipids | |
| OPTIONS I | OPTIONS II |
| Currently taking a statin that is not atorvastatin daily at 20 mg or 40 mg | Currently taking a statin that is not rosuvastatin daily at 10 mg or 20 mg |
Abbreviations: CHD, coronary heart disease; CKD, chronic kidney disease; CV, cardiovascular; CVD, cardiovascular disease; DM, diabetes mellitus; heFH, heterozygous familial hypercholesterolemia; LDL‐C, low‐density lipoprotein cholesterol; LLT, lipid‐lowering therapy; non‐FH, non‐familial hypercholesterolemia; SCORE, systematic coronary risk evaluation; TG, triglycerides.
Randomization
In OPTIONS I, depending on whether they were receiving 20 mg or 40 mg atorvastatin daily at baseline, patients were randomized to 1 of 3 (20 mg atorvastatin daily at baseline) or 1 of 4 (40 mg atorvastatin daily at baseline) treatment arms (Figure 1). In OPTIONS II, patients receiving 10 mg or 20 mg rosuvastatin daily at baseline were randomized to 1 of 3 treatment arms (Figure 1). Screening was stopped once approximately 5% to 10% more patients than specified by the protocol were projected to be randomized for a given baseline statin regimen.
Figure 1.
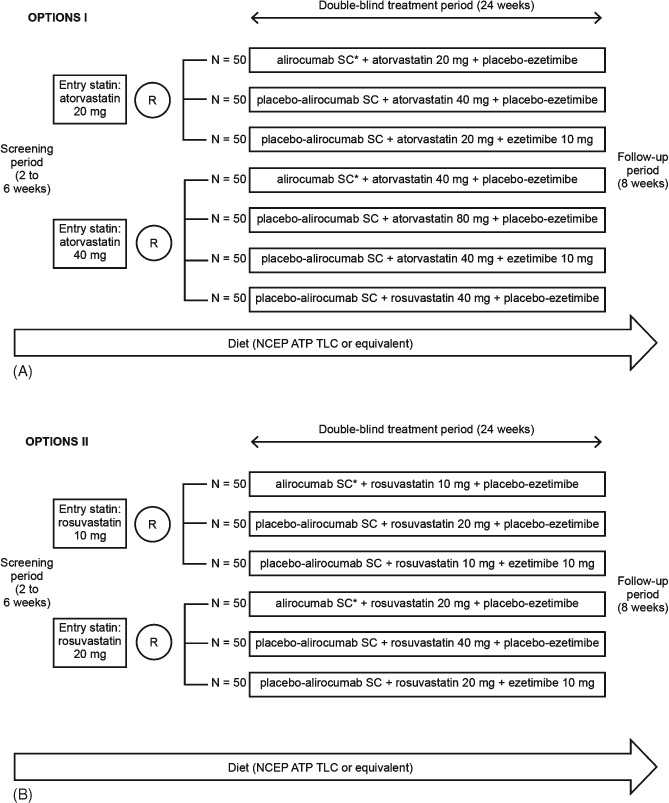
(A) Study design for OPTIONS I. (B) Study design for OPTIONS II. *75 mg SC Q2W with titration (as necessary) to 150 mg SC Q2W. Abbreviations: NCEP ATP III TLC, National Cholesterol Education Program–Adult Treatment Panel III Therapeutic Lifestyle Changes; Q2W, every 2 weeks; R, randomization; SC, subcutaneous.
For each study, and within each baseline statin regimen, randomization was stratified according to whether or not the patient has a history of either myocardial infarction or ischemic stroke. This stratification contributes to balance patient history of these major atherosclerosis‐associated CV events in treatment arms across the clinical trials program.
Study Endpoints and Assessments
The primary endpoint for both OPTIONS I and II is the difference in percent change in calculated LDL‐C from baseline to week 24 (Table 2) in the alirocumab arm vs control arms. The LDL‐C was calculated using the Friedewald formula at screening and at all time points during the double‐blind treatment periods. If TGs exceeded 400 mg/dL (4.52 mmol/L), then the central laboratory reflexively measured LDL‐C (via the β quantification method) rather than calculating it. The LDL‐C was measured (via the β quantification method) at week 0 and week 24.
