

Abstract
Background
US Food and Drug Administration (FDA) set a rigorous standard for defining patient responders in irritable bowel syndrome-C (IBS-C; i.e., FDA's Responder Endpoint) for regulatory approval. However, this endpoint's utility for health-care practitioners to assess clinical response has not been determined. We analyzed pooled IBS-C linaclotide trial data to evaluate clinically significant responses in linaclotide-treated patients who did not meet the FDA responder definition.
Methods
Percentages of FDA non-responders reporting improvement in abdominal pain, bowel function and/or global relief measures were determined using pooled data from two linaclotide Phase 3 IBS-C trials.
Key Results
1602 IBS-C patients enrolled; 34% of linaclotide-treated and 17% of placebo-treated patients met the FDA Responder Endpoint (p < 0.0001). Among FDA non-responders at week 12, 63% of linaclotide-treated patients reported their abdominal pain was at least somewhat relieved, compared with 48% of placebo-treated patients. For stool frequency, 62% of linaclotide-treated patients reported that they were at least somewhat improved at week 12, compared with 46% of placebo-treated patients. For global IBS symptoms, 65% of linaclotide-treated patients reported at least some IBS-symptom relief, 43% reported adequate relief of IBS symptoms, and 57% reported being satisfied with linaclotide treatment, vs placebo rates of 48%, 34%, and 41% respectively.
Conclusions & Inferences
Most linaclotide-treated IBS-C patients who were FDA non-responders reported some improvement in abdominal pain and stool frequency, and global relief/satisfaction. In addition to the FDA Responder Endpoint, differing response thresholds and symptom-specific change from baseline should be considered by clinicians for a complete understanding of clinical response to linaclotide and other IBS-C therapies.
Keywords: abdominal pain, clinical response, complete spontaneous bowel movement, guanylate cyclase type-C receptor, IBS-C
Key Messages
Although the FDA's recommended endpoint for IBS-C trials is useful for determining a medication's suitability for regulatory approval, it may not provide adequate information to clinicians to guide appropriate patient-specific treatment.
The aim of this analysis was to evaluate the performance of the FDA IBS-C Responder Endpoint using data from clinical trials of linaclotide.
Pooled data from two Phase 3 IBS-C clinical trials of linaclotide were used to determine the percentages of FDA endpoint non-responders reporting improvement in abdominal pain, bowel movement frequency, and/or global relief measures.
Clinically meaningful improvement in the key individual IBS-C symptoms of abdominal pain and stool frequency was observed in >60% of linaclotide patients and >45% of placebo patients who did not meet the FDA IBS-C Responder Endpoint.
Introduction
Irritable bowel syndrome (IBS), a chronic gastrointestinal disorder characterized by abdominal pain and/or discomfort with altered bowel movements (BMs), is estimated to affect up to 20% of the US adult population.1 Irritable bowel syndrome negatively affects patients' quality of life and is associated with a substantial economic burden of care due to increased healthcare resource utilization and diminished work productivity.2–5 In addition to abdominal pain and infrequent BMs, IBS with constipation (IBS-C) is characterized by clinically relevant symptoms of abdominal bloating, hard stools, straining, and a sensation of incomplete evacuation.6 Approximately one-third of patients with IBS report symptoms consistent with IBS-C.7
Linaclotide, a first-in-class guanylate cyclase C agonist, was recently approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of IBS-C in adult men and women. Linaclotide stimulates intracellular production of cyclic guanosine monophosphate (cGMP) by binding to guanylate cyclase C receptors on the luminal surface of gastrointestinal epithelial cells.8 The increased cGMP results in chloride and bicarbonate secretion into the gastrointestinal lumen, and, consequently, increased fluid secretion and accelerated intestinal transit.9 Linaclotide has also been shown to reduce visceral hypersensitivity in animal models, which may be related to cGMP modulation of afferent nerve activity in the extracellular space.8,10,11 Two large Phase 3, multicenter, randomized, double-blind, placebo-controlled, parallel-group, clinical trials (Trials 31 and 302) documented the efficacy of oral linaclotide in patients with IBS-C.12,13
The IBS-C responder endpoint recommended in the May 2012 FDA final guidance for industry on the clinical evaluation of products for IBS (‘FDA Responder Endpoint’) was assessed as a primary endpoint in both trials.14 The FDA Responder Endpoint requires patients to have both an improvement of at least 30% in their daily worst abdominal pain and an increase of 1 or more complete spontaneous bowel movement (CSBMs) during the same week for at least 50% of the weeks of treatment. A CSBM is defined as a BM occurring in the absence of laxative, enema, or suppository use during the previous 24 h, with the BM accompanied by the patient self-reporting a feeling of complete evacuation. This endpoint, designed with the intent of better identifying a patient experiencing a clinically meaningful improvement in IBS symptoms, was shown to have high specificity and reasonable sensitivity when it was evaluated using receiver-operator-characteristic-based methods.15 However, because this dichotomous endpoint reduces a patient's level of improvement/relief involving multiple IBS symptoms to a single categorical responder/non-responder status, the FDA Responder Endpoint results in an incomplete clinical understanding of the effect of this treatment.16 Therefore, the objective of these post-hoc analyses of pooled data from both Phase 3 IBS-C linaclotide trials was to evaluate clinical response in linaclotide-treated patients who did not meet the FDA Responder Endpoint (i.e., ‘FDA non-responders’) and to evaluate the distribution of symptom improvement to allow better understanding of clinical efficacy.
