

Abstract
Undecaprenyl pyrophosphate synthase (UPPs) is an essential enzyme in a key bacterial cell wall synthesis pathway. It catalyzes the consecutive condensations of isopentenyl pyrophosphate (IPP) groups on to a trans-farnesyl pyrophosphate (FPP) to produce a C55 isoprenoid, undecaprenyl pyrophosphate (UPP). Here we report the discovery and co-crystal structures of a drug-like UPPs inhibitor in complex with Streptococcus pneumoniae UPPs, with and without substrate FPP, at resolutions of 2.2 and 2.1 Å, respectively. The UPPs inhibitor has a low molecular weight (355 Da), but displays potent inhibition of UPP synthesis in vitro (IC50 50 nM) that translates into excellent whole cell antimicrobial activity against pathogenic strains of Streptococcal species (MIC90 0.4 µg mL−1). Interestingly, the inhibitor does not compete with the substrates but rather binds at a site adjacent to the FPP binding site and interacts with the tail of the substrate. Based on the structures, an allosteric inhibition mechanism of UPPs is proposed for this inhibitor. This inhibition mechanism is supported by biochemical and biophysical experiments, and provides a basis for the development of novel antibiotics targeting Streptococcus pneumoniae.
Keywords: undecaprenyl pyrophosphate synthase, isopentenyl pyrophosphate chain, cell wall, enzyme inhibitor, antibiotic, X-ray crystallography, surface plasmon resonance
Introduction
The search for new antibacterial agents remains an important activity in drug discovery as the threat from emerging antibiotic resistance continues to grow. Efforts to elucidate bacterial genomes and to identify new targets, combined with the ability to produce these targets for screening against large libraries of compounds have been employed extensively to discover new agents to address this threat. Inhibition of bacterial cell wall synthesis has been an important mechanism of action for successful antibacterial agents such as vancomycin and the cephalosporins. Targeting additional critical pathways in this complex process should hold promise for the discovery of new effective antibacterials. One such pathway is the production of UPP, a direct precursor of undecaprenyl phosphate that is an indispensable lipid carrier for bacterial peptidoglycan synthesis.1
UPP is synthesized by the action of UPPs, which catalyzes cis-prenyl chain elongation by condensations of eight IPP groups on to the allylic diphosphate substrate trans-farnesyl pyrophosphate (FPP). This enzyme has been shown to be essential for the growth of an important respiratory tract pathogen Streptococcus pneumoniae,1 and is therefore an attractive target for antibacterial intervention. UPPs is a member of the large cis-prenyltransferases family that catalyzes the production of many linear isoprenoids. These isoprenoids include steroids, terpenes, the side chains of respiratory quinones, carotenoids, natural rubber, the glycosyl carrier lipid, and prenyl proteins.2 They vary in lengths and serve critical functions in many organisms. UPP, the C55 isoprenoid pyrophosphate, is essential in the cross membrane transport of carbohydrates used in bacterial wall synthesis, but not essential in humans, making UPPs an attractive antibacterial target.1 Currently there is no UPPs inhibitor being used as antibacterial agent, but some highly hydrophobic compounds, many containing ionic groups (e.g., bisphosphonates) and/or with high molecular weights (spirohexalines), have been recently reported to inhibit bacterial UPPs.3–5 Most of these compounds have modest in vitro potencies, although some of them do achieve double digit nM IC50 and have cellular activities.3–5
Significant progress in the understanding of the structural basis for the activity of UPPs has been gained through a number of X-ray structures of the enzyme from several different bacterial species.2,4–14 These structures include the apo UPPs from the Gram negative organism E. coli, and in complex with substrates, detergents and bisphosphonate inhibitors.2–10 From Gram positive organisms, structures of the apo UPPs from Micrococcus luteus and the FPP complex from Staphylococcus aureus have been solved.5,11,15 These structures show a large funnel-shaped hydrophobic cleft that is putatively the site for substrate, intermediate and product binding. This binding cleft is formed by two α-helices, H1 and H2, and four β-strands β1–4 lined at the bottom with Ile62, Val105, and Leu137 (E. coli sequence numbering).2 The E. coli and S. aureus UPPs-substrate complex structures show two binding sites for substrates FPP and IPP, respectively, at the top of the binding “funnel”. The binding modes of these substrates as well as a critical catalytic Mg2+ suggested the