Table 2.
Key Study Endpoints (Common to Both Studies)
| Primary endpoint |
| Difference in % change in calculated LDL‐C from baseline to week 24 in the alirocumab vs the control arms in the ITT population, using all LDL‐C values regardless of adherence to treatment (ITT estimand) |
| Key secondary endpoints (unless otherwise noted, all are the difference in % change in lipid parameter in the alirocumab vs control arms): |
| Calculated LDL‐C from baseline to week 24 in the mITT population, using all LDL‐C values during the efficacy treatment period (on‐treatment estimand) |
| Calculated LDL‐C from baseline to week 12 (ITT estimand) |
| Calculated LDL‐C from baseline to week 12 (on‐treatment estimand) |
| Apo B from baseline to week 24 (ITT estimand) |
| Apo B from baseline to week 24 (on‐treatment estimand) |
| Non–HDL‐C from baseline to week 24 (ITT estimand) |
| Non–HDL‐C from baseline to week 24 (on‐treatment estimand) |
| Total cholesterol from baseline to week 24 (ITT estimand) |
| Apo B from baseline to week 12 (ITT estimand) |
| Non–HDL‐C from baseline to week 12 (ITT estimand) |
| Total cholesterol from baseline to week 12 (ITT estimand) |
| Calculated LDL‐C from baseline to week 52 (ITT estimand) |
| Proportion of very high‐CV‐risk patients reaching calculated LDL‐C <70 mg/dL (1.81 mmol/L) or high‐CV‐risk patients reaching calculated LDL‐C <100 mg/dL (2.59 mmol/L) at week 24 (ITT estimand) |
| Proportion of very high‐CV‐risk patients reaching calculated LDL‐C <70 mg/dL (1.81 mmol/L) or high‐CV‐risk patients reaching calculated LDL‐C <100 mg/dL (2.59 mmol/L) at week 24 (on‐treatment estimand) |
| Proportion of patients reaching LDL‐C <70 mg/dL (1.81 mmol/L) at week 24 (ITT estimand) |
| Proportion of patients reaching LDL‐C <70 mg/dL (1.81 mmol/L) at week 24 (on‐treatment estimand) |
| Lp(a) from baseline to week 24 (ITT estimand) |
| HDL‐C from baseline to week 24 (ITT estimand) |
| Fasting TG from baseline to week 24 (ITT estimand) |
| Apo A‐1 from baseline to week 24 (ITT estimand) |
| Lp(a) from baseline to week 12 (ITT estimand) |
| HDL‐C from baseline to week 12 (ITT estimand) |
| Fasting TG from baseline to week 12 (ITT estimand) |
| Apo A‐1 from baseline to week 12 (ITT estimand) |
Abbreviations: Apo, apolipoprotein; HDL‐C, high‐density lipoprotein cholesterol; ITT, intent‐to‐treat; LDL‐C, low‐density lipoprotein cholesterol; Lp(a), lipoprotein(a); mITT, modified intent‐to‐treat; TG, triglycerides.
For both studies, there are 2 baseline statin regimens (atorvastatin 20 mg or 40 mg in OPTIONS I, rosuvastatin 10 mg or 20 mg in OPTIONS II), and there are 3 or 4 treatment arms within each regimen (Figure 1). The primary analysis will compare the difference in percent change in LDL‐C between the alirocumab arm and each of the comparator arms. For example, in OPTIONS I, for the atorvastatin 20‐mg regimen, there will be 2 pairs of comparisons: The alirocumab arm will be compared with the ezetimibe arm, and the alirocumab arm will also be compared with the arm doubling the atorvastatin dose to 40 mg. Key secondary endpoints are summarized in Table 2.