Materials and Methods
Clinical trial design
The two Phase 3 linaclotide clinical trials included a 2-week baseline period, after which patients were randomized to receive either placebo or linaclotide 290 μg once daily during a treatment period of 12 weeks (Trial 31) or 26 weeks (Trial 302).12,13
Study population
Patients were men and women aged 18 years or older who met Rome II criteria for IBS.6 Patients entering the baseline period were required to report <3 spontaneous bowel movements (SBM = a BM occurring in the absence of laxative, enema, or suppository use in the previous 24 h, defined in these trials as the calendar day of the BM or the calendar day before the BM) per week and had 1 or more of the following symptoms for at least 12 weeks, which need not have been consecutive, in the preceding 12 months: (a) straining during >25% of BMs, (b) lumpy or hard stools during >25% of BMs, and (c) a sensation of incomplete evacuation during >25% of BMs. During the baseline period, patients eligible for randomization needed to report an average score of ≥3.0 for daily abdominal pain at its worst on a 0–10 point numeric rating scale (NRS) and an average of ≤5 SBMs per week and <3 CSBMs per week.
Efficacy assessments and endpoints
Each day during the baseline and treatment periods, patients were asked to call into an interactive voice response system (IVRS) to record their IBS-C symptoms. Abdominal pain was measured using an 11-point NRS; the number of BMs and use of rescue medication were reported; and each BM was assessed for sensation of complete bowel emptying (yes/no).
The endpoints for the change-from-baseline analyses were based on a patient's average weekly response during week 12. For abdominal pain, a patient's weekly score, the average of the daily IVRS responses across that week, was used to calculate percent improvement from baseline. Similarly, the weekly rates of SBMs and CSBMs during week 12 were used to calculate these change-from-baseline endpoints.
As mentioned above, an FDA Endpoint Responder14 was a patient who met both of the following criteria in the same week for at least six of the first 12 weeks of the treatment period: (i) an improvement of ≥30% from baseline in the average of the daily worst abdominal pain scores and (ii) an increase of ≥1 CSBM from baseline. In addition, patients were required to complete at least four IVRS calls during the week to qualify as a responder for that week.
In addition to daily rating of symptoms, patient rating of change questions (PRCQs) for abdominal pain relief and SBM and CSBM frequency improvement were asked at each study visit following randomization, while an overall question, degree of relief of IBS symptoms PRCQ, was asked weekly via the IVRS. For abdominal pain relief, patients responded to the following PRCQ: ‘Compared to before you started this study, how would you rate your abdominal pain at its worst during the past 7 days?’; for SBM frequency improvement, patients responded to the following PRCQ: ‘Compared to before you started this study, in the absence of laxative use, how would you rate the frequency of your BMs during the past 7 days?’; for CSBM frequency improvement, patients responded to the following PRCQ: ‘Compared to before you started this study, in the absence of laxative use, how would you rate your frequency of complete BMs (i.e., BMs where you felt like you completely emptied your bowels) during the past 7 days?’; and for degree of relief of IBS symptoms, patients responded to the following PRCQ: ‘Compared to before you started this study, how would you rate your IBS symptoms during the past 7 days?’. For all four PRCQs, the response options were: 1 = Completely relieved/improved, 2 = Considerably relieved/improved, 3 = Somewhat relieved/improved, 4 = Unchanged, 5 = Somewhat worse, 6 = Considerably worse, 7 = As bad as I can imagine. A response of at least ‘somewhat improved’ has been used as an anchor to determine minimal clinical significance.15,17,18 ‘Somewhat improved’ represents the smallest difference in score in these PRCQ measures that patients perceive as beneficial, and that would mandate, in the absence of troublesome side-effects and excessive cost, a change in the patient's management.19
Patients were also asked weekly if they had adequate relief of their IBS symptoms with a yes/no response to the following question: ‘Overall, have you had adequate relief from your IBS symptoms during the past 7 days?’ Treatment satisfaction was assessed at each postrandomization study visit by the following question ‘Overall, how satisfied are you with the study medication's ability to relieve your IBS symptoms?’ Patients selected from the following 5-point ordinal response scale: 1 = Not at all satisfied, 2 = A little satisfied, 3 = Moderately satisfied, 4 = Quite satisfied, 5 = Very satisfied.