catalytic mechanism of UPPs.2–9 The complex structure of E. coli UPPs with the detergent Triton X-100, which enhances the enzyme activity in vitro at low levels,4,13,16 suggested an “open” form of the binding pocket that may accommodate intermediate chain elongation and final product release.4 Based on the location in the E. coli structure and on mutagenesis experiments, Leu137 (L139 in S. pneumoniae UPPs), which sits at the bottom of the hydrophobic cleft, was shown to be involved in limiting product chain length.6 The complex structures of E. coli UPPs with bisphosphonate inhibitors revealed up to four different binding sites at the hydrophobic tunnel, with the first overlapping the FPP site, the second at the IPP site and the fourth near the bottom of the binding tunnel.5 The M. luteus and S. aureus UPPs structures are highly similar to that from E. coli, confirming the structural conservation of UPPs from Gram positive and Gram negative organisms.17
Here we report on the identification and biochemical, biophysical and structural investigation of a novel drug-like UPPs inhibitor, UK-106051, against S. pneumoniae. UK-106051, (3-(2-chlorophenyl)-N-(4-isopropylphenyl-5-methylisoxazole-4-carboxamide, Fig. 1), was identified in a high throughput phosphate release assay using purified recombinant S. pneumoniae UPPs enzyme.18 A direct assay measuring the incorporation of 14C-IPP was also used as a secondary assay to assess inhibitor activity. This UPPs inhibitor displayed both potent in vitro inhibition against the purified enzyme with an IC50 value of 50 nM as well as cell activity (MIC90 = 0.4 µg mL−1) against a collection of clinically important pathogenic S. pneumoniae strains. The first S. pneumoniae UPPs crystal structures, in complex with the inhibitor, define a binding pocket amenable to structure-guided drug design and suggest opportunities for improving inhibitor properties. Such information may provide the basis for the design of effective antibacterial agents against the bacterial UPPs enzyme
Figure 1.
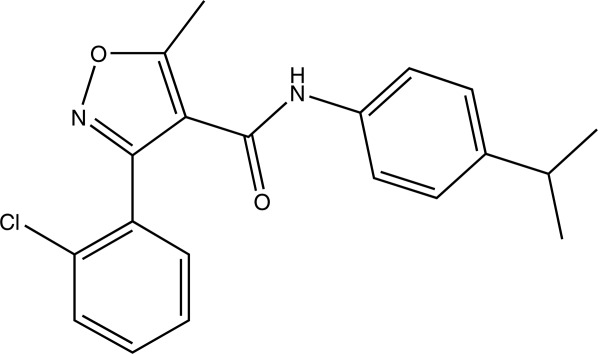
Chemical structure of UK-106051, 3-(2-chlorophenyl)-N-(4-isopropylphenyl-5-methylisoxazole-4-carboxamide), a UPPs inhibitor.
Results
Identification of UK-106051
UPPs inhibitor UK-106051 (Fig. 1) was identified through a high throughput screen of the Pfizer compound collection and subsequently characterized by an inhibition assay and assessed for antibacterial activities. The compound exhibited an in vitro IC50 of 50 nM against purified S. pneumoniae UPPs and a MIC90 of 0.4 µg mL−1 against clinically important pathogenic S. pneumoniae R6 strain,19 and is also active against other S. pneumoniae strains (Table I). With a low molecular weight (355 daltons) and impressive potency, UK-106051 possesses better drug-like properties than previously reported UPPs inhibitors.5,20
Table I.
Antibacterial Activities of UK-106051 Against Streptococcal Pneumoniae Strains (MIC90 in µg mL−1)
| R6 | 02J1046 | 02J1095 | 02J1175 | 02J1229 | 02J1258 |
|---|---|---|---|---|---|
| 0.39 | 6.5 | 2.2 | 12.5 | 1.0 | 0.68 |
Crystal structure of S. pneumonia UPPs-UK-106061 complex
To investigate the inhibition mechanism and the binding mode of UK-106051 to S. pneumoniae UPPs, we carried out crystallographic studies. The S. pneumoniae UPPs enzyme lacking 15 residues from the N-terminus was expressed, purified and co-crystallized with either UK-106051 alone or with the substrate FPP. Crystals of the enzyme with the inhibitor alone grew in the space group C2 and diffracted to 2.2 Å, while crystals of the ternary UPPs–inhibitor–FPP complex grew in the space group P43212 and diffracted to 2.06 Å (Table II).
Table II.
X-Ray Crystallographic Data Collection and Refinement Statistics
| Crystal | UK-106051+FPP | UK-106051 |
|---|---|---|
| Resol (Å) | 50–2.06 | 50–2.2 |
| Space group | P43212 | C2 |
| Unit cell | 91.85, 91.85, 155.5 | 111.5, 131.5, 53.2 |
| 90,90,90 | 90, 102.2, 90 | |
| No. Obs | 503,158 | 107,644 (3116) |
| No. of Refs. | 41,949 (4123) | 32,312 (1521) |
| Rsym (%) | 6.4 (25.5) | 4.4 (47.3) |
| I/σ | 41.9 (6.7) | 24 (1.6) |
| Compl. (%) | 99.9 (99.9) | 84.9 (40.1) |
| Redundancy | 12 | 3.33 (2.05) |
| Rfactor (%) | 20.8 (22.2) | 21.3 |
| Rfree (%) | 24.6 (27.5) | 26.7 |
There are two molecules per asymmetric unit in both crystal forms. Like the UPPs of E. coli, M. luteus, and S. aureus, the two monomers of S. pneumoniae UPPs form a functional dimer [ Fig. 2(A)]. The overall structures of the two monomers in the same complex are highly similar, with a root mean square deviations (rmsd) value of 0.4–0.50 Å after superimposition with all Cα atoms in the monomers, and the dimers of the two complexes are highly similar as well (rmsd = 0.57 Å).