On‐site patient assessments took place at randomization and then at weeks 4, 8, 12, 16, and 24, and at the end‐of‐study visit, week 32.
Safety
Safety events (AEs [including adjudicated CV events categorized as coronary heart disease, death, nonfatal myocardial infarction, fatal and nonfatal ischemic stroke, unstable angina requiring hospitalization, and congestive heart failure requiring hospitalization], injection‐site reactions, laboratory data [blood biochemistry, hematology, and urinalysis], vital signs and electrocardiogram) were assessed throughout the study. The development of anti‐alirocumab antibodies was also monitored.
Inclusion Criteria
In both studies, entry criteria included a lower limit for baseline LDL‐C levels depending on risk category for CVD (Table 1). For patients with heFH, the diagnosis must have been made either by genotyping or clinical criteria. For those patients not genotyped, the clinical criteria were based on either the Simon Broome criteria with criteria for definite heFH18 or the World Health Organization/Dutch Lipid Network criteria with a score of >8 points.19
Exclusion Criteria
The key exclusion criteria were age <18 years; fasting serum TG >400 mg/dL (>4.52 mmol/L) during the screening period; patients receiving a statin that was not atorvastatin 20 mg or 40 mg daily (OPTIONS I) or not rosuvastatin 10 mg or 20 mg daily (OPTIONS II); patients receiving ezetimibe within 4 weeks prior to the screening visit; and uncontrolled endocrine disease known to influence serum lipids (Table 1; for a full list of inclusion and exclusion criteria, see Supporting Information, Supplementary Methods, in the online version of this article).
Study Procedures
Both studies consisted of 3 periods: a 2‐week to 6‐week screening period, a 24‐week double‐blind treatment period, and an 8‐week follow‐up period. The screening period included an intermediate visit during which the patient or caregiver was trained to self‐inject/inject. Patients on a stable atorvastatin (OPTIONS I) or rosuvastatin (OPTIONS II) dose for ≥4 weeks before the screening visit 1 were screened for study eligibility. During screening and at the discretion of the investigator, patients not on a stable dose of atorvastatin (20 mg or 40 mg; OPTIONS I) or rosuvastatin (10 mg or 20 mg; OPTIONS II) for 4 weeks; patients being switched to atorvastatin (OPTIONS I) or rosuvastatin (OPTIONS II); or patients not on a statin but who should have been, according to local guidance, received open‐label atorvastatin (20 mg or 40 mg; OPTIONS I) or rosuvastatin (10 mg or 20 mg; OPTIONS II) for a 4‐week run‐in period between the prescreening visit 1a (day −42) and visit 1. The run‐in dose of atorvastatin or rosuvastatin was based on the judgment of the study physician.
At randomization, treatment kit numbers were allocated using a centralized treatment allocation system, either an interactive voice‐response system or an interactive Web‐response system, depending on the site preference. Study patients, principal investigators, and study‐site personnel were blinded to all randomization assignments throughout the duration of the study. All lipid and antidrug antibody results collected after randomization were masked.
Each eligible patient received a single, subcutaneous, 1‐mL injection by prefilled pen every 2 weeks (Q2W) (aliro‐cumab or placebo‐alirocumab) and 2 oral‐blinded medications daily (a statin [atorvastatin or rosuvastatin] and ezetimibe or placebo‐ezetimibe). The first injection of alirocumab or placebo‐alirocumab was administered at the clinical site (week 0), after study assessments were completed, and as soon as possible after the patient was randomized into the study. The patient/caregiver administered subsequent autoinjections outside of the clinic, at a patient‐preferred location, on weeks 2, 4, 6, 8, and 10.