Safety assessments
At each scheduled study visit, all patients were asked an open-ended question regarding adverse events (AEs). Patients reported AEs by recalling instances since their prior visit. The site investigator assessed all patient-reported AEs and judged each event for severity and relationship to the blinded trial medication. Other safety evaluations included physical examinations, electrocardiogram recordings, vital sign measurements, and standard clinical laboratory tests.
Statistical methods and data analysis
The FDA Responder Endpoint was analyzed using a Cochran–Mantel–Haenszel test, stratified by trial and geographic region, comparing the linaclotide- and placebo-treatment groups. The week 12 percent improvement from baseline for abdominal pain and change from baseline for SBMs and CSBMs between the linaclotide and placebo intent-to-treat (ITT) treatment groups were compared using an analysis of covariance model with fixed-effect terms for treatment group, trial, and geographic region and the corresponding baseline value as a covariate. In addition, the percentage of patients in each treatment group who met or exceeded incrementally increasing threshold levels at week 12 was determined for the abdominal and bowel symptom endpoints. At each of these thresholds, the percentage of linaclotide- and placebo-treated patients meeting or exceeding this improvement threshold was compared via a Fisher's exact test.
To examine the effect of linaclotide on FDA non-responders, the week 12 PRCQs, adequate relief, and treatment satisfaction responder endpoints were summarized for three groups: linaclotide-treated patients who were FDA responders, linaclotide-treated patients who were FDA non-responders, and placebo ITT patients. If the linaclotide-treated patients who were FDA non-responders were experiencing a clinical benefit, the responder rates should be higher than the placebo ITT patients. The patient's abdominal pain and bowel movement frequency PRCQ endpoints were used to mitigate the potential bias of summarizing endpoints using the same abdominal pain and bowel movement frequency scores that define the stratification (i.e., the FDA responder endpoint being used to stratify the results is based on the abdominal pain and bowel movement frequency scores). As the FDA non-responder patient population is based on a postrandomization criterion to determine the population (i.e., whether a patient was an FDA responder based on the 12-week treatment period), 95% confidence intervals (CI) are presented for the responder rates as opposed to a direct p-value based comparison between the FDA non-responders and the placebo ITT groups.
All p values were based on two-sided tests and all confidence intervals were two-sided. Efficacy analyses were based on the ITT Population (all patients in the Safety Population who had at least one postbaseline efficacy measurement for the primary endpoint); safety analyses were based on the Safety Population (all randomized patients who took at least one dose of study drug).
Results
Patient disposition, demographics, and baseline characteristics
A total of 1605 IBS-C patients were included from both Phase 3 IBS-C trials in the pooled Safety Population; 1602 of these patients were included in the ITT Population. The demographics and baseline characteristics of the ITT Population are shown in Table1.
Table 1.
Summary of patient demographic and baseline characteristics (pooled phase 3 ITT population)
| Linaclotide 290 μg (N = 805) | Linaclotide 290 μg FDA responders (N = 271) | Linaclotide 290 μg FDA non-responders (N = 534) | Placebo (N = 797) | |
|---|---|---|---|---|
| Demographic data | ||||
| Age (years), Mean (range) | 44.0 (19–82) | 45.1 (21–78) | 43.4 (19–82) | 43.8 (18–87) |
| ≥65 years, n (%) | 42 (5.2) | 16 (5.9) | 26 (4.9) | 43 (5.4) |
| Gender, n (%) | ||||
| Female | 735 (91.3) | 257 (94.8) | 478 (89.5) | 708 (88.8) |
| Male | 70 (8.7) | 14 (5.2) | 56 (10.5) | 89 (11.2) |
| Race, n (%) | ||||
| White | 629 (78.1) | 225 (83.0) | 404 (75.7) | 611 (76.7) |
| Black | 148 (18.4) | 40 (14.8) | 108 (20.2) | 153 (19.2) |
| Other | 28 (3.5) | 6 (2.2.) | 22 (4.1) | 33 (4.1) |
| BMI, Mean (SD) | 28.0 (6.2) | 28.1 (6.2) | 28.0 (6.2) | 27.7 (6.2) |
| Abdominal and bowel symptoms | ||||
| Abdominal pain*, Mean (SD) | 5.6 (1.7) | 5.7 (1.7) | 5.6 (1.7) | 5.6 (1.7) |
| CSBMs/week, Mean (SD) | 0.2 (0.4) | 0.2 (0.5) | 0.2 (0.4) | 0.2 (0.5) |
| SBMs/week, Mean (SD) | 1.8 (1.4) | 2.0 (1.4) | 1.8 (1.4) | 1.8 (1.4) |
Assessed using an 11-point NRS: 0 = none; 10 = very severe.