Figure 2.
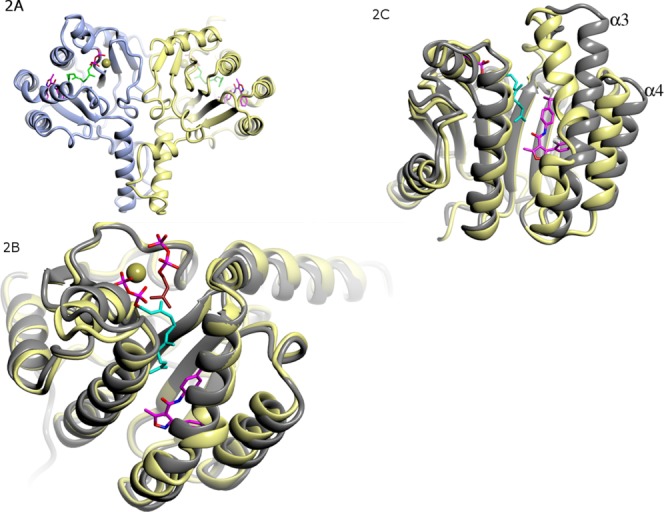
Overall structure of S. pneumoniae UPPs. (A) Dimeric structure of S. pneumoniae UPPs. UPPs is in ribbon diagram, with two protein chains colored yellow and light blue, respectively. Inhibitor UK-106051 (carbon atoms colored magenta) and substrate FPP (carbon atoms colored cyan) are in stick models. N, O, S, and P atoms are colored blue, red, yellow, and brown respectively. (B) Superimposition of the S. pneumoniae UPPs-UK-106051 complex monomer with the closed form E. coli UPPs–FPP complex (gray ribbon, PDB code 1X06) showing good agreement. (C) Superimposition of S. pneumoniae UPPs-UK-106051 complex structure with the open form of E. coli UPPs structure (gray, PDB code 1X09) showing significant conformational differences. Note that α3 has a significant kink near the bound inhibitor, and that both α3 and α4 move closer toward ligands.
The conformation of S. pneumoniae UPPs complexes resembles that of the closed conformation in the M. luteus, E. coli, and S. aureus UPPs–FPP complexes [ Fig. 2(B)], with rmsd deviations of 0.7 Å, 0.8 and 0.9 Å for 169, 152 Cα and 208 backbone atoms in the monomers, respectively. On the other hand, there are significant deviations at α3 and α4 helices between the closed conformation of the S. pneumoniae UPPs complex and the open conformation of the apo E. coli UPPs [ Fig. 2(C)]. In the closed conformation, α3 is significantly kinked near the inhibitor binding site, placing both ends of the helix closer to the ligands [ Fig. 2(C)]. In the closed conformation α4 also moves closer toward the ligands [ Fig. 2(C)].
Inhibitor binding site
In the previously reported E. coli, M. luteus, and S. aureus UPPs structures, a binding cleft containing up to four ligand bindings sites were reported.2,4–13 A very similar ligand binding cleft is also observed in the S. pneumoniae UPPs complex [ Fig. 3(A)]. Both UK-106051 and FPP have well defined electron density at the binding cleft in each UPPs monomer [ Fig. 3(B)]. This binding cleft is defined by the “P-loop” at the entrance and Leu139 (Leu137 in E. coli) at the bottom [ Fig. 3(A)]. FPP binds to the same site as observed in the E. coli, S. aureus, and M. luteus UPPs complex structures, and UK-106051 binds to a site in the middle of this binding cleft. This inhibitor binding site was occupied by a second FPP molecule, a Triton X-100 molecule, or by a bisphosphonate inhibitor in the E. coli or M. luteus UPPs complexes.2,4–13
Figure 3.