At week 12, based on their week 8 LDL‐C level and baseline CV risk, patients randomized to alirocumab may, in a blinded manner, have had their dose increased from 75 mg Q2W to 150 mg Q2W. To maintain blinding, lipid values obtained at week 8 for the purpose of up‐titration occurred in an automated process and were not communicated to investigators or patients. Patients with baseline LDL‐C levels ≥70 mg/dL and documented CVD continued to receive alirocumab 75 mg Q2W if their week 8 LDL‐C level was <70 mg/dL (1.81 mmol/L); otherwise, they were dose up‐titrated to receive alirocumab 150 mg Q2W (also via a 1‐mL autoinjection) if their week 8 LDL‐C level was ≥70 mg/dL (1.81 mmol/L). Patients with baseline LDL‐C levels ≥100 mg/dL without documented CVD continued to receive alirocumab 75 mg Q2W if their week 8 LDL‐C level was <100 mg/dL (2.59 mmol/L); otherwise, they were dose up‐titrated to alirocumab 150 mg Q2W if their week 8 LDL‐C level was ≥100 mg/dL (2.59 mmol/L). Autoinjections continued to be administered at weeks 12, 14, 16, 18, 20, and 22; the last dose of daily oral study drugs was adminis‐tered at week 24.
So as not to interfere with the study drug or serum lipids, the following concomitant LLTs were prohibited from the initial screening visit until the end of the study visit: statins other than atorvastatin and rosuvastatin (provided as blinded medications), ezetimibe (other than that provided as blinded medication), fibrates other than fenofibrate, and red yeast rice products. Lipid‐lowering therapies that were allowed as background therapy throughout the study included fish oils with omega‐3 fatty acids, fenofibrate, bile acid‐binding sequestrants, and niacin. Doses of these medications were stable for ≥4 weeks before screening (≥6 weeks for fenofibrate) and were to remain stable for the duration of the study.
Throughout the studies, patients were asked to follow a stable, National Cholesterol Education Program–Adult Treatment Panel III Therapeutic Lifestyle Changes (NCEP‐ATP III TLC) diet or equivalent diet from screening to the end‐of‐study visit. At the end of the 24‐week treatment period, patients entered an 8‐week follow‐up period.
Statistical Design and Analysis
The planned patient populations were 350 and 300 for OPTIONS I and OPTIONS II, respectively. For efficacy comparisons, it was estimated that a total sample size of 50 patients (OPTIONS I) or 47 patients (OPTIONS II) per arm would have 90% power to detect a difference in means of ≥20% in any one pairwise comparison (ie, alirocumab mean = 50% and control mean = 30%), assuming that the common standard deviation (SD) is 25 and that all 50 or 47 patients, respectively, are evaluable (for full statistical methods, see Supporting Information, Supplementary Methods, in the online version of this article).
In both OPTIONS I and II, the primary efficacy analysis population is the intent‐to‐treat population comprising all randomized patients with ≥1 baseline calculated LDL‐C value available and ≥1 calculated LDL‐C value available at one of the planned time points from weeks 4 and 24, regardless of treatment adherence.
The percent change in calculated LDL‐C from baseline to week 24 will be analyzed using a mixed‐effect model with repeated measures (MMRM) approach to account for missing data (see Supporting Information, Supplementary Methods, in the online version of this article).20, 21 Treatment differences between alirocumab‐treated patients and patients treated with active comparators will be estimated and tested through pairwise comparisons within each baseline statin regimen (see Supporting Information, Supplementary Methods, in the online version of this article). Secondary endpoints will be tested with a similar approach (see Supporting Information, Supplementary Methods, in the online version of this article).
Safety events will be reported using descriptive statistics, based on the safety population (all randomized patients who received ≥1 dose or partial dose of study treatment). The safety analysis will focus on the TEAE period, defined as the time from the first double‐blind dose to 70 days after the last double‐blind dose of the investigational drug.
Results
Three hundred and fifty‐five patients were randomized into OPTIONS I, and 305 patients were randomized into OPTIONS II. Baseline characteristics and lipid parameters are reported in Table 3. Briefly, 65.1% and 61.3% of patients were male in the OPTIONS I and OPTIONS II studies, respectively. The average age was 63 years in OPTIONS I and 61 years in OPTIONS II, and patients had CV risk factors such as coronary heart disease (59.4% and 63.0%), diabetes mellitus (50.1% and 42.0%), and hypertension (78.3% and 72.5%) in OPTIONS I and II, respectively. In OPTIONS I, the average baseline LDL‐C level was 105.1 mg/dL, and in OPTIONS II it was 111.3 mg/dL.