BMI, body mass index; CSBM, complete spontaneous bowel movement; ITT, Intent-to-Treat; SBM, spontaneous bowel movement; SD, standard deviation; NRS, numerical rating scale.
FDA responder endpoint
Across both Phase 3 trials, the FDA Responder Endpoint was met by 271 (33.7%) of 805 linaclotide-treated patients compared with 139 (17.4%) of 797 placebo-treated patients (a treatment difference of 16.2%, 95% CI [12.0%, 20.4%] p < 0.0001). The clinical response of linaclotide using PRCQs on abdominal pain and bowel movement frequency in patients who were FDA non-responders was investigated.
Clinical response in FDA non-responders
Using the PRCQs, greater than 90% of linaclotide-treated patients who were FDA responders reported their abdominal pain or stool frequency (SBM and CSBM) being at least somewhat relieved or improved at week 12 (Fig. 1). For linaclotide-treated patients who were FDA non-responders, 63% (95% CI: 59%, 68%) reported having at least ‘somewhat’ relief of abdominal pain, while similar relief was reported by 48% (95% CI: 44%, 51%) of placebo-treated patients at week 12 (Fig. 1). Among linaclotide-treated FDA non-responders, 52% (95% CI: 47%, 56%) and 62% (95% CI: 58%, 66%) of patients reported having at least ‘somewhat’ improvement in the frequency of CSBMs and SBMs, respectively, at week 12, compared with 39% (95% CI: 36%, 43%) and 46% (95% CI: 42%, 49%) of placebo-treated patients (Fig. 1).
Figure 1.
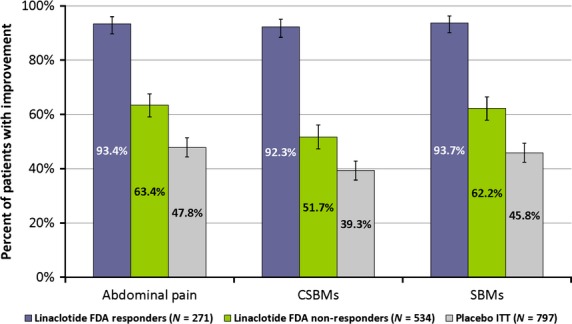
Percentage of patients reporting improvement in abdominal pain, CSBMs, and SBMs at week 12 for linaclotide FDA responders/non-responders and placebo ITT patients (pooled IBS-C phase 3 ITT population; ±95% CI). Patients on linaclotide who were classified as FDA responders and non-responders reported higher rates of being somewhat, considerably, or completely relieved/improved compared to the placebo ITT patients for abdominal pain, CSBMs, and SBMs.
Similarly, among FDA non-responders a greater percentage of linaclotide-treated patients reported improvement in Degree of Relief, Adequate Relief, and Treatment Satisfaction compared with placebo-treated patients at week 12 (Fig. 2). Among linaclotide-treated FDA non-responders at week 12, 65% (95% CI: 61%, 69%) reported at least ‘somewhat’ relief of their IBS symptoms, 43% (95% CI: 39%, 48%) reported having adequate relief of their IBS symptoms, and 57% (95% CI: 52%, 61%) reported being at least moderately satisfied with linaclotide's ability to relieve their IBS symptoms, compared with 48% (95% CI: 44%, 52%), 34% (95% CI: 31%, 37%), and 41% (95% CI: 38%, 45%), respectively, of placebo-treated patients (Fig. 2).
Figure 2.