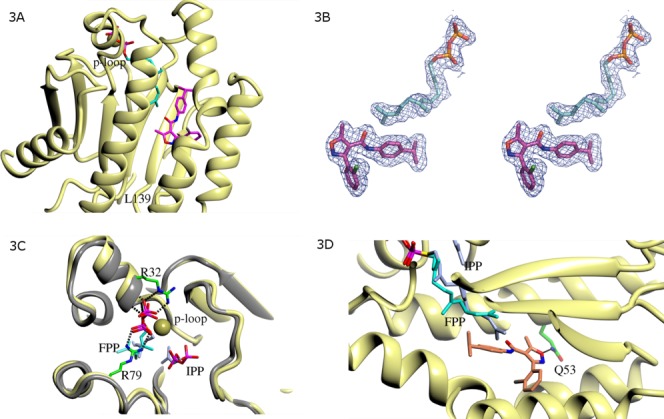
Binding interactions of FPP and UK-106051 with S. pneumoniae UPPs. (A) Hydrophobic ligand binding cleft of S. pneumoniae UPPs. The binding cleft is defined by the p-loop (G29-R32) at the entrance and L139 at the exit. (B) FPP and UK-106051 were ordered in the crystals with well-defined electron density at the binding cleft of S. pneumoniae UPPs. Ligands from the final refined model are shown embedded in the 2Fo-Fc type electron density contoured at 1.2σ. C. FPP at the active site of S. pneumoniae UPPs. Polar interactions with UPPs are shown in dashes. Side chains of Arg32 and Arg79 that interact with FPP are shown (carbon atoms colored green). A bound cadmium ion (golden sphere) was found to coordinate with the pyrophosphate of FPP. For comparison, a composite model of FPP and IPP (carbon atoms colored light blue) bound in the E. coli UPPs complexes (gray ribbon diagram) constructed based on PDB entries ID 1X06 and 1X09 is also shown. (D) UK-106051 binding interactions. UK-106051 contacts the hydrophobic side chains of Met 49, Leu52, Leu102, Ile 109, Leu126, Phe143, Leu 145, and Pro91 (not shown for clarity). The inhibitor also makes a single hydrogen bond (dashes) with the side chain of Gln53. Note the stacking interaction between the farnesyl group of FPP and the isopropylphenyl moiety of UK-106051.
The pyrophosphate of FPP is bound in the same fashion at the active site of S. pneumoniae UPPs as in other UPPs–FPP complex structures,8 stabilized by interactions with the main chain of the P-loop (G29–R32) as well as side chains of Arg32 and Arg79 [ Fig. 3(C)]. One of the bound cadmium ions was found to coordinate with the pyrophosphate of FPP [ Fig. 3(C)].
In the E. coli, S. aureus or M. luteus UPPs-FPP complex, the hydrophobic farnesyl moiety of FPP is in a more extended conformation and would clash with UK-106051 in the S. pneumoniae UPPs complex [ Fig. 3(D)]. Instead, the terminal double bond of the FPP farnesyl group in the S. pneumoniae UPPs complex stacks against the isopropylphenyl moiety of UK-106051 at distances of 3.4–3.8 Å (Fig. 3D). The majority of the protein–inhibitor interactions are hydrophobic, with only a single hydrogen bond present between the oxygen of the isoxazole ring and the side chain of Gln53 [ Fig. 3(D)].
Binding kinetics of UK-106051
Our crystal structures suggested that UK-106051 does not compete with substrate FPP in binding to UPPs, rather, two ligands interact directly. It is possible that UK-106051 and FPP may enhance each other's binding affinity toward UPPs. To evaluate this possibility SPR experiments were employed to study the binding affinity and kinetics of UK-106061 to UPPs. In the absence of FPP, the rate of inhibitor association to S. pneumoniae UPPs (kon) is 1.1 ± 0.03 × 104 M−1 s−1, and the rate of dissociation (koff) is 1.7 ± 0.2 × 10−2 s−1. In the presence of 5 µM FPP (near FPP Km), the association constant for UK-106061 for UPPs is slightly lower than that in the absence of FPP, 5.3 ± 0.75 × 103 M−1 s−1, however, the inhibitor dissociates from the protein much slower with a koff of 3.5 ± 0.5 × 10−4 s−1 in the presence of FPP. This slower off rate results in a 25-fold increase in binding affinity for the compound in the presence of FPP (KD 6.6 ± 0.7 × 10−8 M) compared to the apo protein (KD 1.7 ± 0.3 × 10−6 M) (Table III).
Table III.
Binding Kinetics of UK-106051 with Streptococcal Pneumoniae UPPs (Standard Error from 2 Measurements in Parentheses)
| ka (1 Ms−1) | kd (1 s−1) | KD (M) | |
|---|---|---|---|
| Inhibitor | 1.1 (0.03)104 | 1.7(0.2)10−2 | 1.7(0.3)10−6 |
| +FPP | 5.3 (0.75)103 | 3.5 (0.5)10−4 | 66.0(7)10−9 |
Discussion
In the antibacterial assay, UK-106051 and its related inhibitors were shown to act through a cidal mechanism, with a larger than three log units of reduction in viable bacterial counts observed at 24 h (unpublished data), highlighting the potential significance of inhibiting UPP biosynthesis as an approach in treating bacterial infections. The biosynthesis of the C55 product UPP by UPPs involves the elongation of the C15 FPP through iterative addition of a C5 isoprenyl group. UPPs inhibition can therefore be achieved at any stage of this elongation process. Based on the results here from the biochemical, biophysical and crystallographic experiments, we can conclude that the inhibition of the S. pneumoniae UPPs enzyme by UK-106051 occurs via blocking the first elongation step of the reaction.