Table 3.
Baseline Characteristics and Lipid Parameters
| OPTIONS I, n = 355 | OPTIONS II, n = 305 | |
|---|---|---|
| Characteristic | ||
| Age (SD), y | 62.9 (10.2) | 60.9 (10.3) |
| Male, n (%) | 231 (65.1) | 187 (61.3) |
| Race, n (%) | ||
| White | 306 (86.2) | 256 (83.9) |
| Black | 38 (10.7) | 27 (8.9) |
| Other | 11 (3.1) | 22 (7.2) |
| BMI (SD), kg/m2 | 31.0 (6.4) | 31.3 (6.6) |
| CHD, n (%) | 211 (59.4) | 192 (63.0) |
| DM, n (%) | 178 (50.1) | 128 (42.0) |
| Hypertension, n (%) | 278 (78.3) | 221 (72.5) |
| Current smoker, n (%) | 66 (18.6) | 56 (18.4) |
| Lipid parameters | ||
| LDL‐C (SD), mg/dL | 105.1 (34.1) | 111.3 (39.0) |
| TC (SD), mg/dL | 182.0 (39.0) | 191.1 (44.7) |
| HDL‐C (SD), mg/dL | 48.7 (13.4) | 50.0 (13.1) |
| Fasting TG, median (Q1:Q3), mg/dL | 140.7 (89.0:175.0) | 128.0 (92.0:185.0) |
| Non–HDL‐C (SD), mg/dL | 133.3 (38.8) | 141.1 (43.4) |
| Apo B (SD), mg/dL | 90.8 (23.2) | 95.3 (24.1) |
| Apo A‐1 (SD), mg/dL | 144.2 (23.8) | 146.2 (25.0) |
| Lp(a), median (Q1:Q3), mg/dL | 44.8 (8.0:68.0) | 28.0 (11.0:79.0) |
Abbreviations; Apo, apolipoprotein; BMI, body mass index; CHD, coronary heart disease; DM, diabetes mellitus; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; Lp(a), lipoprotein(a); SD, standard deviation; TC, total cholesterol; TG, triglycerides.
Discussion
Evidence from meta‐analyses and well‐controlled randomized trials of statin therapy show that reducing LDL‐C is associated with a significant reduction in risk of CV events and mortality.1, 22, 23, 24, 25, 26, 27 Despite the availability of statins and other LLTs, many patients with hypercholesterolemia and high CV risk are not achieving LDL‐C levels <100 mg/dL or <70 mg/dL.1, 5, 6, 7, 8, 10, 11
In clinical practice, patients tend to be initiated on low‐ to moderate‐intensity statin therapy (a daily dose that provides an approximate <30% to 30% to 50% LDL‐C reduction, respectively), with up‐titration or switching to a higher‐intensity statin regimen (a daily dose that provides an approximate ≥50% LDL‐C reduction) as necessary for individuals not achieving LDL‐C targets or the expected therapeutic response from the initial dose of statin.12, 13
For high‐risk individuals who require greater LDL‐C lowering than that achieved with statin therapy alone, some guidelines recommend combination of statin therapy with ≥1 other LLT. Guidelines also recommend the use of other LLTs for individuals who are unable to tolerate a statin dose sufficient to reduce LDL‐C to target levels or who are unable to tolerate high‐intensity statin therapy or achieve the desired percent reduction in LDL‐C.1, 12, 13
Despite these guidelines, however, studies conducted in real‐world settings suggest that the percentage of patients achieving recommended LDL‐C levels is not optimal and can be improved.6, 7, 8, 28 For example, an observational study assessing treatment practice patterns in high–CV‐risk patients on statin monotherapy found that although greater LDL‐C reductions were seen in patients who received add‐on ezetimibe therapy or underwent a statin dose up‐titration, approximately 30% of these patients were not achieving LDL‐C levels <70 mg/dL,29 suggesting that currently available treatment options are not sufficient for some higher‐risk patients. Furthermore, recent evidence also suggests doubling the dose to that of a high‐intensity statin may provide minimal benefit from additional LDL‐C lowering.30 For example, it was calculated that increasing the dose from 40 mg to 80 mg of atorvastatin may result in only an approximate 2% reduction in the risk of clinical events.30 Overall, whereas statins have been shown to be efficacious in lowering LDL‐C, patients at high CV risk may benefit from additional, more intensive LLTs to help them achieve their LDL‐C goals.