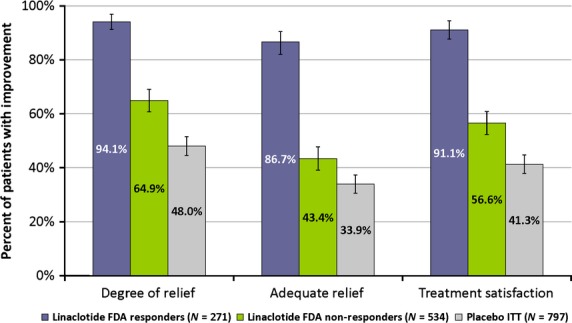
Percentage of patients reporting improvement in degree of relief, adequate relief, and treatment satisfaction at week 12 for linaclotide FDA responders/non-responders and placebo ITT patients (pooled IBS-C phase 3 ITT population; ±95% CI). Patients on linaclotide who were classified as FDA responders and non-responders reported higher rates of relief of IBS symptoms compared with the Placebo ITT patients for the following endpoints: degree of relief of IBS symptoms (somewhat, considerably, or completely relieved), adequate relief of IBS symptoms, and treatment satisfaction (moderately, quite, or very satisfied with the treatment's ability to relieve IBS symptoms).
Distribution of the efficacy endpoints: linaclotide vs placebo
Given that FDA non-responders show improvement in PRCQs for abdominal pain, bowel movement frequency, and global measures of IBS, the FDA Responder Endpoint, as a dichotomous measure, fails to capture the true breadth of clinical response. Therefore, to more completely understand the clinical benefit of linaclotide, a wider distribution of percent improvement in abdominal pain and stool frequency (CSBM and SBM) was independently evaluated for the linaclotide and placebo ITT Populations (Figs3 and 4).
Figure 3.
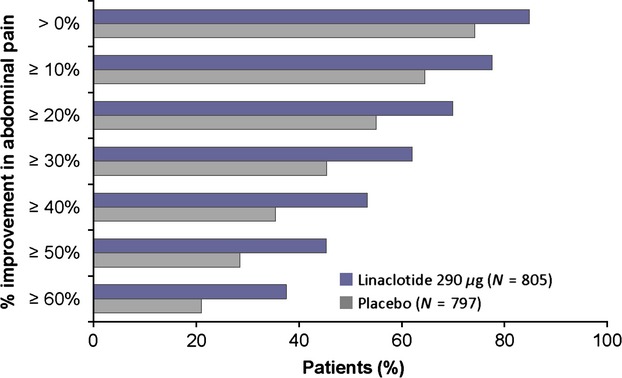
Percentages of patients with specified improvements in abdominal pain at week 12 (pooled IBS-C phase 3 ITT population). At week 12, a greater percentage of patients treated with linaclotide met incremental levels of improvement in abdominal pain compared with placebo patients (p < 0.0001 for linaclotide compared with placebo for each percent level of improvement using Fisher's exact test).
Figure 4.
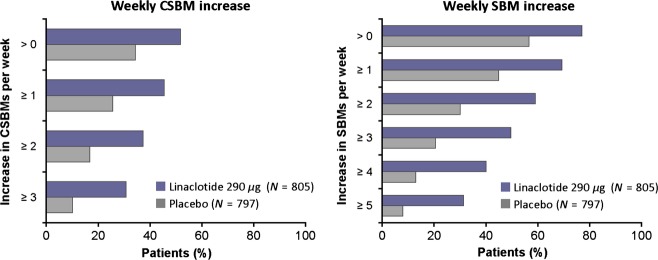
Percentages of patients with specified increases in weekly CSBM frequency and SBM frequency at week 12 (pooled IBS-C phase 3 ITT population). At week 12, a greater percentage of patients treated with linaclotide met incremental levels of increase in weekly CSBM frequency and SBM frequency compared with placebo patients (p < 0.0001 for linaclotide compared with placebo for each level of improvement using Fisher's exact test for both CSBM and SBM frequency).
For all percent reductions (e.g., >10%, >20%, etc.) in abdominal pain, more linaclotide-treated patients reported improvement compared with placebo-treated patients (Fig. 3). For example, 62% of linaclotide-treated patients had an improvement in abdominal pain at week 12 of at least 30% compared with 45% of placebo-treated patients (p < 0.0001, Fig. 3). The mean improvement in abdominal pain at week 12 in linaclotide-treated patients was 42% compared with 26% for placebo-treated patients (p < 0.0001).