The UK-106051 binding site does not overlap with either the FPP or the IPP binding sites observed in other UPPs structures (Fig. 3), yet the compound is a potent inhibitor of the enzymatic reaction (Fig. 4). Interaction observed in the crystal structures between the inhibitor and bound FPP (Fig. 3) suggests a mutual enhancement these two ligands in their binding affinities toward the enzyme. The SPR binding results also agreed with such interpretation (Table III).
Figure 4.
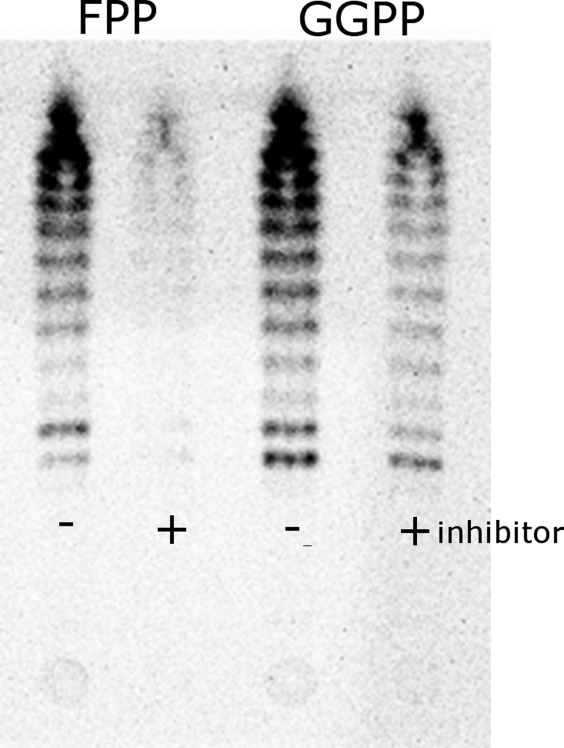
TLC analysis of [14C]IPP incorporation catalyzed by S. pneumoniae UPPs. Samples were collected after 1 h of reaction in the presence or absence of 10 µM UK-106051, with either FPP or GGPP used as substrates.
When compared to the FPP and IPP bound closed conformation of E. coli UPPs structures [ Fig. 3(C)], the binding of UK-106051 does not seem to visibly alter the local conformation of the reaction site of UPPs, but rather forces FPP to adopt a configuration different from when inhibitor is absent [ Fig. 3(C,D)]. In the composite model of E. coli UPPs–FPP–IPP complex, the isoprenyl tail of IPP stacks with the reactive double bond of FPP.21 This stacking interaction should favor the formation of a catalytic reaction complex. In the altered configuration observed in the UK-106051 bound complex, the FFP molecule would clash with a bound IPP molecule at the IPP site. Consistent with this explanation, biophysical investigations of tetramic acid UPPs inhibitors, which appear to occupy the same binding site as UK-106051, showed that FPP and IPP cannot bind simultaneously to UPPs–inhibitor binary complexes.22
Interestingly, in thin layer chromatography experiments when geranylgeranyl pyrophosphate (GGPP, C20) was used as substrate, UK-106051 becomes much less effective in inhibiting the elongation reactions (Fig. 4). In the uninhibited reaction, we detected both the final C55 chain length product and the short chain intermediates in these experiments (Fig. 4). However, in the presence of UK-106051 when FPP was used as substrate, we did not observe accumulation of any short chain intermediates or the final product, indicating UK-106061 appears to inhibit the very first condensation reaction of FPP and IPP (Fig. 4). Apparently, once the first isoprenyl is added to FPP, UK-106051 becomes less effective at inhibiting the remaining steps of the condensation reaction.
UK-106051 stabilizes a closed conformation that is commonly observed with other substrate bound UPPs structures. Such a closed conformation may represent one of the energy minima of the system. Unlike the recently reported much larger UPPs inhibitors, the bindings of which greatly enlarge the binding cleft of UPPs,5 the minimum disruption to the closed conformation of the protein with UK-106051 binding (Figs. 2, 3) may explain the high potency of such a small inhibitor.
Compared to known UPPs inhibitors,5 UK-106051 is more drug-like given its attractive size, ligand efficiency and physical properties. Through structural, biophysical and biochemical studies, we characterized the inhibition mode of UK-106051. Our findings provide a novel chemical scaffold and structural basis for the design of antibiotics against this important target.