A recent study conducted with another proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor, in patients with hypercholesterolemia and on stable, daily, moderate‐intensity (10 mg atorvastatin, 40 mg simvastatin, or 5 mg rosuvastatin) or high‐intensity (80 mg atorvastatin or 40 mg rosuvastatin) statin therapy, assessed the LDL‐C–lowering effect of added evolocumab or ezetimibe.31 Findings showed that the addition of evolocumab resulted in additional LDL‐C lowering.31 The ODYSSEY OPTIONS studies assess the lipid‐lowering efficacy and safety of alirocumab as add‐on therapy in comparison with the current clinical strategies that exist for high–CV‐risk patients whose LDL‐C levels are not sufficiently reduced with the most commonly used atorvastatin or rosuvastatin doses. The studies are designed to answer practical questions facing the clinician: Which clinical strategy provides the best combination of LDL‐C–lowering efficacy and safety/tolerability? Add alirocumab? Increase the statin dose or switch to another statin, or add ezetimibe? The study design also provides flexibility in the alirocumab dosing regimen, which in turn allows for individualized therapy based on the degree of LDL‐C reduction required to achieve the desired treatment response.
Whether additional LDL‐C lowering in patients optimally treated with statins will further reduce CV events is under evaluation in ongoing trials of ezetimibe as well as a number of PCSK9 monoclonal antibodies. The ODYSSEY OUTCOMES trial is evaluating alirocumab in patients with acute coronary syndromes.
In conclusion, the ODYSSEY OPTIONS studies aim to provide further data regarding the efficacy of alirocumab to reduce LDL‐C levels in patients with hypercholesterolemia at high CV risk, in particular those patients who are not achieving specific LDL‐C levels with the most commonly used atorvastatin or rosuvastatin doses. This trial will also evaluate the safety and tolerability of alirocumab in a larger patient population over 24 weeks.
Supporting information
AppendixS1. Supplementary Material
Acknowledgments
The authors acknowledge the study investigators and committee members (see Supporting Information, Investigators, Steering Committee, and Data Committee Acknowledgments in the online version of this article). The authors also acknowledge Joyce Harp for initial crafting of the study protocols.
The ODYSSEY OPTIONS studies are funded by Sanofi and Regeneron Pharmaceuticals, Inc. The design and conduct of the study, as well as analysis of the study data and opinions, conclusions, and interpretation of the data, are the responsibility of the authors. Medical writing support in the preparation of this publication was provided by Betty Thompson, PhD, of Prime Medica and sponsored by Sanofi and Regeneron Pharmaceuticals, Inc. The authors were responsible for all content and editorial decisions and received no honoraria related to the development/presentation of this publication.