Similarly, the percentage of patients with specified increases in weekly CSBM and SBM frequency at week 12 was greater for linaclotide-treated patients compared with placebo-treated patients (Fig. 4). For example, an increase of ≥1 CSBM per week from baseline was reported by 46% of linaclotide-treated patients at week 12 compared with 26% of placebo-treated patients (p < 0.0001, Fig. 4). An increase of ≥2 SBMs per week from baseline was reported by 59% of linaclotide-treated patients at week 12 compared with 30% of placebo-treated patients (P < 0.0001, Fig. 4). The mean increase in weekly CSBM frequency at week 12 among linaclotide-treated patients was 2.2 CSBMs/week compared with an increase of 0.7 in placebo-treated patients (p < 0.0001). The mean increase in weekly SBM frequency at week 12 among linaclotide-treated patients was 3.7 SBMs/week while the increase was 1.0 in placebo-treated patients (p < 0.0001).
Safety results
A total of 440 of 807 linaclotide-treated patients (55%) reported at least one AE over the first 12 weeks of the treatment period compared with 397 of 798 placebo-treated patients (50%). Most AEs were mild or moderate in severity (94% linaclotide; 97% placebo). The incidences of patients with diarrhea (19%), abdominal pain (5%), and flatulence (4%) AEs were greater in the linaclotide group compared with the placebo group (3%, 3%, and 2% respectively). Among these AEs only diarrhea was reported more commonly in linaclotide FDA responders (22%) compared with linaclotide FDA non-responders (17%). None of the patients who reported diarrhea experienced clinically significant sequelae (e.g., orthostatic hypotension or dehydration).
Discussion
For IBS-C, the FDA's guidance for the clinical evaluation of drugs for IBS recommends a combined responder endpoint requiring ≥30% improvement in abdominal pain and an increase of at least 1 CSBM over baseline, both in the same week, for at least 50% of the treatment period. This FDA Responder Endpoint for IBS-C clinical trials represents a rigorous and stringent standard for the evaluation of IBS therapies for regulatory approval. As such, this combined endpoint should be well-equipped to distinguish an efficacious therapeutic agent from a placebo, which is critically important in a field where the placebo-response rate has traditionally been quite high. However, the FDA Responder Endpoint provides only a ‘snapshot’ of the efficacy of an investigational agent. By design, this endpoint paints an incomplete picture of the therapeutic benefit of an agent such as linaclotide, as clinically important individual symptom responses, such as abdominal pain and stool frequency, among others, are reduced to a single binary score. This methodologic limitation is not unique to this particular IBS-C responder endpoint but arises whenever continuous response endpoints are dichotomized.16 Although designed to increase our ability to identify patient ‘responders’, the FDA Responder Endpoint does not address patients who may have clinically important and relevant responses but who do not meet the stringent criteria of the endpoint. Given the chronic non-life-threatening nature of IBS-C, a highly specific and stringent endpoint may be desirable, even if associated with a lower sensitivity, as it ensures there is relative certainty that patients meeting the endpoint have experienced a clinically meaningful benefit. However, it should be acknowledged that there are patients who experience benefit who do not meet the FDA Responder Endpoint. As such, to appropriately assess the ability of a medication to relieve symptoms, it is important that patients' symptom-specific responses are considered in addition to dichotomous responder endpoints. These assessments of individual symptoms are particularly critical in the field of IBS in which symptom expression and symptom intensity vary from patient to patient.
To illustrate this methodological limitation, we evaluated symptom improvement in linaclotide-treated patients who were FDA non-responders. At week 12 linaclotide-treated FDA non-responders were evaluated for patient ratings of change in the key individual symptoms of abdominal pain and CSBMs, the components of the FDA endpoint. In addition, we evaluated the linaclotide-treated FDA non-responders for symptom improvement in SBMs, IBS global endpoints, and treatment satisfaction. For each of these six endpoints, the linaclotide-treated FDA non-responders had a higher rate of improvement than the placebo patients in the ITT Population with non-overlapping 95% CIs (see Figs1 and 2). For example, when considering SBM frequency, 62% of linaclotide-treated FDA non-responders reported improvement, compared with 46% of ITT placebo patients. This result is consistent with SBM frequency results in the overall ITT population, which showed that a greater percentage of linaclotide-treated patients met SBM frequency improvement thresholds when compared with placebo-treated patients (Fig. 4). These results indicate that linaclotide-treated patients who were classified as FDA non-responders are experiencing improvement in their IBS symptoms above what is seen in the placebo group.