Materials and Methods
Protein expression and purification
Streptococcus pneumoniae UPPs (Ala16 to Val258 of GenePept accession AAK99044) was cloned by PCR into a pET28(a) derivative as a His tag fusion with a thrombin cleavage site. Expression was performed in BL21 Gold λΔΕ3 cells (Stratagene) and Luria broth supplemented with 0.2% glucose at 37°C to an OD600 = 1.0 and then induced with 50 µM isopropyl β-d-thiogalactoside for 16 h before harvesting.
Cells expressing the construct were lysed in buffer A (50 mM Tris–HCl, 300 mM NaCl, 4 mM β-mercaptoethanol, EDTA-free protease inhibitors (Roche Biochemicals cat # 1 873 580; 1 tab/50 mL), 1 mM PMSF, 3 µg mL−1 DNAase, 10 mM MgCl2, pH 8.0) using a Branson Sonifier 450. The lysate was clarified by centrifugation at 43,000g for 90 min. The lysate supernatant was purified by Ni-NTA chromatography. Lysate was diluted with buffer B (50 mM Tris–HCl, 300 mM NaCl, 4 mM β-mercaptoethanol, EDTA-free protease inhibitors, 1 mM PMSF, pH 8.0) plus 10 mM imidazole, pH 8.0, and loaded on to the column. After wash, the bound proteins were eluted using a gradient of 0–50% buffer B (buffer A + 0.3M Imidazole, pH 8.0). Ni-NTA eluted fractions were characterized using 10% SDS-PAGE. Fractions containing UPPs were pooled and dialyzed against buffer B to reduce imidazole to 10 mM. Ni-NTA-pooled fractions were digested with thrombin to cleave the His-tag and loaded onto a second Ni-NTA column to remove the tag. Flow through from the second Ni-NTA column was concentrated and loaded on to a high load 16/60 Superdex S-200 size exclusion column (Amersham) equilibrated with buffer C (50 mM Tris–HCl, 300 mM NaCl, 8 mM DTT, pH 7.5).
UPPs inhibition assay
Purified S. pneumoniae UPPs was used in a high throughput screen assay to identify potential inhibitors. The assay couples UPPs activity with an inorganic pyrophosphatase to produce orthophosphate, which can be measured using malachite green.16 The reaction runs for 60–90 min at room temperature with a 30 µL reaction mixture (in 384-well format) containing 100 mM Tris–HCL (pH 7.5), 0.2 mM MgCl2, 0.05% Triton X-100, 30 µM IPP, 5 µM FPP, and 0.01 U yeast inorganic pyrophosphatase.
Antibacterial activity assay
Broth cultures of several S. pneumoniae strains were examined for a reduction in viable counts after exposure to inhibitor at a series of different concentrations. Samples were taken and tittered for viable counts at 0, 4, 8, and 24 h. MIC90 values were calculated based on kinetic kill curves generated.
Crystallization of UPPs complexes
The purified S. pneumoniae UPPs at 1.0 mg mL−1 containing 200 µM UK-106051 was concentrated to 6 mg mL−1. Crystallization using the hanging-drop vapor diffusion method at 22°C resulted in large crystals, with 0.1M Tris–HCl, pH 8.5, 1.75M (NH4)2SO4, 0.2M Li2SO4, 3.5% 1,2,3 heptanetriol as the precipitant. The protein at 1.0 mg mL−1 containing 200 µM UK-106051 plus 33 µM FPP was concentrated to 6 mg mL−1. Crystallization by the hanging-drop vapor diffusion method at 22°C resulted in large crystals using 0.1M sodium acetate, pH 4.6, 0.2M CdCl2, 30% PEG 400 as the precipitant.
X-ray structure determination
Crystals were flash-cooled in liquid nitrogen or in a 100K N2 cool stream before data collections. X-ray crystallographic data were collected either in house (UPPs–inhibitor complex) with a Raxis-II detector mounted on a FRE X-ray generator (MSC, TX), or at the synchrotron source (IMCA-17ID, Advanced Photon Source, Chicago) (UPPs–inhibitor–FPP complex) at −173°C. Data were processed with program suite HKL200017 and further calculations were done with CCP4 program package.18 Structure of UPPs–inhibitor–FPP complex was solved using molecular replacement method with program Amore19 using E. coli UPPs–FPP complex structure as search model (PDB code 1UEH), with water molecules and ligands removed. Rigid body and maximum-likelihood refinements with program REFMAC20 were carried out on initial models. Model rebuilding, ligand recognition, and fitting into electron density were carried out with the graphic program XtalView.21 Cross validation on was maintained throughout refinements by setting 5% reflections as the reference reflections.22
Surface plasmon resonance
Surface Plasmon Resonance (SPR) binding measurements were performed using a Biacore 3000 instrument. The UPPs protein was first minimally biotinylated using sulfo-NHS-LC-LC-biotin (Pierce) and processed to remove excess biotin. The biotinylated protein was then captured on to streptavidin sensors (Sensor Chip SA, GE Healthcare) for the binding experiments. The binding experiments were carried out in 10 mM Tris, pH 8, 300 mM NaCl, 1 mM MgCl2, 3% DMSO both with and without 5 µM FPP supplemented in the buffer at 25°C. Compound was serially diluted typically from 10 µM in threefold dilutions and run in duplicate. Data were processed using Scrubber (BioLogic Software) to zero, align, reference and correct for excluded volume effects. Data fitting was performed using BiaEval software (GE Healthcare).