Jennifer G. Robinson is employed by a university that has received research funds from Amarin, Amgen, AstraZeneca, Daiichi‐Sankyo, Esperion, Genentech/Hoffmann‐La Roche, GlaxoSmithKline, Merck, Regeneron/Sanofi, and Zinfandel/Takeda and is a consultant for Amgen, Merck, Pfizer, Sanofi, and Regeneron. Helen M. Colhoun is a consultant or on an advisory panel for Pfizer, Sanofi Aventis, Regeneron, Novartis, and Eli Lilly, has received research support from Roche, Pfizer, Eli Lilly, Boehringer Ingelheim, and AstraZeneca, has participated in a lecture/speakers bureau and received honorarium from Pfizer, and is a shareholder in Roche. Harold Bays (in the past 12 months) has served as a consultant and/or speaker for Amarin, Amgen, AstraZeneca, Bristol‐Myers Squibb, Catabasis, Daiichi‐Sankyo, Eisai, Isis, Merck, Novartis, Omthera, VIVUS, and WPU, and his research site has received research grants from Alere, Amarin, Amgen, Ardea, AstraZeneca, Boehringer Ingelheim, Bristol‐Myers Squibb, California Raisin Board, Catabasis, Eisai, Elcelyx, Eli Lilly, Esperion, Forest, Gilead, Given, GlaxoSmithKline, High Point Pharmaceuticals LLC, Hoffmann‐La Roche, Home Access, Janssen, Merck, Metabolex, Nektar, Novartis, Novo Nordisk, Omthera, Orexigen, Pfizer, Pronova, Regeneron, Takeda, TIMI, Transtech Pharma, Trygg, VIVUS, and WPU Pharmaceuticals. Peter Jones is a member of advisory boards for Merck and Atherotech, and has received honoraria as a consultant for Sanofi/Regeneron and Amgen. Yunling Du and Stephen Donahue are employees of Regeneron Pharmaceuticals, Inc. Corinne Hanotin is an employee of Sanofi.
The authors have no other funding, financial relationships, or conflicts of interest to disclose.
References
- 1. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S1–S45. [DOI] [PubMed] [Google Scholar]
- 2. Anderson TJ, Gregoire J, Hegele RA, et al. 2012 update of the Canadian Cardiovascular Society guidelines for the diagnosis and treatment of dyslipidemia for the prevention of cardiovascular disease in the adult. Can J Cardiol. 2013;29:151–167. [DOI] [PubMed] [Google Scholar]
- 3. Expert Dyslipidemia Panel , Grundy SM. An International Atherosclerosis Society Position Paper: global recommendations for the management of dyslipidemia. J Clin Lipidol. 2013;7:561–565. [DOI] [PubMed] [Google Scholar]
- 4. Perk J, De Backer G, Gohlke H, et al. European Guidelines on cardiovascular disease prevention in clinical practice (version 2012): the Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Atherosclerosis. 2012;223:1–68. [DOI] [PubMed] [Google Scholar]
- 5. Jones PH, Nair R, Thakker KM. Prevalence of dyslipidemia and lipid goal attainment in statin‐treated subjects from 3 data sources: a retrospective analysis. J Am Heart Assoc. 2012;1:e001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Banegas JR, López‐García E, Dallongeville J, et al. Achievement of treatment goals for primary prevention of cardiovascular disease in clinical practice across Europe: the EURIKA study. Eur Heart J. 2011;32:2143–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kotseva K, Wood D, De Backer G, et al. Cardiovascular prevention guidelines in daily practice: a comparison of EUROASPIRE I, II, and III surveys in eight European countries. Lancet. 2009;373:929–940. [DOI] [PubMed] [Google Scholar]
- 8. Park JE, Chiang CE, Munawar M, et al. Lipid‐lowering treatment in hypercholesterolaemic patients: the CEPHEUS Pan‐Asian survey. Eur J Prev Cardiol. 2012;19:781–794. [DOI] [PubMed] [Google Scholar]
- 9. European Society of Cardiology. EUROASPIRE IV reveals success and challenges in secondary prevention of CVD across Europe . http://www.escardio.org/about/press/press‐releases/esc13‐amsterdam/Pages/euroaspire‐iv‐success‐challenges‐secondary‐prevention‐CVD‐europe.aspx. Published September 3, 2013. Accessed March 25, 2014.