These findings illustrate the limitations of using a dichotomous endpoint to simply classify an IBS-C patient as a ‘Responder’ or ‘Non-responder’. The majority of linaclotide-treated patients who were classified as non-responders using the FDA Responder Endpoint reported the degree of improvement in their abdominal pain and/or stool frequency as at least ‘somewhat relieved’ at week 12. These results highlight the difference between a patient responding to treatment and a patient being classified as a responder. Thus, while the FDA Responder Endpoint for IBS trials appears to accurately distinguish an efficacious therapeutic agent from placebo, the use of a dichotomous responder endpoint may not accurately convey the scope of clinical improvements on the hallmark symptoms of IBS-C (abdominal pain and BM infrequency). While responder endpoints may be useful for assessing if a treatment warrants FDA marketing approval, they provide limited information for the clinician to make treatment decisions specific to the individual patient's IBS-C symptoms and may significantly underestimate a patient's true clinical response.
There are some important limitations to these analyses. First, the use of PRCQs, which require patients to rate their symptoms in relation to their status at baseline, are susceptible to potential recall bias. However, improvement trends similar to those observed for the PRCQs were seen for adequate relief and treatment satisfaction (parameters that are not measured relative to baseline). Second, this post-hoc analysis was conducted in patients meeting Rome II criteria for IBS-C, and may not sufficiently represent patients with less-severe symptoms. Lastly, it should be noted that the number of male patients in these trials (composing only 5–11% of the analysis subpopulations) is lower than IBS incidence rates in males observed in epidemiologic studies.7
Acknowledgments
Chris O'Dea of Ironwood Pharmaceuticals and Kristen Quinn, PhD of KnowledgePoint 360 Group provided medical writing support for this manuscript.
Funding
These trials were funded by Forest Research Institute and Ironwood Pharmaceuticals Inc.
Disclosure
Drs. Brian E. Lacy (BEL) and Anthony J. Lembo (AJL) have served as paid consultants to Ironwood Pharmaceuticals and Forest Research Institute. Drs. James E. MacDougall, Steven J. Shiff, Caroline B. Kurtz, Mark G. Currie, and Jeffrey M. Johnston are employees of Ironwood Pharmaceuticals (JEM, CBK, MGC, JMJ) or Forest Research Institute (SJS) and also own stock and shares in their respective companies.
Author Contribution
Analyses were designed by JEM; Data were interpreted by BEL, AJL, JEM, SJS, CBK, MGC, and JMJ; BEL and AJL wrote the draft manuscript; All authors provided critical review of the manuscript; All authors approved the submitted version of the manuscript.
References
- 1.Brandt LJ, Chey WD, Foxx-Orenstein AE, Schiller LR, Schoenfeld PS, Spiegel BM, Talley NJ, Quigley EM, et al. An evidence-based position statement on the management of irritable bowel syndrome. Am J Gastroenterol. 2009;104(Suppl. 1):S1–35. doi: 10.1038/ajg.2008.122. [DOI] [PubMed] [Google Scholar]
- 2.Akehurst RL, Brazier JE, Mathers N, O'Keefe C, Kaltenthaler E, Morgan A, Platts M, Walters SJ. Health-related quality of life and cost impact of irritable bowel syndrome in a UK primary care setting. PharmacoEconomics. 2002;20:455–62. doi: 10.2165/00019053-200220070-00003. [DOI] [PubMed] [Google Scholar]
- 3.Dibonaventura M, Sun SX, Bolge SC, Wagner JS, Mody R. Health-related quality of life, work productivity and health care resource use associated with constipation predominant irritable bowel syndrome. Curr Med Res Opin. 2011;27:2213–22. doi: 10.1185/03007995.2011.623157. [DOI] [PubMed] [Google Scholar]
- 4.Longstreth GF, Wilson A, Knight K, Wong J, Chiou CF, Barghout V, Frech F, Ofman JJ. Irritable bowel syndrome, health care use, and costs: a U.S. managed care perspective. Am J Gastroenterol. 2003;98:600–7. doi: 10.1111/j.1572-0241.2003.07296.x. [DOI] [PubMed] [Google Scholar]