Thin layer chromatography assay
To visualize the elongated isoprenyl chains, 5 µM FPP was incubated with 50 μM [14C]IPP (Biotrend, FL) and S. pneumoniae UPPs. Alternatively, geranylgeranyl pyrophosphate (GGPP, C20) was also tested as substrate. The reaction was quenched with 10 mM EDTA to stop the reaction after 1 h. For the inhibition reaction, 10 µM UK-106051 was added. The 1-butanol extractable reaction products were separated using thin layer chromatography S and visualized by a bioimaging analyzer (Molecular Dynamics Storm, GMI, Minnesota).
Glossary
- FPP
farnesyl pyrophosphate
- GGPP
geranylgeranylpyrophosphate
- IPP
isopentyl diphosphate
- UPP
undecaprenyl pyrophosphate
- UPPs
undecaprenyl pyrophosphate synthase
- UK-106051
3-(2-chlorophenyl)-N-(4-isopropylphenyl-5-methylisoxazole-4-carboxamide)
References
- 1.Apfel CM, Takacs B, Fountoulakis M, Stieger M, Keck W. Use of genomics to identify bacterial undecaprenyl pyrophosphate synthetase: Cloning, expression, and characterization of the essential uppS gene. J Bacteriol. 1999;181:483–492. doi: 10.1128/jb.181.2.483-492.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogura K, Koyama T. Enzymatic aspects of isoprenoid chain elongation. Chem Rev. 1998;98:1263–1276. doi: 10.1021/cr9600464. [DOI] [PubMed] [Google Scholar]
- 3.Inokoshi J, Nakamura Y, Hongbin Z, Uchida R, Nonaka K-i, Masuma R, Tomoda H. Spirohexalines, new inhibitors of bacterial undecaprenyl pyrophosphate synthase, produced by Penicillium brasilianum FKI-3368. J Antibiotics. 2013;66:37–41. doi: 10.1038/ja.2012.83. [DOI] [PubMed] [Google Scholar]
- 4.Pan J-J, Chiou S-T, Liang PH. Product distribution and pre-steady state kinetic analysis of Eschericia coli undecaprenyl pyrophosphate synthase reaction. Biochemistry. 2000;39:10936–10942. doi: 10.1021/bi000992l. [DOI] [PubMed] [Google Scholar]
- 5.Zhu W, Zhang Y, Sinko W, Hensler ME, Olson J, Molohon KJ, Lindert S, Cao R, Li K, Wang K, Wang Y, Liu Y-L, Sankovsky A, de Oliveira CAF, Mitchell DA, Nizet V, McCammon JA, Oldfield E. Antibacterial drug leads targeting isoprenoid biosynthesis. Proc Natl Acad Sci USA. 2013;110:123–128. doi: 10.1073/pnas.1219899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ko TP, Chen YK, Robinson H, Tsai PC, Gao YG, Chen AP, Wang AH, Liang PH. Mechanism of product chain length determination and the role of a flexible loop in Escherichia coli undecaprenyl-pyrophosphate synthase catalysis. J Biol Chem. 2001;276:47474–47482. doi: 10.1074/jbc.M106747200. [DOI] [PubMed] [Google Scholar]
- 7.Chang S-Y, Chen Y-K, Wang AH-J, Po-Huang L. Identification of the active conformation and the importance of length of the flexible loop 72–83 in regulating the conformational change of undecaprenyl pyrophosphate synthase. Biochemistry. 2003;42:14452–14459. doi: 10.1021/bi035283x. [DOI] [PubMed] [Google Scholar]
- 8.Chang S-Y, Ko T-P, Chen AP-C, Wang AH-J, Liang P-H. Substrate binding mode and reaction mechanism of undecaprenyl pyrophosphate synthase deduced from crystallographic studies. Protein Sci. 2004;13:971–978. doi: 10.1110/ps.03519904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo R-T, Ko T-P, Chen AP-C, Kuo C-J, Wang AH-J, Po-Huang L. Crystal structures of undecaprenyl pyrophosphate synthase in complex with magnesium, isopentenyl pyrophosphate, and farnesyl thiopyrophosphate: roles of the metal ion and conserved residues in catalysis. J Biol Chem. 2005;280:20762–20774. doi: 10.1074/jbc.M502121200. [DOI] [PubMed] [Google Scholar]