- 10. Stein EA, Strutt K, Southworth H, et al. Comparison of rosuvastatin versus atorvastatin in patients with heterozygous familial hypercholesterolemia. Am J Cardiol. 2003;92:1287–1293. [DOI] [PubMed] [Google Scholar]
- 11. Pijlman AH, Huijgen R, Verhagen SN, et al. Evaluation of cholesterol lowering treatment of patients with familial hypercholesterolemia: a large cross‐sectional study in The Netherlands. Atherosclerosis. 2010;209:189–194. [DOI] [PubMed] [Google Scholar]
- 12. Reiner Z, Catapano AL, De Backer G, et al. ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the Management of Dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J. 2011;32:1769–1818. [DOI] [PubMed] [Google Scholar]
- 13. Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110:227–239. [DOI] [PubMed] [Google Scholar]
- 14. Waters DD, Brotons C, Chiang CW, et al. Lipid treatment assessment project 2: a multinational survey to evaluate the proportion of patients achieving low‐density lipoprotein cholesterol goals. Circulation. 2009;120:28–34. [DOI] [PubMed] [Google Scholar]
- 15. Stein EA, Gipe D, Bergeron J, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low‐density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380:29–36. [DOI] [PubMed] [Google Scholar]
- 16. Roth EM, McKenney JM, Hanotin C, et al. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med. 2012;367:1891–1900. [DOI] [PubMed] [Google Scholar]
- 17. McKenney JM, Koren MJ, Kereiakes DJ, et al. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;59:2344–2353. [DOI] [PubMed] [Google Scholar]
- 18. Scientific Steering Committee on behalf of the Simon Broome Register Group . Risk of fatal coronary heart disease in familial hypercholesterolaemia. BMJ. 1991;303:893–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. World Health Organization. Familial Hypercholesterolaemia (FH): Report of a second WHO consultation . http://whqlibdoc.who.int/hq/1999/WHO_HGN_FH_CONS_99.2.pdf. Published 1999. Accessed March 13, 2014.
- 20. Siddiqui O, Hung HM, O'Neill R. MMRM vs. LOCF: a comprehensive comparison based on simulation study and 25 NDA datasets. J Biopharm Stat. 2009;19:227–246. [DOI] [PubMed] [Google Scholar]
- 21. National Research Council, Panel on Handling Missing Data in Clinical Trials . The Prevention and Treatment of Missing Data in Clinical Trials. Washington, DC: The National Academies Press; 2010. [PubMed] [Google Scholar]
- 22. Taylor F, Huffman MD, Macedo AF, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013;1:CD004816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C‐reactive protein. N Engl J Med. 2008;359:2195–2207. [DOI] [PubMed] [Google Scholar]
- 24. Pedersen TR, Faergeman O, Kastelein JJ, et al. High‐dose atorvastatin vs usual‐dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA. 2005;294:2437–2445. [DOI] [PubMed] [Google Scholar]
- 25. Cholesterol Treatment Trialists' (CTT) Collaborators , Mihaylova B, Emberson J, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta‐analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352:1425–1435. [DOI] [PubMed] [Google Scholar]
- 27. Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–1504. [DOI] [PubMed] [Google Scholar]
- 28. Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Toth PP, Foody JM, Tomassini JE, et al. Therapeutic practice patterns related to statin potency and ezetimibe/simvastatin combination therapies in lowering LDL‐C in patients with high‐risk cardiovascular disease. J Clin Lipidol. 2014;8:107–116. [DOI] [PubMed] [Google Scholar]
- 30. Sniderman A, Thanassoulis G, Couture P, et al. Is lower and lower better and better? A re‐evaluation of the evidence from the Cholesterol Treatment Trialists' Collaboration meta‐analysis for low‐density lipoprotein lowering. J Clin Lipidol. 2012;6:303–309. [DOI] [PubMed] [Google Scholar]
- 31. Robinson JG, Nedergaard BS, Rogers WJ, et al. Effect of evolocumab or ezetimibe added to moderate‐ or high‐intensity statin therapy on LDL‐C lowering in patients with hypercholesterolemia: the LAPLACE‐2 randomized clinical trial. JAMA. 2014;311:1870–1882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
AppendixS1. Supplementary Material