- 5.Hillilä MT, Färkkilä NJ, Färkkilä MA. Societal costs for irritable bowel syndrome–a population based study. Scand J Gastroenterol. 2010;45:582–91. doi: 10.3109/00365521003637211. [DOI] [PubMed] [Google Scholar]
- 6.Longstreth GF, Thompson WG, Chey WD, Houghton LA, Mearin F, Spiller RC. Functional bowel disorders. Gastroenterology. 2006;130:1480–91. doi: 10.1053/j.gastro.2005.11.061. [DOI] [PubMed] [Google Scholar]
- 7.Saito YA, Schoenfeld P, Locke GR. The epidemiology of irritable bowel syndrome in North America: a systematic review. Am J Gastroenterol. 2002;97:1910–5. doi: 10.1111/j.1572-0241.2002.05913.x. [DOI] [PubMed] [Google Scholar]
- 8.Bryant AP, Busby RW, Bartolini WP, Cordero EA, Hannig G, Kessler MM, Pierce CM, Solinga RM, et al. Linaclotide is a potent and selective guanylate cyclase C agonist that elicits pharmacological effects locally in the gastrointestinal tract. Life Sci. 2010;86:760–5. doi: 10.1016/j.lfs.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 9.Andresen V, Camilleri M, Busciglio IA, Grudell A, Burton D, McKinzie S, Foxx-Orenstein A, Kurtz CB, et al. Effect of 5 days linaclotide on transit and bowel function in females with constipation-predominant irritable bowel syndrome. Gastroenterology. 2007;133:761–8. doi: 10.1053/j.gastro.2007.06.067. [DOI] [PubMed] [Google Scholar]
- 10.Busby RW, Bryant AP, Bartolini WP, Cordero EA, Hannig G, Kessler MM, Mahajan-Miklos S, Pierce CM, et al. Linaclotide, through activation of guanylate cyclase C, acts locally in the gastrointestinal tract to elicit enhanced intestinal secretion and transit. Eur J Pharmacol. 2010;649:328–35. doi: 10.1016/j.ejphar.2010.09.019. [DOI] [PubMed] [Google Scholar]
- 11.Eutamene H, Bradesi S, Larauche M, Theodorou V, Beaufrand C, Ohning G, Fioramonti J, Cohen M, et al. Guanylate cyclase C-mediated antinociceptive effects of linaclotide in rodent models of visceral pain. Neurogastroenterol Motil. 2010;22:312–22. doi: 10.1111/j.1365-2982.2009.01385.x. [DOI] [PubMed] [Google Scholar]
- 12.Rao S, Lembo AJ, Shiff SJ, Lavins BJ, Currie MG, Jia XD, Shi K, MacDougall JE, et al. A 12-week, randomized, controlled trial with a 4-week randomized withdrawal period to evaluate the efficacy and safety of linaclotide in irritable bowel syndrome with constipation. Am J Gastroenterol. 2012;107:1714–25. doi: 10.1038/ajg.2012.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chey WD, Lembo AJ, Lavins BJ, Shiff SJ, Kurtz CB, Currie MG, MacDougall JE, Jia XD, et al. Linaclotide for irritable bowel syndrome with constipation: a 26-week, randomized, double-blind, placebo-controlled trial to evaluate efficacy and safety. Am J Gastroenterol. 2012;107:1702–12. doi: 10.1038/ajg.2012.254. [DOI] [PubMed] [Google Scholar]
- 14.U.S.Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Guidance for Industry: Irritable Bowel Syndrome - Clinical Evaluation of Drugs for Treatment. 2012. Available at: www.fda.gov/downloads/Drugs/Guidances/UCM205269.pdf. (accessed December 2013) [Google Scholar]
- 15.Macdougall JE, Johnston JM, Lavins BJ, Nelson LM, Williams VS, Carson RT, Shiff SJ, Shi K, et al. An evaluation of the FDA Responder Endpoint for IBS-C clinical trials: analysis of data from linaclotide Phase 3 clinical trials. Neurogastroenterol Motil. 2013;25:481–6. doi: 10.1111/nmo.12089. [DOI] [PubMed] [Google Scholar]
- 16.Uryniak T, Chan ISF, Fedorov VV, Jiang Q, Oppenheimer L, Snapinn SM, Teng C-H, Zhang J. Responder analyses- A PhRMA position paper. Stat Biopharm Res. 2011;3:476–87. [Google Scholar]
- 17.Crosby RD, Kolotkin RL, Williams GR. Defining clinically meaningful change in health-related quality of life. J Clin Epidemiol. 2003;56:395–407. doi: 10.1016/s0895-4356(03)00044-1. [DOI] [PubMed] [Google Scholar]
- 18.McLeod LD, Coon CD, Martin SA, Fehnel SE, Hays RD. Interpreting patient-reported outcome results: US FDA guidance and emerging methods. Expert Rev Pharmacoecon Outcomes Res. 2011;11:163–9. doi: 10.1586/erp.11.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaeschke R, Singer J, Guyatt GH. Measurement of health status. Ascertaining the minimal clinically important difference. Control Clin Trials. 1989;10:407–15. doi: 10.1016/0197-2456(89)90005-6. [DOI] [PubMed] [Google Scholar]