- 10.Liang PH, Ko TP, Wang AH. Structure, mechanism and function of prenyltransferases. Eur J Biochem/FEBS. 2002;269:3339–3354. doi: 10.1046/j.1432-1033.2002.03014.x. [DOI] [PubMed] [Google Scholar]
- 11.Chang SY, Ko TP, Liang PH, Wang AH. Catalytic mechanism revealed by the crystal structure of undecaprenyl pyrophosphate synthase in complex with sulfate, magnesium, and triton. J Biol Chem. 2003;278:29298–29307. doi: 10.1074/jbc.M302687200. [DOI] [PubMed] [Google Scholar]
- 12.Guo RT, Cao R, Liang PH, Ko TP, Chang TH, Hudock MP, Jeng WY, Chen CK, Zhang Y, Song Y, Kuo CJ, Yin F, Oldfield E, Wang AH. Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc Natl Acad Sci USA. 2007;104:10022–10027. doi: 10.1073/pnas.0702254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keenan MV, Allen J, Charles M. Characterization of undecaprenyl pyrophosphate synthetase from Lactobacillus plantarum. Arch Biochem Biophys. 1974;161:375–383. doi: 10.1016/0003-9861(74)90318-x. [DOI] [PubMed] [Google Scholar]
- 14.Guo R-T, Cao R, Liang P-H, Ko T-P, Chang T-H, Hudock MP, Jeng W-Y, Chen CK-M, Zhang Y, Song Y, Kuo C-J, Yin F, Oldfield E, Wang AH-J. Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc Natl Acad Sci USA. 2007;104:10022–10027. doi: 10.1073/pnas.0702254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ammirati M, Pandit J. 2002. Crystal structure of Staphylococcus undecaprenyl pyrophosphate synthase and uses thereof. US 10/688,167.
- 16.Allen CM, Muth JD. Lipid activation of undecaprenyl pyrophosphate synthetase from Lactobacillus plantarum. Biochemistry. 1977;16:2908–2915. doi: 10.1021/bi00632a017. [DOI] [PubMed] [Google Scholar]
- 17.Fujihashi M, Zhang Y-W, Higuchi Y, Li X-Y, Koyama T, Miki K. Crystal structure of cis-prenyl chain elongating enzyme, undecaprenyl diphosphate synthase. Proc Natl Acad Sci USA. 2001;98:4337–4342. doi: 10.1073/pnas.071514398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Webb MR. A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc Natl Acad Sci USA. 1992;89:4884–4887. doi: 10.1073/pnas.89.11.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoskins J, Alborn WE, Jr, Arnold J, Blaszczak LC, Burgett S, DeHoff BS, Estrem ST, Fritz L, Fu DJ, Fuller W, Geringer C, Gilmour R, Glass JS, Khoja H, Kraft AR, Lagace RE, LeBlanc DJ, Lee LN, Lefkowitz EJ, Lu J, Matsushima P, McAhren SM, McHenney M, McLeaster K, Mundy CW, Nicas TI, Norris FH, O'Gara M, Peery RB, Robertson GT, Rockey P, Sun PM, Winkler ME, Yang Y, Young-Bellido M, Zhao G, Zook CA, Baltz RH, Jaskunas SR, Rosteck PR, Jr, Skatrud PL, Glass JI. Genome of the bacterium Streptococcus pneumoniae strain R6. J Bacteriol. 2001;183:5709–5717. doi: 10.1128/JB.183.19.5709-5717.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson TW, Dress KR, Edwards M. Using the Golden Triangle to optimize clearance and oral absorption. Bioorg Med Chem Lett. 2009;19:5560–5564. doi: 10.1016/j.bmcl.2009.08.045. [DOI] [PubMed] [Google Scholar]
- 21.Chang S-Y, Ko T-P, Liang P-H, Wang AH-J. Catalytic mechanism revealed by the crystal structure of undecaprenyl pyrophosphate synthase in complex with sulfate, magnesium, and triton. J Biol Chem. 2003;278:29298–29307. doi: 10.1074/jbc.M302687200. [DOI] [PubMed] [Google Scholar]
- 22.Lee LV, Granda B, Dean K, Tao J, Liu E, Zhang R, Peukert S, Wattanasin S, Xie X, Ryder NS, Tommasi R, Deng G. Biophysical investigation of the mode of inhibition of tetramic acids, the allosteric inhibitors of undecaprenyl pyrophosphate synthase. Biochemistry. 2010;49:5366–5376. doi: 10.1021/bi100523c. [DOI] [PMC free article] [PubMed] [Google Scholar]